

Abstract
Birt-Hogg-Dubé (BHD) syndrome is a rare autosomal dominant condition caused by mutations in the FLCN gene and characterized by benign hair follicle tumors, pneumothorax, and renal cancer. Folliculin (FLCN), the protein product of the FLCN gene, is a poorly characterized tumor suppressor protein, currently linked to multiple cellular pathways. Autophagy maintains cellular homeostasis by removing damaged organelles and macromolecules. Although the autophagy kinase ULK1 drives autophagy, the underlying mechanisms are still being unraveled and few ULK1 substrates have been identified to date. Here, we identify that loss of FLCN moderately impairs basal autophagic flux, while re-expression of FLCN rescues autophagy. We reveal that the FLCN complex is regulated by ULK1 and elucidate 3 novel phosphorylation sites (Ser406, Ser537, and Ser542) within FLCN, which are induced by ULK1 overexpression. In addition, our findings demonstrate that FLCN interacts with a second integral component of the autophagy machinery, GABA(A) receptor-associated protein (GABARAP). The FLCN-GABARAP association is modulated by the presence of either folliculin-interacting protein (FNIP)-1 or FNIP2 and further regulated by ULK1. As observed by elevation of GABARAP, sequestome 1 (SQSTM1) and microtubule-associated protein 1 light chain 3 (MAP1LC3B) in chromophobe and clear cell tumors from a BHD patient, we found that autophagy is impaired in BHD-associated renal tumors. Consequently, this work reveals a novel facet of autophagy regulation by ULK1 and substantially contributes to our understanding of FLCN function by linking it directly to autophagy through GABARAP and ULK1.
Keywords: autophagy, BHD, FLCN, GABARAP, MAP1LC3B, SQSTM1, ULK1
Introduction
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved process where intracellular lipid and protein components are broken down to replenish cellular energy and amino acid supplies. Autophagy also removes protein aggregates, redundant macromolecules, and dysfunctional organelles that, if not efficiently recycled, contribute to cell stress and consequently disease.1 For example, autophagy plays both pro- and anti-oncogenic roles in cancer development (for a review see ref. 1), while defects result in age-related cardiomyopathy2 and can lead to marked neurodegeneration.3,4 Autophagy involves sequestering cytoplasmic material in double-membrane vesicles known as autophagosomes, which subsequently fuse with lysosomes to form autolysosomes. Once fusion occurs, lysosomal hydrolases degrade sequestered material allowing permeases to transport amino acids and lipids into the cytoplasm for use in either biosynthesis or the generation of energy (for a review see ref. 5).
Yeast screens uncovered more than 30 autophagy-related (ATG) genes,6 many of which are recruited to the phagophore assembly site, a structure involved in nucleation/expansion of the phagophore. Atg8 is conjugated to phosphatidylethanolamine and selectively incorporated into autophagosomes, making it a commonly used autophagy marker. Mammals have 2 Atg8 ortholog subfamilies, the MAP1LC3 (microtubule-associated protein 1 light chain 3; commonly called LC3) subgroup, and the GABARAP (GABA(A) receptor-associated protein) and GABARAPL2 (GABA(A) receptor-associated protein-like 2) subfamily. Both mammalian Atg8 ortholog subfamilies are modified by PE conjugation, localize to autophagosomes,7 and are essential for autophagy. Current evidence indicates that MAP1LC3 and GABARAP act at different stages of autophagosome formation.8
ULK1 (unc-51 like autophagy activating kinase 1; a mammalian ortholog of Atg1) acts at the most upstream step of autophagy.9 ULK1 is a serine/threonine kinase that functions within a complex containing ATG13, RB1CC1 (RB1-inducible coiled-coil 1), and ATG101 to drive autophagosome formation.10-13 This kinase complex is positively regulated by many internal ULK1-mediated phosphorylation events, including ULK1 autophosphorylation.10-12,14 In addition, when energy and nutrients are plentiful, the mechanistic target of rapamycin complex 1 (MTORC1) promotes cell growth in part by inhibiting autophagy via phosphorylation of ULK1.10,11 Conversely, during energy and nutrient stress when cell growth is not feasible, PRKAA2/AMPK (protein kinase, AMP-activated, α 2 catalytic subunit) interacts with and phosphorylates ULK1 to enhance autophagy.15-18 Downstream ULK1 substrates are gradually being identified. AMBRA1 was the first ULK1 substrate identified that is integral to the autophagy machinery19 but is not a component of the ULK1-ATG13-RB1CC1 complex. More recently, ULK1 phosphorylation of ATG9 has been identified as an important step during expansion of the phagophore.20 Additionally, a further molecular link has been identified, whereby active ULK1 can directly phosphorylate BECN1/Beclin 1, allowing the proautophagy PIK3C3/VPS34 (phosphatidylinositol 3-kinase, catalytic subunit type 3) complexes to promote autophagy induction and autophagosome maturation.21 ULK1 is known to indirectly impact autophagy via phosphorylation of both PRKAA222 and RPTOR/Raptor within MTORC1.23,24
Mutations in FLCN are responsible for Birt-Hogg-Dubé (BHD) syndrome (MIM #135150), characterized by benign hair follicle tumors, pneumothorax, cysts, and renal cancer.25 BHD is a ciliopathy and FLCN is localized at primary cilia.26 Interestingly, a compromised ability to activate autophagy has been hypothesized to underlie some ciliopathies,27 raising the possibility that autophagy may be altered in BHD syndrome. In support of this autophagy connection, FLCN has recently been shown to localize to lysosomes and modulate nutrient sensing through the RRAG (Ras-related GTP binding) small G proteins.28-30 Given these findings, we wanted to ascertain whether autophagy is compromised in BHD syndrome. Our study uncovers a link between autophagy and BHD syndrome, revealing that FLCN is an important component of the autophagy machinery and that the FLCN complex is modulated by ULK1.
