

Summary
More than half of human cancers have aberrantly upregulated phosphoinositide signals; yet how phospholipid signals are controlled during tumorigenesis is not fully understood. We report here that TIPE3 (TNFAIP8L3) is the transfer protein of phosphoinositide second messengers that promote cancer. High-resolution crystal structure of TIPE3 shows a large hydrophobic cavity that is occupied by a phospholipid-like molecule. TIPE3 preferentially captures and shuttles two lipid second messengers, i.e., phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate, and increases their levels in the plasma membrane. Importantly, human cancers have markedly upregulated TIPE3 expression. Knocking out TIPE3 diminishes tumorigenesis whereas enforced TIPE3 expression enhances it in vivo. Thus, the function and metabolism of phosphoinositide second messengers are controlled by a specific transfer protein during tumorigenesis.
Keywords: TNFAIP8L3; PtdIns(4,5)P2; PtdIns(3,4,5)P3; Lipid Signaling; Signal Transduction; Cancer
Introduction
Tumorigenesis is a multi-step process orchestrated by several classes of molecules that control cell death and/or growth. The TNFAIP8 (tumor necrosis factor-alpha-induced protein 8, or TIPE) family of proteins is recently described regulators of tumorigenesis and inflammation (Kumar et al., 2004; Sun et al., 2008). TNFAIP8 promotes tumor metastasis and is a risk factor for non-Hodgkin's lymphoma in humans and for bacterial infection in mice (Ahn et al., 2010; Zhang et al., 2012). Similarly, TIPE2 (TNFAIP8L2) regulates both carcinogenesis and inflammation, and its germ line deletion leads to fatal inflammatory diseases (Gus-Brautbar et al., 2012; Sun et al., 2008; Wang et al., 2012). TIPE1 has been reported to regulate cell death (Hitomi et al., 2008), whereas the role of TIPE3 has not yet been described. How TNFAIP8 family regulates tumorigenesis is largely unknown.
Phosphoinositides constitute approximately 1% of lipids in the eukaryotic plasma membrane, but play a major role as signaling molecules in both normal and tumor cells. There are seven phosphoinositides, which are generated by phosphorylation of the inositol ring of phosphatidylinositol (PtdIns) on the third, fourth, and/or fifth hydroxyl groups (Fruman et al., 1998). Phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] plays key signaling roles (Kwiatkowska and Sobota, 1999; McLaughlin et al., 2002). It is the precursor of at least three second messengers: inositol 1,4,5-trisphosphate [Ins(1,4,5)P3] and diacylglycerol (DAG) generated through hydrolysis by phospholipase C (PLC) isoforms, and PtdIns(3,4,5)P3 generated through phosphorylation by phosphoinositide-3 kinases (PI3Ks). More than half of human cancers have aberrantly upregulated PtdIns(3,4,5)P3 or PtdIns(4,5)P2 signals. While somatic mutations of PTEN, Ras, and the α catalytic subunit of PI3K (PIK3CA) account for many cases of the phosphoinositide signaling upregulation, other mechanisms of the dysregulation are being discovered as well (Vivanco and Sawyers, 2002; Yuan and Cantley, 2008). In this report, we describe a mammalian transfer protein for PtdIns(4,5)P2 and PtdIns(3,4,5)P3 and examine its role in phosphoinositide signaling.
Results
TIPE3 is markedly up-regulated in four types of human cancers, and in cells treated with growth factors
TIPE3 is expressed in a wide range of organs but its expression levels vary greatly (Figure S1A). High levels of TIPE3 mRNA were detected in murine uterus and developing embryos, whereas little or no TIPE3 was detected in lymphoid organs. Moderate expression of TIPE3 was observed in many organs including intestine, lung, brain, bladder, and colon (Figure S1A). Similarly, murine and human cell lines have varying levels of TIPE3 expression (Figure S1, B and C). Interestingly, when treated with platelet-derived growth factor (PDGF), serum-starved NIH3T3 cells and primary murine embryonic fibroblasts (MEFs) both markedly upregulated TIPE3 expression in a time-dependent manner (Figure 1A).
Figure 1. Upregulation of TIPE3 expression correlates with tumorigenesis.
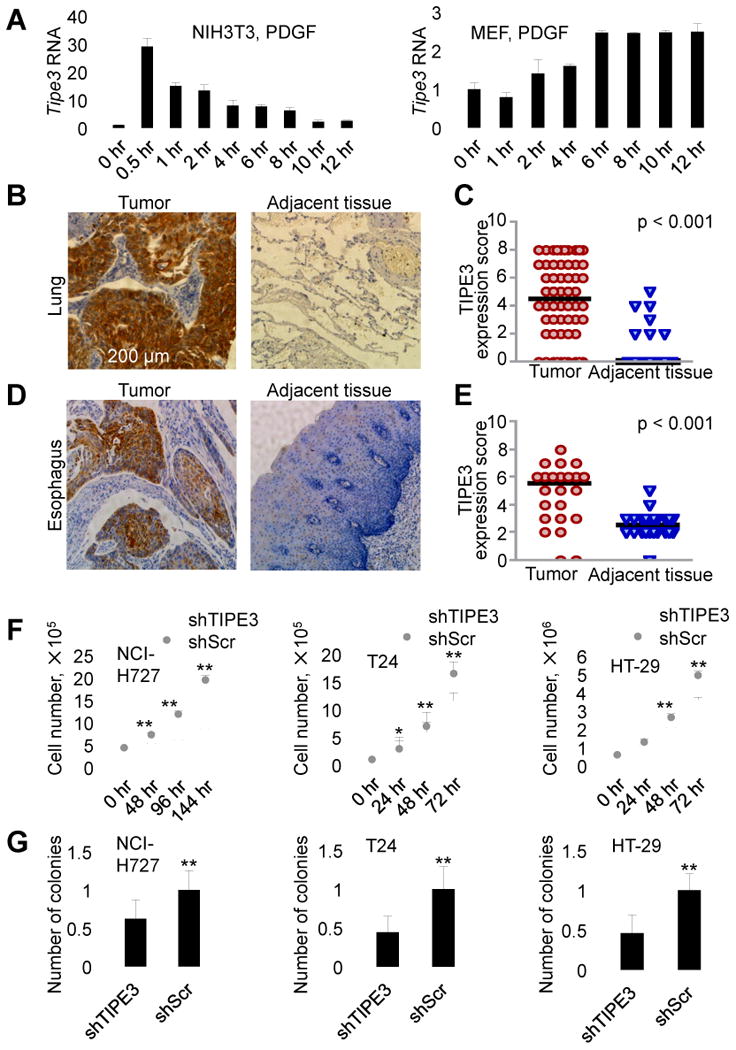
(A) RT-PCR analyses of TIPE3 mRNA levels in the NIH3T3 fibroblast cell line (left panel) and C57BL/6 murine embryonic fibroblasts (MEF, right panel). Cells were first serum-starved for 12 hr and then stimulated with 20 (left panel) and 50 (right panel) ng/ml of PDGF for the indicated times before being collected for the RNA analyses. The expression levels of TIPE3 in NIH3T3 (right panel) and MEF (left panel) at 0 hr were set as 1. (B-E) TIPE3 upregulation in human cancers. Immunohistochemistry analyses of TIPE3 protein expression in tumors and their adjacent normal tissues from patients diagnosed with lung cancer (B and C) and esophageal cancer (D and E). TIPE3-positive cells are shown in brown (B and D). Quantitation of the TIPE3 expression in tumors and adjacent tissues of 60 patients with lung cancer (C) and 24 patients with esophageal cancer (E) was performed as described in Supplemental Experimental Procedures. Each data point represents a TIPE3 expression score of patients. The horizontal bars represent the medians. (F) Cell growth analysis of human NCI-H727, T24, and HT-29 cells stably expressing either shTIPE3 or short hairpin scrambled (shScr) RNA (control) over the indicated times. (G) Soft agar colony formation of NCI-H727, T24, and HT-29 cells expressing either shTIPE3 or shScr. Numbers of colonies formed by cells expressing shScr were set as 1. For (A), (F), and (G), values represent means ± SD, * p < 0.05, and ** p < 0.01; h denotes hr. The experiments were repeated at least three times with similar results. See also Figure S1.
To determine TIPE3 expression in human tumors, we studied, by immunohistochemistry, tumor samples from 60 lung cancer patients and 24 esophageal cancer patients. Marked upregulation of TIPE3 protein was observed in the majority of tumor sections compared with adjacent non-tumor sections from the same patients (Figure 1, B-E, and Figure S1, D and E). Additionally, we also detected significant TIPE3 upregulation in human cervical cancer (70 patients) and colonadenocarcinoma (82 patients) (data not shown). These results indicate that, similar to other TIPE proteins, TIPE3 may be involved in tumorigenesis.
