

Abstract
Non‐small cell lung cancer (NSCLC) is the leading cause of cancer death. Systematically characterizing miRNAs in NSCLC will help develop biomarkers for its diagnosis and subclassification, and identify therapeutic targets for the treatment. We used next‐generation deep sequencing to comprehensively characterize miRNA profiles in eight lung tumor tissues consisting of two major types of NSCLC, squamous cell carcinoma (SCC) and adenocarcinoma (AC). We used quantitative PCR (qPCR) to verify the findings in 40 pairs of stage I NSCLC tissues and the paired normal tissues, and 60 NSCLC tissues of different types and stages. We also investigated the function of identified miRNAs in lung tumorigenesis. Deep sequencing identified 896 known miRNAs and 14 novel miRNAs, of which, 24 miRNAs displayed dysregulation with fold change ≥4.5 in either stage I ACs or SCCs or both relative to normal tissues. qPCR validation showed that 14 of 24 miRNAs exhibited consistent changes with deep sequencing data. Seven miRNAs displayed distinctive expressions between SCC and AC, from which, a panel of four miRNAs (miRs‐944, 205‐3p, 135a‐5p, and 577) was identified that cold differentiate SCC from AC with 93.3% sensitivity and 86.7% specificity. Manipulation of miR‐944 expression in NSCLC cells affected cell growth, proliferation, and invasion by targeting a tumor suppressor, SOCS4. Evaluating miR‐944 in 52 formalin‐fixed paraffin‐embedded SCC tissues revealed that miR‐944 expression was associated with lymph node metastasis. This study presents the earliest use of deep sequencing for profiling miRNAs in lung tumor specimens. The identified miRNA signatures may provide biomarkers for early detection, subclassification, and predicting metastasis, and potential therapeutic targets of NSCLC.
Keywords: Lung cancer, microRNA, Biomarkers, Therapy, Deep sequencing
Highlights
Using deep sequencing to characterize miRNAs in lung tumor tissues.
Validating the findings in additional tumor tissues.
Investigating the function of identified miRNAs in vitro and in vivo.
The miRNAs provide markers for classification and outcome of lung cancer.
The identified miRNAs provide potential therapeutic targets of lung cancer.
1. Introduction
Non‐small‐cell lung cancer (NSCLC) is the leading cause of cancer death worldwide. The 5‐year survival rate for patients with advanced stages of NSCLC is 14%, whereas patients with stage I NSCLC who received treatments is 83% (Siegel et al., 2013). Thus, finding early stage NSCLC and developing new therapeutic targets for its treatment may reduce the mortality (Siegel et al., 2013). Furthermore, approximately 50% NSCLC will relapse within 5 years after initial surgery, and hence have a poor prognosis (Naruke et al., 1988). Therefore, the ability to identify NSCLC patients with lymph node metastasis or recurrence after surgical resection who would benefit from adjuvant therapies, while sparing patients without metastasis from unnecessary cytotoxic chemotherapy, will also reduce the death rate (Winton et al., 2005). Clinical‐pathological staging system has reached the limit of the usefulness for predicting lymph node metastasis (Grondin and Liptay, 2002). In addition, NSCLC is divided into two major histological subtypes: squamous cell carcinoma (SCC) and adenocarcinoma (AC) (Travis et al., 2011). Differentiating between SCC and AC is increasingly becoming more important, because different treatments have diverse therapeutic or adverse effects depending on the histological type (Mukhopadhyay and Katzenstein, 2011). For example, the epidermal growth factor receptor (EGFR) inhibitors are more effective in AC than in SCC (Hirsch et al., 2008). The antivascular endothelial growth factor agents are contraindicated in SCC, as they often cause pulmonary hemorrhages (Azzoli et al., 2011; Lebanony et al., 2009). Histological examination of biopsy and surgical specimens is a major approach for subtyping of NSCLC. Recently, several methods including immunohistochemistry and gene expression profiling have been investigated to assist subtyping of NSCLC (Lebanony et al., 2009; Wolff et al., 2007). However, numerous factors often yield conflicting results in subtyping of NSCLC (Lebanony et al., 2009; Wolff et al., 2007). Altogether, it is clinically imperative to develop new biomarkers that can improve early detection, subclassification, predicting recurrence of NSCLC, as well as identify novel molecular targets for its treatment.
miRNAs are involved in various biologic and pathologic processes by regulating more than 30% of protein‐coding genes (Ambros, 2003). miRNAs show aberrant expression patterns and functional abnormalities in human cancers, and thus provide potential biomarkers and novel drug targets of malignances (Lu et al., 2005; Tili et al., 2007). For example, quantitative real‐time PCR (qRT‐PCR) analysis of miRNAs including miR‐205 in clinical specimens may present an approach for finding early stage NSCLC and distinguishing SCC from AC (Bishop et al., 2010; Landi et al., 2010; Lebanony et al., 2009; Raponi et al., 2009; Xing et al., 2010; Yanaihara et al., 2006; Yu et al., 2010). We and others have found that miRNAs are reliably measurable by qRT‐PCR in sputum and plasma (Shen et al., 2014, 2011, 2010). We have also shown that combined analysis of two miRNAs (miR‐31 and miR‐210) in sputum could yield 65.2% sensitivity and 89.7% specificity for lung cancer diagnosis (Shen et al., 2014).
qRT‐PCR and hybridization‐based microarray platforms have been used to identify lung cancer‐associated miRNA aberrations (Shen and Jiang, 2012). Yet these technologies only measure relative abundant and known miRNA sequences, and have limited capacity in identifying novel miRNAs whose aberrations are associated with lung cancer. Next‐generation deep sequencing has emerged as a powerful tool for global miRNA analysis. Its advantages over the current techniques include pooling of samples for high‐throughput purposes, a wide detectable expression range, analyzing expression of all annotated miRNAs, and detecting novel miRNAs (Schee et al., 2013). Hu et al. (2010) used Solexa sequencing to evaluate miRNA profiling in serum of patients with stages I to IIIa NSCLC. Levels of four serum‐based miRNAs (miR‐486, miR‐30d, miR‐1 and miR‐499) were significantly associated with overall survival. Using SOLiD transcriptome sequencing of miRNAs in peripheral blood of lung cancer patients, Keller et al. (2011) identified 32 annotated and seven unknown miRNAs that were altered in the blood specimens of cancer patients. However, there is no report of using next‐generation deep sequencing for systematically characterizing miRNA transcriptome of primary lung tumor tissues. This is important research, because deep sequencing‐based miRNA profiling directly from clinically defined and histologically confirmed surgical tumor tissues will be disease‐specific. Characterization of microRNA transcriptome in lung tumor tissues by next‐generation deep sequencing may help develop strategies to improve NSCLC diagnosis and treatment.
This study aimed to use next‐generation sequencing to globally characterize aberrations of miRNAs in surgical lung tumors with the following three objectives: 1) comprehensively profiling miRNA expression patterns in stage I NSCLC to have miRNA signatures of the disease; 2) verifying the findings in additional NSCLC tissues using qPCR and identifying miRNAs that could distinguish SCC from AC; 3) evaluating whether the miRNA could provide potential therapeutic targets for lung cancer by functionally analyzing the role of a newly identified NSCLC‐associated miRNA.
2. Materials and methods
2.1. Patients and clinical specimens
Surgical specimens were obtained from 108 lung cancer patients who had either a lobectomy or a pneumonectomy. Tumor tissues were intraoperatively dissected from the surrounding lung parenchyma. Paired normal lung tissues were obtained from the same patients at an area distant from their tumors. The samples were immediately snap‐frozen in liquid nitrogen and then stored at −80 °C until use. Simultaneously, the resected specimens were processed for histopathological assessment and classified based on WHO classification of tumors of the lung (Brambilla et al., 2001). Cryostat microtome and hematoxylin‐eosin stained slides were evaluated for tumor content by experienced pathologists (median tumor content in the samples was 60%). The demographic, histopathological, and clinical characteristics of the cancer patients are summarized in Supplementary Table 1. Furthermore, formalin‐fixed paraffin‐embedded (FFPE) lung tumor tissues of 52 SCC patients were also collected. All variants, including age, sex, stage and lymph node metastasis, were obtained from clinical and pathologic records (Supplementary Table 2). None of the patients had received preoperative adjuvant chemotherapy or radiotherapy before they received treatments.
