

Abstract
Sessile marine mussels must “dry” underwater surfaces before adhering to them. Synthetic adhesives have yet to overcome this fundamental challenge. Previous studies of bio-inspired adhesion have largely been performed under applied compressive forces but these are poor predictors of an adhesive’s ability to spontaneously penetrate surface hydration layers. In a force-free approach to measuring molecular-level interaction via the surface water diffusivity, different mussel foot proteins were found to have differential abilities to evict hydration layers from the surfaces—a necessary step for adsorption and adhesion. It was anticipated that Dopa would mediate dehydration given its efficacy forbio-inspired wet adhesion. Instead, hydrophobic side-chains are found to be a critical component in bringing about protein-surface intimacy. This is the first direct measurement of interfacial water dynamics during force-free adsorptive interactions at solid surfaces, and offers guidance for engineering wet adhesives and coatings.
Keywords: ESR spectroscopy, dynamic nuclear polarization, hydrophobic effect, mussel foot proteins, wet adhesion
Man-made adhesive performance and water are fundamentally in conflict.[1] The presence of stable hydration layers around both the adhesive polymer and surface results in strong repulsive hydration forces that undermine adhesion. A renewed appreciation of wet adhesion among sessile marine organisms, however, is invigorating efforts to better reconcile the two.[2] Understanding the adaptive mechanisms by which mussels, for example, overcome repulsive hydration forces to adhere to any type of surface underwater could enable the design of a new generation of surface-drying wet adhesives that have the potential to influence applications ranging from enhancing biomedical implants to underwater coatings.[3,4] While recognizing that actual mussel adhesion involves a complex choreography at multiple length scales encompassing protein chemistry, interfacial energy, and plaque mechanics, we believe that a reductionist approach to how adhesive protein scope with surface hydration could be implemental for improving the performance of wet adhesive polymers.
Mussels attach to solid surfaces by a holdfast structure known as the byssus (Figure 1), which consists of a bundle of threads, each equipped with a distal adhesive plaque. The plaques contain at least eight different types of mussel foot proteins (Mfps) that collectively determine the adhesive properties of the plaque. Most Mfps are intrinsically disordered in aqueous solution,[5] and all contain post-translationally modified 3,4-dihydroxyphenylalanine (Dopa), though to different degrees (range 0.1 to 30 mol%).[3] Studies using either the atomic force microscope (AFM) or surface forces apparatus (SFA) have suggested that under compressive contact, Dopa plays a critical role in the adhesion of Mfps to wet surfaces.[6,7]
Figure 1.

(a) A mussel (Mytilus californianus) attached to the substratum by a byssus - essentially a bundle of adhesive-tipped threads shown in. (b) Cartoon of one of the adhesive tips or plaques in a) enlarged to show the approximate location of known Mfps. (c) Hopp and Woods mean hydropathy values of proteins per amino acid using ExPaSy tools. Hopp and Woods hydropathy[8] was selected over others because it is based on experimentally determined partition coefficients of amino acids including Dopa.[9]
AFM or SFA measurements, however, can less easily assess Mfp’s ability to spontaneously “dry” a surface, because the applied forces themselves effectively break through or disrupt the repulsive hydration layer. Conversely, an experimental approach that operates under force-free conditions can assess how well Mfps or synthetic polymers interact with hydrated surfaces based on their intrinsic properties, and offer an experimentally unexplored, yet critical, perspective to wet adhesion. Given the apparent wet adhesive ability of Dopa and the molecular diversity of Dopa-containing mussel adhesive proteins, we pose the following questions: Do the protein constituents released by the mussel foot “dry” the surface before adhering to it? How important is Dopa in different Mfps for overcoming repulsive hydration forces ubiquitously present on wet surfaces?
