

Abstract
Accumulation of β-amyloid (Aβ) and resultant inflammation are critical pathological features of Alzheimer disease (AD). Microglia, a primary immune cell in brain, ingests and degrades extracellular Aβ fibrils via the lysosomal system. Autophagy is a catabolic process that degrades native cellular components, however, the role of autophagy in Aβ degradation by microglia and its effects on AD are unknown. Here we demonstrate a novel role for autophagy in the clearance of extracellular Aβ fibrils by microglia and in the regulation of the Aβ-induced NLRP3 (NLR family, pyrin domain containing 3) inflammasome using microglia specific atg7 knockout mice and cell cultures. We found in microglial cultures that Aβ interacts with MAP1LC3B-II via OPTN/optineurin and is degraded by an autophagic process mediated by the PRKAA1 pathway. We anticipate that enhancing microglial autophagy may be a promising new therapeutic strategy for AD.
Keywords: Alzheimer disease, NLRP3, autophagy, microglia, β-amyloid
Introduction
Alzheimer disease (AD) is a devastating neurodegenerative disease that is characterized by the widespread loss of neurons and synapses and the progressive loss of memories. The extracellular accumulation of β-amyloid (Aβ) in senile plaques is thought to be one of the irrefutable pathological findings of AD and is regarded as the primary causative factor that results in neurodegeneration.1 Apoetm3(APOE*4)Mae, the major genetic risk factor for late onset AD, is suggested to cause a defect in the clearance of Aβ.2,3 Microglia are the resident macrophages in the brain that constantly survey the neural environment for pathogens, foreign materials, and apoptotic cells; hence, microglia are considered the principal immuno-effector phagocyte in the brain.4 Microglia not only degrade Aβ but also secrete various inflammatory cytokines after activation by Aβ.5
Autophagy is the catabolic process that sequesters cytoplasm, including aberrant organelles and macromolecules, into double-membrane vesicles and delivers it to lysosomes for degradation and the eventual recycling of the resulting macromolecules.6 We speculated that the autophagic processes of microglia might be involved in the degradation of extracellular amyloid fibrils and wondered if microglial autophagy is also important for the regulation of Aβ-mediated inflammation. Despite the fact that glia are much more abundant in the brain and were recently identified as much more important in the regulation of synaptic activity and the maintenance of homeostasis of brain,2,7 most previously published studies have focused on neuronal autophagy and few studies have focused on the glial autophagy, such as that performed by microglia and astrocytes. Notably, the relationship between microglia and autophagy in AD has not been elucidated.
Recent genome-wide association studies (GWAS) have reported CR1 (complement component [3b/4b] receptor 1 [Knops blood group]), CD33, and TREM2 (triggering receptor expressed on myeloid cells 2) as the risk genes for late onset AD.8-10 This points toward a dysregulation of the immune system and inflammation as potential genetic basis for late onset AD. Activated microglia in senile plaques has pro-inflammatory phenotype linked with neurotoxicity.11 Moreover, Aβ fibrils activate the NLRP3 inflammasome resulting in release of pro-inflammatory cytokine IL1B (interleukin 1, β) in microglia5 and NLRP3 inflammasome is activated in the brains of AD patients.12
In this study, we showed for the first time that the degradation of extracellular Aβ fibrils by microglia is dependent on autophagic processes. Phagocytosed Aβ interacts with MAP1LC3B-II via OPTN/optineurin and is degraded by an autophagic process mediated by the STK11/LKB1 (serine/threonine kinase 11)-PRKAA1/AMPKα (protein kinase, AMP-activated, α 1 catalytic subunit) pathway. Autophagy is also important for the regulation of Aβ-mediated NLRP3 inflammasome activation, thereby affecting neuronal survival.
Results
Induction of autophagy in microglia by extracellular Aβ fibrils
The BV2 microglial cell line and primary mouse microglia were treated with FITC-labeled Aβ fibrils (fAβ), as previously reported,13 in order to observe the uptake and degradation of fAβ by microglia. After 2 h of incubation, the FITC-Aβ signals appeared in the inside microglial cell and the signals are decreased in a time-dependent manner (Fig. S1D and S1E), indicating the uptake and degradation of fAβ by microglia, as previously reported.14,15 To further confirm the uptake and degradation of extracellular fAβ by microglia, the intracellular Aβ and extracellular Aβ levels were examined using western blot analysis. The level of intracellular Aβ and extracellular Aβ levels were also decreased in both BV2 and primary microglia in a time-dependent manner (Fig. S1F). These data show that extracellular fAβ was first internalized and then degraded by microglia, similar to previous studies.14-16
To determine the relationship between Aβ and autophagic vacuoles, the BV2 microglial cell line and primary mouse microglia were treated with FITC-fAβ for 2 h and stained with anti-MAP1LC3B specific antibody. The number of MAP1LC3B dots was increased following fAβ treatment (Fig. 1A), suggesting that autophagy was induced in microglia by extracellular fAβ. Interestingly, internalized fAβ appeared in both vacuolar and punctated forms (Fig. 1A), which were colocalized with the MAP1LC3B dots representing the autophagic vacuoles. These findings indicate a close association between autophagic vacuoles and Aβ in microglia.

Figure 1. fAβ degradation depends on the fAβ-induced autophagic processes. (A) The BV2 microglial cell line and primary mouse microglia treated with FITC-labeled Aβ fibrils (1 μM) and then stained with MAP1LC3B (red). The representative images show that the number of MAP1LC3B dots is increased and colocalized with FITC-Aβ in FITC-Aβ-treated BV2 cells and primary microglia. Scale bar: 10 μm. (B) The BV2 microglial cell line and primary mouse microglia were treated with Aβ fibrils (1 μM) for 2 h. Whole cell lysates were collected at the indicated time points and analyzed by immunoblotting for ATG5–12, MAP1LC3B, and ACTB (loading control). The levels of the ATG12–ATG5 complex and MAP1LC3B-II were increased in the Aβ fibril-treated BV2 and primary microglia. (C) The BV2 microglia cell line was treated with 25 nM bafilomycin A1 for 30 min before 1 μM Aβ fibrils. After 2 h, whole cell lysates were collected and analyzed using immunoblotting for MAP1LC3B and ACTB (loading control). The level of the MAP1LC3B-II is increased in the Aβ fibril-treated BV2 cells and more in BV2 cells cotreated with Aβ and bafilomycin A1.
Next, the BV2 microglial cell line and primary mouse microglia were treated with fAβ for 2 h, and the levels of MAP1LC3B-II, a lipidated MAP1LC3B that is a reliable marker of autophagic vacuoles, and the ATG12–ATG5 complex, which is necessary for the elongation of autophagosomal membranes, an indicator of the induction of autophagy,17 were investigated. The MAP1LC3B-II and ATG12–ATG5 complex levels were increased in both BV2 and primary microglia (Fig. 1B) and MAP1LC3B-II levels were more increased when maturation of autophagosome was blocked by bafilomycin A1 (Fig. 1C), indicating that autophagy was induced in the microglia by extracellular fAβ.