Results
Loss of FLCN affects SQSTM1 expression
SQSTM1 (sequestome 1) is an established marker of autophagy, which associates with autophagosomes and is degraded during autophagy.31 Of interest, SQSTM1 is often amplified in renal cell carcinoma.32 Given that BHD syndrome predisposes patients to renal cell carcinoma, we analyzed whether FLCN loss could enhance SQSTM1 protein levels. Previously, it was observed that tumor initiation via cystogenesis occurred within Flcn+/− mice upon loss of Flcn in renal proximal tubule cells.33 Therefore, we utilized human renal proximal tubule (HK2) cells with stable FLCN knockdown for our studies. We observed higher endogenous SQSTM1 protein expression in FLCN-deficient cells compared with control cells, which was more pronounced under normal growth conditions (Fig. 1A), implying FLCN may play a role in basal autophagy, but may be dispensable for acute starvation-induced autophagy. We also observed a similar pattern in flcn−/− mouse embryonic fibroblasts (MEFs) (Fig. S1A). We then re-expressed FLCN in FLCN-deficient HK2 cells under normal growth conditions and coexpressed HA-SQSTM1 to specifically measure autophagy in the transfected cells. As indicated by reduced SQSTM1, re-expression of FLCN restored a higher level of basal autophagy (Fig. 1B). Immunohistochemistry revealed elevated SQSTM1 protein levels in a BHD patient renal tumor (with a c.499C > T mutation encoding a truncated FLCN mutant, pGln167X) when compared with unaffected tissue (Fig. 1C). Collectively, these data reveal that the protein expression of SQSTM1 is negatively regulated by FLCN.
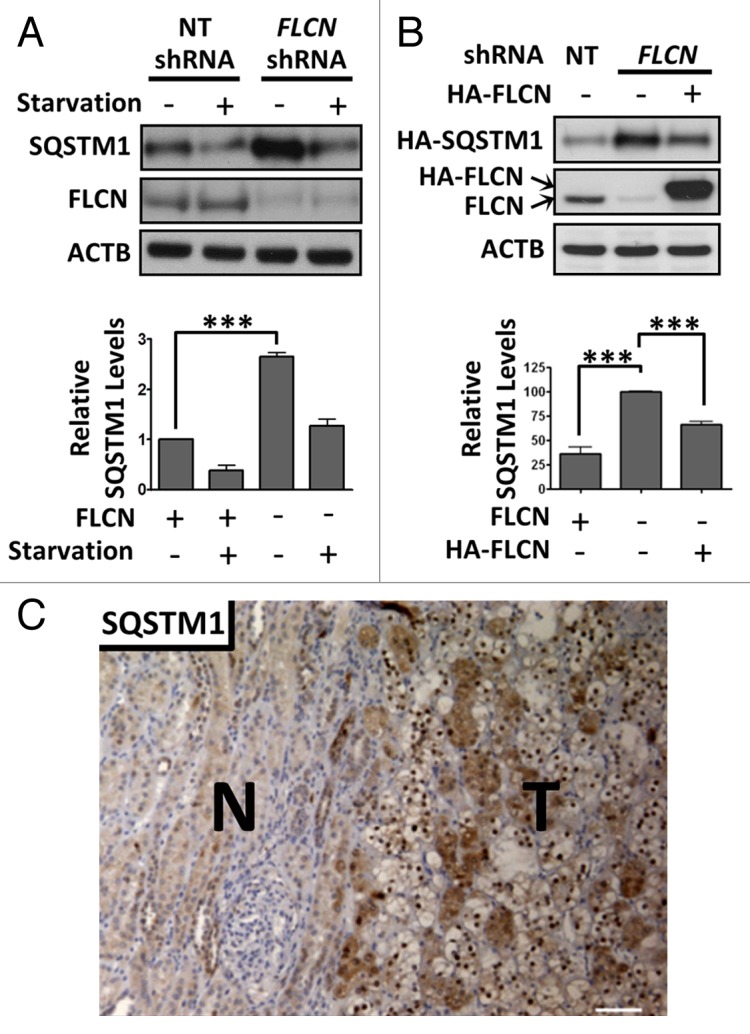
Figure 1. SQSTM1 levels are elevated in FLCN-deficient cells and BHD-tumor derived tissue. (A) Control HK2 cells (non-target (NT) shRNA) and those with stable knockdown of FLCN were starved for 4 h in KRB or grown in normal media and SQSTM1 levels were analyzed. Data are mean ± SEM of 3 independent experiments. (B) HA-FLCN was re-expressed (with coexpressed HA-SQSTM1) in FLCN knockdown HK2 cells and HA-SQSTM1 levels were analyzed by western blot. Data are mean ± SEM of 3 independent experiments. ***P < 0.001. (C) A tumor sample (T) showing mixed histology of clear and chromophobe cells, and surrounding normal tissue (N) from a BHD patient was stained for SQSTM1. Scale bar: 100 μm.
FLCN-deficient cells exhibit impaired autophagy
We wanted to examine whether this elevation of SQSTM1 protein might be due to autophagy defects in the FLCN-deficient cells. We assessed autophagic flux using a vector expressing tandem red and green fluorescent protein-tagged MAP1LC3B (RFP-GFP-LC3), which works on the principle that the GFP signal is less stable in the acidic environment of the lysosome than the RFP signal.34 Of the cells displaying multiple puncta under normal growth conditions, we detected proportionally fewer red puncta in the FLCN knockdown HK2 cells (Fig. 2A and graphed in panel below), indicating impaired maturation of autophagosomes (reduced fusion of autophagosomes with lysosomes), and therefore reduced autophagic flux. This fits with our finding of SQSTM1 accumulation due to lack of degradation in lysosomes. Interestingly, in both HK2 cells (Fig. 2B) and HEK293 cells (Fig. 2C) FLCN knockdown causes a reduction in MAP1LC3B-II conversion following chloroquine treatment. As perturbation of autophagosome maturation would be expected to allow an accumulation of MAP1LC3B-II, this suggests that autophagosome synthesis may also be modestly impaired in FLCN-deficient cells. While these data supports a positive role for FLCN in both autophagosome formation and autophagic flux, some FLCN-deficient cells do contain many autophagosomes (Fig. 2A), while still exhibiting impaired autophagic flux. Analysis of another mammalian Atg8 ortholog, GABARAP, which has a potential role in regulating the sealing process needed for autophagosome maturation,8 revealed enhanced endogenous GABARAP expression in both FLCN-deficient HK2 and HEK293 cells (Fig. 2B and C). This elevation of GABARAP protein was not due to enhanced GABARAP gene expression, as mRNA levels were comparable between the cell lines (Fig. S1B). Overall, these data indicate an impaired autophagy pathway operates in the absence of FLCN, along with altered GABARAP processing.

Figure 2. FLCN is a positive driver of autophagy. (A) HK2 control and FLCN knockdown cells were transfected with the ptfMAP1LC3B vector, fixed, and examined by confocal microscopy. Representative maximal Z-projection images of cells showing the RFP-GFP-MAP1LC3B puncta are shown in the upper panel. Scale bar: 10 μm. Red and yellow puncta were scored across 3 independent experiments (at least 30 cells per cell line in total) and are plotted below the images, mean ± SEM (B) Control HK2 cells and those with stable knockdown of FLCN were treated with 100 μM chloroquine (CQ) for the indicated times. Samples were probed for conversion of MAP1LC3B (graphed in panel below, mean ± S.E.M.) and GABARAP expression. (C) As for (B) but in HEK293 cells with transient knockdown of FLCN expression. For all graphs *P < 0.05, **P < 0.01.