2.2. RNA isolation
We used a mirVana miRNA Isolation Kit (Ambion, Austin, TX) to isolate RNA containing small RNA from the tissue specimens as described in our previous study (Wang et al., 2014; Xing et al., 2010; Yu et al., 2010). We used a dual beam UV spectrophotometer (Eppendorf AG, Hamburg, Germany) to determine the purity and concentration of RNA from OD260/280 readings. We utilized the RNA 6000 Nano Lab‐on‐a‐Chip kit and the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA) to determine RNA integrity. Only RNA extracts with RNA integrity number values ≥ seven underwent further deep sequencing analysis.
2.3. Deep sequencing
We generated small RNA libraries for deep sequencing as described in previous reports (Hamfjord et al., 2012; Jima et al., 2010; Kozubek et al., 2013; Lang et al., 2012; Osanto et al., 2012; Rohr et al., 2013; Schee et al., 2013). Briefly, we ligated 3′ RNA adapter to 1 μg of RNA. We reverse‐transcribed the doubly ligated, purified RNA using 150 U of Superscript II (Invitrogen, Carlsbad, CA) and RT primer, (5′‐ATTGATGGTGCCTAC AG‐3′) to produce cDNA libraries. cDNA was amplified by using PCR with the following primers: (forward) 5′‐GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T‐3′; and (reverse) 5′‐CAA GCA GAA GAC GGC ATA CGA GCT CTT CCG ATC TAT TGA TGG TGC CTA CAG‐3′. We applied an Illumina® Genome Analyzer IIx (Illumina, San Diego, CA) to sequence the RNA libraries. For data analysis, we first applied Illumina's software packages (SCS2.9/RTA1.9 and Off‐line Basecaller v1.9) to filter the reads. The average length of Illumina sequencer reads was 36 nucleotide and greater than the average size of microRNAs (19–25 nucleotide), because the reads might contain part of the 3′‐adaptor at the end of the sequence. We used Novoalign (V2.08.01 Novocraft 2010; www.novocraft.com) to cut all reads at the 3′ end to remove the adapter sequences. After adaptor trimming, reads were aligned to the human genome build 19 (hg19) using Bowtie version 0.12.3 (Langmead et al., 2009). Reads that aligned with zero mismatches and were between 19 and 25 nucleotides were used as input for miRDeep (Friedlander et al., 2008) (Perdomo et al., 2013) to forecast novel microRNA loci. The number of reads that overlapped with each known microRNA from miRBase v16 (Kozomara and Griffiths‐Jones, 2011) or that overlap with each novel microRNA from miRDeep was counted by using BEDTools v2.9.0 (Quinlan and Hall, 2010). To normalize read counts, we first calculated the normalization factor for all samples by dividing the total number of reads. Second, we aligned the number of reads to the genome, which permitted various hits. Third, we mapped the number of reads to annotated mature miRNAs. We generated the normalized expression values for each miRNA by dividing the read count of the miRNA with the according normalization factor. Once normalized, miRNAs with read counts less than 10 across all samples were set to zero. We selected the data set that was normalized against annotated miRNAs for the remaining analysis.
2.4. Quantitative real‐time PCR (qRT‐PCR)
qRT‐PCR was performed to determine expression of miRNAs as described in our previous studies (Shen et al., 2014). Briefly, 100 ng RNA from each sample was reversely transcribed by a T100 thermal cycler (Applied Biosystems, Foster City, CA) with TaqMan miRNA Reverse Transcription Kit and miRNA‐specific stem‐loop primers (Applied BioSystems). Information of the oligo sequences of miRNAs for RT reactions and the associated thermocycler parameters could be found on the website of Life Technologies Corporation (Life Technologies Corporation, Frederick, MD). Each RT reaction included 50 nM stem‐loop RT primer, 1× RT buffer, 0.25 mM each of dNTPs, and 3.33 U/μl MultiScribe reverse transcriptase in a total volume of 15 μL. For PCR analysis, the 20 μl PCR reaction included RT product, 1× TaqMan® Universal PCR Master Mix (Applied Biosystems), the corresponding primers, and Taqman probe for the target genes. qPCR was carried out on an Applied BioSystems 7900HT thermocycler (Applied Biosystems) at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. PCR data was analyzed using the RQ Manager software (Applied Biosystems) with automatic Ct setting for assigning baseline and threshold for Ct determination. All tests were performed in triplicates.
2.5. Pathway analysis
We performed pathway analysis as previously described (Kozubek et al., 2013; Lang et al., 2012; Schee et al., 2013). Briefly, TargetScan, PicTar, and miRBase were used in combination to identify potential gene targets of the miRNAs. The resulting data was then uploaded into DNA intelligent analysis (DIANA) software for pathway identification by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
2.6. Cell culture
Human NSCLC cell lines, including H520 (SCC), H292 (SCC), SK‐MES‐1 (SCC), and CALU‐1(lung epidermoid carcinoma), were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained at 37 °C in a humidified air atmosphere containing 5% carbon dioxide in RPMI1640 (Biofluids, Rockville, MD), F12 (Biofluids) or Dulbecco's Modified Eagle's Media (Biofluids) supplemented with 10% fetal bovine serum (Biofluids).
2.7. Increasing or reducing expression of miR‐944 in NSCLC cells
To force expression of miR‐944 in cancer cells, cells were transfected with precursor molecules mimicking miR‐944 or scrambled sequences that were designed and made by Ambion (Ambion, Austin, TX) by using Lipofectamine 2000 (Invitrogen, Grand Island, NY). To decrease expression of miR‐944, an inhibitor of miR‐944 or negative inhibitor control that were also designed and made by Ambion was transfected into cancer cells using HiPerFect transfection reagent (Qiagen, Valencia, CA). Experiments were repeated at least three times.
2.8. siRNA silencing of SOCS4
Sequences of siRNA specifically against SOCS4 (si‐SOCS4) were designed and made by Santa Cruz Biotechnology (Santa Cruz Biotechnology, Dallas, TX) with a catalog number of sc‐63050. Transfections were performed using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's protocol with si‐SOCS4 or scrambled sequences. At least three independent experiments were carried out.
2.9. Methylthiazol tetrazolium (MTT), BrdU incorporation, Annexin V, and transwell assays
MTT assay was performed as previously described (Wang et al., 2014). Briefly, after transfection, cells were plated in 96‐well plates, and the cells viability was assessed in ten replicates. BrdU incorporation assay was carried out as previously described (Wang et al., 2014). Briefly, cells were plated on coverslips, and BrdU was added for 12 h. Immunocytofluorescence was performed on cells with mouse anti BrdU antibody (DAKO, Carpinteria, CA), and the fluorochrome conjugated secondary antibody against mouse Ig (Invitrogen). DAPI was used to counterstain the nuclei. Immunostained cells were analyzed under fluorescent microscope (Leica, Germany). Annexin V assay was performed as previously described (Wang et al., 2014). Briefly, cells were stained with annexin V‐FITC and PI using the ANNEXIN V‐FITC Kit (Beckman Coulter, Boulevard Brea, CA) for flow cytometric analysis. The apoptotic index was calculated as the percentage of annexin V+/PI‐cells. Transwell assay was performed as briefly described below: after transfection, cells were plated in medium without serum in the top chamber of a transwell (Corning, NY). The bottom chamber contained standard medium with 10% FBS. Cells that had migrated to the lower surface of the membrane were fixed with formalin, stained with crystal violet, and photographed under microscope. Cell numbers were counted under a light microscope. All experiments were carried out at least three times.