Aβ degradation in microglia by autophagic processes
Because we observed a close association between autophagy and internalized fAβ in microglia (Fig. 1), we investigated the possibility that autophagic processes are necessary for the clearance of Aβ from microglia. The MAP1LC3B and ATG7 protein levels were effectively decreased by siRNA-targeting Map1lc3B and Atg7 (Fig. 2A and B). The BV2 microglial cell line and primary mouse microglia were transfected with siMap1lc3B and siAtg7 for 24 h, then treated with fAβ or FITC-fAβ for the next 24 h in order to observe Aβ degradation. Western blot analysis demonstrates that the level of Aβ in microglia was higher following siMap1lc3B and siAtg7 transfection compared with scrambled siRNA (siCTL) transfection (Fig. 2A and B), indicating that Aβ still remains at higher concentrations and does not degrade in siMap1lc3B- and siAtg7 transfected microglia. Downregulation of MAP1LC3B and ATG7 did not affect expression levels of Aβ-degrading enzyme insulin degrading enzyme (IDE) (Fig. S4), suggesting that changes in Aβ level may be not due to IDE but due to autophagy. The FITC-Aβ signal was also higher in microglia that had been transfected with siMap1lc3b compared with siCTL (Fig. 2C), further confirming that autophagic processes are necessary for the degradation of fAβ in microglia.

Figure 2. Interactions between Aβ, MAP1LC3B, and OPTN in microglia. (A and B) Western blots show that the levels of MAP1LC3B and ATG7 are decreased in the indicated siRNAs-transfected cells. Immunoblots of Aβ in BV2 and primary microglia transfected with the indicated siRNAs and treated with Aβ fibrils (1 μM) for 24 h show high levels of Aβ in siMap1lc3b- and siAtg7-transfected microglia compared with control (siCTL). The bar graphs show the densitometric quantification of the immunoblot bands. Each graph shows the band densities of the immunoreactive proteins as a percentage of CTL (100%). Data are presented as the means ± SEM of 3 independent experiments and were analyzed using the Student t test. *P < 0.05, **P < 0.01, ***P < 0.005 vs. control. (C) The BV2 microglial cell line and primary murine microglia were transfected with the siMap1lc3b and treated with FITC-labeled Aβ fibrils (1 μM) for 24 h. The representative images show that FITC-Aβ was increased in the siMap1lc3b-transfected microglia. (D) After fractionation of BV2 cells into cytosol and membranous organelles, western blots show that Aβ is present not only in the cytosol but also in the membranous organelles. GAPDH is used as a marker for cytosol. EEA1 (early endosomal antigen 1), RAB7 and LAMP1 (lysosomal-associated membrane protein 1) were used as a marker for membranous organelles. (E) BV2 microglia were treated with Aβ fibrils (1 μM) for 2 h. Aβ fibril-treated and control cells were immunoprecipitated with the Aβ antibody (4G8), and, subsequently, coimmunoprecipitation with MAP1LC3B-II was assessed by western blot analysis with MAP1LC3B antibodies. MAP1LC3B-II was confirmed to coimmunoprecipitate with Aβ. (F) The interactions between Aβ and MAP1LC3B were visualized in Aβ fibril-treated cells by staining with the Duolink assay and proximity probes directed against Aβ (6E10) and MAP1LC3B, which was followed by ligation. The hybridization probes were labeled with Alexa 555 (red). Note that red puncta are apparent in fAβ-treated microglia, indicating interactions between Aβ and MAP1LC3B-II in the autophagic vacuoles. The bar graphs show the number of puncta per cells (n = 20). Scale bar: 10 μm. Data are presented as the means ± SEM of 3 independent experiments and were analyzed using the Student t test. ***P < 0.005 vs. control. (G) Western blots show that the level of OPTN is decreased in the siOptn-transfected cells. Immunoblots of Aβ in BV2 and primary microglia transfected with siOptn and treated with Aβ fibrils (1 μM) for 24 h show high levels of Aβ in siOptn-transfected microglia compared with siCTL. The bar graphs show the densitometric quantification of the immunoblot bands. Each graph shows the band densities of the immunoreactive proteins as a percentage of CTL (100%). Data are presented as the means ± SEM of 3 independent experiments and were analyzed using the Student t test. **P < 0.01 vs. control.
Optineurin is important for autophagic clearance of Aβ
Given the fact that autophagic processes are necessary for the degradation of fAβ in microglia, we wondered whether Aβ could be exposed to cytosol and how Aβ is recognized by the autophagic machinery and how autophagic vacuoles are generated around Aβ. After fractionation of cells into the cytosol and the membraneous organelles, Aβ was detected not only in the fractions of membraneous organelles but also in the fractions of cytosol (Fig. 2D), indicating that Aβ fibrils could be exposed to cytosol. The proteins that are degraded by autophagy form complexes with MAP1LC3B and become the targets of autophagic vacuoles via the actions of molecular linkers such as SQSTM1 (sequestosome 1), OPTN (optineurin), and NBR1 (neighbor of BRCA1 gene 1).18 Hence, we speculated that MAP1LC3B could interact with Aβ via molecular linkers. To identify these interactions, we treated MAP1LC3B- or OPTN-transfected microglia with fAβ for 2 h and performed immunoprecipitation using 4G8 antibodies, which recognizes Aβ, and immunoblotted with MAP1LC3B or the OPTN and 6E10 antibodies, which also recognize Aβ. Interestingly, MAP1LC3B-II and OPTN were coimmunoprecipitated with Aβ (Fig. 2E), suggesting that lipidated MAP1LC3B and OPTN made a complex with Aβ and probably recruited Aβ to the autophagic vacuoles. The interaction of MAP1LC3B with Aβ was further confirmed by the protein–protein interaction assay with Duolink in situ PLA probe analysis.19,20 The Cy3 signal, which appears when Aβ interacts with MAP1LC3B, was increased in fAβ-treated microglia (Fig. 2E), further confirming the interactions between Aβ and MAP1LC3B. Interestingly, the Cy3 signal appeared in both vacuolar and punctated forms (Fig. 2F), indicating that this interaction occurs in and around autophagic vacuoles. Western blot analysis demonstrates that the level of Aβ in microglia still remains at higher concenterations when OPTN was downregulated by siRNA targetging Optn (siOptn) (Fig. 2G), indicating that Aβ does not degrade in siOptn-transfected microglia, further suggesting that Aβ interacts with MAP1LC3B via OPTN.
Involvement of PRKAA1 in the autophagic degradation of Aβ fibrils by microglia
Next, we investigated the signaling pathways that are involved in fAβ-induced autophagy in microglia. The levels of Ser428-phosphorylated STK11, which is required for the phosphorylation of PRKAA1, and Thr172-phosphorylated PRKAA1, which is a known inducer of autophagy,21 were increased in fAβ-treated BV2 and primary microglia (Fig. 3A), suggesting the involvement of STK11-PRKAA1 activation in the induction of autophagy. When PRKAA1 protein level was decreased by siPrkaa1, Aβ does not degrade in siPrkaa1-transfected microglia compared with siCTL. This Aβ accumulation was decreased when PRKAA1 expression was rescued with silent-mutant Prkaa1 (Fig. 3B). Therefore, we investigated how PRKAA1 regulates the autophagic degradation of fAβ in microglia with a PRKAA1 activators (AICAR and metformin) and an PRKAA1 inhibitor (compound C). Both AICAR and metformin increased MAP1LC3B lipidation, indicating these activators induced autophagy (Fig. S5). We found that both PRKAA1 activators enhanced the degradation of intracellular Aβ, while compound C inhibited Aβ degradation (Fig. 3C), suggesting the involvement of the STK11-PRKAA1 pathway in the autophagic degradation of Aβ. In AICAR- and metformin-treated microglia, the FITC-Aβ signal intensity was lower than in microglia that had been treated with FITC-fAβ alone, but these effects were reversed by treatment with compound C (Fig. 3D). This was further confirmed by the findings indicating that the enhancement of Aβ degradation by PRKAA1 activators was abolished when autophagy was disabled by siMap1lc3b and siAtg7 (Fig. 3E and F). These data indicate that the STK11-PRKAA1 pathway mediates the microglial autophagic processes required for extracellular fAβ degradation.