FLCN interacts with GABARAP in the presence of FNIP1 and 2
The FLCN binding partner, FNIP1, was recently shown to interact with GABARAP.35 Combined with our finding of enhanced GABARAP levels in FLCN-deficient cells, we wished to explore this connection further. Unbiased GABARAP interaction mass spectrometry was performed, and we identified 8 high-confidence interacting proteins (Fig. 3A), including both FNIP1 and FLCN (FLCN peptide identification shown in Fig. S2A). We confirmed the FLCN-GABARAP interaction using an in vitro binding assay and could detect endogenous FLCN interaction with bacterially generated recombinant GABARAP protein (Fig. 3B). FLCN interacted more strongly with GABARAP than MAP1LC3B, a member of the other Atg8 ortholog subfamily (Fig. 3C), suggesting enhanced specificity for GABARAP family members. Interestingly, the interaction between FLCN and GABARAP in vivo in mammalian cells was not detectable unless FNIP1/2 was also present. FNIP2 especially was able to potently enhance the interaction (Fig. 3D). The interaction in mammalian cells was clearly specific for GABARAP, and not MAP1LC3B. We further confirmed this stronger interaction of GABARAP to FLCN-FNIP1/2 by using in vitro binding assays with recombinant GABARAP protein (Fig. S2B). Immunofluorescence analysis revealed colocalization of FLCN with GABARAP at punctate structures when coexpressed with FNIP2 (Fig. 3E). We could also detect endogenous FLCN and endogenous GABARAP in a complex with immunoprecipitated myc-FNIP2 (Fig. 3F). As observed by other groups, overexpression of FNIP2 was necessary to detect binding of endogenous components of the complex.29 Overall, these data reveal that FLCN-FNIP interacts with the autophagy machinery via GABARAP.

Figure 3. FLCN interacts with GABARAP, which is enhanced in the presence of FNIP1/2. (A) A network of GABARAP interactors, as determined by mass spectrometry. (B) GST alone or GST-GABARAP was used as bait, and bound endogenous FLCN was detected by western blot. (C) Bacterially expressed GST, GST-MAP1LC3B, and GST-GABARAP was used as bait for lysates with or without overexpression of HA-FLCN. Following GST purification, bound HA-FLCN was detected by western blot. (D) V5-FLCN, HA-MAP1LC3B or HA-GABARAP were expressed in HEK293 cells with FNIP proteins where indicated. Following an HA immunoprecipitation, V5-FLCN was detected by western blot. Total blots represent 30% of IP input. (E) MDCK cells were transfected with EGFP-FLCN-WT, myc-FNIP2 and HA-GABARAP. Cells were stained with mouse-anti-HA and polyclonal FNIP2 antibody. Scale bar: 20 μm. (F) Myc-FNIP2 was expressed and immunoprecipitated from HEK293 cells. Endogenous FLCN and GABARAP bound to FNIP2 were detected by western blotting. Total blots represent 1% of IP input.
FLCN does not function upstream of ULK1
ULK1 is a key activator of the autophagy cascade and a known GABARAP interactor.36 We observed a weak interaction between V5-tagged ULK1 and untagged FLCN (Fig. 4A), which suggests that FLCN might influence autophagy at the level of ULK1. This FLCN-ULK1 interaction was markedly enhanced when ULK1 contained a kinase-inactivating mutation (K46I, referred to as kinase-dead). In a reciprocal experiment, immunoprecipitated HA-FLCN similarly showed a more robust interaction with kinase-dead ULK1 (Fig. S3A).

Figure 4. Expression of wild-type ULK1 induces phosphorylation of FLCN. (A) Untagged FLCN was coexpressed with V5-tagged wild-type (WT) or kinase-dead (KD) ULK1 as indicated in HEK293 cells, and subjected to V5 immunoprecipitation. FLCN bound to ULK1 was detected by western blotting. Total blots represent 40% of IP input. (B) Incorporation of [32P] into HA-FLCN in vivo was determined in the presence and absence of ULK1. (C) An in vitro kinase assay was performed using wild-type (WT) or kinase-dead (KD) ULK1 against purified ATG13 and FLCN. Phosphorylation was determined by [32P] incorporation.
To determine whether this observed FLCN-ULK1 interaction had a cellular function, we analyzed whether ULK1 activity could be modulated by FLCN knockdown. To do this, we examined the comparative activity of both PRKAA2 and MTORC1, as these signaling pathways are known to impact autophagy through ULK1 phosphorylation.10-12,15-17 PRKAA2 activates autophagy via phosphorylation of ULK1 at Ser555,15 whereas Ser758 phosphorylation of ULK1 by MTORC1 is inhibitory and appears to modulate ULK1-PRKAA2 interaction.16,17 Both PRKAA2 and MTORC1 have also been previously linked to FLCN.28-30,37 In control HK2 cells, starvation potently induced the phosphorylation of ULK1 at the PRKAA2-mediated site (Ser555), while growth media induced the phosphorylation of the MTORC1 site (Ser758) (Fig. S3B). Importantly, we did not notice any marked differences in the levels of ULK1 phosphorylation upon FLCN knockdown, except a modest elevation in ULK1 phosphorylation at Ser758 under starvation conditions. Through ULK1 kinase assays, we further confirmed that endogenous ULK1 activity was not significantly impacted upon loss of FLCN (Fig. S3C).
ULK1 induces FLCN phosphorylation
Unlike wild-type ULK1, kinase-dead ULK1 is predominantly found in a larger 1.2-MDa complex, suggesting that autophosphorylation as well as substrate phosphorylation is necessary for normal interaction dynamics between ULK1 and substrates.38 As wild-type ULK1 appears to weakly interact with FLCN compared with kinase-dead ULK1 (Fig. 4A; Fig. S3A), this transient interaction might be modulated through ULK1 phosphorylation. To determine whether ULK1 can induce FLCN phosphorylation, we [γ-32P]-orthophosphate radiolabeled HEK293 cells expressing HA-FLCN in vivo and determined [γ-32P]-incorporation into HA-FLCN in the presence or absence of ULK1. We observed that overexpressed ULK1 potently induced FLCN phosphorylation in vivo (Fig. 4B), implying that ULK1 might function upstream of FLCN. To test whether ULK1 can directly phosphorylate FLCN, we performed in vitro ULK1 kinase assays toward FLCN and a known ULK1 substrate, ATG13.10,11 We found that wild-type ULK1 robustly phosphorylated both FLCN and ATG13 in vitro, whereas no phosphorylation was detected with kinase-dead ULK1 (Fig. 4C). These data reveal that FLCN phosphorylation is induced by active ULK1 overexpression.