Overexpressing TIPE3 promotes tumorigenesis whereas knocking out TIPE3 inhibits it
To explore the potential role of TIPE3 in tumorigenesis, we used knockdown, knockout, and overexpression approaches. Transfection with a short hairpin RNA (shRNA) that targets TIPE3 (shTIPE3) significantly reduced TIPE3 expression in three human cancer cell lines: NCI-H727 lung carcinoma cells (with KRasV12 and PIK3CAE545 mutations), T24 bladder carcinoma cells (with HRasV12 mutation), and HT-29 colorectal adenocarcinoma cells (with PIK3CAT449 and BRAFV600E mutations) (Figure S1, F and G). This was associated with significant reduction in total cell numbers (Figure 1F) and compromised ability of the cells to form colonies in soft agar (Figure 1G). Conversely, when overexpressed, TIPE3 significantly increased the numbers of 293T cells with HRasV12 or PIK3CAE545 mutation (Figure S2A) and of HRasV12-transformed NIH3T3 cells (NIH3T3-HRasV12) (Figure 2A). TIPE3 overexpression also increased the size of the colonies formed in soft agar (Figure 2B and Figure S2B), and accelerated cell cycle progression as evidenced by a reduced population of G0/G1 cells and increased S/G2/M cells (Figure 2C). These results indicate that TIPE3 promotes tumor cell growth in vitro.
Figure 2. TIPE3 promotes tumorigenesis, cell growth, and cell survival in vitro and in vivo.
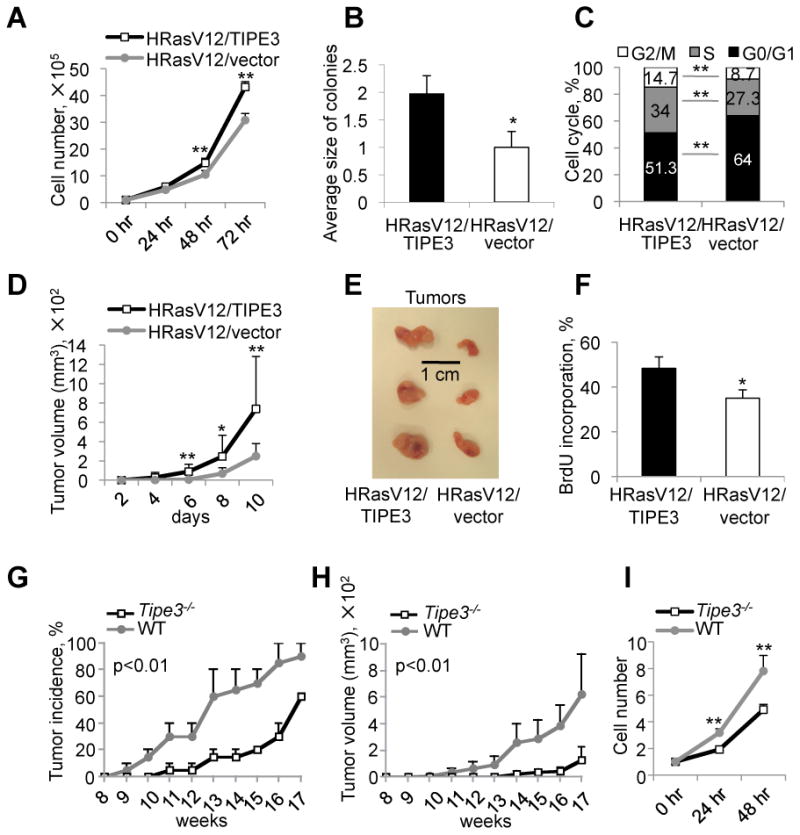
(A-C) Analyses of cell growth (A), relative colony size in soft agar (B), and cell cycle progression (C) of NIH3T3-HRasV12 cells stably transfected with either TIPE3-Flag or empty vector. For (B), number of colonies formed by NIH3T3-HRasV12 cells transfected with empty vector was set as 1. The experiments were repeated at least three times with similar results. Values represent means ± SD. (D-F) TIPE3 promotes tumor growth in a murine xenograft model. Nude mice (n=14) were injected subcutaneously with NIH3T3-HRasV12 cells stably transfected with either TIPE3-Flag or empty vector. Tumor volume was determined over a course of 10 days (D). At day 10, tumors were excised and photographed (E). In vivo cell proliferation was analyzed by BrdU incorporation. The percentages of BrdU-positive cells in tumor sections are shown (F). Results are representative of two independent experiments. Values represent means ± SD. (G-I) Tipe3 deficiency reduces tumor growth in mice. Tipe3-/- mice and wild-type (WT) controls (n=15), 7 weeks of age, were injected subcutaneously with 3-methylcholanthrene. Tumor incidence was examined for up to 17 weeks (G). Data are pooled from two independent experiments. Tumor volume was monitored for up to 17 weeks (H). Results are representative of two independent experiments. Values represent means ± SEM; the p values shown are for differences after week 11. Growth of Tipe3-/- and wild-type fibrosarcoma cells over the indicated times (I). Data are pooled from three independent Tipe3-/- and two independent wild-type fibrosarcoma cell cultures established from different 3-methylcholanthrene-induced tumors. The cell numbers at 0 hr was set as 1. The experiments were repeated four times with similar results. Values represent means ± SD. For all panels, * p<0.05, and ** p<0.01; h denotes hr, d denotes days. See also Figure S2.
To examine the effect of TIPE3 on tumorigenesis in vivo, we first injected NIH3T3-HRasV12 cells with or without TIPE3 overexpression into nude mice (nu/nu). Strikingly, both tumor onset and tumor growth were significantly accelerated in the TIPE3 group (Figure 2D). By day 10 following tumor cell injection, tumors of the TIPE3 group were twice as large as those of the control group (p<0.05, n=14) (Figure 2E and Figure S2C). The difference in tumor size was most likely due to enhanced proliferation of the TIPE3-overexpressing cells because increased bromodeoxyuridine (BrdU) incorporation was observed in tumor sections of the TIPE3 group (Figure 2F and Figure S2D). Cell death, as determined by cleaved (active) caspase-3 staining, may also play a role, because 0.86% cells in TIPE3-transfected tumors underwent cell death compared to 1.94% cells in the control tumors (Figure S2E).
Next, we studied carcinogen-induced tumorigenesis in the recently generated Tipe3 knockout (Tipe3-/-) mice (Figure S2F). Under pathogen-free conditions, these mice developed normally and did not exhibit noticeable signs of spontaneous diseases during the first three months of their lives (data not shown). However, following subcutaneous injection with the carcinogen 3-methylcholanthrene, Tipe3-/- mice exhibited a markedly delayed skin tumor onset and reduced tumor size in comparison with wild-type mice (Figure 2, G and H). As expected, Tipe3-/- fibrosarcoma cells grew much slower than WT controls in the culture (Figure 2I).
The unique N-terminal region of TIPE3 is essential for its effect on cell growth and survival
Unlike other members of the TNFAIP8 family that were capable of inducing cell death, TIPE3 did not reduce cell viability in the culture (Figure S2, A and G). To the contrary, when overexpressed in NIH3T3 cells, TIPE3 was able to increase the percentage of S phase cells even in the absence of the oncogenic Ras (Figure 3A). Consistent with this observation, TIPE3 drives cap-dependent protein synthesis required for G1–S transition. Co-transfection of TIPE3-expressing vector and green fluorescence protein (GFP)-expressing vector that contained a cap-dependent translation initiation site significantly increased the expression of GFP (Figure 3B). The TIPE3 effect was less apparent in cells transfected with a GFP-expressing vector that contained an internal ribosome entry site (IRES) (Figure S3A). The effect of TIPE3 on cap-dependent GFP expression was significantly reduced by the PI3K inhibitor LY294002 and/or the MEK-ERK pathway inhibitor PD98059 (Figure S3B) indicating that TIPE3 may play a role in the regulation of the PI3K-AKT and MEK-ERK signaling pathways.
Figure 3. The N-terminal region of TIPE3 is essential for its ability to promote cell growth and survival.