Figure 3. STK11-PRKAA1 activates autophagic degradation of Aβ fibrils by microglia. (A) The BV2 microglial cell line and primary mouse microglia were treated with Aβ fibrils (1 μM) for 2 h. Whole cell lysates were collected and analyzed by immunoblotting with indicated antibodies. The levels of p-STK11 and p-PRKAA1 (Thr172) were increased in fAβ-treated microglia. (B) The BV2 microglial cell line was transfected with the indicated siRNAs and Prkaa1 silent-mutant and treated with Aβ fibrils (1 μM) for 24 h. Immunoblots of 6E10 and ACTB in BV2 that were transfected with the indicated siRNAs and treated with Aβ fibrils (1 μM) for 24 h. High levels of Aβ were observed in siPrkaa1-transfected microglia compared with siCTL. (C) BV2 microglial cells were preincubated with AICAR, metformin, or compound C for 30 min before Aβ fibrils (1 μM) were added. Whole cell lysates were collected 24 h later, and then the lysates were analyzed by immunoblotting for 6E10 and ACTB (loading control). Lower levels of Aβ were observed in AICAR- and metformin-treated microglia, but higher levels of Aβ were observed in compound C-treated microglia. (D) The BV2 microglial cell line and primary mouse microglia were preincubated with AICAR, metformin, or compound C for 30 min before FITC-labeled Aβ fibrils (1 μM) were added. The cells were observed 24 h later. Representative images show that FITC-labeled Aβ was lower in AICAR- and metformin-treated microglia but higher in compound C-treated cells. Scale bar: 10 μm. (E and F) Immunoblots of primary microglia cell lysates demonstrating that the decrease in Aβ by AICAR or metformin was abolished in siMap1lc3b- and siAtg7 transfected microglia. The bar graphs show the densitometric quantification of the immunoblot bands. Each graph shows the band densities of the immunoreactive proteins as a percentage of the indicated group. Data are presented as the means ± SEM of 3 independent experiments and were analyzed using the Student t test. **P < 0.01, **P < 0.005 vs. control.
Increased inflammation following the disruption of microglial autophagy
NLRP3 inflammasomes are involved in the innate immune response to Aβ5 and coupled with PYCARD (PYD and CARD domain containing) and CASP1/CASPASE 1).22 It was recently reported that autophagy regulates the inflammatory response in macrophages and that dysfunction of autophagy aggravates the inflammatory response;23 therefore, we speculated that the impairment of autophagic processes in microglia during Aβ treatment may aggravate inflammatory responses. Hence, primary murine microglia were transfected with siMap1lc3b and siAtg7 and treated with fAβ for 8 h in order to determine if dysfunction of autophagy aggravates fAβ-induced inflammation. The cleavage of CASP1 and the oligomerization of PYCARD, which are necessary for the activation of NLRP3 inflammasomes, were increased in fAβ-treated microglia and aggravated when autophagy was downregulated by siMap1lc3b and siAtg7 (Fig. 4A and B). This suggests that the activation of NLRP3 inflammasomes by fAβ and the aggravation of NLRP3 inflammasome signaling occur after the impairment of autophagy.

Figure 4. Increased inflammation following the disruption of microglial autophagy. (A and B) Primary mouse microglia were transfected with the indicated siRNAs and treated with Aβ fibrils (1 μM). Levels of cleaved CASP1 and PYCARD oligomerization were increased in microglia following the addition of fAβ; these levels were even higher following the knockdown of Map1lc3b and Atg7. The bar graphs show the densitometric quantification of the immunoblot bands. Each graph displays the band densities of the indicated immunoreactive protein as expressed as a percentage of the siCTL controls (100%). Data are presented as the means ± SEM of 3 independent experiments and were analyzed using the Student t test. ***P < 0.005 vs. control. (C–E) Primary mouse microglia were transfected with the indicated siRNAs and preactivated with LPS (1 ng/mL) for 3 h before the Aβ fibrils (1 μM) were added. The levels of IL1B and TNF were measured in the supernatant fractions of the microglia 24 h later using ELISA. (F–H) Primary mouse microglia were transfected with the indicated siRNAs and preactivated with LPS were treated with AICAR or metformin 30 min before the Aβ fibrils (1 μM) were added. The IL1B level was measured in the supernatants of the microglia 24 h later using ELISA. The decrease in IL1B by AICAR or metformin was not observed following the knockdown of Map1lc3b or Atg7. Data are presented as the means ± SEM of 3 independent experiments and were analyzed using the Student t test. **P < 0.01, ***P < 0.005 vs. control.
CASP1 cleaves proinflammatory cytokines, such as IL1B, to their active forms that are then released from cells.24 The release of IL1B into the media, as measured by ELISA, was significantly decreased by siNlrp3 and siPycard (Fig. 4C) and increased in fAβ-treated microglia when autophagy was impaired by siMap1lc3b and siAtg7 (Fig. 4D); however, TNF/TNFα (tumor necrosis factor) was not significantly affected by siMap1lc3b (Fig. 4E), suggesting that autophagy plays a role in the regulation of fAβ-induced NLRP3 inflammasome activation. In contrast, treatment of autophagy activators (AICAR and metformin) decreased IL1B release (Fig. 4F); however, the effects of these activators on IL1B release were abolished when autophagy was impaired by siMap1lc3b and siAtg7 (Fig. 4G and H). These findings further confirm the role of autophagy in the regulation of fAβ-induced NLRP3 inflammasome activation.
Aggravation of neuronal damage following the disruption of microglial autophagy
Because inflammatory mediators could induce neuronal damage,5 we speculated that fAβ-induced inflammation in microglia may be harmful to neurons, especially when microglial autophagy is hampered. Hence, primary murine microglia were transfected with siMap1lc3b and treated with fAβ for 24 h, and the conditioned media was then transferred to primary cortical neurons. Immunocytochemical analysis with MAP2, a dendrite marker, demonstrated that the dendrites of the neurons that had been transferred with fAβ-treated microglia media were retracted (Fig. 5A), suggesting that these neurons underwent degeneration induced by the inflammatory mediators that are released by fAβ-treated microglia, as previously reported.5 Dendrite length was further decreased in the neurons that had been treated with culture media of fAβ-treated microglia when its autophagy was impaired by siMap1lc3b (Fig. 5B), suggesting that the impairment of autophagy may aggravate fAβ-induced microglial inflammation and neuronal damage. Interestingly, the conditioned media obtained from metformin- and AICAR-treated microglial culture reduced dendritic damage (Fig. 5B), suggesting that autophagic enhancement may reduce fAβ-induced microglial inflammation and neuronal damage. The effects of metformin and AICAR were abolished by treatment with siMap1lc3b (Fig. 5), further confirming that these effects were mediated by microglial autophagy. These data of neuronal health regulated by microglial autophagy were further confirmed by TUBB3 (tubulin, β 3 class III) staining and phase-contrast images (Fig. S7A and S7B).