2.10. 3′ untranslated region (UTR) luciferase reporter assay
Luciferase reporter assay was performed as described in our previous report (Wang et al., 2014). Briefly, sequences from putative miR‐944 binding sites in the 3′ UTR of SOCS4 were synthesized and ligated into the pGL3‐REPORT vector (Promega Corporation, Madison, WI). The following primers were used to amplify the 3′ UTR of SOCS4: Forward, CTCTCAACCTGCAAGAGCTAAA and Reverse, GCCCTGTTTCTGGGATAGTAAA. The amplified fragment was cloned into pGL3 luciferase report vector at Mlu I and Xho I sites. Cancer cells were co‐transfected with firefly luciferase‐3′‐UTR (pGL3‐ SOCS4, 500 ng) and pRL‐TK vector (Promega) along with a miR‐944 mimic or control (Ambion). Firefly luciferase and Renilla luciferase were measured by using synergy TM HT microplate reader (BioTek, Winooski, VT). Luciferase activities were normalized to Renilla luciferase activity.
2.11. Western blotting
Western blot analysis was performed as previously described (Wang et al., 2014). Briefly, blots were incubated with a primary mouse monoclonal antibody, SOCS4 (Santa Cruz Biotechnology). Antibody for β‐actin (Santa Cruz Biotechnology) was used as a control. The blots were then re‐probed with secondary antibody and visualized by the ECL system (GE Healthcare Life Sciences, Piscataway, NJ).
2.12. Statistical analysis
We used hierarchical clustering to visualize expression patterns of data created from deep sequencing analysis. Using the log2 method, we transformed the normalized expression values and carried out unsupervised two‐way hierarchical clustering by using Euclidean distance and weighted average linkage to concurrently group miRNAs and specimens. The differences of results between groups were analyzed by using Student t test when there were only two groups, or evaluated by one‐way ANOVA when there were more than two groups. We constructed the receiver‐operator characteristic (ROC) curve and the area under ROC curve (AUC) value by numerical integration of the ROC curve. We used the AUC to determine the maximum sensitivity and specificity of each miRNA at which to distinguish different types of NSCLC, yielding corresponding optimal thresholds for defining levels of the miRNAs. Spearman's correlation analysis was used to determine associations between miRNA expression and clinical characteristics of patients. A p value less than 0.05 was considered statistically significant.
3. Results
3.1. Differentially expressed miRNAs in stage I NSCLC specimens versus corresponding normal lung tissues
Four stage I AC, four stage I SCC tissues, and their paired normal tissue specimens were successfully sequenced by using Illumina Genome Analyzer II. The number of raw reads obtained per specimen ranged from 21,703,452 to 43,663,118 (average = 29,485,315). From the raw reads, an average of 26,070,789 reads (ranging from 18,407,811 to 38,455,007) were filtered and mapped to human genome as shown in Figure 1A. 896 mature miRNAs were successfully annotated in miRBase5, and thus considered as known miRNAs. Furthermore, the two pairs of SCC and normal lung tissues and two pairs of AC and normal lung tissues were deep sequenced twice, designated as run 1 and run 2. The generated data sets from the separate runs were compared. As shown in Figure 1B, measured miRNA expressions in the replicates were highly correlated (all p < 0.0001), demonstrating that the deep sequencing approach could produce robust results.
Figure 1.
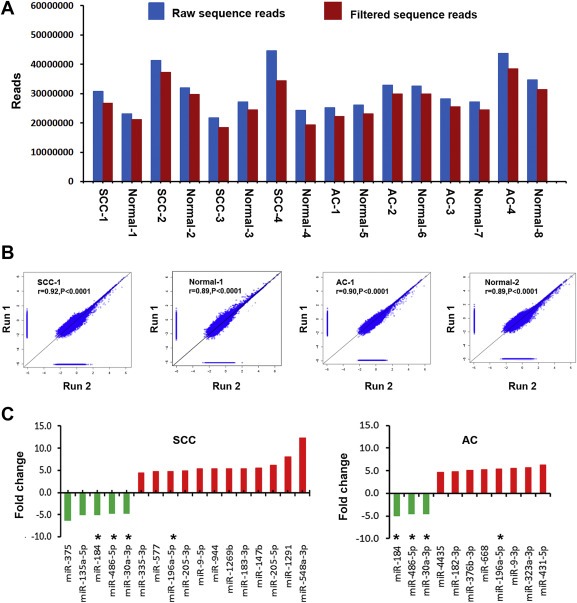
Global deep sequencing analysis of miRNAs in non‐small cell lung cancer (NSCLC) tissue and normal lung tissue specimens. A, an average of 29,485,315 raw sequence reads (blue bars) per specimen were identified by Illumina high‐throughput sequencing. From the raw reads, an average of 26,070,789 filtered sequence reads (red bars) were obtained per specimen. B, expression of miRNAs measured by deep sequencing across duplicates (run 1 and run 2) for squamous cell carcinomas (SCCs) and adenocarcinomas (ACs) and their matched normal lung tissues. A close correlation was found from two different experiments (run 1 and run 2) on the same samples (r values ranged from 0.89 to 0.92, all p < 0.0001). The figure only shows results from one SCC and one AC and their paired normal lung tissues. C, twenty‐four differentially expressed miRNAs with ≥ 4.5 fold changes in NSCLC vs. normal tissues. Up‐regulated miRNAs (red bars) are shown above the x‐axis, whereas down‐regulated miRNAs (green bars) below the x‐axis. Left, 17 miRNAs with dysregulation in stage I SCC tissues. Right, 11 miRNAs had dysregulation in stage I AC tissues. *, four miRNAs (miRs‐196a‐5p, 30a‐3p, 486‐5p, and 184) displayed altered expressions in both SCC and AC. Therefore, there are a total of 24 miRNAs showing dysregulation in the NSCLC specimens. More information about changes of the miRNAs can be found in Table 1
Based on a 2.5‐fold change cut‐off value, 40 and 28 known miRNAs were identified to show statistically different expression levels between the stage I SCC and AC specimens and theirs corresponding normal tissues, respectively (all p ≤ 0.001) (Table 1). With stricter cut‐off criteria (a fold change ≥ 4.5), 17 miRNAs displayed considerable expression difference between the SCC and normal tissues (all p ≤ 0.001): 12 were up‐regulated and five down‐regulated (Figure 1C). Eleven miRNAs exhibited substantial difference between the AC and normal tissues: eight were up‐regulated and three down‐regulated (Figure 1C). Furthermore, four miRNAs (miRs‐196a‐5p, 30a‐3p, 486‐5p, and 184) displayed altered expressions in both SCC and AC (Figure 1C). Altogether, based on the 4.5‐fold change cut‐off, a total of 24 miRNAs (Figure 1C) had dysregulation in either stage I AC or SCC or the both, and thus underwent further analysis in the current study.
Table 1.
Up‐regulated and down‐regulated miRNAs in stage I SCC and AC tissues defined by deep sequencing.