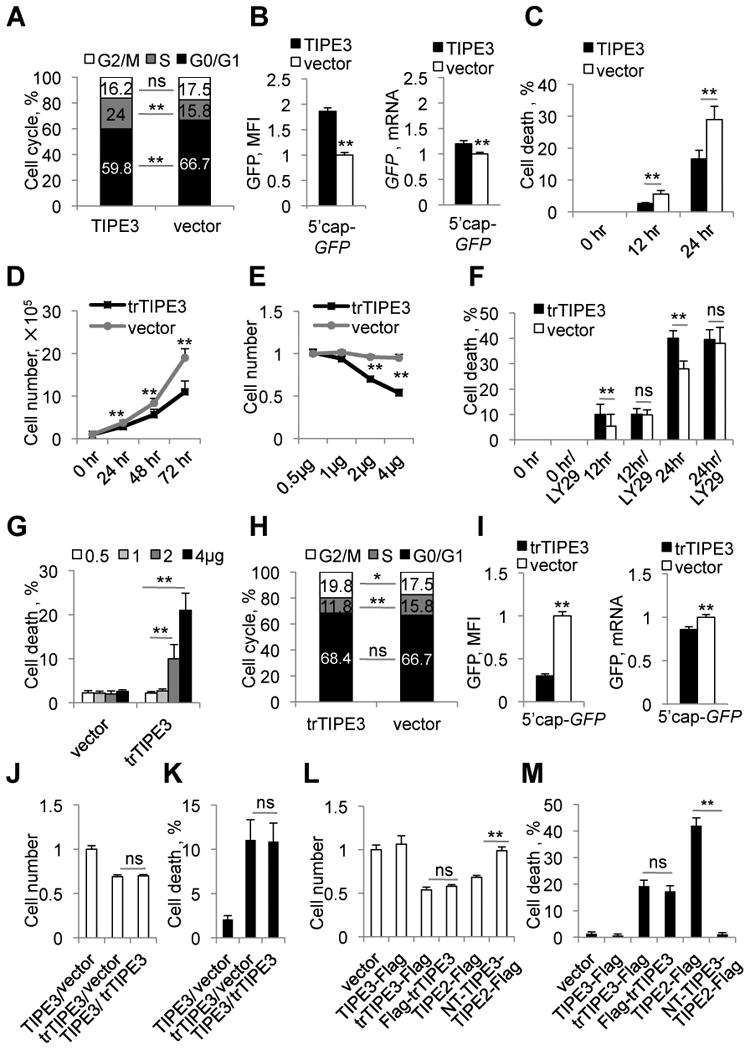
(A) Cell cycle analysis of NIH3T3 cells stably transfected with either TIPE3-Flag or empty vector. (B) 293T cells were co-transfected with pEGFP-C3 (5′cap-GFP, for cap-dependent translation) and either TIPE3-Flag-expressing or empty vector. The relative levels of GFP protein expression 32 hr after transfection were quantified by measuring the cellular mean fluorescence intensity (MFI) (left panel), whereas the relative levels of GFP mRNA were determined by real-time PCR (right panel). Protein and mRNA levels of cells transfected with empty vector were set as 1. (C) NIH3T3 cells stably transfected with either TIPE3-Flag or empty vector were cultured in serum-free medium for the indicated times, and cell death was assessed by trypan blue staining. (D) Cell growth of NIH3T3 cells stably transfected with either trTIPE3-Flag or empty vectors over indicated times. (E) Relative numbers of 293T cells transfected with the indicated amounts of trTIPE3-Flag plasmid or empty vector were determined 32 hr after transfection. Number of 293T cells transfected with 0.5 μg of empty vector was set as 1. (F) NIH3T3 cells stably transfected with either trTIPE3-Flag or empty vectors were cultured in serum-free medium with or without LY29 (LY29004) for the indicated times. Cell death was assessed by trypan blue staining. (G) Cell death of 293T cells transfected with the indicated amounts of trTIPE3-Flag plasmid or empty vector were assessed by trypan blue staining. (H) Cell cycle analysis of NIH3T3 cells stably transfected with either trTIPE3-Flag or empty vectors. (I) 293T cells were co-transfected with pEGFP-C3 and either trTIPE3-Flag-expressing or empty vectors. The relative levels of GFP protein expression 16 hr after transfection were quantified by measuring cellular MFI, whereas the relative levels of the GFP mRNA expression were determined by real-time PCR. Protein and mRNA levels of cells transfected with empty vector were set as 1. (J and K) 293T cells were co-transfected with equal amounts (2 μg) of the following plasmids as indicated: empty vectors, TIPE3-Flag- or trTIPE3-Flag-expressing vectors. Relative cell numbers were determined (J), and the degree of cell death (K) was assessed by trypan blue staining 32 hr later. For (J), number of 293T cells transfected with empty vector was set as 1. (L and M) 293T cells were transfected with one of the following plasmids as indicated: empty vector, TIPE3-Flag, trTIPE3-Flag, Flag-trTIPE3, TIPE2-Flag, NT-TIPE3-TIPE2-Flag (TIPE2-Flag fused with the NT region of TIPE3). Relative cell numbers were determined (L), and the degree of cell death (M) was assessed by trypan blue staining 32 hr later. For (L), number of 293T cells transfected with empty vector was set as 1. Y-axis values represent means ± SD; ns denotes not significant; * p<0.05, and ** p< 0.01; h denotes hr. The experiments were performed in duplicates and repeated at least three times with similar results. See also Figure S3.
As stated above, TIPE3 overexpression reduced the percentage of dying tumor cells in nude mice. In vitro, a similar effect was observed in cultured NIH3T3 cells. TIPE3 significantly decreased serum deprivation-induced death of NIH3T3 cells as assessed by trypan blue staining (Figure 3C) and cleaved caspase-3 staining (Figure S3C), and the addition of the PI3K inhibitor LY294002 abolished this effect (Figure S3, C and D).
All TIPE family proteins contain a highly conserved TIPE2 homology (TH) domain consisting of seven α helixes (Figure S3E) (Zhang et al., 2009). TIPE3 has a unique N-terminal (NT) sequence of 19 amino acids, designated as the NT region here, which is not seen in other members of the TIPE family. Hypothesizing that the TIPE3 NT region is important for its unique ability to promote cell growth and survival, we investigated the effect of deleting it. A truncated TIPE3 variant lacking amino acids 2-20 (trTIPE3) not only lost the function of TIPE3, but appeared to exert a dominant negative effect. Overexpression of trTIPE3 significantly reduced cell growth, decreased the number of S-phase cells, enhanced cell death, and reduced cap-dependent translation (Figure 3, D-I). The trTIPE3 effect on serum starvation-induced cell death was not further enhanced by LY294002, suggesting that trTIPE3 might inhibit the PI3K pathway (Figure 3F). Importantly, when co-transfected into the same cells, the inhibitory effects of trTIPE3 on cell growth and death prevailed over those of WT TIPE3, indicating that trTIPE3 may function in a dominant negative fashion (Figure 3, J and K). It is to be noted that location of the Flag-tag did not affect TIPE3 functions (Figure 3, L and M). TIPE2, another member of the TIPE family that exhibits similar activities as trTIPE3 in cell growth and death, completely lost its function when fused to the NT region of TIPE3 (Figure 3, L and M).
TIPE3 promotes activation of the PI3K-AKT and MEK-ERK pathways
TIPE3 overexpression in NIH3T3 cells substantially enhanced AKT phosphorylation, whereas trTIPE3 overexpression reduced it, and similar effects were seen for phosphorylation of ERK and p70S6K (Figure 4A and Figure S4A). Both AKT and ERK regulate cyclin D1 levels and thus influence cell cycle G1-S transition. Cyclin D1 levels were elevated in NIH3T3 cells transfected with TIPE3 and decreased in cells transfected with trTIPE3 (Figure 4A and Figure S4A). Additionally, TIPE3 enhanced HRasV12-induced phosphorylation of AKT and ERK in NIH3T3 cells (Figure 4A and Figure S4A).
Figure 4. TIPE3 promotes the activation of PI3K-AKT and MEK-ERK pathways.
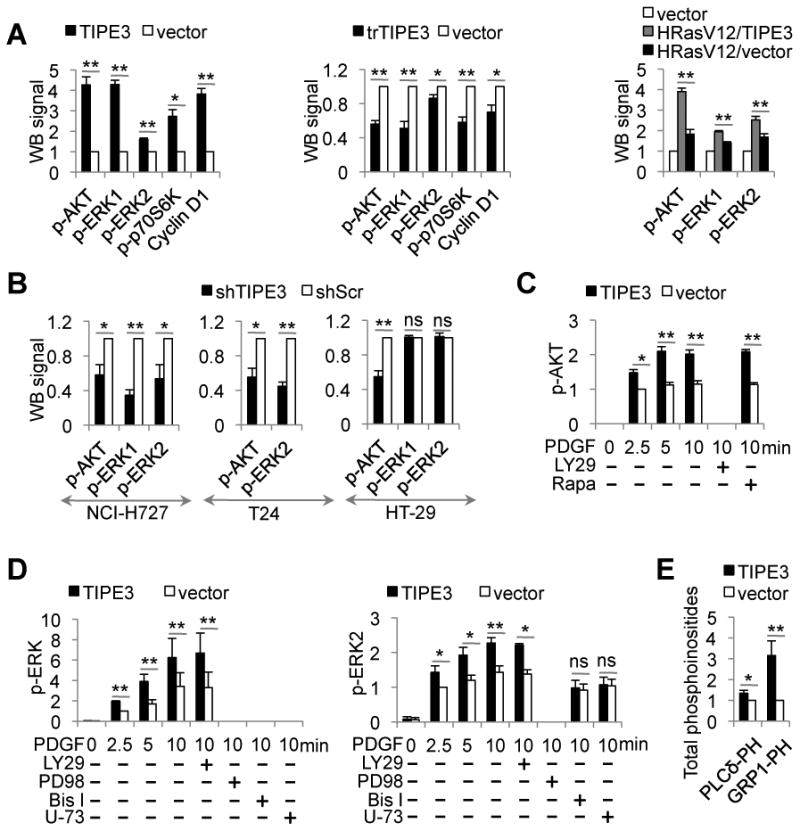
(A) Whole cell lysates were prepared from NIH3T3 cells stably transfected with TIPE3-Flag, trTIPE3-Flag, or empty vectors (left two panels), or TIPE3-Flag and/or HRasV12 vectors (right panel). Western blot (WB) was performed using antibodies against the indicated proteins. The densitometric quantification of protein signals was made using ImageJ software. Signals of cells transfected with empty vector were set as 1. (B) Whole cell lysates were prepared from NCI-H727, T24, and HT-29 cells stably expressing either shTIPE3 or shScr. Western blot was performed using antibodies against the indicated proteins. Signals of cells treated with shScr were set as 1. (C and D) NIH3T3 cells stably transfected with either TIPE3-Flag or empty vectors were serum-starved, and then stimulated with 40 ng/ml of PDGF for the indicated times, with or without the following inhibitors: LY29 (LY29004), Rapa (Rapamycin), PD98 (PD98059), Bis I (Bisindolylmaleimide I), or U-73 (U-73122). Lysates of these cells were used for Western blot with antibodies against the indicated proteins. Signals of cells transfected with vector 2.5 min after PDGF stimulation were set as 1. (E) Cellular levels of PtdIns(4,5)P2 and PtdIns(3,4,5)P3 in NIH3T3 cells stably transfected with either TIPE3-Flag or empty vector were estimated by protein-lipid overlay assay with GST-PLCδ-PH and GST-GRP1-PH domains as described in Supplemental Experimental Procedures. Signals of cells transfected with empty vector were set as 1. Y-axis values represent means ± SD; ns denotes not significant; * p<0.05, and ** p< 0.01. p- indicatedes phosphorylated. The experiments were performed at least three times with similar results. See also Figure S4.