We address these questions by studying the diffusion dynamics of water hydrating a solid surface that is suspended in bulk water. The perturbation of the surface water diffusion dynamics by macromolecules in solution is a measure of their ability to break through the solid surface’s hydration layer to adsorb to the surface. These measurements are enabled by a spectroscopic approach termed Overhauser dynamic nuclear polarization (ODNP) relaxometry, which quantifies the diffusion dynamics of surface local water.[10,11] Specifically, we investigate the molecular intimacy between Mfps and the surface of spin-labeled polystyrene (SLPS) and silica (SLSiO2) nanobeads suspended in water. We propose that those Mfps that exhibit the greatest initial intimacy with a given solid surface are the initial facilitators sic vanguards of wet adhesion under force-free conditions, paving the way for other Mfp proteins to finalize adhesive bond formation and sealing. Three different Mfp families were analyzed in this study – Mfp-1, Mfp-3, and Mfp-5 (see SI). Mfp-3 is an interfacial protein encompassing two electrophoretically distinguishable polymorphic families, i.e., the slow (S)- and fast (F)- moving isoforms.[12] Based on the Hopp and Woods hydropathy index,[8] Mfp-3S (Dopa 10–15 mol%) is significantly more hydrophobic than Mfp-3F, Mfp-1, and Mfp-5 (Figure 1c).
ODNP exploits a 1H NMR-signal enhancement of water at 0.35 Tesla by driving electron spin resonance (ESR) saturation at its Larmor frequency of ~10 GHz and detecting 1H NMR signal amplification at ~15 MHz. Only 1H in fast moving H2O molecules, relative to ~10 GHz, will induce concerted electron-1H spin flipflops that give rise to 1H NMR signal enhancement, and in this way be exploited to quantify local water diffusivity near nitroxide radical-based spin labels, commonly used for ESR analysis (SI).[10,11] The motion of hydration water is expressed with a translational diffusion correlation time (τsurface-water) and represents the time needed for water to diffuse across a distance b (typically 5–15 Å), depending on the local diffusion coefficient (D) according to τ = b2/D.[10,11,13] Crucially, ODNP, when combined with NMR relaxometry, can separate contributions of freely, diffusively, translating hydration water (kσ, picosecond timescale) from those of bound water (klow, nanosecond timescale).[14] Protein adsorption will decrease the diffusivity of the surface hydration water due to increased molecular collision, reflected in decreased kσ, and in instances of strong adsorption, also in increased klow. We further apply ESR line-shape analysis to obtain the rotational diffusivity of the spin label itself in the given local environment.[15]
In order to capture interfacial interactions by ODNP and ESR, we prepared SLPS beads (d~50 nm) (SI). An acidic solution of pH 3.0 was chosen to prevent Dopa oxidation and better mimic the delivery conditions of the Mfps in mussels.[6,7] ESR line-shape analysis confirms the presence of a single population of slow and anisotropically moving spin labels with an average rotational correlation time of τR = 4.2 ns, contrasted to the isotropically moving free spin labels with τR = 20 ps (Figure 2a and S1). Although, PS is considered hydrophobic, the benzene rings on the PS bead surface are reported to form hydrogen bonds with water molecules thereby increasing wettability. [16]The ODNP-derived τsurface-water on SLPS surfaces was found to be 335 ps (Figure 2b), a >10-fold retardation compared with that of bulk water (τ= 33 ps).[11] This considerably slower diffusion baseline implies that water is constrained within the hydration layer of the PS surface; the magnitude of retardation suggests spin labels may be partially obscured by a soft polymer coat, consistent with the somewhat immobilized spin label motion derived from ESR line-shapes.
Figure 2.
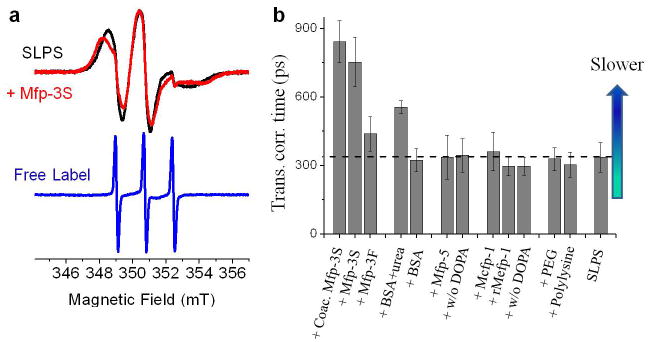
(a) ESR spectra of SLPS before (black) and after addition of Mfp-3S (slow) (red) in MES buffer, pH 3.0 and 20°C. ESR spectrum of free 4-carboxy TEMPO in MES buffer and pH 3.0 (blue). (b) Translational correlation time (τsurface-water) of hydration water at the surface of SLPS before and after addition of (2.5 mg/mL) Mfp-3S, Mfp-3F (fast), 5 M urea induced BSA, Mfp-5, rMfp-5 without Dopa, rMefp-1, rMefp-1 without DOPA, 4.5 kDa PEG, poly-L-lysine in MES buffer, pH 3.0 and coacervated Mfp-3S in MES buffer, pH 5.5. The horizontal dashed line marks the reference τ value of the bare SLPS surface. Higher τ values reflect slower hydration water motion. Error bar represents the standard deviation.