Figure 5. Aggravation of neuronal damage after the disruption of microglial autophagy. (A) Primary mouse microglia were transfected with the indicated siRNAs, preactivated with LPS, and treated with Aβ fibrils (1 μM) for 24 h. The conditioned media was transferred to the cortical neurons, and these neurons were stained with antibody specific to MAP2 (microtubule-associated protein 2). The representative images show that these neurons degenerated when fAβ-treated microglia media was used to treat these neurons; these effects were more pronounced following the knockdown of MAP1LC3B. (B) Bar graphs showing the dendrite lengths of the treated neurons (n = 100). Scale bar: 20 μm. Data are presented as the mean ± SEM of 3 independent experiments and were analyzed using the Student t test. **P < 0.01 vs. control.
Increased inflammation in the neural tissues of fAβ-injected Atg7flox/flox /Lyz2-Cre mice
The role of microglial autophagy in Aβ degradation and inflammation was further examined in vivo using Atg7flox/flox/Lyz2-Cre mice, in which Atg7 had been depleted specifically in microglia, and their wild-type counterparts, Atg7flox/flox mice. We stereotaxically injected 1 μL of 100 μM fAβ into the hippocampi of Atg7flox/flox/Lyz2-Cre mice and Atg7flox/flox mice. Mice were sacrificed 3 d after fAβ injection, and the level of IL1B in the hippocampus was examined using ELISA. IL1B levels were higher in fAβ-injected Atg7flox/flox mice than PBS-injected mice, and were even higher in fAβ-injected Atg7flox/flox/Lyz2-Cre mice (Fig. 6A). Cleaved CASP1 was concomitantly increased in fAβ-injected Atg7flox/flox/Lyz2-Cre mice (Fig. 6B), further confirming the role of microglial autophagy plays in the in vivo regulation of fAβ-induced NLRP3 inflammasome activation, similar to that observed in the microglial cultures (Fig. 4). Microglia were recruited around the injection sites and demonstrated morphological changes, including swollen and shortened processes in Aβ-injected Atg7flox/flox and Atg7flox/flox/Lyz2-Cre mice (Fig. 6C; Fig. S8). Immunohistochemical analysis of the brain tissues confirmed the in vitro data (Fig. 1). Some microglia demonstrated Aβ in both the hippocampus and cortex of Aβ-injected Atg7flox/flox mice, and this finding was even more apparent in fAβ-injected Atg7flox/flox/Lyz2-Cre mice (Fig. 6D–F; Fig. S8). These findings suggest that the intracellular clearance of Aβ from microglia is impaired in Atg7flox/flox/Lyz2-Cre mice. Dendrites were damaged more in fAβ-injected Atg7flox/flox/Lyz2-Cre mice (Fig. 6G), as was the case with neuron cultures (Fig. 5).

Figure 6. Increased inflammation in the hippocampi of Aβ-injected Atg7flox/flox /Lyz2-Cre mice. (A) Brain extracts of stereotaxically injected Atg7flox/flox/Lyz2-Cre mice and Atg7flox/flox mice were diluted in PBS (1:40) and 1% BSA for examination of IL1B using ELISA. The bar graphs show the concentrations of IL1B. IL1B was increased by the greatest amount in fAβ-injected Atg7flox/flox/Lyz2-Cre mice. The data are presented as the mean ± SEM of 17 samples and were analyzed using the Student t test. ***P < 0.005 vs. fAβ-injected Atg7flox/flox mice. (B) Extracts were analyzed by immunoblotting for CASP1. The bar graphs show the densitometric quantification of the immunoblot bands. Each graph shows the band densities of the immunoreactive proteins as expressed as a percentage of the PBS-injected Atg7flox/flox mice (100%). The level of cleaved CASP1 was increased in fAβ-injected Atg7flox/flox/Lyz2-Cre mice. Data are shown as the mean ± SEM of 13 samples and were analyzed using the Student t test. ***P < 0.005. (C–E) Brain sections of stereotaxically injected Atg7flox/flox/Lyz2-Cre mice and Atg7flox/flox mice were double-immunostained with AIF1 (red) and 6E10 (green). Representative images showing that microglia in the hippocampi of fAβ-injected Atg7flox/flox/Lyz2-Cre and Atg7flox/flox mice are associated with Aβ. (F) Bar graphs showing the number of Aβ-positive microglia. A higher number of microglia, including those with Aβ, were observed in fAβ-injected Atg7flox/flox/Lyz2-Cre mice than fAβ-injected Atg7flox/flox mice. Data are shown as the mean ± SEM of 17 samples and were analyzed using the Student t test. **P < 0.01, ***P < 0.005 vs. fAβ-injected Atg7flox/flox mice. (G) Brain sections of stereotaxically injected Atg7flox/flox/Lyz2-Cre mice and Atg7flox/flox mice were immunostained with a MAP2 (microtubule-associated protein 2) antibody (red) to examine the dendritic damages. Shortening and fragmentation of dendrites are the most apparent in fAβ-injected Atg7flox/flox/Lyz2-Cre mice.
Increased SQSTM1 and OPTN in AD brains
SQSTM1 or OPTN, the adaptor proteins to link MAP1LC3B and proteins to be degraded, have been used to monitor autophagy flow.11 Hence, to monitor them in AD brains, we examined the level of adaptor proteins in 8 temporal cortexes of AD patients and 7 temporal cortexes of age-matched controls (Table 1). The levels of SQSTM1 and OPTN were increased in AD brains compared with controls (Fig. 7), suggesting that autophagy flow was inhibited in AD.
Table 1. Human medial temporal gyrus samples used in this study.
| Diagnosisa | Sex | Age | Braakb | Amyloidc | PMDd | pH | Weight |
|---|---|---|---|---|---|---|---|
| Alzheimer disease | M | 85 | 5 | C | 07:10 | 6.13 | 1020 |
| Alzheimer disease | M | 65 | 6 | C | 08:50 | 6.88 | 1057 |
| Alzheimer disease | M | 65 | 5 | C | 05:50 | 6.36 | 1355 |
| Alzheimer disease | M | 65 | 5 | C | 07:20 | 6.47 | 1173 |
| Alzheimer disease | M | 87 | 5 | C | 06:10 | 6.14 | 1047 |
| Alzheimer disease | M | 67 | 5 | C | 04:10 | 6.40 | 1252 |
| Alzheimer disease | M | 70 | 6 | C | 04:50 | 6.95 | 1040 |
| Alzheimer disease | M | 82 | 5 | C | 05:15 | 6.34 | 1182 |
| Non-demented control | M | 73 | 0 | 0 | 24:45 | ? | 1267 |
| Non-demented control | M | 71 | 1 | 0 | 07:40 | 6.20 | 1150 |
| Non-demented control | M | 87 | 1 | A | 10:20 | 6.32 | 1256 |
| Non-demented control | M | 80 | 0 | 0 | 07:15 | 5.80 | 1331 |
| Non-demented control | M | 84 | 1 | A | 05:35 | 6.98 | 1337 |
| Non-demented control | M | 82 | 1 | 0 | 05:10 | 6.75 | 1087 |
| Non-demented control | M | 78 | 1 | 0 | < 17:40 | 6.52 | 1125 |
a Medial temporal gyri from 8 AD patients and 7 age- and sex-matched controls were provided by the Netherlands Brain Bank. bBraak stage based on neurofibrillary tangles was 5 or 6 in AD cases, and 0 or 1 in controls.25cBraak stage based on amyloid plaques was C in AD cases, and 0 or A in controls.25dTissue preparation time from death is displayed as postmortem delay (PMD).