Through mass spectrometry, we identified multiple ULK1-mediated phosphorylation events toward FLCN in cells. Within the C terminus of FLCN, we observed 3 new phosphorylation sites (Ser406, Ser537 and Ser542), which were unique to FLCN when coexpressed with wild-type ULK1 but not kinase-dead ULK1 (Fig. 5A–C). The Ser406 signal was not localized, but the closest alternative candidate phosphorylation site is Ser407, which is not as well conserved between species (Fig. 5D). The other 2 localized ULK1 sites, Ser537 and Ser542, are well conserved between species (Fig. 5D). Additional ULK1-mediated phosphorylation sites were observed in the linker region of FLCN (Ser316 and Thr317), but these residues are poorly conserved among species (Fig. S4). A structural model showing the 3 best conserved ULK1-mediated phosphorylation sites of FLCN was generated from the recently determined C-terminal crystal structure of FLCN (Fig. 5E).39 From the crystal structure [PDB Id: 3V42], we observed that all 3 ULK1 phosphorylation sites are solvent exposed to the surrounding environment, making them accessible for phosphorylation.

Figure 5. Mass spectrometry reveals 3 ULK1-mediated phosphorylation sites on FLCN. (A–C) Mass spectrometry (LC-MS/MS) was used to determine the phosphorylated residues of FLCN coexpressed with ULK1. (D) A multispecies alignment of FLCN proteins using Clustal Omega shows that the Ser406, Ser537 and Ser542 phosphorylation sites are well conserved between species. (E) Cartoon representation of the mapped phosphorylation sites on the crystal structure of the FLCN C-terminal domain (PDB Id: 3V42). The insets show a closer view of the serine residues, which are represented as sticks.
ULK1 modulates the FLCN-GABARAP complex
To determine whether FLCN phosphorylation by ULK1 overexpression had a functional consequence, we analyzed the FLCN-FNIP2-GABARAP complex in the presence of ULK1. We found that expression of wild-type ULK1 impaired the interaction of GABARAP with FLCN-FNIP2, whereas this interaction remained intact in the presence of kinase-dead ULK1 (Fig. 6A). In agreement with the literature,36 ULK1 was also found in the GABARAP immunoprecipitates. In support of the finding that ULK1 modulates the FLCN-GABARAP interaction, knockdown of endogenous ULK1 expression by shRNA under normal growth conditions markedly strengthened the interaction between FLCN-FNIP2 and GABARAP (Fig. 6B). Collectively, these data imply that the kinase activity of ULK1 is required for FLCN-FNIP2 dissociation from GABARAP. To determine whether the 3 identified phosphorylation sites in FLCN were important for modulating the interaction, we tested the in vitro binding of both wild type and a triple serine-to-alanine FLCN mutant (3A) to bacterially expressed GST-GABARAP or GST-MAP1LC3B as bait. Loss of these phosphorylation sites modestly enhanced binding of FLCN to GABARAP and MAP1LC3B (Fig. 6C). As observed previously (Fig. 3C), FLCN preferentially bound to GABARAP. However, when we tested the strength of FLCN(3A)-GABARAP binding in mammalian cells, we found that ULK1 expression could still cause disassembly of the complex (data not shown). This suggests that additional ULK1-mediated phosphorylation events (i.e., additional sites within FLCN, FNIP2 and/or GABARAP) further regulate formation of the FLCN-FNIP2-GABARAP complex in cells. However, we observed slight impairment in the ability of the FLCN(3A) mutant to drive autophagy in cells, as determined by a modest repression of SQSTM1 expression when compared with wild-type FLCN (Fig. 6D).

Figure 6. ULK1 modulates the FLCN-GABARAP interaction. (A) V5-FLCN and myc-FNIP2 bound to HA-GABARAP in the presence or absence of wild-type (WT) or kinase-dead (KD) V5-ULK1 were determined by immunoprecipitating HA-GABARAP and detecting bound proteins by western blot. Total blots represent 20% of IP input. (B) Cells were transfected with control or ULK1 shRNA, along with V5-FLCN, myc-FNIP2, and HA-GABARAP and grown in complete DMEM. Following an HA immunoprecipitation, V5-FLCN and myc-FNIP2 were detected by western blot. Total blots represent 20% of IP input. The graphs show relative binding of V5-FLCN to HA-GABARAP and myc-FNIP2 to HA-GABARAP as determined by densitometry across 4 independent experiments, mean ± SEM *P < 0.05. (C) Bacterially expressed GST, GST-MAP1LC3B or GST-GABARAP was used as bait for lysates containing HA-FLCN (WT or 3A mutant). Following GST purification, bound HA-FLCN was detected by western blot. GST loading controls are shown for each pulldown. (D) Wild-type (WT) FLCN, or the serine-to-alanine (3A) FLCN mutant, were re-expressed in FLCN-deficient HK2 cells, along with HA-SQSTM1. HA-SQSTM1 levels were determined by western blot and densitometry of SQSTM1 levels from 3 independent experiments are plotted in the graph, mean ± SEM *P < 0.05, ***P < 0.001.
Patient tumors show autophagy defects
To determine whether our findings translated to clinical samples, we analyzed SQSTM1 protein levels along with the Atg8 ortholog family members, GABARAP and MAP1LC3B, in a BHD patient renal tumor containing 2 mutated copies of FLCN (a c.499C > T mutation (encoding a truncated FLCN mutant, pGln167X) in one allele and deletion of exon 6 in the other allele).40 We found that SQSTM1 and GABARAP proteins were elevated in both chromophobe and clear cell sections of the tumor when compared with normal control kidney tissue, with slight elevation of MAP1LC3B (Fig. 7A). The observation of raised MAP1LC3B and SQSTM1 protein levels could indicate a blockage in the autophagic pathway, preventing proper autophagic flux. We next determined whether FLCN mutants exhibited altered association with ULK1 and GABARAP. We tested ULK1 interaction with a panel of BHD-patient derived mutations (curated in the Leiden Open Variation Database). We discovered that the C-terminal truncating mutations (Y463X and H429X) interacted more avidly with ULK1 than either wild-type FLCN or a BHD patient-derived point mutation, K508R (Fig. 7B). This suggests that the extreme C terminus of FLCN does not bind directly to ULK1 and may play a role in dissociation from ULK1. In contrast, these truncation mutants of FLCN show impaired binding to GABARAP (Fig. 7C). Additionally, the mutants were not able to repress SQSTM1 levels as efficiently as wild-type FLCN (Fig. 7D). Collectively, our data suggest that FLCN functions as a positive modulator of autophagy, where loss of FLCN impairs basal autophagy, both in vitro and in the disease setting.