Next, the surface hydration dynamics were tracked following the addition of various Mfps (2.5 mg/mL) (Figure 2b), as well as bovine serum albumin (BSA), 4.5 kDa polyethylene glycol (PEG) and poly-L-lysine. In the presence of native Mcfp-1, recombinant Mefp-1 with Dopa, recombinant Mefp-1 without Dopa, native Mfp-5, recombinant Mfp-5 without Dopa, PEG and poly-L-lysine, the τsurface-water and the kσ or klow values do not change beyond the margin of error, nor do their ESR spectra (Figure 2S), signifying that none are achieving significant intimacy with the SLPS surface. Mfp-1 and Mfp-5, with or without Dopa, are positively charged at pH 3 and display chiefly hydrophilic domains (Figure 1c). Apparently, the strong hydration shell surrounding these proteins, as well as hydrophilic PEG and cationic poly-L-lysine, prevents them from approaching the SLPS surface under force-free conditions, irrespective of whether DOPA functionalities are present or not.
In contrast, addition of hydrophobic Mfp-3S at pH 3 resulted in significant retardation of surface water, with τsurface-water increasing from 335 ps to 752 ps (Figure 2b). This was independently corroborated by observing kσ and klow values, showing the increased contributions from slow, bound, hydration water, and decreased contributions from fast, diffusing, hydration water, implying significant protein adsorption to the SLPS surface (Figure S3).
Mfp-3S is unique among the Mfps in that it self-coacervates at higher pH.[17] Coacervated Mfp-3S retards the τsurface-water from 335 ps to 842 ps at pH 5.5, which resembles Mfp-3S in solution (Figure 2a and S1). We propose that Mfp-3S, in essence, breaks down the repulsive hydration layer of the SLPS surfaces by exposing a hydrophobic face (containing tryptophan and tyrosine) that is weakly hydrated, and thus interacts more aggressively with hydrated polymer surfaces before adsorbing to them (Figure 3). This localized interaction seems relatively unaffected by whether the Mfp-3S is delivered as a dilute solution or as a concentrated fluidic coacervate, suggesting strong intrinsic Mfp-3S attraction to surfaces. In contrast, highly polar Mfp-3F weakly, but measurably, retards the τsurface- water from 335 ps to 438 ps even though Mfp-3F should be stably hydrated like Mfps-1 and -5. This anomalous behavior may be due to a higher content of flexible amino acid residues in Mfp-3Fmaking it slightly more interaction-prone[18] or to homologous hydrophobic tryptophan-containing sequences, e.g. GWN/G and PWP, found in both Mfp-3F (3 per protein) and Mfp-3S (4 per protein). Of particular interest is a comparison of results obtained with Mfps and BSA. BSA is frequently used as a “standard protein” and a blocking agent to prevent nonspecific hydrophobic binding of antigens and antibodies to nontarget surfaces. In the presence of native BSA (2.5 mg/mL), the SLPS surface water diffusivity remains unaltered. However, partially unfolded BSA (exposed to 5 mM urea) moderately slows the water dynamics on the SLPS surface from 335 ps to 555 ps (Figure 2b), which implies that initially buried hydrophobic segments may be recruited to facilitate interaction with the SLPS surface. Still, Mfp-3S interacts much more intimately with SLPS surfaces than unfolded BSA, with its effect comparable with that of 12-fold concentrated BSA (Figures S4–5).
Figure 3.

Mfps adhesion to surfaces requires breaking through the hydration layers. Under force-free solution conditions, the strong hydration layers (azure) surrounding Mfp-3F and the PS surface pose a double hydration barrier to the adhesive interactions between these structures. In contrast, the weak hydration layers (light blue) of Mfp-3S facilitate the intimate approach between Mfp-3S and the PS surface, by readily evicting the initially surface-bound hydration water molecules, and subsequently mediating hydrophobic interactions as depicted in the zoom-in circle.