On the other hand, TIPE3 knockdown human NCI-H727 and T24 carcinomas cells and Tipe3 knockout murine fibrosarcoma cells showed significantly decreased phosphorylation of both AKT and ERK (Figure 4B and Figure S4, B and C). In HT-29 cells, TIPE3 knockdown impaired AKT activation, but did not affect ERK activation.
To gain more insight into TIPE3-mediated regulation of AKT and ERK activation, we treated NIH3T3 cells with PDGF in the absence or presence of overexpressed TIPE3. PDGF markedly increased the levels of phosphorylated AKT and ERK, which were further augmented by TIPE3 (Figure 4, C and D, and Figure S4, D and E). The PI3K inhibitor LY294002 completely blocked AKT phosphorylation, whereas the mTOR inhibitor rapamycin had no effect (Figure 4C and Figure S4D), indicating that TIPE3 amplifies PI3K-dependent activation of AKT. However, LY294002 did not change the level of ERK phosphorylation, whereas PD98059, a MEK inhibitor, abolished ERK phosphorylation entirely, with or without TIPE3 overexpression. Interestingly, U-73122, an inhibitor of PLC, and bisindolylmaleimide I, an inhibitor of PKC, overpowered the effect of TIPE3 on ERK phosphorylation (Figure 4D and Figure S4E).
TIPE3 regulates phosphoinositide metabolism and signaling
We observed significant increases in the total levels of PtdIns(4,5)P2 in TIPE3-overexpressing cells using three approaches: protein-lipid overlay assay with PLCδ-PH domain that specifically binds PtdIns(4,5)P2 (Figure 4E), immunofluorescence confocal microscopy of fixed cells with anti-PtdIns(4,5)P2 antibody (Figure S4, F and G), and immunoblotting with an anti-PtdIns(4,5)P2 antibody (Figure S4H). Similarly, the total pool of PtdIns(3,4,5)P3 was also markedly increased (∼4 fold) in TIPE3-overexpressing cells (Figure 4E and Figure S4H). Consistent with these results, significant reductions in the PtdIns(4,5)P2 and PtdIns(3,4,5)P3 cellular levels were observed in Tipe3 knockout murine fibrosarcoma cells as compared to wild-type cells (Figure S4I). The effect of over-expressed TIPE3 was dependent on the NT region since the trTIPE3 that lacks it decreased PtdIns(4,5)P2 and PtdIns(3,4,5)P3 levels (Figure S4J).
To visualize changes in PtdIns(4,5)P2 and PtdIns(3,4,5)P3 levels following stimulation of cells with PDGF, we performed confocal microscopy of fixed cells using GFP-tagged PH domains. In untreated NIH3T3 cells, PLCδ-PH-GFP, which selectively binds PtdIns(4,5)P2, was localized predominantly to the plasma membrane (Figure 5A and Figure S5A). PDGF stimulation (for 5.5 min) significantly reduced membrane-bound but increased cytoplasmic PLC5-PH-GFP signals, consistent with PI3K- and PLC-induced PtdIns(4,5)P2 depletion. Remarkably, TIPE3 overexpression significantly increased the membrane-bound PLCδ-PH-GFP signal in both untreated and PDGF-treated cells, whereas trTIPE3 overexpression greatly decreased it (Figure 5A and Figure S5A). Importantly, TIPE3 overexpression completely eliminated the PDGF-induced reduction of the membrane-bound PLCδ-PH-GFP signal (vector vs. TIPE3 groups) indicating that PtdIns(4,5)P2 depletion was not significant in the presence of overexpressed TIPE3. The GRP1 and AKT PH domains selectively bind PtdIns(3,4,5)P3 and PtdIns(3,4,5)P3/PtdIns(3,4)P2, respectively (Ferguson et al., 2000). In untreated NIH3T3 cells, AKT-PH-GFP was localized primarily to the cytoplasm, and weakly to the nucleus and plasma membrane; GRP1-PH-GFP was localized prominently to the nucleus and weakly to the cytoplasm (Figure 5, B and C, and Figure S5, B and C). A few minutes after PDGF stimulation, both AKT-PH-GFP and GRP1-PH-GFP showed a noticeable translocation to the plasma membrane. TIPE3 significantly increased the levels of plasma membrane-bound AKT-PH-GFP and GRP1-PH-GFP in both untreated and PDGF-stimulated cells. These TIPE3 effects were dependent on the NT region since the trTIPE3 that lacks it had the opposite effects (Figure 5, B and C, and Figure S5, B and C).
Figure 5. TIPE3 regulates lipid metabolism and signaling.
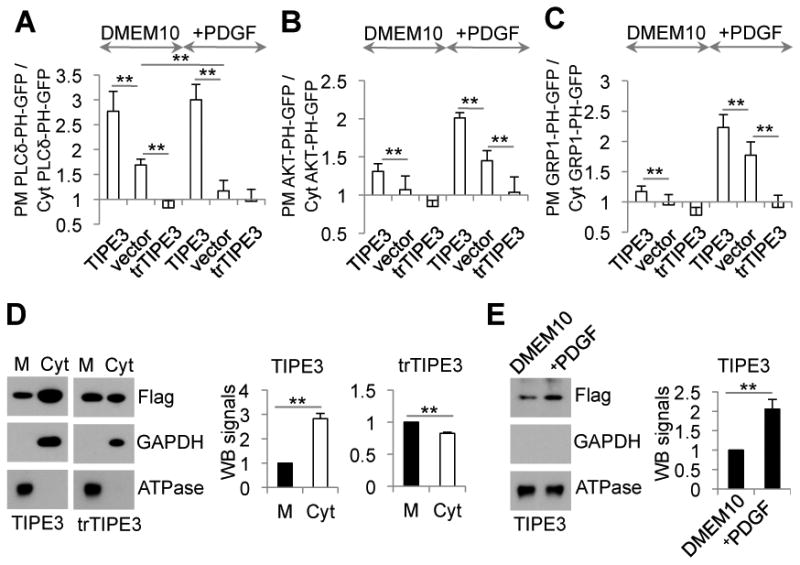
(A-C) NIH3T3 cells were co-transfected with (A) PLCδ-PH-GFP, (B) AKT-PH-GFP, or (C) GRP1-PH-GFP and one of the following plasmids as indicated: pRK5 vector (vector), pRK5-TIPE3-Flag (TIPE3), and pRK5-trTIPE3-Flag (trTIPE3). Thirty-two hr after the transfection, cells were cultured in DMEM with 10% FCS (DMEM10, A-C), or in DMEM10 with 40 ng/ml of PDGF for 5.5 min (+PDGF, A), or serum-starved for 4 hr and then cultured in DMEM containing 40 ng/ml of PDGF for 7.5 min (+PDGF, B and C) followed by fixation. Cells expressing the Flag-tagged proteins were selected by immunofluorescence microscopy with an anti-Flag antibody. Ratios of GFP fluorescence intensity on plasma membrane (PM) over that in the cytoplasm (Cyt) of the same cells are calculated based on line fluorescence intensity profiles. (D) Membrane (M) and cytoplasmic (Cyt) proteins fractions were prepared from NIH3T3 cells stably transfected with TIPE3-Flag and trTIPE3-Flag. Western blot was performed using antibodies against the indicated proteins. Signals of TIPE3 or trTIPE3 in membrane fractions were set as 1. (E) NIH3T3 cells stably transfected with TIPE3-Flag were cultured in DMEM with 10% of FCS (DMEM10) or serum-starved for 4 hr and then cultured in DMEM containing 40 ng/ml of PDGF for 7.5 min (+PDGF). Membrane protein fractions (M) of these cells were analyzed by Western blot using antibodies against the indicated proteins. Signals of TIPE3 in membrane fractions of cells cultured in DMEM10 were set as 1. For (A-E), values represent means ± SD; **p<0.01. The experiments were repeated at least three times with similar results. See also Figure S5.