Figure 7. Patient tumors and patient-derived FLCN mutations show autophagy defects. (A) Kidney tumor tissues from a BHD patient showing mixed histology of clear cell and chromophobe cells were stained for SQSTM1, GABARAP and MAP1LC3B and compared with normal kidney. Scale bar: 50 μm. (B) HA-FLCN (wild-type or patient-derived mutants) was coexpressed with V5-tagged wild-type (WT) ULK1 in HEK293 cells, and subjected to V5 immunoprecipitation. FLCN bound to ULK1 was detected by western blotting. Total blots represent 40% of IP input. (C) HA-GABARAP was coexpressed with untagged-FLCN (wild-type or patient-derived mutants) and myc-FNIP2 in HEK293 cells, and subjected to HA immunoprecipitation. FLCN bound to GABARAP was detected by western blotting. Total blots represent 5% of IP input. (D) Untagged FLCN (wild type or mutants) was re-expressed (with coexpressed HA-SQSTM1) in FLCN knockdown HK2 cells and HA-SQSTM1 levels were analyzed by western blot. Data are mean ± SEM of 5 independent experiments. *P < 0.05, **P < 0.01.
Discussion
In this study, we discovered that ULK1 modulates the interaction of FLCN-FNIP2 with the autophagy component, GABARAP. Our analysis reveals that multiple residues within FLCN are phosphorylated in an ULK1-dependent manner. We also uncovered that FLCN plays a positive role in autophagy, where loss of FLCN leads to impaired autophagic flux.
The binding partners of FLCN, FNIP1, and FNIP2, have previously been connected to B-cell development,41 autophagy via GABARAP,35 and the induction of apoptosis following DNA-base mispairing.42 We reveal that FNIP2 (and to a lesser extent FNIP1) enhances FLCN-GABARAP binding, implying that FLCN functions as a complex with FNIP proteins to regulate autophagy. Lack of this functional complex due to FLCN mutations in BHD syndrome could explain the impaired autophagy observed in tumor tissue from BHD syndrome patients.
GABARAP subfamily members appear to function downstream of phagophore membrane elongation in a step coupled to dissociation of the ATG12-ATG5-ATG16L1 complex8 and have been hypothesized to recruit and anchor the ULK1 complex on phagophores.36,43 ULK1, along with its binding partners, ATG13 and RB1CC1, contain LC3-interacting region (LIR) motifs which preferentially bind to the GABARAP subfamily of Atg8 ortholog proteins.36,43 As FLCN appears to have a preference for binding GABARAP over MAP1LC3B, it is likely that FLCN modulates autophagy through GABARAP-dependent processes. Additionally, FNIP2 has a potential LIR motif (Fig. S5), while FNIP1 does not, which might explain why FNIP2 further enhances the FLCN-GABARAP interaction.
We detected elevated SQSTM1 protein levels, as well as GABARAP and MAP1LC3B, in BHD kidney tumors compared with normal kidney tissue. Interestingly, upregulation of SQSTM1 is observed in several cancers, including glioblastoma multiforme,44 colorectal cancer,45 and hepatocellular carcinomas,46 while genomic amplification of the SQSTM1 gene is seen in some clear cell renal cell carcinomas.32 SQSTM1 is also overabundant in breast cancer,47 where its expression level correlates with poorer disease-free survival.48 Similarly, elevated SQSTM1 is associated with poor prognosis in lung cancer patients.49 It is known that genetic inactivation of Atg7 in mice leads to SQSTM1 accumulation and generation of ubiquitin-positive inclusions in the liver. This then sequentially leads to accumulation of nuclear factor, erythroid 2-like 2, enhanced cellular stress, and hepatotoxicity.50 Sustained SQSTM1 expression has also been linked to tumorigenesis via elevated levels of reactive oxygen species and DNA damage51 in addition to enhanced cellular migration and invasion.44 A similar mechanism could operate in FLCN-deficient kidney cells, whereby inactivation of FLCN causes autophagy deficiencies and elevated SQSTM1 levels, and the resulting cellular stress could promote tumor development.
Recently, both SQSTM1 and FLCN have been linked to amino acid sensing through the RRAG proteins and MTORC1. SQSTM1 was found to bind the RRAG proteins to favor formation of the active RRAG heterodimer, thereby helping activate MTORC1 at the lysosome.52 Three publications have now linked the FLCN-FNIP complex to the RRAG at the lysosome, linking FLCN to amino acid-dependent MTORC1 signaling.28-30 We did not detect substantially altered MTORC1 signaling following FLCN loss in our cell line model under normal or starved conditions, while other publications have found enhanced MTORC1 activity in animal models lacking FLCN, at least in certain cell types.33,53 It appears that MTORC1 signaling in the context of FLCN-deficiency is cell-type dependent and can also alter during tumor development in response to accumulation of other genetic mutations.
BHD syndrome has lately been reported to be a ciliopathy,26 as alteration to FLCN levels can cause changes to the onset of ciliogenesis. Changes in FLCN levels are also associated with disruption of planar cell polarity and dysregulation of the canonical WNT signaling pathway. As a compromised ability to activate the autophagic response may be an underlying feature in some ciliopathies,27 it is possible that there is also an association between cilia and autophagy in BHD syndrome. For instance, impaired autophagy could be a contributing factor to ciliary defects and renal cyst formation in BHD syndrome patients.
While our work helps refine our understanding of FLCN by revealing that FLCN interacts with components integral to autophagy, it is important to highlight that FLCN function is not just restricted to autophagy or the lysosome. For instance, FLCN also interacts with plakophilin-4 involved in desmosomal and adherens junctions.54,55 Additionally, multiple pathways that drive cancer progression can become dysregulated when FLCN expression is lost, including defects in TGFB1 (transforming growth factor, β 1)-mediated signaling,56,57 enhanced HIF1A (hypoxia inducible factor 1, α subunit [basic helix-loop-helix transcription factor]) activity58 and TFE3 (transcription factor binding to IGHM enhancer 3) activity.59 Therefore, FLCN appears to play a broader ‘housekeeping’ role in the cell and is likely to be a fundamental player in autophagy and cellular homeostasis outside the disease setting. Although further studies are required, it seems reasonable to assume that impaired autophagy upon loss of FLCN expression contributes in part to cancer progression in BHD patients.