| SCC | AC | ||||
|---|---|---|---|---|---|
| miRNAs | Fold‐change (tumor/normal) | p Value | miRNAs | Fold‐change (tumor/normal) | p Value |
| miR‐548a‐3p | 12.308824 | 0.00012743 | miR‐431‐5p | 6.317018 | 2.91617E‐06 |
| miR‐1291 | 8.060843 | 2.43E‐23 | miR‐323a‐3p | 5.698014 | 2.542E‐05 |
| miR‐205‐5p | 6.173350 | 1.56E‐15 | miR‐9‐3p | 5.518930 | 6.29421E‐05 |
| miR‐147b | 5.515700 | 1.20E‐07 | miR‐196a‐5p | 5.452727 | 9.32211E‐06 |
| miR‐183‐3p | 5.404806 | 4.68E‐06 | miR‐668 | 5.260027 | 0.000104731 |
| miR‐1269b | 5.401679 | 1.08E‐06 | miR‐376b‐3p | 5.118238 | 0.000128737 |
| miR‐944 | 5.342495 | 2.26E‐11 | miR‐182‐3p | 4.804702 | 0.000568362 |
| miR‐9‐5p | 5.304565 | 1.85E‐12 | miR‐4435 | 4.583124 | 0.000231087 |
| miR‐205‐3p | 4.829734 | 0.000325 | miR‐21‐5p | 4.485680 | 3.45658E‐05 |
| miR‐196a‐5p | 4.771010 | 9.80E‐10 | miR‐196a‐3p | 4.482990 | 9.99902E‐06 |
| miR‐577 | 4.687445 | 8.85E‐06 | miR‐1185‐1‐3p | 4.430215 | 0.000608887 |
| miR‐335‐3p | 4.500000 | 3.45E‐05 | miR‐431‐3p | 4.404831 | 0.000671589 |
| miR‐21‐5p | 4.480231 | 1.11E‐06 | miR‐1197 | 4.388206 | 0.000327892 |
| miR‐3609 | 4.463120 | 2.85E‐12 | miR‐323b‐3p | 4.369117 | 0.000691047 |
| miR‐1910 | 3.899553 | 7.82E‐05 | miR‐9‐5p | 4.366718 | 2.97523E‐05 |
| miR‐149‐5p | 3.842863 | 6.61E‐07 | miR‐487a | 4.332483 | 0.000695979 |
| miR‐873‐3p | 3.679904 | 3.11E‐05 | miR‐380‐3p | 4.303067 | 0.000425358 |
| miR‐758‐3p | 3.335272 | 0.000736 | miR‐577 | 4.005734 | 0.001037253 |
| miR‐301b | 3.309103 | 2.99E‐05 | miR‐410 | 3.966207 | 0.000198251 |
| miR‐708‐5p | 3.191239 | 5.37E‐06 | miR‐192‐5p | 2.789685 | 0.0001095 |
| miR‐31‐5p | 3.180813 | 1.36E‐05 | miR‐451a | −2.526477 | 0.001015833 |
| miR‐3676‐5p | 3.091173 | 0.00013327 | miR‐30a‐5p | −2.851723 | 1.60505E‐05 |
| miR‐31‐3p | 3.063404 | 0.00025179 | miR‐486‐3p | −3.131728 | 2.68134E‐05 |
| miR‐224‐5p | 2.979244 | 6.61E‐05 | miR‐490‐3p | −4.131157 | 0.000875453 |
| miR‐873‐5p | 2.923541 | 0.00189254 | miR‐30a‐5p | −4.453866 | 1.12345E‐05 |
| miR‐708‐3p | 2.865219 | 0.00017849 | miR‐30a‐3p | −4.516783 | 2.34568E‐05 |
| miR‐183‐5p | 2.793837 | 0.00015804 | miR‐486‐5p | −4.538656 | 1.12345E‐05 |
| miR‐409‐5p | 2.726459 | 0.00017722 | miR‐184 | −4.934109 | 4.68414E‐05 |
| miR‐449c‐5p | −2.502744 | 0.00114347 | |||
| miR‐3614‐5p | −2.530638 | 0.00047691 | |||
| miR‐223‐5p | −2.646799 | 7.93E‐05 | |||
| miR‐29b‐1‐5p | −2.868420 | 0.00031147 | |||
| miR‐30c‐2‐3p | −3.077404 | 3.17E‐05 | |||
| miR‐486‐3p | −3.338364 | 2.76E‐07 | |||
| miR‐138‐1‐3p | −3.407908 | 0.00095397 | |||
| miR‐30a‐3p | −4.656785 | 1.25E‐05 | |||
| miR‐486‐5p | −4.689655 | 1.23E‐07 | |||
| miR‐184 | −5.019975 | 3.00E‐07 | |||
| miR‐135a‐5p | −5.067272 | 5.70E‐06 | |||
| miR‐375 | −6.320925 | 4.08E‐18 | |||
SCC, squamous cell carcinoma; AC, adenocarcinoma.
Based on a 2.5‐fold change cut‐off value, 40 and 28 miRNAs show different level between stage I SCC or AC and normal tissues.
Based on a 4.5‐fold change cut‐off, 17 and 11 miRNAs (bold font) display dysregulation in either SCC or AC as shown in Figure 1C.
Using a miRDeep score cut‐off of 10, we predicted 14 novel miRNA candidates (Supplementary Table 3), as they met criteria of stem‐loop secondary structure of miRNA precursor and had not been reported before. However, these potential novel miRNAs did not show significant differential expression between normal and tumor tissues, and thus were not further processed in this study for validation.
3.2. qRT‐PCR validation of the differential expression of miRNAs
To confirm the deep sequencing results, we used qRT‐PCR to assess expression of the 24 miRNAs in 40 stage I NSCLC tissues consisting of 20 SCCs and 20 ACs, and the paired normal lung tissues. Of the 24 miRNAs, 22 genes had <35 Ct value. However, two (miRs‐668 and 4435) had >35 Ct value in 60% tissue specimens. The results suggested that the two miRNAs were not reliably measured by qRT‐PCR in the tissue specimens, and thus excluded from further analysis. Expression level of each of the remaining 22 miRNA was calculated using comparative Ct method, in which Ct value of each miRNA was normalized in relation to that of all 22 miRNAs together using the equation 2 − ΔΔCt. Of the miRNAs, 14 exhibited significantly different level in stage SCC, or AC, or the both compared with normal lung tissues (All p < 0.05) (Table 2). In addition, all the 14 miRNA displayed changes by qRT‐PCR in the same direction as by deep sequencing analysis, although the magnitude of changes differed between the two methods. The validation data created from an independent set of tissue specimens using a different approach implies that the 14 miRNAs are signatures of stage I NSCLC.
Table 2.
Expression levels of 14 miRNAs in 40 stage I NSCLC and normal lung tissues determined by qRT‐PCR.
| MiRNAs | Mean ± in normal tissues | Mean ± in SCC tissues | p | Mean ± in AC tissues | p |
|---|---|---|---|---|---|
| miR‐577 | 0.01451 ± 0.003321 | 0.2515 ± 0.08886 | 0.019322136 | 0.5677 ± 0.1608 | 0.0016452 |
| miR‐9‐3p | 0.5848 ± 0.07936 | 1.145 ± 0.9148 | 0.181212245 | 4.406 ± 1.676 | 0.0214848 |
| miR‐9‐5p | 1.259 ± 0.3149 | 16.49 ± 4.287 | 0.001063353 | 24.80 ± 8.250 | 0.0047478 |
| miR‐196a‐5p | 2.537 ± 0.5663 | 10.70 ± 2.803 | 0.00690936 | 9.641 ± 3.711 | 0.0499931 |
| miR‐184 | 3.914 ± 0.8641 | 0.2541 ± 0.06159 | 0.000144289 | 0.4388 ± 0.08817 | 0.0005477 |
| miR‐182‐3p | 0.04403 ± 0.006379 | 0.1417 ± 0.03219 | 0.006178624 | 0.2706 ± 0.05945 | 0.0004249 |
| miR‐205‐5p | 23.95 ± 5.303 | 308.1 ± 58.16 | 2.01882E‐05 | 13.73 ± 3.180 | 0.1164496 |
| miR‐147b | 0.1172 ± 0.02934 | 0.3063 ± 0.05407 | 0.003199587 | 0.1709 ± 0.03255 | 0.2283051 |
| miR‐183‐3p | 2.646 ± 0.2470 | 5.881 ± 1.235 | 0.010504173 | 2.890 ± 0.5396 | 0.6832877 |
| miR‐944 | 0.03953 ± 0.004614 | 0.1930 ± 0.03721 | 0.000300373 | 0.05165 ± 0.01756 | 0.4988552 |
| miR‐135a‐5p | 22.25 ± 5.083 | 1.779 ± 0.5716 | 0.000281447 | 5.075 ± 1.148 | 0.0034008 |
| miR‐205‐3p | 0.03880 ± 0.02018 | 0.3976 ± 0.09866 | 0.001312184 | 0.02559 ± 0.008623 | 0.5675627 |
| miR‐486‐5p | 228.0 ± 41.94 | 13.15 ± 8.543 | 1.24895E‐05 | 9.225 ± 1.659 | 1.812E‐05 |
| miR‐30a‐3p | 350.9 ± 53.31 | 20.22 ± 7.034 | 3.54479E‐07 | 26.16 ± 4.965 | 1.494E‐06 |
NSCLC, non‐small cell lung cancer; SCC, squamous cell carcinoma; AC, adenocarcinoma; SD, standard deviation.