We also carried out parallel measurements on silica nanobeads suspended in buffered water solution (see SI). The ODNP-derived τsurface-water value is 357 ps, confirming that the spin label intimately experiences the retarded surface hydration water, whereas the spin label τR = 2.5 ns shows surface immobilization. Notably, ODNP and ESR-derived surface dynamics did not change upon addition of any of the tested adhesive proteins or polymers e.g. Mfps, BSA, P188, beyond the margin of error, signifying that none achieved intimacy with the silica surface (Figures S6–7). Quartz Crystal Microbalance (QCM-D) measurements confirm that there is no measurable spontaneous adsorption of Mfp-3S (20 μg/ml) from solution onto silica surfaces, independently verifying the ODNP results (Figure S8). Other hydrophilic surfaces, such as surfaces of unilamellar liposomes freely suspended in solution, were also tested for spontaneous surface adsorption. Mfp-3S did not adsorb to any of the hydrophilic membrane surfaces tested (Figure S9).
Silica surfaces are surrounded by much stronger hydration barriers, to the extent that Mfps studied by SFA and AFM require an applied pressure to adhere to silica.[18,19] Notably, silica surfaces are employed in size exclusion chromatography for protein separation, as permanent interactions between proteins and silica surfaces are weak, further supporting the notion that spontaneous adsorption of proteins to silica surfaces is hindered by the strongly repulsive silica hydration layer. That mussels have no problem sticking to silica based glass[20] surfaces appears to contradict this prediction. However, sessile mollusks have multiple options to counteract strong surface hydration: these include high foot pressure derived from suction[21], the opportunity to piggyback on microbial biofilms already adsorbed to surfaces, and perhaps even the existence of additional Mfp-3 variants specifically adapted silica surfaces. The emerging theme is that wet bio-adhesion may require imposed “clamping” forces except when surface water is effectively evicted by a molecular agent.
The findings presented here are consistent with the dominant biological role of hydrophobic interactions, which are considered the strongest non-covalent interactions between a polar/weakly polar molecules in physiological saline media. For example, recent SFA measurements show that all three hydrophilic Mfps (Mfp-1, Mfp-3F and Mfp-5) adhere 10 times more strongly to hydrophobic surfaces than to hydrophilic surfaces.[22] We suggest that this is because, in general, “dry” contact surfaces are prerequisites for achieving strong adhesion underwater and are facilitated at hydrophobic interfaces whose hydration layers are less strongly attracted to the surfaces, hence cost less energy to remove once forces bring the adsorbates into contact. Our studies support the interpretation that the “drying” ability of Mfps relies on constituent hydrophobic side chains, not their DOPA functionalities, and is dramatically dampened when approaching a highly repulsive hydration layer as found on silica surfaces compared to modestly hydrophilic PS surfaces.
Interestingly, Mfp-3S is abundant in plaque footprints harvested from Plexiglas™ surfaces according to in situ analysis by MALDI-TOF mass spectrometry.[12] We propose that mussels squirt out Mfp-3S onto polymer surfaces as a “molecular vanguard” in anticipation of adhesive action in order to prepare a surface by breaking down the hydration layer barriers (Figure 3). Specific Mfps adapted for highly hydrophilic surfaces, such as silica, have yet to be identified. Once the molecular vanguards have achieved intimacy with a wet surface, stronger and more lasting adhesive interactions can be introduced by recruiting the Dopa-rich Mfp-3F and Mfp-5. Our results suggest that hydrophobic molecular vanguards (whose solubility is maintained by strategically placed hydrophilic residues) weaken surface hydration forces, enhance the adhesive-surface encounter, more reliably and readily achieve a dry adhesion interface, and pave the way for stronger and lasting adhesion underwater.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (NSF) through the MRSEC Program DMR-1121053 (MRL-UCSB) for all authors. This work utilized the MRL Central Facilities supported by the MRSEC Program of the NSF under DMR-1121053; a member of the NSF-funded Materials Research Facilities Network (www.mrfn.org). S.H. and Y.A. acknowledge support by the 2012 NIH Innovator Award, and J.H.W. acknowledges support from the U.S. National Institutes of Health (R01 DE018468) and the Human Frontiers of Science Program