Although slightly concentrated at the plasma membrane, TIPE3 protein was distributed throughout the cytoplasm of untreated cells (Figure 5D and Figure S5, A-C). Upon PDGF activation, TIPE3 protein translocated from cytoplasm to plasma membrane, yielding a clear appearance of plasma membrane localization (Figure 5E and Figure S5, A-C). No such PDGF-induced effect was seen for the trTIPE3 protein, although a fraction of the protein was also localized to the plasma membrane (Figure 5D and Figure S5, A-C).
To determine whether altering TIPE3 subcellular localization changes the phosphoinositide distribution, we fused TIPE3 with GFP to force it going into the nucleus as previously described (Seibel et al., 2007). We found that TIPE3 was excluded from the nucleus whereas GFP and the TIPE3-GFP fusion protein were present in the nucleus, indicating that GFP confers the ability for TIPE3 to enter this organelle (Figure S5, D-F). Importantly, we observed strong PtdIns(4,5)P2 staining in the nuclei of all cells expressing TIPE3-GFP, but not in those of cells expressing TIPE3 or GFP alone, or non-transfected cells (p<0.0001) (Figure S5, D-F).
The crystal structure of TIPE3 reveals a lipid-binding scaffold
To help elucidate the molecular mechanism by which TIPE3 functions, we sought to determine its crystal structure. We crystallized the full-length conserved TH-domain, i.e., a trTIPE3 that lacks amino acids 2-20, in the space group I222 (PDB ID: 4Q9V). The structure was determined by molecular replacement and the atomic model was refined at 2.3 Å resolution (Figure 6A and Table S1).
Figure 6. The crystal structure of the TH-domain of human TIPE3.
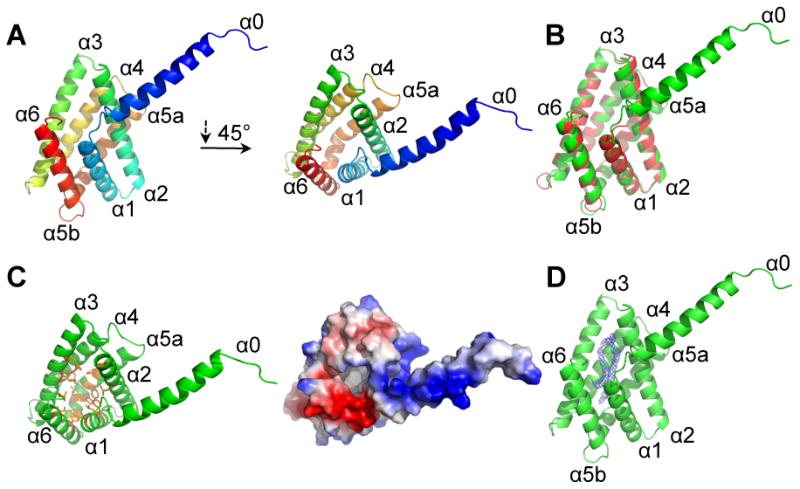
(A) Cartoon presentation of TH-domain of human TIPE3 (trTIPE3, residues 21-204). Helices are rainbow-colored with α0 in blue and α6 in red. (B) Cartoon representation of superposition of human TIPE3 (residues 21-204, shown in green) with the human TIPE2 (residues 24-184, shown in red) structures. (C) TIPE3 contains a centrally located hydrophobic cavity. (Left panel) TIPE3 is shown in green cartoon representation; amino acids with hydrophobic side chains that line the cavity are shown in orange. (Right panel) TIPE3 eletrostatic surface potential: blue, positive; red, negative. (D) TIPE3 is shown in green cartoon representation. The two long connected tubes of 2Fo-Fc electron density found in TIPE3 cavity is shown in blue mesh, contoured at 1.5σ. See also Table S1 and Figure S6.
The structure of the crystallized TIPE3 consists of an N-terminal α0 helix, and six additional helices (α1-α6) that are similar to those observed for TIPE2 (residues 24-184, PDB ID: 3F4M) (Zhang et al., 2009). The helices α1-α6 of TIPE3 can be aligned to those of TIPE2 with a root-mean-squared deviation (RMSD) of 0.574 Å over 128 aligned Cα atoms (Figure 6B). The most prominent feature of the TH fold is the presence of a large centrally located hydrophobic cavity (Figure 6C), which, with 20 Å in depth and 10 Å in diameter, was thought to be the binding site for a lipophilic molecule. Similar to TIPE2 (Zhang et al., 2009), the hydrophobic cavity in TIPE3 is occupied by two tubes of electron density (each measuring approximately 20 Å in length), which are connected at the surface opening of the cavity (Figure 6D). The shape and size of the electron density are reminiscent of two aliphatic tails of a phospholipid, although the head group cannot be identified due to poor electron density. Importantly, positive ion electrospray ionization mass spectrometry of lipids co-purified with trTIPE3 from bacteria revealed two major peaks: 653 mass-to-charge and 683 mass-to-charge. The molecular mass of the first peak is indicative of 16:0/16:1 phosphatidc acid (PA), whereas that of the second peak is indicative of 16:0/18:0 or 16:0/18:1 PA. Therefore, it is likely that PAs are the phospholipids that occupy the hydrophobic cavity of trTIPE3 expressed in bacteria (which do not possess phosphoinositides).
Within the same asymmetric unit, helix α0 from one TIPE3 molecule forms a coiled coil with that of another TIPE3 molecule, constituting a homodimer (Figure S6A). These two α-helices interact with each other mainly through van der Waals contacts. In addition, the α0 helix from one TIPE3 molecule also interacts with the α1 helix of its partner molecule. Notably, truncation of the α0 helix exhibits no significant effect on the elution profile of TIPE3 on gel filtration, suggesting that the observed TIPE3 dimer is likely an artifact of crystallization at high protein concentrations.
TIPE3 binds to phosphoinositides through its TH domain, and this binding is essential for its function
Next, we examined whether affinity-purified TIPE3 exhibits any binding specificity for the various eukaryotic lipid molecules in the protein-lipid overlay assay (Figure S6, B-D). Of the 22 different types of eukaryotic lipids tested, TIPE3 appeared to predominantly interact with PtdIns(4,5)P2, PtdIns(3,5)P2, PtdIns(3,4)P2, PtdIns4P, PtdIns(3,4,5)P3, and PA (Figure S6, C and D, and data not shown). The trTIPE3, as well as TIPE2, TIPE1, and TNFAIP8 proteins interacted with the same set of lipids, indicating that the highly conserved TH domain, but not the NT region, is responsible for the observed lipid binding activity (Figure S6, B, C, and E). By contrast, the PLCδ-PH domain (GST-PLCδ-PH) interacted only with PtdIns(4,5)P2 in the same assay (Figure S6C). PLCδ-PH binds to the PtdIns(4,5)P2 head-group, inositol-1,4,5-trisphosphate (Ins(1,4,5)P3), and addition of 15 μM free Ins(1,4,5)P3 to the overlay assay was sufficient to prevent PLCδ-PH binding to nitrocellulose-bound PtdIns(4,5)P2 (Figure S6C). However, adding free Ins(1,4,5)P3 had no effect on TIPE3-binding to PtdIns(4,5)P2, suggesting that the mode of interaction is different from that between PLCδ-PH and PtdIns(4,5)P2 (Figure S6C). This is consistent with our structural data, which identifies an anchoring role for the acyl chains of phospholipid (Figure 6D).
To test the importance of the hydrophobic cavity in the TIPE3 TH domain, we replaced leucine 60, positioned in the center of this pocket, with the much bulkier tryptophan to yield the TIPE3 60W mutant (Figure S7A). A similar approach was described to test the importance of the hydrophobic cavity in orphan nuclear hormone receptors hypothesized to bind phosphoinositides (Krylova et al., 2005; Sablin et al., 2003). TIPE3 60W mutant showed markedly reduced ability to bind phosphoinositide (Figure S7B). This finding is consistent with the structure-based proposition that the acyl chains of phosphoinositide are positioned inside the TH domain cavity, allowing the negatively charged head group to form electrostatic interactions with the neighboring positively charged side chains that line the opening of the cavity (Figure 6, C and D). Supporting this analysis, a TIPE3 4Q mutant, in which lysines 33, 34, 38, and 42 were all replaced with glutamines, and a TIPE3 2Q mutant, in which arginines 93 and 109 were replaced with glutamines, both exhibited marked reductions in phosphoinositide binding (Figure S7, A and B). By contrast, a TIPE3 3Q mutant – in which glutamine replaces lysines 76, 79, and 83 that lie on a positive-charged surface distant from the cavity opening – showed wild-type levels of phosphoinositide binding in the overlay assay (Figure S7, A and B).