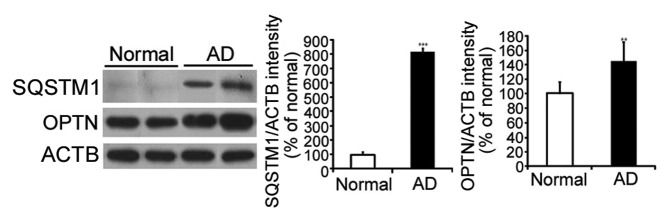
Figure 7. Increase of SQSTM1 and OPTN in AD brain. Representative images of immunoblots from 8 samples of AD temporal cortex and 7 age- and sex-matched control samples. SQSTM1 (sequestosome 1) and OPTN (optineurin) increased in AD brains compared with controls. **P < 0.01, ***P < 0.005 vs. control.
Discussion
In this study, we first demonstrated that autophagic processes are important for the degradation of extracellular Aβ fibrils by microglia. MAP1LC3B-II and optineurin bind to Aβ and is supposed to recruit Aβ into the autophagic vacuoles for degradation (Fig. 8). Enhancement of autophagy via PRKAA1 activation by metformin or AICAR increases the degradation of Aβ fibrils. Aβ-induced NLRP3 inflammasome activation and IL1B release were aggravated by impaired autophagy, which further induced neuronal damage. Our results suggest the important role that microglial autophagy plays in the clearance of extracellular Aβ fibrils and the regulation of Aβ-induced inflammation, thereby affecting neuronal viability.

Figure 8. Role of microglial autophagy in Aβ degradation and NLRP3 inflammasome regulation. (A) Autophagy in microglia is involved in the degradation of extracellular Aβ fibrils and the regulation of Aβ-induced NLRP3 inflammasomes. (B) The impairment of autophagy in microglia results in increased inflammation and subsequent neuronal damage. The enhancement of autophagy by a PRKAA1 activator, such as metformin, may reduce Aβ burden and inflammation.
Autophagy is a catabolic process that degrades a cell’s own components, such as organelles or proteins, by capturing debris in double-membrane autophagosomes and fusing them with lysosomes. Notably, autophagic processes have recently been found to be involved in the degradation of not only a cell’s own components, but also extracellular materials such as pathogens and dead cells.26 In this study, we found that extracellular Aβ fibrils are degraded by autophagic processes in microglia (Fig. 2). Extracellular Aβ deposits are an irrefutable hallmark of AD.27 Thus, it has been suggested that the removal of Aβ plaques is beneficial for the prevention of AD, and various attempts to reduce the burden of Aβ plaques have been attempted.3,28,29 This study adds new mechanistic insights into how the degradation of extracellular Aβ fibrils occurs via microglial autophagy.
Our biochemical fractionation results showed that Aβ can be exposed to cytosol in microglia after phagocytosis (Fig. 2D). During the fractionation procedure, Aβ might leak out of the endolysosomal system into the cytosol. However, this concern can be alleviated by the evidence obtained in intact cells that Aβ interacts with cytosolic factors (Fig. 2E and F). Amyloid fibrils can interact directly with membrane and cause membrane leakage and elicit cellular damage.30-33 Hence, we suspect that these phenomena could occur in the microglia after phagocytosis, which may explain the mechanism for the exposure of Aβ fibrils to the cytosol and the need for the autophagy machinery for its removal.
The substrates of autophagic degradation are recruited to autophagic vacuoles via the interactions between the substrates, i.e., MAP1LC3B-II (a phagophore and autophagosomal membrane protein) and some adaptor molecules.18 Various adaptor molecules bind to both protein aggregates and MAP1LC3B via LC3-interacting motifs (LIR),34 leading to the degradation of protein aggregates via autophagic processes. Mitochondria and some bacteria have also been reported as the targets of autophagosomes for degradation.35,36 Adaptor protein SQSTM1 recognizes the polyubiquitinated complexes associated with bacteria or mitochondria and interacts with MAP1LC3B-II.35,37 Other adaptor proteins, such as CALCOCO2/NDP52, yeast Atg32, BNIP3L/Nix, and OPTN, also form complexes with substrates and MAP1LC3B-II in order to target autophagic vacuoles for degradation.18 Here, we found that Aβ in microglia forms complexes with MAP1LC3B-II and OPTN by immunoprecipitation (Fig. 2E), suggesting that Aβ is also targeted by autophagic vacuoles via interactions with OPTN and MAP1LC3B-II. Evaluation using the Duolink assay also confirmed the interactions between Aβ and MAP1LC3B-II, which further indicates that Aβ interacts with MAP1LC3B-II in autophagic vacuoles because it appeared in punctated forms.
Autophagy is activated by the STK11-PRKAA1 pathway.38 STK11 and PRKAA1 were activated in fAβ-treated microglia (Fig. 3A), which was also correlated with autophagosome formation. PRKAA1 activation by AICAR or metformin increased Aβ degradation, and PRKAA1 inhibition with compound C reduced Aβ degradation (Fig. 3C). This indicates that the autophagic degradation of Aβ fibrils by microglia is dependent on the PRKAA1 pathway. PRKAA1 activity has been reported to decrease with age,39 and PRKAA1 deficiency increases Aβ generation.40 Some molecules have been reported to decrease Aβ levels via the PRKAA1 pathway. Quercetin (HPD), a flavonoid found in green and black teas, decreases Aβ accumulation in murine brain tissue via the activation of PRKAA1.41 LEP (leptin), a metabolism-regulating hormone, reduces Aβ production in neuronal cultures in an PRKAA1-dependent manner.42 Our results also suggest that PRKAA1 activation in microglia could degrade Aβ fibrils via autophagic pathways. Recent studies have also reported that metformin induces not only PRKAA1-dependent autophagy but also the phagocytosis of Aβ fibrils by acidifying the lysosomal and endosomal compartments of microglia.7
Activated microglia in senile plaques demonstrate high expression levels of IL1B,43 and higher concentrations of IL1B are typically found in the cerebrospinal fluid of AD patients.44 Here, we show that Aβ fibrils activate the NLRP3 inflammasomes that are involved in the innate immune response, thereby increasing IL1B release as previously reported.5 Autophagy is important for the regulation of the innate immune responses of NLRP3 inflammasomes.23 We also show in our current study that Aβ-induced NLRP3 inflammasome activation and IL1B release are aggravated by impaired autophagy (Fig. 4). This suggests that autophagy is not only important for the degradation of Aβ, but also for the regulation of Aβ-induced NLRP3 inflammasome activation. Moreover, Aβ-induced inflammatory mediators from microglia damaged neurons, and this inflammatory response is controlled by microglial autophagy (Fig. 5; Fig. S7). AICAR and metformin may demonstrate neuroprotective effects by enhancing Aβ degradation and regulating NLRP3 inflammasomes via the autophagic processes of microglia. Recently, it has been reported that NLRP3 is activated in the brains of AD patients and the microglia of APP-PS1 mice and thereby is contributing to the pathology of AD.12 Microglia isolated from the brains of AD patients shows reduced level of BECN1 (Beclin 1, autophagy related).45 Thus, we strongly suspect that microglial autophagy is impaired in the brains of AD patients (Fig. 7), which may contribute to NLRP3 inflammasome activation in AD brains.