Materials and Methods
Cell culture
Stable FLCN knockdown in HK2 cells was previously described.25 Flcn+/+ and flcn−/− MEF cells were gifted by Prof Arnim Pause (McGill University, Canada) derived from mice described previously.33 All cell lines were cultured in DMEM (Life Technologies, 11995065) supplemented with 10% (v/v) fetal calf serum (Life Technologies, 10270-106), 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies, 15070-063). Lipofectamine 2000 transfection was used unless otherwise stated and performed according to the manufacturer’s protocol (Life Technologies, 11668019). Cells were harvested 24 to 36 h post-transfection. Experiments were performed under normal growth conditions, unless otherwise stated. For complete starvation, cells were washed twice in phosphate-buffered saline (Sigma Aldrich, P4417) and incubated in Kreb’s Ringer buffer (KRB) (20 mM HEPES, pH 7.4, 115 mM NaCl, 5 mM KCl, 10 mM NaHCO3, 2.5 mM MgCl2, 2.5 mM CaCl2) for 4 h.
Plasmids
V5-ULK1 wild type and kinase-dead (K46I) and GST-ATG13 have been described previously.23 HA-FLCN was generated in the pN3HA backbone (a kind gift from Dr Sylvia Neumann, The Scripps Research Institute, San Diego, CA, USA) and untagged-FLCN in the pcDNA3.1 vector. GST-FLCN in pDEST27 (Life Technologies, 11812013) and V5-FNIP1 in pcDNA3.1/nV5-DEST (Life Technologies, 12290010) were generated using the Gateway system (Life Technologies). HA-FNIP1 was a kind gift from Dr Laura Schmidt (National Institutes of Health, Bethesda, MD, USA) and myc-FNIP2 was a kind gift from Dr O Hino (Juntendo University School of Medicine, Tokyo, Japan).60 HA-SQSTM1 (plasmid #28027)61 and ptfMAP1LC3B vector (plasmid #21074)34 were from Addgene. MAP1LC3B and GABARAP (from pDONR [12535035]35) were cloned into pDEST15 (Life Technologies, 11802014) or pcDNA-HA, respectively. Mutations were introduced using the QuikChange Site-directed mutagenesis kit (Stratagene, Agilent Technologies, 200521).
Antibodies
Anti-HA antibody (11867423001) was purchased from Roche. Anti-ACTB (4967), phospho-ULK1 Ser555 (5869) and Ser758 (6888) and total ULK1 (4773) antibodies were from Cell Signaling Technology. Anti-V5 (46-0705) was from Life Technologies. Anti-Myc clone 9E10 antibody (M5546) was from Sigma-Aldrich and anti-GST antibody (05-782) was from Merck Millipore. Anti-FLCN was gifted from Prof Arnim Pause (McGill University, Canada). Anti-SQSTM1/p62 C-terminal antibody (GP62-C/DS-160211) was from Progen Biotechnik GmbH. Anti-MAP1LC3B antibody for immunofluorescence and immunohistochemistry was from Novus Biologicals (NB100-2220), and for western blotting from Nanotools (0260-100/LC3-2G6). GABARAP antibody (AP1821a) was from Abgent.
shRNA knockdown
JetPEI transfection mixtures (VWR International, 101-10N) containing 2 μg of scrambled shRNA or ULK1 shRNA (MISSION shRNA 1-1064slcl, Sigma) were prepared according to the manufacturer’s protocol (Polyplus-transfection) and reverse transfected into HEK293 cells. Following incubation at 37 °C for 24 h, plates were then transfected with V5-FLCN, myc-FNIP2 and HA-GABARAP using the JetPEI protocol (forward transfection) and incubated for a further 28 h prior to lysis.
Immunoprecipitation, GST-affinity isolation, and western blotting
Cells were lysed in BHD lysis buffer (20 mM Tris, pH 7.5, 135 mM NaCl, 5% [v/v] glycerol, 50 mM NaF, 0.1% [v/v] Triton X-100, plus protease inhibitors [cOmplete Tablets, Mini EDTA-fee, Roche Diagnostics, 04693159001]), centrifuged and the protein quantified using Bradford reagent (Sigma-Aldrich, B6916). Anti-HA and anti-V5 coupled to protein G-Sepharose beads (GE Healthcare Life Sciences, 17-0618-05) were used to immunoprecipitate HA and V5-tagged proteins as appropriate. Immunoprecipitates were washed 3 times in lysis buffer and resuspended in NuPAGE LDS sample buffer (Life Technologies, 1386564). Samples for GST-affinity isolation were lysed in Buffer B (40 mM HEPES, pH 7.5, 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM β-glycerophosphate, 50 mM NaF, 1.5 mM Na3VO4, 0.3% [w/v] CHAPS [Thermo Scientific, 28300], plus protease inhibitors) and incubated with glutathione-Sepharose beads (GE Healthcare Life Sciences, 17-0756-01). Beads were washed 3 times in lysis buffer, and GST-tagged proteins were eluted using 10 mM glutathione. Western blotting was performed as previously described.23 Blots shown are representative of at least 3 independent experiments.
ULK1 kinase assay
Cell lysates from nutrient-replete or starved HK2 cells were immunoprecipitated using anti-ULK1 antibody (J.T. Murray, Queen’s University, Belfast, UK). Immunocomplexes were captured using protein G-Sepharose (GE Healthcare Life Sciences, 17-0618-05), washed 3 times in Low Salt Buffer (50 mM Tris-Cl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 0.3% [w/v] CHAPs, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 0.27 M sucrose, 0.1% [v/v] 2-mercaptoethanol) and twice in Assay Buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% β-mercaptoethanol, 25 mM β-glycerophosphate, 100 μM orthovanadate). In vitro ULK1 kinase assays against GST-FLCN and GST-ATG13 (purified from HEK293 cells), were performed using immunoprecipitated V5-tagged ULK1. The assay mix of immunoprecipitated ULK1, 10 mM MgAc, substrate (3 μg MBP, GST-FLCN or GST-ATG13 as required) and 100 μM [32P] ATP in Assay Buffer was incubated at 30 °C for 10 min, then quenched with NuPAGE LDS sample buffer (Life Technologies, 1386564) and subjected to SDS-PAGE. Relative levels of [32P]-incorporation were determined by autoradiography.