Importantly, the TIPE3 mutants that showed impaired phosphoinositide binding also had markedly reduced TIPE3 functions. Compared with wild-type TIPE3, the TIPE3 60W, 2Q, and 4Q mutants had significantly reduced capacities to promote cap-dependent translation of GFP (Figure S7C), whereas TIPE3 3Q (which retains phosphoinositide binding) did not – although all mutants were expressed at similar levels as the wild-type TIPE3 (Figure S7, C and D). As expected, TIPE3 4Q had no detectable effects on ERK activation or PtdIns(4,5)P2 levels and might even reduce the levels of AKT phosphorylation and PtdIns(3,4,5)P3 (Figure S7, E and F). Introduction of a 4Q-equivalent mutation in trTIPE3 abolished its ability to bind phosphoinositide (Figure S7B). Moreover, 4Q, 2Q, and 60W mutations in trTIPE3 reduced its ability to inhibit cell proliferation and to induce cell death (Figure S7, G-I), whereas the 3Q mutation had no such effect.
TIPE3 can act as a PtdIns(4,5)P2 and PtdIns(3,4,5)P3 transfer protein
Although the TH fold is unique, the lipid-binding mode by the TIPE family of proteins is reminiscent of lipid-binding mode by lipid transfer proteins (Prinz, 2010). We wondered whether phosphoinositide-loaded TIPE3 might exert its effects in part by transferring phosphoinositides to the plasma membrane and that the dominant negative trTIPE3 might promote cell death by reducing the membrane pool of PtdIns(4,5)P2 and PtdIns(3,4,5)P3. Consistent with this notion, trTIPE3-induced cell death could be partially reversed by overexpressing a myristoylated variant of phosphatidylinositol-4-phosphate 5-kinase (PIP5K) that leads to elevated plasma membrane PtdIns(4,5)P2 (Figure S7J) (Volpicelli-Daley et al., 2010; Wang et al., 2008). Neither a constitutively active PI3K (PI3KCAE545) nor HRasV12 had any detectable effect on trTIPE3-induced cell death (Figure S7J).
To determine whether wild-type TIPE3 protein might have properties consistent with a phosphoinositide transfer protein, we examined whether it could recognize PtdIns(4)P, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 present in a lipid bilayer, and extract the lipid from the membrane. We purified recombinant TIPE3 and TIPE3 4Q from mammalian cells (Figure S6B) and assessed their binding to small unilamellar vesicles (SUVs) containing 100% phosphatidylcholine (PC), 10% PtdIns(4)P, 10% PtdIns(4,5)P2 or 10% PtdIns(3,4,5)P3 in a PC background using a well-established centrifugation-based assay (Kavran et al., 1998; Lee and Lemmon, 2001). TIPE3 sedimented efficiently with vesicles containing 10% phosphoinositides with the following order of preference: PtdIns(3,4,5)P3 > PtdIns(4,5)P2 > PtdIns(4)P (Figure 7, A and B). Consistent with the results of the overlay assay, TIPE3 4Q showed significantly reduced binding to PtdIns(4,5)P2- and PtdIns(3,4,5)P3-containing vesicles compared with wild-type TIPE3; more than 60% of the 20 μM TIPE3 and only ∼30% of 20 μM TIPE3 4Q were bound to vesicles when the concentration of vesicles was 1000 μM (Figure 7, A and B). Lack of binding specificity of TIPE3 4Q to PtdIns(4)P, PtdIns(4,5)P2 and PtdIns(3,4,5)P3-containing vesicles suggests that lysines in the α0 helix may form electrostatic interactions with 5- and 3-phosphate groups of PtdIns(4,5)P2 and PtdIns(3,4,5)P3. By contrast, only background levels of TIPE3 and TIPE3 4Q sedimented with 100% PC vesicles. A negative control protein (trypsin inhibitor) showed no binding to either type of vesicles (Figure 7, A and B).
Figure 7. TIPE3 is a phosphoinositide transfer protein.
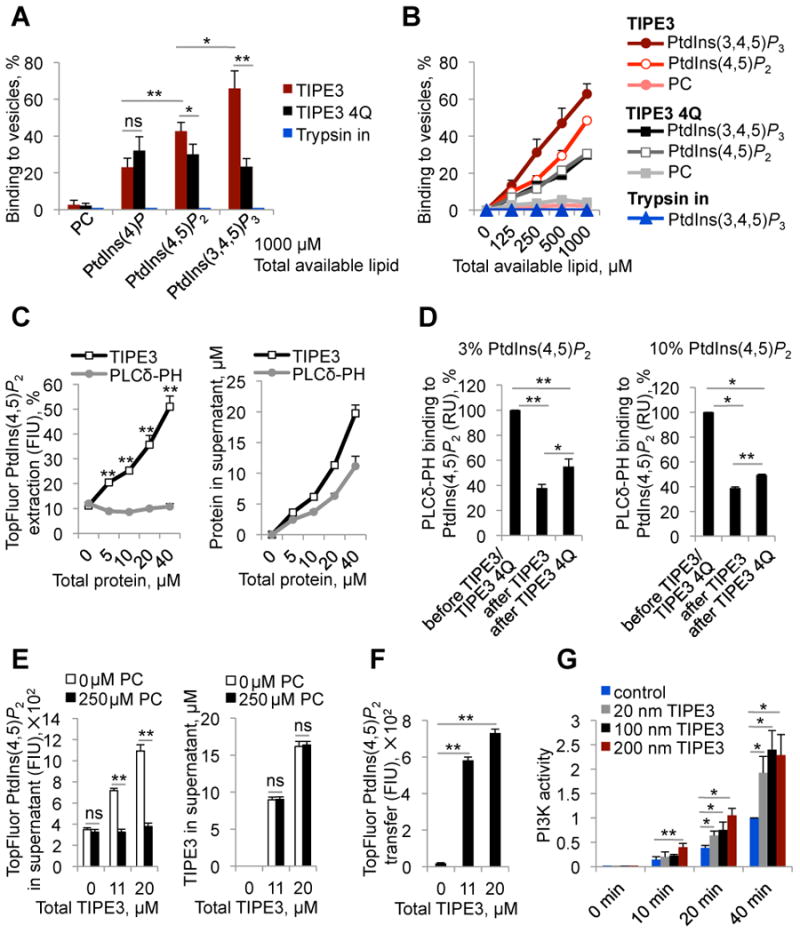
(A and B) The sedimentation-based binding assay was used to determine the percentages of TIPE3, TIPE3 4Q, and control protein trypsin inhibitor (Trypsin in) bound to vesicles containing 10% PtdIns(4)P+90% PC [10% PtdIns(4)P], 10% PtdIns(3,4,5)P3+90% PC [10% PtdIns(3,4,5)P3], 10% PtdIns(4,5)P2+90% PC [10% PtdIns(4,5)P2], or 100% PC as indicated. Proteins were used at a concentration of 20 μM. (C) The sedimentation-based extraction assay was used to determine (i) the percentages of TopFluorPtdIns(4,5)P2 extracted from 100 μM 20% TopFluorPtdIns(4,5)P2+80% PC vesicles and transferred to supernatant by TIPE3 and PLCδ-PH (left panel), and (ii) fractions of TIPE3 and PLCδ-PH proteins not bound to the 20% TopFluorPtdIns(4,5)P2+80% PC vesicles (right panel). (D) In the SPR-based assay, 3% (left panel) and 10% (right panel) PtdIns(4,5) P2-containing vesicles were immobilized on L1 sensor chips, and the binding signal obtained with a saturating injection of GST-PLCδ-PH (5 μM) was measured before and after TIPE3 or TIPE3 4Q (both at 40 μM) had been flowed over the sensor chip. GST-PLCδ-PH binding to PtdIns(4,5)P2-containing surfaces before TIPE3 or TIPE3 4Q exposure is set as 100% and GST-PLCδ-PH binding to PtdIns(4,5) P2-containing surfaces after TIPE3 or TIPE3 4Q exposure is presented as the percentage reduction. (E) The sedimentation-based transfer assay was used to measure the depletion of TopFluorPtdIns(4,5)P2 from soluble TIPE3-TopFluorPtdIns(4,5)P2 in supernatants (left panel) and fractions of TIPE3 remaining in supernatants (right panel) after incubation with 100% PC vesicles or buffer alone. (F) The sedimentation-based transfer assay was used to monitor the transfer of TopFluorPtdIns(4,5)P2 from soluble TIPE3-TopFluorPtdIns(4,5)P2 in supernatants to 100% PC vesicles. (G) Time course of PI3K (p110α/p85α)-mediated phosphorylation of PtdIns(4,5)P2 in the absence or presence of TIPE3 at indicated concentrations. The levels of PtdIns(3,4,5)P3 were measured by protein-lipid overlay assay with GST-GRP1-PH domain. PI3K activity determined at 40 min in the absence of TIPE3 was set to 1. FIU, fluorescence intensity units; RU, resonance units. Values represent means ± SD; ns denotes not significant; * p<0.05, and ** p<0.01. For (A-F), the experiments were repeated at least three times with similar results. For (G), the experiments were performed in triplicates and repeated two times with similar results. See also Figure S7.