To the best of our knowledge, our results demonstrate for the first time the novel role that microglial autophagy plays in Aβ degradation and its effects on NLRP3 inflammasome-associated AD. The key role played by microglial autophagy in Aβ clearance and Aβ-mediated inflammation suggests that a therapeutic strategy that enhances microglial autophagy (e.g., the administration of drugs such as metformin) may effectively interfere with the progression of AD.
Materials and Methods
Cell culture
BV2 cell line
The BV2 murine microglial cell line (a gift from professor Onyou Hwang, University of Ulsan College of Medicine) was cultured in 100-mm dishes (Thermo Fisher Scientific, Inc., 172958) in culture medium (Dulbecco’s modified Eagle’s medium [DMEM]/high glucose [Life Technologies, Inc., 21013024] with 5% fetal bovine serum [FBS; Life Technologies, Inc., 10082147] and 1% PenStrep [Life Technologies, Inc., 15140122]).
Primary murine microglia
Microglia were isolated from mixed primary glial cultures that were obtained from the cerebral cortexes of 3-d-old mice. Briefly, cerebral cortices were dissected in Ca2+- and Mg2+-free HBSS (Life Technologies, Inc., 14185652) and incubated in a 0.125% trypsin solution for 10 min at 37 °C. The resulting cell suspensions were diluted in complete media (DMEM-F12 [Life Technologies, Inc., 11320033] with 10% FBS, 10% horse serum [HS; Life Technologies, Inc., 16050114], 1 mM L-glutamine [Life Technologies, Inc., 25030], and 1% PenStrep [Life Technologies, Inc., 15140122]) and cultured at 37 °C in a humidified 5% CO2 atmosphere for 14 d. Any pure floating microglia were collected from the mixed glial cultures by shaking the flask at 0.4 g for 12 h at 37 °C on orbital shaker (KE011, KOMA biotech inc.). Lipopolysacharide (LPS) was from Sigma (L3024).
Neuron culture
Primary cultures of rat cortical neurons were prepared from brains of embryonic d 16 pups. Briefly, cerebral cortices were dissected in calcium- and magnesium-free Hank’s balanced salt solution and incubated with a 0.125% trypsin solution for 10 min at 37 °C. The resulting cell suspensions were diluted in neurobasal medium supplemented with B27 components (GibcoBRL, 17054044) and plated onto poly-d-lysine- (50 μg/ml, Sigma, P0899) and laminin (1 μg/ml, GibcoBRL, 23017015)-coated plates or coverslips.
Preparation of Aβ fibrils
Amyloid β-protein1–42 (Aβ1–42) or FITC-amyloid β-protein1–42 (FITC-Aβ1–42) was purchased from Bachem (H-1368, M-2585) and dissolved in DMSO to a final concentration of 500 μM and stored at −80 °C until use. In order to maintain fibrillary conditions, conditioning medium (DMEM-F12 with 10% FBS, 10% or DMEM with 5% FBS) was added in order to bring the peptides to a final concentration of 50 μM. This solution was then incubated for 24 h at 37 °C. Fibrillization of Aβ was confirmed using thioflavin S (Sigma, T1892). Thioflavin S was added to the fibrillar Aβ solution to a final concentration of 20 μM. The signal was viewed using Leica DM IRB, followed images were processed using Metamorph and measured at excitation of 405 nm and emission of 535 nm in a spectrofluorometer VICTOR X4 (PerkinElmer, Massachusetts, USA).
Western blot analysis
Western blots were done according to general methods with denaturating and reducing conditions. Blots were incubated for 16 h at 4 °C with the following primary antibodies: anti-Aβ (6E10; 1:1000; Covance, SIG-39340), anti-MAP1LC3B (1:1000; Sigma, L7543), anti-ATG12 (1:1000; Cell Signaling Technology, 4180), anti-Ser428-phosphorylated STK11 (p-STK11 [Ser428], 1:1000; Cell Signaling Technology, 3482), anti-Thr172-phosphorylated PRKAA1 (p-PRKAA1 [Thr172], 1:1000; Cell Signaling Technology, 2531), anti-PRKAA1 (1:1000; Cell Signaling Technology, 2532), anti-CASP1 (p10, 1:1000; Santa Cruz, SC-514), anti-PYCARD (1:1000; Santa Cruz, SC-22514), and ACTB (1:1000; Sigma, A5441). Densitometric quantification of the bands was performed using Image J (Image Processing and Analysis in Java, National Institutes of Health).
Transfection of plasmids and siRNA
The BV2 and primary microglia cultures were transfected with plasmids or with scrambled siRNA (siCTL), siMap1lc3b, siAtg7, siPrkaa1, and siOptn using Lipofectamine 2000 (Invitrogen, 11668-019) as a manufacturer’s guide. For the rescue of PRKAA1 expression under siPrkaa1 transfection, The Prkaa1 wild-type construct was silent-mutated to ignore the siRNA target effect. The siRNA-target sequence, 5′-gaaatgtgtg caaatctaatt-3′, was replaced by 5′-gagatgtgcg caaacctaatt-3′ (mutations are indicated with bold font). Silent mutations were introduced with site-direct mutagenesis using Pfu ultra HF (Agilent, 600380-51) and were confirmed by sequencing.
Subcellular fractionation
Fractionation of cells into cytosol and membranous organelles were done as described previously.46 The BV2 cultures were treated with fAβ 1 µM for 1 h and removed media followed by washing with cold PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4). The cells were collected by centrifugation at 1500 rpm for 5 min and permeabilized in 0.015% digitonin (Calbiochem, 300410) for 5 min on ice (total). Following centrifugation at 800 g for 5 min, supernatant fractions (cytosol) were transferred to new tubes and pellet fractions (membranous organelles) were resuspended with 1% SDS (Bio-Rad Laboratories, Inc., 161-0418) in 100 mM TRIS-HCl pH 8.0. Each fraction was boiled in SDS sample buffer (62.5 mM TRIS-HCl, pH 6.8, 1% SDS, 2.5% glycerol, 0.5% β-mercaptoethanol, and bromophenol blue) and analyzed by western blots.
Protein–protein interaction assay
The interactions between Aβ peptides and autophagosomes were assessed using Duolink II detection reagents orange (Olink Bioscience, DUO92007), as instructed by the manufacturer. Briefly, cells were grown on glass coverslips and fixed for 30 min at room temperature in 4% paraformaldehyde and 0.1 M phosphate buffer, pH 7.4, and the cell membranes were permeabilized by incubation for 30 min in 0.05 M Tris buffer, pH 7.4, containing 0.1% Triton X-100, 2% bovine serum albumin (Millipore, 82-100-6), and 2% normal horse serum (GibcoBRL, 26050-088). Cultures were incubated overnight at 4 °C in primary anti-6E10 (1:100; Covance) and anti-MAP1LC3B antibody solution (1:200; Sigma), then washed and incubated in PLA probe plus and minus solution (Olink Bioscience, 92002-0100, 92004-0100) for 1 h at 37 °C. The cultures were washed and incubated with Ligation-Ligase solution (Olink Bioscience, 82027, 82009) for 30 min at 37 °C, then incubated with amplification-polymerase solution (Olink Bioscience, 82028, 82101) for 100 min at 37 °C. The culture was then mounted using Duolink II and DAPI and viewed using an Zeiss Axio Observer Z1 microscope (Carl Zeiss, Oberkochen, Germany). Images were processed using the axiovision 4.8.2 .