3.3. Associations between miRNA expression and histopathological parameters of NSCLC
To substantiate the findings, expressions of the 14 miRNAs were assessed using additional 60 NSCLC tissues with different stages and histological types. In line with the above findings, all the 14 miRNA showed dysregulation in either SCC or AC specimens or both relative to normal tissues (All p < 0.05) (Supplementary Table 4). Furthermore, seven miRNAs (miRs‐577, 205‐5p, 147b, 183‐3p, 944, 135a‐5p, and 205‐3p) displayed distinctive expression levels in SCC versus AC (All p < 0.05) (Figure 2A). Of the seven miRNAs, expression levels of miRs‐205‐5p, 944, 205‐3p, 147b, and 183‐3p were significantly higher, whereas miRs‐577 and 135a‐5p were lower in SCC relative to AC (Figure 2A). In addition, the ROC curve analysis showed that the individual miRNAs displayed AUC values ranging from 0.76 to 0.91 in distinguishing the two major different types of NSCLC (all ≤ 0.0001) (Supplementary Table 5). Interestingly, the four miRNAs (miRs‐944, 205‐3p, 135a‐5p, and 577) used in combination could differentiate SCC from AC with 0.95 AUC (Figure 2B). Consequently, combined use of the four miRNAs yielded 93.3% sensitivity and 86.7% specificity in distinguishing SCC from AC. In addition, high expression levels of three miRNAs (miRs‐577, 183‐3p, and‐944) were correlated with advanced stages of SCC tumors (p = 0.02, p = 0.03, and p = 0.04, respectively). miR‐577 had a lower expression level in stage I SCCs compared with stage I ACs when analyzed in the 40 stage I NSCLC specimens (Table 2 and Figure 2A). The finding was confirmed in the 60 NSCLC tissues with different stages and histological types. Furthermore, the miR‐577 expression level was correlated with advanced stages SCCs rather than ACs when analyzed in the 60 NSCLC tissues. However, there was no association of changes of the miRNAs with the age, gender, and race of the NSCLC patients (All p > 0.05).
Figure 2.
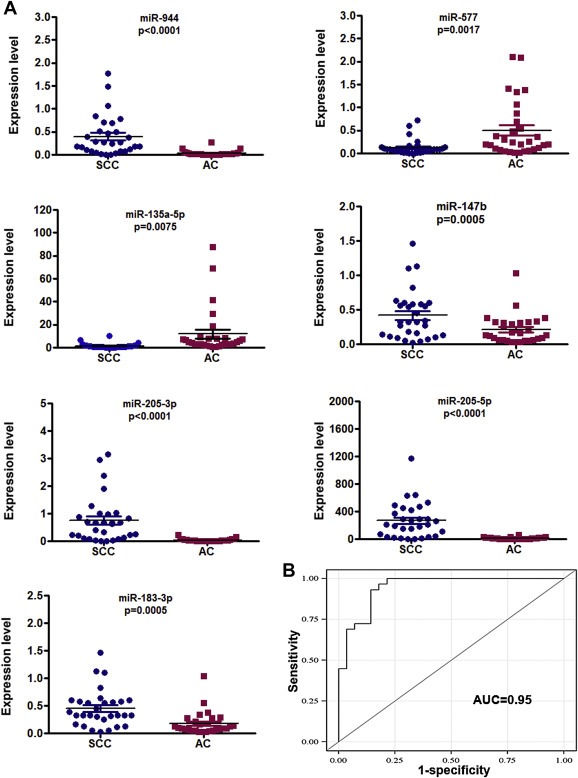
Seven miRNAs show different expression levels between SCC and AC determined by qRT‐PCR. A, expressions of miR‐944, 205‐3p, miR‐205‐5p, miR‐147b, and miR‐183‐3p were higher, whereas miR‐577 and miR‐135a‐5p were lower in SCC compared to AC. B, four miRNAs (miRs‐944, 205‐3p, 135a‐5p, and 577) used in combination produced an AUC of 0.95.
3.4. Involved pathways of the NSCLC‐associated miRNAs
Pathway analysis by integrating TargetScan, PicTar, amiRBase, and DIANA created a rank‐ordered list of 20 pathways with the most significant gene‐enrichment (Supplementary Table 5), in which the 14 miRNAs might be involved. Importantly, the pathways in cancer and NSCLC were at the top of the list. Particularly, within the NSCLC pathway, multiple miRNAs were predicted to target cancer genes. For example, miRs‐577, 9‐5p, 205‐5p could target E2F, whereas miRs‐30a‐3p, 944, and 486‐5p might target CDK4 and 6 (Supplementary Figure 1). In addition, one single miRNA could target several crucial genes. For instance, miR‐486‐5p regulated RASSF, Forkhead, CDK4/6, and CyclinD1 (Supplementary Figure 1). Furthermore, miR‐944 could target suppressor of cytokine signaling (SOCS), which is an inducible feedback inhibitor in regulation of EGFR signaling. Altogether, the combined effect of the miRNA signatures would contribute to tumorigenesis of NSCLC through regulating these important cancer biologic pathways.
3.5. Dysregulation of miR‐944 contributes to tumorigensis of NSCLC by targeting SOCS4
Because miR‐944 was one of the miRNA that displayed the highest expression level, and its role in NSCLC carcinogenesis remained unclear, we performed functional analysis to determine whether dysregulation of the miRNA might contribute to lung tumor. We forced expression of miR‐944 in two NSCLC lines (H520 and CALU‐1) that endogenously had a low level of the miRNA. As shown in Figure 3, ectopic expression of miR‐944 raised growth rate of the cancer cells determined by MTT assay. Furthermore, BrdU incorporation analysis suggested that cells transfected with miR‐944 mimic had a higher proliferative level than in cells treated with scrambled sequence control (p < 0.05) (Figure 3B). In addition, ectopic expression miR‐944 provoked the migratory and invasive abilities of the cells determined by Transwell assay (Figure 3C). Therefore, upregulation of miR‐944 promoted cell growth, proliferation, migration and invasion. However, after transfection with a miR‐944 inhibitor, SK‐MES‐1 and H292 cells that endogenously had a high miR‐944 expression level displayed down‐regulation of miR‐944. As a result, the cells with reduced miR‐944 expression exhibited lower cell growth, proliferative and invasive abilities, and a higher level of apoptosis, compared with cells treated with control (Supplementary Figure 2). Taken together, these gain‐ and loss‐of‐function analyses indicate that miR‐944 may have oncogenic function in pathogenesis of NSCLC.
Figure 3.