Contributor Information
Dr. Yasar Akdogan, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA, 93106, USA Fax: (+) 1 805 8934120İzmir Institute of Technology, Department of Materials Science and Engineering, İzmir, 35430 Turkey
Dr. Wei Wei, Department of Molecular, Cell & Development Biology, University of California Santa Barbara, Santa Barbara, CA, 93106 USA
Dr. Kuo-Ying Huang, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA, 93106, USA Fax: (+) 1 805 8934120
Dr. Yoshiyuki Kageyama, Department of Chemistry, Faculty of Science, Hokkaido University Sappora 060-0810 Japan
Eric W. Danner, Department of Molecular, Cell & Development Biology, University of California Santa Barbara, Santa Barbara, CA, 93106 USA
Dusty R. Miller, Department of Molecular, Cell & Development Biology, University of California Santa Barbara, Santa Barbara, CA, 93106 USA
Nadine R. Martinez Rodriguez, Department of Molecular, Cell & Development Biology, University of California Santa Barbara, Santa Barbara, CA, 93106 USA
Prof. Dr. J. Herbert Waite, Department of Molecular, Cell & Development Biology, University of California Santa Barbara, Santa Barbara, CA, 93106 USA
Prof. Dr. Songi Han, Email: songi@chem.ucsb.edu, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA, 93106, USA Fax: (+) 1 805 8934120
References
- 1.Adams ME. Int J Adhesion Adhesives. 3:68–69. [Google Scholar]
- 2.Stewart RJ, Ransom TC, Hlady V. J Polymer Sci B. 49:757–771. doi: 10.1002/polb.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Annu Rev Mater Res. 41:99–132. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sedo J, Saiz-Poseu J, Busque F, Ruiz-Molina D. Adv Mater. 25:653–701. doi: 10.1002/adma.201202343. [DOI] [PubMed] [Google Scholar]
- 5.Hwang DS, Waite JH. Prot Sci. 21:1689–1695. doi: 10.1002/pro.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu J, Wei W, Danner E, Ashley RK, Israelachvili JN, Waite JH. Nat Chem Biol. 7:588–590. doi: 10.1038/nchembio.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu J, Wei W, Danner E, Israelachvili JN, Waite JH. Adv Mat. 23:2362–2366. doi: 10.1002/adma.201003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopp TP, Woods KR. Proc Natl Acad Sci USA. 78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nozaki Y, Tanford C. J Biol Chem. 245:1648–1652. [PubMed] [Google Scholar]
- 10.Armstrong BD, Han S. J Am Chem Soc. 131:4641–4647. doi: 10.1021/ja809259q. [DOI] [PubMed] [Google Scholar]
- 11.Franck JM, Pavlova A, Scott JA, Han S. Prog Nucl Magn Reson. 74:33–56. doi: 10.1016/j.pnmrs.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao H, Robertson NB, Jewhurst SA, Waite JH. J Biol Chem. 11090:2006. doi: 10.1074/jbc.M510792200. [DOI] [PubMed] [Google Scholar]
- 13.Hwang LP, Freed JH. J Chem Phys. 63:4017–4025. [Google Scholar]
- 14.Hussain S, Franck JM, Han S. Angew Chem Int Ed. 52:1953–1958. doi: 10.1002/anie.201206147. [DOI] [PubMed] [Google Scholar]
- 15.Januszyk K, Fleissner MR, Atchabahian L, Shieh FK, Altenbach C, Martin SL, Guo F, Hubbell WL, Clubb RT. Prot Sci. 20:1231–1243. doi: 10.1002/pro.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tretinnikov ON. Langmuir. 16:2751–2755. [Google Scholar]
- 17.Wei W, Tan Y, Rodriguez NM, Yu J, Israelachvili JN, Waite JH. Acta Biomaterialia. 10:1663–1670. doi: 10.1016/j.actbio.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Q, Danner E, Waite JH, Israelachvili JN, Zeng H, Hwang DS. J R Soc Interface. 10:20120759. doi: 10.1098/rsif.2012.0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank BP, Belfort G. Biotechnol Prog. 18:580–586. doi: 10.1021/bp010140s. [DOI] [PubMed] [Google Scholar]
- 20.Crisp DJ, Walker G, Young GA, Yule AB. J Coll Interf Sci. 104:40–50. [Google Scholar]
- 21.Smith A. J Exp Biol. 161:151–169. [Google Scholar]
- 22.Yu J, Kan Y, Rapp M, Danner E, Wei W, Das S, Miller DR, Chen Y, Waite JH, Israelachvili JN. Proc Natl Acad Sci USA. 110:15680–15685. doi: 10.1073/pnas.1315015110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.