Having established that TIPE3 could bind to vesicles containing PtdIns(4,5)P2, we next asked whether the protein could extract fluorescently labeled phosphoinositide from these vesicles, and effectively solubilize it. When increasing concentrations of TIPE3 were incubated for 1 hour with 100 μM vesicles containing 20% TopFluorPtdIns(4,5)P2 and 80% PC, substantial amounts of TopFluorPtdIns(4,5)P2 fluorescence was detected in the supernatant after ultracentrifugation – whereas equivalent experiments with the PLCδ-PH domain showed no significant fluorescence in the supernatant (Figure S6B and Figure 7C). This result suggests that TIPE3 can effectively remove PtdIns(4,5)P2 from the vesicles and solubilize it in the aqueous phase – extracting up to 50% of the total available PtdIns(4,5)P2 at a protein concentration of 40 μM. Size-exclusion chromatographic analysis of the supernatants revealed a single peak corresponding to TIPE3 monomer, ruling out the possibility that TIPE3/vesicle complexes remain in the supernatants (data not shown). Supporting its dominant negative effect, trTIPE3 did not remove TopFluorPtdIns(4,5)P2 after binding to 20% TopFluorPtdIns(4,5)P2-containing vesicles; ∼10% of 20μM trTIPE3 remained in supernatant (Figure S7K). Similar to trTIPE3, TIPE2 strongly bound to vesicles containing 20% PtdIns(4,5)P2 and 80% PC, with only ∼15% remained unbound (data not shown). Importantly, neither TIPE3 nor TIPE3 4Q was able to bind and extract TopFluorPC from 100 μM vesicles containing 20% TopFluorPC and 80% PC used as control (Figure S7K).
We further investigated the ability of TIPE3 to extract PtdIns(4,5)P2 from membranes using a surface plasmon resonance (SPR)-based assay. PtdIns(4,5)P2-containing vesicles (3% and 10%) were immobilized on L1 sensor chips, and the binding signal obtained with a saturating injection of PLCδ-PH was measured before and after TIPE3 or TIPE3 4Q had been flowed over the sensor chip. A significant reduction (more than 60%) in PLCδ-PH binding at saturation was detected after TIPE3 had been exposed to the sensor chip (Figure 7D), indicating that TIPE3 extracts PtdIns(4,5)P2 from the immobilized vesicles. TIPE3 4Q also appeared to extract PtdIns(4,5)P2 from the immobilized membranes, but to a reduced extent – consistent with our other studies of this mutant (Figure 7D).
As discussed above, TIPE3 overexpression appears to increase both total and plasma membrane levels of PtdIns(4,5)P2 in NIH3T3 cells. To determine whether TIPE3 is capable of inserting extracted PtdIns(4,5)P2 into a lipid bilayer, we analyzed transfer of TopFluorPtdIns(4,5)P2 from the soluble TIPE3/TopFluorPtdIns(4,5)P2 complexes detected in the supernatant in Figure 7C to 100% PC vesicles. We collected supernatants from mixtures of TIPE3 with 100 μM TopFluorPtdIns(4,5)P2-containing vesicles that had been subjected to ultracentrifugation, and incubated them with vesicles containing 250 μM 100% PC for 1 h at room temperature. The mixtures were then re-centrifuged as described (Lee and Lemmon, 2001), and the level of TopFluorPtdIns(4,5)P2 fluorescence remaining in the supernatant was measured. Addition of vesicles containing 100% PC alone led to a substantial reduction in fluorescence corresponding to presumed TIPE3/TopFluorPtdIns(4,5)P2 complexes in the supernatant (Figure 7E). This was accompanied by a dramatic increase in fluorescence intensity associated with the vesicle pellet (Figure 7F), indicating that TopFluorPtdIns(4,5)P2 was transferred into the PC vesicles. The concomitant loss of TopFluorPtdIns(4,5) P2-derived fluorescence from the supernatant and its acquisition in the pellet was not accompanied by any change in TIPE3 protein concentrations in the supernatant, arguing that TIPE3 has transferred the labeled PtdIns(4,5)P2 to the PC vesicles (Figure 7E).
TIPE3 can promote the generation of PtdIns(3,4,5)P3 by PI3K
To examine if TIPE3 could promote the phosphorylation of PtdIns(4,5)P2 by PI3K(p110α/p85α), we performed in vitro membrane capture (enzymatic) assay for the PI3K in the absence or presence of increasing amounts of TIPE3 (Knight et al., 2007). SUVs containing 10% PtdIns(4,5)P2 in a PC background were employed in this assay. We found that TIPE3 indeed stimulated PI3K activity in a concentration-dependent manner (Figure 7G). At 20 min after the initiation of the PI3K assay, we observed 3 times more PtdIns(3,4,5)P3 in reactions containing 200 nM TIPE3, 2 times more PtdIns(3,4,5)P3 in reactions containing 100 nM TIPE3, and 1.65 times more PtdIns(3,4,5)P3 in reactions containing 20 nM TIPE3 than those of control PI3K reactions lacking TIPE3 (BSA was used as a control protein). Finally, we examined if TIPE3 associated with PI3K. We immunoprecipitated endogenous p110α, p110β, and p85 from NIH3T3 cells overexpressing TIPE3, with or without PDGF treatment, and found that TIPE3 did not co-immunoprecipitate with these PI3K proteins. In addition, we did not detect an association of TIPE3 with the oncogenic mutant of p110α, E545K, when both proteins were overexpressed in 293T cells.
Discussion
Phosphatidylinositol transfer proteins (PITPs) play essential roles in phospholipid signaling, and their dysregulation leads to a variety of disorders including cancer and degenerative diseases (Nile et al., 2010). It is unclear whether and to what degree currently known PITPs or PITP-like proteins can transfer phosphoinositides. It was reported that the yeast Kes1/Osh4p protein (de Saint-Jean et al., 2011) and the human α-TTP protein (Kono et al., 2013) that belongs to the Sec14p-like family might transfer PtdIns4P and PtdIns(4,5)P2, respectively, but not other phosphoinositides. The mammalian nuclear receptor SF-1 and LRH-1 proteins of the NR5A family were also found to accommodate phosphoinositides within their large hydrophobic cavities (Krylova et al., 2005), but whether they can transfer these lipids are unknown.
Results reported here argue that the TH-domain of TIPE proteins can function as a lipid transfer domain, and that TIPE3 is a lipid transfer protein for two second messengers, PtdIns(4,5)P2 and PtdIns(3,4,5)P3. Crystal structure and mutagenesis analyses revealed that the acyl chains of lipid is likely accommodated in the hydrophobic cavity formed by the α1-α6 helixes of the TIPE3 TH-domain. The negatively charged head group of phosphoinositides is likely involved in electrostatic interactions with positively charged amino acids of the α0 helix (that might function as a flexible “lid”) and those located at the entrance to the hydrophobic cavity. Structure determination of the full-length TIPE3 bound to phosphoinositides is required to provide additional insight into this issue.
Our results support a dual role model for TIPE3 in phosphoinositide signaling and metabolism. TIPE3 may regulate the spatial and temporal distribution of lipid second messengers in response to external or internal stimuli by sequestrating PtdIns(4,5)P2 and PtdIns(3,4,5)P3 from lipid bilayer and shuttling them through aqueous phase. Additionally, TIPE3 may also function as a lipid-presenting protein to enhance the activity of phosphoinositide-modifying enzymes such as PI3K. A similar function has been reported for phosphatidylinositol transfer proteins such as Sec14, the major yeast PITP protein that regulates phosphoinositide signaling and metabolism by presenting phosphatidylinositol to PI4K (Bankaitis et al., 2010; Lev, 2010; Prinz, 2014; Schaaf et al., 2008). Consistent with this view, TIPE3 physically translocates from cytoplasm to plasma membrane upon cell activation by PDGF or upon cell transformation by the oncogenic HRasV12 (data not shown). Upregulation of TIPE3 expression in PDGF-stimulated cells and cancer cells may positively feed back to cell activation through the aforementioned mechanism.
The TIPE3 TH domain is highly homologous to TH domains of other members of the TIPE family. The domain is also highly conserved through evolution, found in vertebrates, invertebrates such as mosquitoes and fruit fly, and unicellular eukaryotes such as choanoflagellate, Dictyostelium, and Entamoeba. This raises the question as to whether all TH domains are involved in regulating lipid signaling. Our studies indicate that all murine TIPE family proteins bind phosphoinositides, and in addition to TIPE3, TIPE2 regulates the PtdIns(3,4,5)P3 signaling pathway. However, the functions of TH domains may be regulated by other sequences present in the protein. For example, as reported here, the TIPE3 NT region, which is not shared by other members of the family, is essential for the effects seen on cell growth and survival; TIPE3 lacking the NT region functions in a dominant negative fashion. Similarly, some TH-containing proteins of the unicellular eukaryotes possess N-terminal PH domains, which likely affect their functions. Therefore, TH-containing proteins may play diverse roles depending on the non-TH sequences that they also possess. Thus, despite their high sequence homology in the TH domains, TIPE2 and TIPE3 appear to play distinct roles. TIPE2 functions as a negative regulator of neoplastic cell growth, and its expression is downregulated during liver carcinogenesis. The tumor suppressor activity of TIPE2 is likely dependent on its interaction with Rac1 and Rgl/RalGDS molecules (Gus-Brautbar et al., 2012; Wang et al., 2012). However, no such interaction could be found for TIPE3 (data not shown). Therefore, TIPE2 and TIPE3 appear to have distinct molecular mechanisms.