Immunoprecipitation
Whole cell lysates of fAβ-treated or untreated cells were incubated with an 1.5 μg of the anti-4G8 antibody (Covance, SIG-39220) at 4 °C overnight followed by additional incubation with 100 μL of protein G-agarose beads (GE Healthcare Life Sciences, 17-0712-01) for 1 h at 4 °C. The immunoprecipitates were collected by centrifuge (6000 rpm for 1 min at 4 °C) and washed 2 times with lysis buffer (0.1% Triton X-100 in PBS). The samples were boiled in SDS sample buffer and processed by western blots.
IL1B-release enzyme-linked immunosorbent assay
The cultured cell supernatants were diluted (1:40), and the murine brain extracts were assayed for IL1B using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems) according to the manufacturer’s instructions. Briefly, the supernatant fractions and recombinant murine IL1B as standard were mixed with murine IL1B capture antibodies (4 µg/mL) and coated on 96-well ELISA plate. After 2 h of incubation at room temperature, the plates were washed 3× with a wash buffer (0.05% Tween 20 [Bio-Rad Laboratories, Inc., 170-6531] in PBS; pH 7.4) and the murine IL1B biotinylated detection antibody solution (0.4 µg/mL) was added. After 2 h of incubation at room temperature, the plates were washed 3× with wash buffer (0.05% Tween 20 in PBS; pH 7.4) and streptavidin HRP was added to each well. The plates were incubated for 20 min at room temperature, then visualized in the dark using a substrate solution for reaction with HRP and a microtiter plate reader MAX190 (Molecular Devices, California, USA) calibrated to 450 nm.
Immunocytochemistry
Immunocytochemistry was done according to a general method. Primary antibodies were anti-TUBB3 (1;100; Millipore, MAB1637) and MAP2 antibody solution (1:100; Millipore, MAB5622), followed by a cyanine 3 fluorescent dye-conjugated secondary antibody (1:500; Jackson Laboratories, 115-165-006). Cells were stained with DAPI and mounted using DAKO fluorescence mounting medium. Fluorescent images were taken with a Zeiss Axio Observer Z1 microscope and processed using the axiovision version 4.8.2.
Long-lived protein degradation assay
The overall procedure was performed as previously described47 with minor modifications. In brief, BV2 cells were plated in a 12-well dish with a density of 60,000 cells per plate and incubated with complete medium supplied with 0.2 mCi/ml of L-[14C]valine for 18 h. After incubation, the cells were 3 times washed with PBS and subsequently incubated with fAβ several times as indicated (6, 9, and 12 h) in HBSS supplemented with 10 mM valine (Sigma, V0500). After fAβ treatment, the medium was collected and precipitated with trichloroacetic acid (TCA; Sigma, T0699) added to a final 10% concentration. After centrifugation for 10 min at 2000 rpm at 4 °C, 500 μl of the supernatant fraction was collected and its radioactivity was measured by liquid scintillation counting. Cells were washed twice with 10% TCA and dissolved in 0.2 M NaOH (Sigma, S5881). 500 μl of lysate was used for liquid scintillation counting. The rate of long-lived protein degradation was calculated from the ratio of the radioactivity in medium to the radioactivity in cell lysate.
Human tissues
Medial temporal gyri from 8 AD patients and 7 age- and sex-matched controls were provided by the Netherlands Brain Bank (Table 1). Pathological staging of AD was based on the Braak stages.
Animals
Lyz2-Cre transgenic mice were purchased from Jackson Laboratories, and Atg7flox/flox mice were provided by Masaaki Komatsu of the Tokyo Metropolitan Institute of Medical Science (Tokyo, Japan). To generate microglia Atg7-deficient mice, Atg7flox/flox mice were bred with Lyz2-Cre transgenic mice. Animals were maintained at a constant ambient temperature (22 ± 1 °C) with a 12 h:12 h light-dark cycle and free access to water and food. All procedures were approved by the Institutional Animal Care and Use Committee of the Asan Institute for Life Sciences in Seoul, Korea. Each mouse was housed in a different cage.
Stereotaxic injection
Four-mo-old male or female Atg7flox/flox/Lyz2-Cre mice and Atg7flox/flox mice were anesthetized with 0.02 mL lumpun:zoletil (prepared at a 1:3 ratio) and placed in a stereotaxic apparatus (HARVARD apparatus, Massachusetts, USA). Mice were stereotaxically injected with 1 μL of 100 μM fAβ that was delivered into the hippocampus dentate gyrus (0.2 μL/min); the injection was administered 2.3 mm posterior to the bregma, 1.5 mm lateral to the midline, and 2.0 mm below the dura. After the injection was administered, the needle was maintained in place for 5 min before it was slowly extracted.
Immunohistochemistry
The paraffinized hippocampal sections were subsequently deparaffinized and permeabilized by incubation for 30 min in 0.05 M Tris buffer, pH 7.4, containing 0.1% Triton X-100, 2% bovine serum albumin, and 2% normal horse serum. The sections were incubated overnight at 4 °C in primary anti-6E10 (Covance, SIG-39340) and AIF1 (Iba-1) antibody solution (1:100; Wako, 019-19741), then washed and incubated with fluorescein isothiocyanate-conjugated anti-mouse (Jackson Laboratories, 115-095-006) and cyanine 3 fluorescent-conjugated anti-rabbit (Jackson Laboratories, 111-165-006) secondary antiserum (1:500). Cells were stained with DAPI (Life Technologies, Inc., D1306) and mounted using DAKO fluorescence mounting medium (DAKO North America, Inc., S3023). Fluorescence images were taken by using a Zeiss LSM710 confocal microscope (Carl Zeiss, Oberkochen Germany) and processed using the soft confocal Zen.
Quantitative analysis of neurite outgrowth
Low-resolution images (10× objective magnification) of the coverslipped MAP2-immunostained neurons, which were acquired from 15 different fields of view per sample, were collected. The neurite lengths of all the neurons in each image were measured using Metamorph software (Universal Imaging Corporation).
Quantitative analysis of microglia, including Aβ
Three brain sections were collected from different areas of the stereotaxic injection site of each mouse. These sections were double-immunostained with anti-Aβ (6E10; green) and anti-microglia (AIF-1; red) antibodies. Low-resolution images (10× objective magnification) of the stained brain sections were acquired, and both green and red signal-positive cells were counted in each image.