Immunofluorescence and image analysis
HK2 cells were transfected with RFP-GFP-LC3 for 24 h, fixed with paraformaldehyde, and imaged under oil immersion at 20 °C using a Leica TCS SPE confocal laser scanning fluorescence microscope, using Leica software (Leica Microsystems B.V., Rijswijk, 2288 ED Netherlands). Confocal images were stacked and merged using ImageJ v1.43 software. Puncta were counted manually across multiple fields of view from > 10 cells per condition, over 3 independent experiments. The proportion of each type of puncta was calculated and plotted. For colocalization experiments, MDCK cells were transfected with pEGFP-FLCN-WT, myc-FNIP2 and HA-GABARAP using MetaFectene Pro (Biontex Laboratories, T040-1.0). After 46 h, cells were washed twice in PBS and starved in KRB containing 4.5 g/l glucose. Cells were stained with mouse-anti-HA (Cell Signaling Technology, 2367) and polyclonal FNIP2 antibody (Abnova, PAB16703) directed against AA117–131 of human FNIP2. Secondary antibodies were goat-anti-rabbit-Alexa568 (Life Technologies, A11036) and goat-anti-mouse-Cy5 (Southern Biotech, 1034-15), with DAPI counterstain.
Binding of FLCN to GST-baits
Bacterially expressed GST, GST-MAP1LC3B, and GST-GABARAP were purified using glutathione beads, washed in BHD lysis buffer and then incubated with lysate from HEK293 cells transfected with HA-FLCN (WT or 3A), HA-FNIP1 or myc-FNIP2. Beads were washed 3 times in lysis buffer, and GST-tagged proteins eluted using 10 mM glutathione.
In vivo radiolabeling
Transfected HEK293 cells were incubated in phosphate-free medium containing 0.2 mCi [32P]-orthophosphate (PerkinElmer, NEX063002MC) for 4 h. These cells were harvested using BHD lysis buffer. HA-FLCN was immunoprecipitated with anti-HA antibody bound to Protein G-Dynabeads (Life Technologies, 10007D) and washed in lysis buffer.
Mass spectrometry
GST-FLCN was purified from HEK293 cells coexpressing wild-type or kinase-dead ULK1. Samples were separated by SDS-PAGE and excised bands were subjected to a modified in-gel trypsin digestion procedure.62 Eluted peptides were subjected to electrospray ionization prior to mass spectrometry in an LTQ-Orbitrap instrument (Thermo Fisher Scientific Inc., Waltham, MA, USA 02451). Peptide sequences were determined by matching protein or translated nucleotide databases with the acquired fragmentation pattern by Sequest (ThermoFinnigan).63 Modification of 79.9663 mass units to serine, threonine and tyrosine was included in the database searches to determine phosphopeptides and manually inspected to ensure confidence.
Structural modeling
Crystal structure coordinates used in the current manuscript are as deposited in the protein data bank39 (PDB Id: 3V42). The model was generated using Pymol.
Immunostaining of patient tumor
Tissue sections were processed using standard methodology.26 Sections were incubated with antibodies for MAP1LC3B, GABARAP, and SQSTM1 antibody (BD Bioscience, 610832) in 3% (w/v) bovine serum albumin (Sigma-Aldrich, A7906) overnight at 4 °C.
GABARAP interaction network
GABARAP interaction mass spectrometry was performed and analyzed as described previously,35 with the exception that autophagy interaction network baits (33 thereof) were stably expressed using the MSCV NTAP system in A549 non-small cell lung carcinoma cell lines.
Densitometry and statistical analysis
Densitometry was performed using ImageJ v1.43 software. The Student t test or one-way ANOVA followed by LSD post-hoc testing (as appropriate) were used for statistical analysis, with P < 0.05 taken to be significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Mass spectrometry was performed by Steve Gygi and Ross Tomaino at the Taplin Biological Mass Spectrometry Facility, Harvard Medical School, Boston, USA. This work was supported by the Myrovlytis Trust (to ART, EAD, SS, and RKN); Association for International Cancer Research (Career Development Fellowship No.06-914/915 to ART, Grant 11-0687 to MvS, BJC, and ART); National Institute for Social Care and Health Research (Wales Gene Park); FindACure and Cancer Research UK development fund (to ART), Cancer Research UK Career Development Fellowship (C20685/A12825 to SW); the Deutsche Forschungsgemeinschaft (BE 4685/1-1 to CB); the Dutch Cancer Society KWF (2009-4352 to MvS and BJC); the Annadal foundation (to MvS and BJC); GROW School for Oncology and Developmental Biology (to TC); the McClay Trust (to ER); and the British Heart Foundation (to JTM).