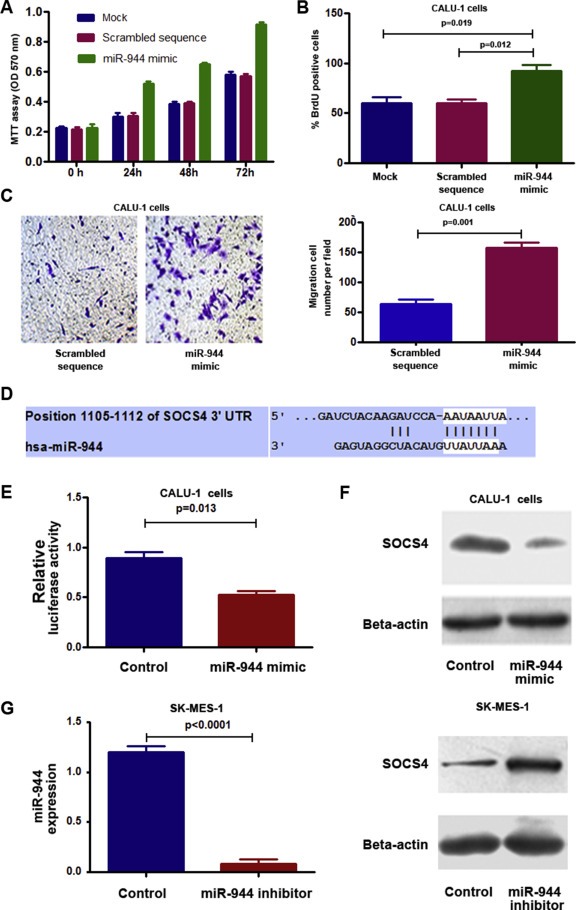
Dysregulation of miR‐944 contributes to tumorigensis of NSCLC by targeting SOCS4. A, forced expression of miR‐944 in CALU‐1 cells increased cell growth. B, ectopic expression of miR‐944 in CALU‐1 cells augmented cell proliferation. C, ectopic expression of miR‐944 in CALU‐1 promoted cell invasion and migration. The experiments were performed in two cell lines, CALU‐1 and H520, and produced similar data. Figures A–C only showed the results from CALU‐1 cells. D, a miR‐944 target site within 3′‐UTR of SOCS4 was predicted by bioinformatic algorithms. E, a luciferase reporter assay in CALU‐1 cells showed that luciferase activity of SOCS4‐3’UTR was inhibited by increased miR‐944 expression. F, Consequently, protein expression of SOCS4 was reduced. G, SK‐MES‐1 cells that had a high endogenous miR‐944 level after transfection with miR‐944 inhibitor showed a lower miR‐944 level determined by qRT‐PCR (left), but a higher SOCS4 level compared with cells treated with control (right). All data were obtained from three independent experiments and shown as mean ± SD.
To elucidate the mechanisms responsible for the oncogenic potential of miR‐944, we used bioinformatics analysis to identify its target genes. SOCS4, a major member of SOCS, was identified as one of the candidate targets, because miR‐944 potentially bound to the 3′‐UTR region of SOCS4 (Figure 3D). We, therefore, performed luciferase reporter assay to determine whether SOCS4 could be regulated by miR‐944. As shown in Figure 3E, a miR‐944 mimic diminished SOCS4 3′UTR‐associated luciferase reporter translation. Furthermore, transfection of CALU‐1 cells with the miR‐944 mimic generated a reduction of SOCS4 protein content (Figure 3F). In contrast, transfection of SK‐MES‐1 and H292 cells with a miR‐944 inhibitor produced an increased protein level of SOCS4 (Figure 3G). Altogether, miR‐944 may bind to the 3′‐UTR sequences of SOCS4, and hence inhibit its expression through posttranscriptional regulation.
Furthermore, SK‐MES‐1 cells transfected with miR‐944 inhibitor displayed a higher expression level of SOCS4 compared with the cells transfected with both miR‐944 inhibitor and si‐SOCS4 (Supplementary Figure 3A). Interestingly, the cells transfected with miR‐944 inhibitor showed lower cell growth, proliferative and invasive capabilities compared with the cells with both miR‐944 inhibitor and si‐SOCS4 (Supplementary Figure 3B–C). The observations suggest that the effects of reduced miR‐944 expression on the inhibition of NSCLC cell growth, migration, and invasion could be diminished by reduced expression of SOCS4. Therefore, SOCS4 may play a vital role in lung tumorigenesis mediated by miR‐944.
3.6. Upregulation of miR‐944 is associated with advanced stage and lymph node metastasis of SCC
To determine the clinical pathologic significance of the miR‐944 aberration, we evaluated its expression in FFPE specimens of 52 SCC tissues (Supplementary Table 2). Supporting the above finding in frozen tissues, miR‐944 expression level in the tumor specimens increased with advanced stages of SCC (p = 0.036) (Supplementary Table 2). Furthermore, miR‐944 expression was significantly higher in SCCs that displayed lymph node metastasis than in SCCs that did not (p = 0.002) (Supplementary Table 2). Therefore, the elevated miR‐944 expression is related to the progression and metastasis of SCC.
4. Discussion
The biomarkers that can augment current diagnostic approaches for NSCLC subclassification will significantly benefit personalized therapy of lung cancer (Hirsch et al., 2008). Lebanony et al. (2009) found that the assessment of miR‐205 expression in surgical tissues could efficiently separate SCC from non‐SCC lung cancer. On the contrary, Visecovo et al. showed that analysis of miR‐205 was not able to discriminate SCC from AC (Del Vescovo et al., 2011). Our deep sequencing analysis data was consistent with the results of Lebanony's study. Importantly, we further identified two members (miRs‐205‐3p and 205‐5p) of the miR‐205 family that were unique signatures for SCC. In addition to the miRNA‐205 members, we also discovered another five miRNAs that showed different level between SCC and AC. Furthermore, combined analysis of four (miRs‐944, 205‐3p, 135a‐5p, and 577) of the miRNAs produced 93.3% sensitivity and 86.7% specificity in distinguishing SCC from AC. In addition, qRT‐PCR validation revealed that high expressions of miRs‐577, 183‐3p, and 944 were more related to advanced stages of SCC compared with stage I SCC. Moreover, miR‐944 expression was associated with lymph node status, implying that the miRNA may be a predictor for progression and metastasis of SCC. Therefore, this study demonstrated the power of deep sequencing for the identification of NSCLC‐associated miRNAs and biomarkers for classification and predicting prognosis of NSCLC. As miRNAs are stable in various types of specimens, a clinical assay based on the miRNAs may be developed in future as a small and cost‐effective platform to assist differential diagnosis of NSCLC and predicting metastasis of the malignancy. Nevertheless, undertaking a large and prospective study in large case and control cohorts to comprehensively determine whether the miRNA could be useful in optimizing the treatment strategies for NSCLC is required.
Our study also reveals that the miRNA signatures of NSCLC may involve in pathways of cancer, particularly NSCLC. Among the predicted targets of the miRNAs in this NSCLC pathway, numerous vital genes participate in several important signaling pathways including ErbB, PI3K‐Akt, MAPK, cell cycle, Calcium, and p53. Dysregulation of the miRNAs might function as oncogenes or tumor suppressors, thereby contributing to tumor initiation and progression. Modulation of miRNA expression holds great hope for potential cancer therapy (Lu et al., 2005; Tili et al., 2007). Therefore, our prediction that the differentially expressed miRNAs have the capability to target multiple components in these critical pathways makes them promising molecular targets for the treatment of NSCLC.
One of the advantages of next‐generation deep‐sequencing technology over conventional procedures is that it allows the discovery of miRNAs that have not been related with malignancies by the traditional techniques. Using deep sequencing, we identify a set of differentially expressed miRNAs whose dysregulation has not well been documented in lung tumorigenesis. For example, we show that aberrations of miRs‐147b and 944 are associated with lung tumorigenesis. miR‐944 is of particular interest, because our function annotation of target genes indicated that it tends to target genes involved in regulation of SOCS‐EGFR signaling. Indeed, our experimental investigation further confirms that miR‐944 may have oncogenic function in pathogenesis of NSCLC, as its dysregulation contributes to NSCLC cell growth, proliferation and invasion by targeting SOCS4, a tumor suppressor (Kobayashi et al., 2012). SOCS4 can bind to the EGFR and suppresses this receptor signaling, and thus suppresses EGFR‐driven tumorigenesis mechanisms (Kario et al., 2005). Furthermore, we discover 14 novel miRNA candidates, supporting that deep sequencing is a valuable tool to identify novel miRNAs of as yet unknown biological function. Therefore, our data lays a solid basis for future investigations aiming to confirm novel miRNAs and characterize the role of the miRNAs in lung cancer pathogenesis.