Statistical analysis
When data distribution was assumed to be normal, data are presented as the mean ± SEM and were analyzed using the 2-tailed Student t test for the bar graphs. The Mann-Whitney test was used to analysis of non-normally distributed data. A P value < 0.05 was considered statistically significant. All experiments were successfully replicated 3 times and all replicates were biological. The sample sizes were not determined a priori but are justified by significance testing and experience from previous studies. Randomization was acquired naturally during the experiments and no blinding was done and there are no excluding data.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Professors Ki-Up Lee and Eun-Joo Chang for critically reading the manuscript and Professor Masaaki Komatsu for providing the Atg7fl/fl mouse and Sang-Wook Kang for helping the subcellular fractionation experiments and for helpful discussions and Professor Onyou Hwang for giving BV2 cell line and Professor Heuiran Lee for giving mCherry-GFP-MAP1LC3B construct. This work was supported by grants from the National Research Foundation of Korea (2008-0062286), the Basic Science Research Program through the National Research Foundation of Korea and the Ministry of Education, Science, and Technology (2012R1A1A1039173), and the Asan Institute for Life Sciences, Seoul, Korea (2012-522). We thank the Netherlands Brain Bank (NBB) for supplying human brain materials and also thank the donors and their relatives for the kind gift of human brains for neuropathological studies.
Glossary
Abbreviations:
- Aβ
β-amyloid
- AD
Alzheimer disease
- atg
autophagy-related
- Baf.A1
bafilomycin A1
- BECN1
Beclin 1, autophagy related
- BNIP3L
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like
- CALCOCO2
calcium-binding and coiled-coil domain-containing protein 2
- CASP
caspase
- Cy3
cyanine 3
- CR1
complement component (3b/4b) receptor 1
- ELISA
enzyme-linked immunosorbent assay
- FITC
fluorescein isothiocyanate
- GWAS
genome-wide association studies
- IDE
insulin-degrading enzyme
- IL1B
interleukin 1, beta
- LIR
LC3-interacting motifs
- Lyz2
lysozyme 2
- MAP1LC3B
microtubule-associated proteins 1A/1B light chain 3B
- MAP2
microtubule-associated protein 2
- NBR1
neighbor of BRCA1 gene 1
- NLRP3
NLR family, pyrin domain containing 3
- OPTN
optineurin
- PRKAA1
protein kinase, AMP-activated, alpha 1 catalytic subunit
- PYCARD
PYD and CARD domain containing
- SQSTM1
sequestosome 1
- STK11
serine/threonine kinase 11
- TNF
tumor necrosis factor
- TREM2
triggering receptor expressed on myeloid cells 2
- TUBB3
tubulin, beta 3 class III
References
- 1.Giménez-Xavier P, Francisco R, Platini F, Pérez R, Ambrosio S. LC3-I conversion to LC3-II does not necessarily result in complete autophagy. Int J Mol Med. 2008;22:781–5. [PubMed] [Google Scholar]
- 2.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–95. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hemming ML, Patterson M, Reske-Nielsen C, Lin L, Isacson O, Selkoe DJ. Reducing amyloid plaque burden via ex vivo gene delivery of an Abeta-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Med. 2007;4:e262. doi: 10.1371/journal.pmed.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labuzek K, Liber S, Gabryel B, Adamczyk J, Okopień B. Metformin increases phagocytosis and acidifies lysosomal/endosomal compartments in AMPK-dependent manner in rat primary microglia. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:171–86. doi: 10.1007/s00210-009-0477-x. [DOI] [PubMed] [Google Scholar]
- 8.Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One. 2012;7:e50976. doi: 10.1371/journal.pone.0050976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohn TT. The triggering receptor expressed on myeloid cells 2: “TREM-ming” the inflammatory component associated with Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:860959. doi: 10.1155/2013/860959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malpass K. Alzheimer disease: functional dissection of CD33 locus implicates innate immune response in Alzheimer disease pathology. Nat Rev Neurol. 2013;9:360. doi: 10.1038/nrneurol.2013.119. [DOI] [PubMed] [Google Scholar]
- 11.Bodles AM, Barger SW. Cytokines and the aging brain - what we don’t know might help us. Trends Neurosci. 2004;27:621–6. doi: 10.1016/j.tins.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiala M, Liu PT, Espinosa-Jeffrey A, Rosenthal MJ, Bernard G, Ringman JM, Sayre J, Zhang L, Zaghi J, Dejbakhsh S, et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci U S A. 2007;104:12849–54. doi: 10.1073/pnas.0701267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer’s amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–8. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- 15.Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, Maxfield FR. Degradation of fibrillar forms of Alzheimer’s amyloid beta-peptide by macrophages. Neurobiol Aging. 2008;29:707–15. doi: 10.1016/j.neurobiolaging.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majumdar A, Capetillo-Zarate E, Cruz D, Gouras GK, Maxfield FR. Degradation of Alzheimer’s amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol Biol Cell. 2011;22:1664–76. doi: 10.1091/mbc.E10-09-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 18.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 20.Choi HK, Choi KC, Yoo JY, Song M, Ko SJ, Kim CH, Ahn JH, Chun KH, Yook JI, Yoon HG. Reversible SUMOylation of TBL1-TBLR1 regulates β-catenin-mediated Wnt signaling. Mol Cell. 2011;43:203–16. doi: 10.1016/j.molcel.2011.05.027. [DOI] [PubMed] [Google Scholar]
- 21.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 23.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J Cell Mol Med. 2008;12(6A):2255–62. doi: 10.1111/j.1582-4934.2008.00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 26.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arendt T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:167–79. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- 28.Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–72. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 29.Wang A, Das P, Switzer RC, 3rd, Golde TE, Jankowsky JL. Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-beta immunotherapy. J Neurosci. 2011;31:4124–36. doi: 10.1523/JNEUROSCI.5077-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butterfield SM, Lashuel HA. Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew Chem Int Ed Engl. 2010;49:5628–54. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 31.Stefani M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010;277:4602–13. doi: 10.1111/j.1742-4658.2010.07889.x. [DOI] [PubMed] [Google Scholar]
- 32.Hebda JA, Miranker AD. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from type II diabetes. Annu Rev Biophys. 2009;38:125–52. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- 33.Milanesi L, Sheynis T, Xue WF, Orlova EV, Hellewell AL, Jelinek R, Hewitt EW, Radford SE, Saibil HR. Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc Natl Acad Sci U S A. 2012;109:20455–60. doi: 10.1073/pnas.1206325109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–96. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 36.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–21. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Terpstra EJ. Ubiquitin receptors and protein quality control. J Mol Cell Cardiol. 2013;55:73–84. doi: 10.1016/j.yjmcc.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 39.Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Won JS, Im YB, Kim J, Singh AK, Singh I. Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun. 2010;399:487–91. doi: 10.1016/j.bbrc.2010.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu J, Wu DM, Zheng YL, Hu B, Zhang ZF, Shan Q, Zheng ZH, Liu CM, Wang YJ. Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J Pathol. 2010;222:199–212. doi: 10.1002/path.2754. [DOI] [PubMed] [Google Scholar]
- 42.Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380:98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, 3rd, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–5. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blum-Degen D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- 45.Lucin KM, O’Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79:873–86. doi: 10.1016/j.neuron.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang SW, Yoon SY, Park JY, Kim DH. Unglycosylated clusterin variant accumulates in the endoplasmic reticulum and induces cytotoxicity. Int J Biochem Cell Biol. 2013;45:221–31. doi: 10.1016/j.biocel.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 47.Pattingre S, Petiot A, Codogno P. Analyses of Galpha-interacting protein and activator of G-protein-signaling-3 functions in macroautophagy. Methods Enzymol. 2004;390:17–31. doi: 10.1016/S0076-6879(04)90002-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.