Glossary
Abbreviations:
- ATG
autophagy-related
- BHD
Birt-Hogg-Dubé
- FLCN
folliculin
- FNIP
folliculin-interacting protein
- GABARAP
GABA(A) receptor-associated protein
- GABARAPL2/GATE-16
GABA(A) receptor-associated protein-like 2
- GST
glutathione S-transferase
- KRB
Kreb’s Ringer buffer
- LIR
LC3-interacting region
- MAP1LC3
microtubule-associated protein 1 light chain 3
- MBP
myelin basic protein
- MTORC1
mechanistic target of rapamycin complex 1
- PRKAA2
protein kinase, AMP-activated, alpha 2 catalytic subunit
- RB1CC1
RB1-inducible coiled-coil 1
- RRAG
Ras-related GTP binding
- SQSTM1
sequestome 1
- ULK1
unc-51 like autophagy activating kinase
References
- 1.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 2.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–6. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 3.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 4.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 5.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, Debnath J, Deretic V, Elazar Z, Eskelinen EL, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7:1273–94. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–12. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 8.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5:649–62. doi: 10.4161/auto.5.5.8249. [DOI] [PubMed] [Google Scholar]
- 14.Dorsey FC, Rose KL, Coenen S, Prater SM, Cavett V, Cleveland JL, Caldwell-Busby J. Mapping the phosphorylation sites of Ulk1. J Proteome Res. 2009;8:5253–63. doi: 10.1021/pr900583m. [DOI] [PubMed] [Google Scholar]
- 15.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011;108:4788–93. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440:283–91. doi: 10.1042/BJ20101894. [DOI] [PubMed] [Google Scholar]
- 19.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–68. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53:471–83. doi: 10.1016/j.molcel.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–50. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, Campbell DG, Wesselborg S, Alessi DR, Stork B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 23.Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7:737–47. doi: 10.4161/auto.7.7.15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212–21. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157–64. doi: 10.1016/S1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 26.Luijten MN, Basten SG, Claessens T, Vernooij M, Scott CL, Janssen R, Easton JA, Kamps MA, Vreeburg M, Broers JL, et al. Birt-Hogg-Dube syndrome is a novel ciliopathy. Hum Mol Genet. 2013;22:4383–97. doi: 10.1093/hmg/ddt288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pampliega O, Orhon I, Patel B, Sridhar S, Díaz-Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194–200. doi: 10.1038/nature12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol. 2013;202:1107–22. doi: 10.1083/jcb.201307084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini DM. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52:495–505. doi: 10.1016/j.molcel.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martina JA, Diab HI, Lishu L, Jeong-A L, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7:ra9. doi: 10.1126/scisignal.2004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudon V, Sabourin S, Dydensborg AB, Kottis V, Ghazi A, Paquet M, Crosby K, Pomerleau V, Uetani N, Pause A. Renal tumour suppressor function of the Birt-Hogg-Dubé syndrome gene product folliculin. J Med Genet. 2010;47:182–9. doi: 10.1136/jmg.2009.072009. [DOI] [PubMed] [Google Scholar]
- 34.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 35.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem. 2012;287:39275–90. doi: 10.1074/jbc.M112.378109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, Esposito D, Gillette WK, Hopkins RF, 3rd, Hartley JL, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552–7. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan EYW, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–71. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nookala RK, Langemeyer L, Pacitto A, Ochoa-Montaño B, Donaldson JC, Blaszczyk BK, Chirgadze DY, Barr FA, Bazan JF, Blundell TL. Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol. 2012;2:120071. doi: 10.1098/rsob.120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menko FH, Johannesma PC, van Moorselaar RJ, Reinhard R, van Waesberghe JH, Thunnissen E, Houweling AC, Leter EM, Waisfisz Q, van Doorn MB, et al. A de novo FLCN mutation in a patient with spontaneous pneumothorax and renal cancer; a clinical and molecular evaluation. Fam Cancer. 2013;12:373–9. doi: 10.1007/s10689-012-9593-8. [DOI] [PubMed] [Google Scholar]
- 41.Baba M, Keller JR, Sun HW, Resch W, Kuchen S, Suh HC, Hasumi H, Hasumi Y, Kieffer-Kwon KR, Gonzalez CG, et al. The folliculin-FNIP1 pathway deleted in human Birt-Hogg-Dubé syndrome is required for murine B-cell development. Blood. 2012;120:1254–61. doi: 10.1182/blood-2012-02-410407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim TH, Fujikane R, Sano S, Sakagami R, Nakatsu Y, Tsuzuki T, Sekiguchi M, Hidaka M. Activation of AMP-activated protein kinase by MAPO1 and FLCN induces apoptosis triggered by alkylated base mismatch in DNA. DNA Repair (Amst) 2012;11:259–66. doi: 10.1016/j.dnarep.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 43.Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012;31:3691–703. doi: 10.1038/emboj.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, Dinsdale D, Condorelli F, Brandner S, Campanella M, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32:699–712. doi: 10.1038/onc.2012.111. [DOI] [PubMed] [Google Scholar]
- 45.Ren F, Shu G, Liu G, Liu D, Zhou J, Yuan L, Zhou J. Knockdown of p62/sequestosome 1 attenuates autophagy and inhibits colorectal cancer cell growth. Mol Cell Biochem. 2014;385:95–102. doi: 10.1007/s11010-013-1818-0. [DOI] [PubMed] [Google Scholar]
- 46.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–84. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 2003;22:2322–33. doi: 10.1038/sj.onc.1206325. [DOI] [PubMed] [Google Scholar]
- 48.Luo RZ, Yuan ZY, Li M, Xi SY, Fu J, He J. Accumulation of p62 is associated with poor prognosis in patients with triple-negative breast cancer. Onco Targets Ther. 2013;6:883–8. doi: 10.2147/OTT.S46222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–6. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 51.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–46. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hasumi Y, Baba M, Ajima R, Hasumi H, Valera VA, Klein ME, Haines DC, Merino MJ, Hong SB, Yamaguchi TP, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A. 2009;106:18722–7. doi: 10.1073/pnas.0908853106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medvetz DA, Khabibullin D, Hariharan V, Ongusaha PP, Goncharova EA, Schlechter T, Darling TN, Hofmann I, Krymskaya VP, Liao JK, et al. Folliculin, the product of the Birt-Hogg-Dube tumor suppressor gene, interacts with the adherens junction protein p0071 to regulate cell-cell adhesion. PLoS One. 2012;7:e47842. doi: 10.1371/journal.pone.0047842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nahorski MS, Seabra L, Straatman-Iwanowska A, Wingenfeld A, Reiman A, Lu X, Klomp JA, Teh BT, Hatzfeld M, Gissen P, et al. Folliculin interacts with p0071 (plakophilin-4) and deficiency is associated with disordered RhoA signalling, epithelial polarization and cytokinesis. Hum Mol Genet. 2012;21:5268–79. doi: 10.1093/hmg/dds378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hong SB, Oh H, Valera VA, Stull J, Ngo DT, Baba M, Merino MJ, Linehan WM, Schmidt LS. Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-beta signaling. Mol Cancer. 2010;9:160. doi: 10.1186/1476-4598-9-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cash TP, Gruber JJ, Hartman TR, Henske EP, Simon MC. Loss of the Birt-Hogg-Dubé tumor suppressor results in apoptotic resistance due to aberrant TGFβ-mediated transcription. Oncogene. 2011;30:2534–46. doi: 10.1038/onc.2010.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Preston RS, Philp A, Claessens T, Gijezen L, Dydensborg AB, Dunlop EA, Harper KT, Brinkhuizen T, Menko FH, Davies DM, et al. Absence of the Birt-Hogg-Dubé gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene. 2011;30:1159–73. doi: 10.1038/onc.2010.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong SB, Oh H, Valera VA, Baba M, Schmidt LS, Linehan WM. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS One. 2010;5:e15793. doi: 10.1371/journal.pone.0015793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takagi Y, Kobayashi T, Shiono M, Wang L, Piao X, Sun G, Zhang D, Abe M, Hagiwara Y, Takahashi K, et al. Interaction of folliculin (Birt-Hogg-Dubé gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene. 2008;27:5339–47. doi: 10.1038/onc.2008.261. [DOI] [PubMed] [Google Scholar]
- 61.Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, Zhu M, Zhong Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy. 2010;6:614–21. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 63.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.