Of 24 miRNAs that displayed dysregulations in NSCLC discovered by deep sequencing, 11 did not exhibit changes assessed by qRT‐PCR. The inconsistence between the two approaches for assessing miRNA expression is not unusual as demonstrated by previous studies of miRNAs using microarray, qRT‐PCR, and high‐throughput sequencing (Hamfjord et al., 2012; Pradervand et al., 2010; Schee et al., 2013). One of the reasons underlying the observation might be that qRT‐PCR has lower sensitivity than deep sequencing, and hence is not able to measure the difference of some miRNA between normal and tumor tissues. Furthermore, the different proportions of the tumor cells in the specimens tested by deep sequencing from those in the specimens assessed by qRT‐PCR could also lead to the inconsistent results between the two techniques.
There are some potential limitations of using next‐generation sequencing in clinical practice, including the high cost and requiring computational infrastructure for data analysis and interpretation (Chugh and Dittmer, 2012). To translate the lung cancer‐miRNA signatures identified by next‐generation sequencing into clinical settings, more cost‐effective techniques are needed. qPCR is one of the most commonly used techniques that can estimate expression levels of miRNAs in clinical specimens. However, qPCR has two major challenges for the assessment of miRNAs. First, qPCR uses endogenous small RNAs as controls, by which CT values for miRNA targets are normalized and referenced across samples. However, none of endogenous genes has been widely accepted as a standard control for qPCR. Second, the sensitivity of qPCR for the detection of low‐copy‐number miRNAs is not high enough. For instance, miRs‐668 and 4435 are undetectable by qPCR in the present study. The emerging digital PCR is a straightforward technique for precise, direct, and absolute quantification of low‐copy‐nucleic acids without requiring control genes (Li et al., 2014; Ma et al., 2013). We have recently demonstrated that digital PCR is a robust tool for quantitative assessment of miRNAs in sputum and plasma, and has a higher sensitivity to detect low abundant miRNAs compared with qPCR (Li et al., 2014; Ma et al., 2013). We will evaluate if digital PCR can directly quantify the low‐copy‐number miRNAs defined by next‐generation sequencing whose changes are the hallmark of lung cancer.
In sum, this study presents the earliest use of deep sequencing for comprehensively profiling miRNAs in primary lung tumor specimens. Our comprehensive survey of differentially expressed miRNAs not only confirms some existing findings, but discovers dysregulated miRNAs that have not been found in lung carcinogenesis, thereby providing new signatures of early stage NSCLC. Furthermore, by performing qRT‐PCR, we validate miRNA signatures that can distinguish malignant from normal specimens, and identify a small panel of miRNAs that can efficiently differentiate SCC from AC and a miRNA for predicting lymph node status of SCC. The identified miRNAs may provide promising biomarkers to improve early detection and subclassification and monitoring metastasis of NSCLC in future. Moreover, successfully determining an oncogenic role of miR‐944 further demonstrates the power of deep sequencing in identification for cancer‐related miRNAs, which may provide novel therapeutic targets of NSCLC.
Disclosure
The authors declare no conflict of interest.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
This work was supported in part by NCI R01CA161837, VA merit Award I01 CX000512, LUNGevity/Upstage Foundation Early Detection Award, and University of Maryland Cancer Epidemiology Alliance Seed Grant (F.J.).
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.03.019.
Ma Jie, Mannoor Kaiissar, Gao Lu, Tan Afang, Guarnera Maria A., Zhan Min, Shetty Amol, Stass Sanford A., Xing Lingxiao, Jiang Feng, (2014), Characterization of microRNA transcriptome in lung cancer by next‐generation deep sequencing, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.03.019.
References
- Ambros, V. , 2003. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 113, 673–676. [DOI] [PubMed] [Google Scholar]
- Azzoli, C.G. , Patel, J.D. , Krug, L.M. , Miller, V. , James, L. , Kris, M.G. , Ginsberg, M. , Subzwari, S. , Tyson, L. , Dunne, M. , 2011. Pralatrexate with vitamin supplementation in patients with previously treated, advanced non-small cell lung cancer: safety and efficacy in a phase 1 trial. J. Thorac. Oncol. 6, 1915–1922. [DOI] [PubMed] [Google Scholar]
- Bishop, J.A. , Benjamin, H. , Cholakh, H. , Chajut, A. , Clark, D.P. , Westra, W.H. , 2010. Accurate classification of non-small cell lung carcinoma using a novel microRNA-based approach. Clin. Cancer Res. 16, 610–619. [DOI] [PubMed] [Google Scholar]
- Brambilla, E. , Travis, W.D. , Colby, T.V. , Corrin, B. , Shimosato, Y. , 2001. The new World Health Organization classification of lung tumours. Eur. Respir. J. 18, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Chugh, P. , Dittmer, D.P. , 2012. Potential pitfalls in microRNA profiling. Wiley Interdiscip. Rev. RNA. 3, 601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vescovo, V. , Cantaloni, C. , Cucino, A. , Girlando, S. , Silvestri, M. , Bragantini, E. , Fasanella, S. , Cuorvo, L.V. , Palma, P.D. , Rossi, G. , 2011. miR-205 expression levels in nonsmall cell lung cancer do not always distinguish adenocarcinomas from squamous cell carcinomas. Am. J. Surg. Pathol. 35, 268–275. [DOI] [PubMed] [Google Scholar]
- Friedlander, M.R. , Chen, W. , Adamidi, C. , Maaskola, J. , Einspanier, R. , Knespel, S. , Rajewsky, N. , 2008. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 26, 407–415. [DOI] [PubMed] [Google Scholar]
- Grondin, S.C. , Liptay, M.J. , 2002. Current concepts in the staging of non-small cell lung cancer. Surg. Oncol. 11, 181–190. [DOI] [PubMed] [Google Scholar]
- Hamfjord, J. , Stangeland, A.M. , Hughes, T. , Skrede, M.L. , Tveit, K.M. , Ikdahl, T. , Kure, E.H. , 2012. Differential expression of miRNAs in colorectal cancer: comparison of paired tumor tissue and adjacent normal mucosa using high-throughput sequencing. PLoS One. 7, e34150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, F.R. , Spreafico, A. , Novello, S. , Wood, M.D. , Simms, L. , Papotti, M. , 2008. The prognostic and predictive role of histology in advanced non-small cell lung cancer: a literature review. J. Thorac. Oncol. 3, 1468–1481. [DOI] [PubMed] [Google Scholar]
- Hu, Z. , Chen, X. , Zhao, Y. , Tian, T. , Jin, G. , Shu, Y. , Chen, Y. , Xu, L. , Zen, K. , Zhang, C. , 2010. Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancer. J. Clin. Oncol. 28, 1721–1726. [DOI] [PubMed] [Google Scholar]
- Jima, D.D. , Zhang, J. , Jacobs, C. , Richards, K.L. , Dunphy, C.H. , Choi, W.W. , Au, W.Y. , Srivastava, G. , Czader, M.B. , Rizzieri, D.A. , 2010. Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood. 116, e118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kario, E. , Marmor, M.D. , Adamsky, K. , Citri, A. , Amit, I. , Amariglio, N. , Rechavi, G. , Yarden, Y. , 2005. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J. Biol. Chem. 280, 7038–7048. [DOI] [PubMed] [Google Scholar]
- Keller, A. , Backes, C. , Leidinger, P. , Kefer, N. , Boisguerin, V. , Barbacioru, C. , Vogel, B. , Matzas, M. , Huwer, H. , Katus, H.A. , 2011. Next-generation sequencing identifies novel microRNAs in peripheral blood of lung cancer patients. Mol. Biosyst. 7, 3187–3199. [DOI] [PubMed] [Google Scholar]
- Kobayashi, D. , Nomoto, S. , Kodera, Y. , Fujiwara, M. , Koike, M. , Nakayama, G. , Ohashi, N. , Nakao, A. , 2012. Suppressor of cytokine signaling 4 detected as a novel gastric cancer suppressor gene using double combination array analysis. World J. Surg. 36, 362–372. [DOI] [PubMed] [Google Scholar]
- Kozubek, J. , Ma, Z. , Fleming, E. , Duggan, T. , Wu, R. , Shin, D.G. , Dadras, S.S. , 2013. In-depth characterization of microRNA transcriptome in melanoma. PLoS One. 8, e72699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara, A. , Griffiths-Jones, S. , 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, (Database issue) D152–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi, M.T. , Zhao, Y. , Rotunno, M. , Koshiol, J. , Liu, H. , Bergen, A.W. , Rubagotti, M. , Goldstein, A.M. , Linnoila, I. , Marincola, F.M. , 2010. MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin. Cancer Res. 16, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, M.F. , Yang, S. , Zhao, C. , Sun, G. , Murai, K. , Wu, X. , Wang, J. , Gao, H. , Brown, C.E. , Liu, X. , 2012. Genome-wide profiling identified a set of miRNAs that are differentially expressed in glioblastoma stem cells and normal neural stem cells. PLoS One. 7, e36248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. , Salzberg, S.L. , 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebanony, D. , Benjamin, H. , Gilad, S. , Ezagouri, M. , Dov, A. , Ashkenazi, K. , Gefen, N. , Izraeli, S. , Rechavi, G. , Pass, H. , 2009. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J. Clin. Oncol. 27, 2030–2037. [DOI] [PubMed] [Google Scholar]
- Li, N. , Ma, J. , Guarnera, M.A. , Fang, H. , Cai, L. , Jiang, F. , 2014. Digital PCR quantification of miRNAs in sputum for diagnosis of lung cancer. J. Cancer Res. Clin. Oncol. 140, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Getz, G. , Miska, E.A. , Alvarez-Saavedra, E. , Lamb, J. , Peck, D. , Sweet-Cordero, A. , Ebert, B.L. , Mak, R.H. , Ferrando, A.A. , 2005. MicroRNA expression profiles classify human cancers. Nature. 435, 834–838. [DOI] [PubMed] [Google Scholar]
- Ma, J. , Li, N. , Guarnera, M. , Jiang, F. , 2013. Quantification of plasma miRNAs by digital PCR for Cancer diagnosis. Biomark Insights. 8, 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay, S. , Katzenstein, A.L. , 2011. Subclassification of non-small cell lung carcinomas lacking morphologic differentiation on biopsy specimens: utility of an immunohistochemical panel containing TTF-1, napsin A, p63, and CK5/6. Am. J. Surg. Pathol. 35, 15–25. [DOI] [PubMed] [Google Scholar]
- Naruke, T. , Goya, T. , Tsuchiya, R. , Suemasu, K. , 1988. Prognosis and survival in resected lung carcinoma based on the new international staging system. J. Thorac. Cardiovasc. Surg. 96, 440–447. [PubMed] [Google Scholar]
- Osanto, S. , Qin, Y. , Buermans, H.P. , Berkers, J. , Lerut, E. , Goeman, J.J. , van Poppel, H. , 2012. Genome-wide microRNA expression analysis of clear cell renal cell carcinoma by next generation deep sequencing. PLoS One. 7, e38298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomo, C. , Campbell, J.D. , Gerrein, J. , Tellez, C.S. , Garrison, C.B. , Walser, T.C. , Drizik, E. , Si, H. , Gower, A.C. , Vick, J. , 2013. MicroRNA 4423 is a primate-specific regulator of airway epithelial cell differentiation and lung carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 110, 18946–18951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradervand, S. , Weber, J. , Lemoine, F. , Consales, F. , Paillusson, A. , Dupasquier, M. , Thomas, J. , Richter, H. , Kaessmann, H. , Beaudoing, E. , 2010. Concordance among digital gene expression, microarrays, and qPCR when measuring differential expression of microRNAs. Biotechniques. 48, 219–222. [DOI] [PubMed] [Google Scholar]
- Quinlan, A.R. , Hall, I.M. , 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raponi, M. , Dossey, L. , Jatkoe, T. , Wu, X. , Chen, G. , Fan, H. , Beer, D.G. , 2009. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 69, 5776–5783. [DOI] [PubMed] [Google Scholar]
- Rohr, C. , Kerick, M. , Fischer, A. , Kuhn, A. , Kashofer, K. , Timmermann, B. , Daskalaki, A. , Meinel, T. , Drichel, D. , Borno, S.T. , 2013. High-throughput miRNA and mRNA sequencing of paired colorectal normal, tumor and metastasis tissues and bioinformatic modeling of miRNA-1 therapeutic applications. PLoS One. 8, e67461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schee, K. , Lorenz, S. , Worren, M.M. , Gunther, C.C. , Holden, M. , Hovig, E. , Fodstad, O. , Meza-Zepeda, L.A. , Flatmark, K. , 2013. Deep sequencing the MicroRNA transcriptome in colorectal Cancer. PLoS One. 8, e66165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Jiang, F. , 2012. Applications of MicroRNAs in the diagnosis and prognosis of lung Cancer. Expert Opin. Med. Diagn. 6, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Liao, J. , Guarnera, M.A. , Fang, H. , Cai, L. , Stass, S.A. , Jiang, F. , 2014. Analysis of MicroRNAs in sputum to improve computed tomography for lung Cancer diagnosis. J. Thorac. Oncol. 9, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Liu, Z. , Todd, N.W. , Zhang, H. , Liao, J. , Yu, L. , Guarnera, M.A. , Li, R. , Cai, L. , Zhan, M. , 2011. Diagnosis of lung cancer in individuals with solitary pulmonary nodules by plasma microRNA biomarkers. BMC Cancer. 11, 374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R. , Naishadham, D. , Jemal, A. , 2013. Cancer statistics, 2013. CA Cancer J. Clin. 63, 11–30. [DOI] [PubMed] [Google Scholar]
- Tili, E. , Michaille, J.J. , Gandhi, V. , Plunkett, W. , Sampath, D. , Calin, G.A. , 2007. miRNAs and their potential for use against cancer and other diseases. Future Oncol. 3, 521–537. [DOI] [PubMed] [Google Scholar]
- Travis, W.D. , Brambilla, E. , Noguchi, M. , Nicholson, A.G. , Geisinger, K.R. , Yatabe, Y. , Beer, D.G. , Powell, C.A. , Riely, G.J. , Van Schil, P.E. , 2011. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 6, 244–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Tian, X. , Han, R. , Zhang, X. , Wang, X. , Shen, H. , Xue, L. , Liu, Y. , Yan, X. , Shen, J. , 2014. Downregulation of miR-486-5p contributes to tumor progression and metastasis by targeting protumorigenic ARHGAP5 in lung cancer. Oncogene. 33, (9) 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton, T. , Livingston, R. , Johnson, D. , Rigas, J. , Johnston, M. , Butts, C. , Cormier, Y. , Goss, G. , Inculet, R. , Vallieres, E. , 2005. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N. Engl. J. Med. 352, 2589–2597. [DOI] [PubMed] [Google Scholar]
- Wolff, A.C. , Hammond, M.E. , Schwartz, J.N. , Hagerty, K.L. , Allred, D.C. , Cote, R.J. , Dowsett, M. , Fitzgibbons, P.L. , Hanna, W.M. , Langer, A. , 2007. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 25, 118–145. [DOI] [PubMed] [Google Scholar]
- Xie, Y. , Todd, N.W. , Liu, Z. , Zhan, M. , Fang, H. , Peng, H. , Alattar, M. , Deepak, J. , Stass, S.A. , Jiang, F. , 2010. Altered miRNA expression in sputum for diagnosis of non-small cell lung cancer. Lung Cancer. 67, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, L. , Todd, N.W. , Yu, L. , Fang, H. , Jiang, F. , 2010. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod. Pathol. 23, 1157–1164. [DOI] [PubMed] [Google Scholar]
- Yanaihara, N. , Caplen, N. , Bowman, E. , Seike, M. , Kumamoto, K. , Yi, M. , Stephens, R.M. , Okamoto, A. , Yokota, J. , Tanaka, T. , 2006. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 9, 189–198. [DOI] [PubMed] [Google Scholar]
- Yu, L. , Todd, N.W. , Xing, L. , Xie, Y. , Zhang, H. , Liu, Z. , Fang, H. , Zhang, J. , Katz, R.L. , Jiang, F. , 2010. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int. J. Cancer. 127, 2870–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data