Hyper-activation of PtdIns(3,4,5)P3 signaling is reported in more than 50% of human cancers. Somatic gene mutations of the signaling molecules such as PTEN and PI3K are found in many of these cancers. Our findings reveal a mechanism of hyper-activating PtdIns(3,4,5)P3 signaling pathway in cancer cells by TIPE3, a phosphoinositide transfer protein. Therefore, TIPE3 may represent a therapeutic target for treating malignant diseases.
Experimental Procedures
Mice
Male athymic nude mice (nu/nu) and wild-type C57BL/6 mice were purchased from Jackson Laboratories. Tipe3+/- C57BL/6 mice (Tnfaip8l3tm1a(KOMP)Mbp) were purchased from Knockout Mouse Project Repository (Davis, California). Mice were housed in the University of Pennsylvania Animal Care Facilities under pathogen-free conditions. All animal procedures were preapproved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and all experiments conform to the relevant regulatory standards.
Human samples
Human non-small cell lung cancer arrays containing 60 tumor samples and their corresponding adjacent normal tissues, and human esophageal carcinoma arrays containing 24 tumor samples and their adjacent normal tissues were purchased from Outdo Biotech Co., Shanghai, China. Human colon cancer and cervical cancer microarrays were purchased from Xi'an Alena Biotech CO., Shanxi, China. All samples have been de-identified prior to analysis and are therefore considered exempt. All human study procedures were preapproved by the Institutional Review Board of the Shandong University.
Crystallization, data collection, and structure determination
The human trTIPE3 (residues 21-204) was purified as described in Supplemental Experimental Procedures. Crystals of trTIPE3 were grown at 18°C using the hanging-drop vapor-diffusion method by mixing 1 μl of protein (10 mg/ml in 25 mM Tris-HCl, 1 M NaCl, pH 8.0) with 1 μl of reservoir solution (0.1 M MES6.7, 1.95 M (NH4)2SO4). The crystals appeared in two days and grew to full size in a week. The 2.3 Å native data were collected at Shanghai Synchrotron Radiation Facility beamline BL17U. The data sets were integrated and scaled using the HKL2000 package. Further processing was performed using the CCP4 suite. The structure was solved by molecular replacement using PHASER, manually refined with COOT and PHENIX. Data collection and refinement statistics are listed in Table S1. All the figures were prepared using PyMOL software.
Database ID
The TIPE3 structure data were deposited in the Protein Data Bank (PDB) with the ID code of 4Q9V.
Other Experimental Procedures including in vivo tumorigenesis studies, human tumor studies, cell culture, protein purification, protein crystallization, protein-lipid overlay assay, and phosphoinositide binding, extraction, and transfer assays are available in Supplementary Information.
Supplementary Material
Highlights.
TIPE3 is a transfer protein for phosphoinositide second messengers.
TIPE3 is markedly upregulated in human cancer cells.
TIPE3 has a large hydrophobic cavity that is occupied by a phospholipid.
Tipe3 knockout mice have reduced tumorigenesis.
Significance.
More than half of human cancers have aberrantly upregulated phosphoinositide signals; yet how lipid signals are controlled in cancer cells is not fully understood. The TNFAIP8 family of proteins is recently described risk factors for human cancer although its mechanisms of action are largely unknown. We report here that TIPE3 (TNFAIP8L3) is the transfer protein of two lipid second messengers, PtdIns(4,5)P2 and PtdIns(3,4,5)P3, and is hijacked by cancer cells to cause malignant transformation. This finding explains why normal cells can control their phospholipid signals but cancer cells can't, a phenomenon widely recognized, but poorly understood. Therefore, TIPE3 may represent a therapeutic target for treating malignant diseases.
Acknowledgments
We thank Thomas Porturas, George Luo, Qingguo Ruan, Derek Johnson, Yael Gus-Brautbar, Xiaohong Liang, Fumin Shi for conceptual inputs and technical assistance. We are grateful to Jeannine Mendrola for providing purified PLCδ-PH, and Katarina Moravcevic for providing purified GST-PLCδ-PH and technical assistance. This work was funded by National Institutes of Health, USA (AI-077533, AI-050059, and GM-085112 to YHC), National Natural Science Foundation of China (projects 31130002 and 31021002 to YS), and the Ministry of Science and Technology (grant numbers 2009CB918801 to YS and 2011CB503906 to LZ).
Footnotes
Supplementary Information is linked to the online version of the paper at www.Cell.com/Cancer-Cell.
Author Contributions: S.A.F. and Y.H.C. conceived the study and wrote the paper. S.A.F. designed and performed the experiments, and analyzed the data. C.L.O., A.V., T.C., Z.W., and H.S. were involved in the design or execution of several experiments. M.A.L. made suggestions for the design of the lipid biochemical studies and contributed to manuscript preparation. X.L., L.Z., J.C., and S.L. designed and performed the human tumor studies. J.W. and Y.S. solved the crystal structure of TIPE3 and contributed to manuscript preparation. Y.H.C. oversaw the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Yigong Shi, Email: ygshi@tsinghua.edu.cn.
Youhai H. Chen, Email: yhc@mail.med.upenn.edu.
References
- Ahn SH, Deshmukh H, Johnson N, Cowell LG, Rude TH, Scott WK, Nelson CL, Zaas AK, Marchuk DA, Keum S, et al. Two genes on A/J chromosome 18 are associated with susceptibility to Staphylococcus aureus infection by combined microarray and QTL analyses. PLoS Pathog. 2010;6:e1001088. doi: 10.1371/journal.ppat.1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Mousley CJ, Schaaf G. The Sec14 superfamily and mechanisms for crosstalk between lipid metabolism and lipid signaling. Trends Biochem Sci. 2010;35:150–160. doi: 10.1016/j.tibs.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin G. Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J Cell Biol. 2011;195:965–978. doi: 10.1083/jcb.201104062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, Lemmon MA. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Molecular cell. 2000;6:373–384. doi: 10.1016/s1097-2765(00)00037-x. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Gus-Brautbar Y, Johnson D, Zhang L, Sun H, Wang P, Zhang S, Chen YH. The anti-inflammatory TIPE2 is an inhibitor of the oncogenic Ras. Mol Cell. 2012;45:610–618. doi: 10.1016/j.molcel.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavran JM, Klein DE, Lee A, Falasca M, Isakoff SJ, Skolnik EY, Lemmon MA. Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. The Journal of biological chemistry. 1998;273:30497–30508. doi: 10.1074/jbc.273.46.30497. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Feldman ME, Balla A, Balla T, Shokat KM. A membrane capture assay for lipid kinase activity. Nat Protoc. 2007;2:2459–2466. doi: 10.1038/nprot.2007.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono N, Ohto U, Hiramatsu T, Urabe M, Uchida Y, Satow Y, Arai H. Impaired alpha-TTP-PIPs interaction underlies familial vitamin E deficiency. Science. 2013;340:1106–1110. doi: 10.1126/science.1233508. [DOI] [PubMed] [Google Scholar]
- Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Kumar D, Gokhale P, Broustas C, Chakravarty D, Ahmad I, Kasid U. Expression of SCC-S2, an antiapoptotic molecule, correlates with enhanced proliferation and tumorigenicity of MDA-MB 435 cells. Oncogene. 2004;23:612–616. doi: 10.1038/sj.onc.1207123. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska K, Sobota A. Signaling pathways in phagocytosis. Bioessays. 1999;21:422–431. doi: 10.1002/(SICI)1521-1878(199905)21:5<422::AID-BIES9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Lee A, Lemmon MA. Analysis of phosphoinositide binding by pleckstrin homology domain from dynamin. Methods Enzymol. 2001;329:457–468. doi: 10.1016/s0076-6879(01)29107-1. [DOI] [PubMed] [Google Scholar]
- Lev S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol. 2010;11:739–750. doi: 10.1038/nrm2971. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annual review of biophysics and biomolecular structure. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- Nile AH, Bankaitis VA, Grabon A. Mammalian diseases of phosphatidylinositol transfer proteins and their homologs. Clin Lipidol. 2010;5:867–897. doi: 10.2217/clp.10.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA. Lipid trafficking sans vesicles: where, why, how? Cell. 2010;143:870–874. doi: 10.1016/j.cell.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA. The lipid trade. Nat Rev Mol Cell Biol. 2014;15:79. doi: 10.1038/nrm3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablin EP, Krylova IN, Fletterick RJ, Ingraham HA. Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1. Mol Cell. 2003;11:1575–1585. doi: 10.1016/s1097-2765(03)00236-3. [DOI] [PubMed] [Google Scholar]
- Schaaf G, Ortlund EA, Tyeryar KR, Mousley CJ, Ile KE, Garrett TA, Ren J, Woolls MJ, Raetz CR, Redinbo MR, et al. Functional anatomy of phospholipid binding and regulation of phosphoinositide homeostasis by proteins of the sec14 superfamily. Mol Cell. 2008;29:191–206. doi: 10.1016/j.molcel.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibel NM, Eljouni J, Nalaskowski MM, Hampe W. Nuclear localization of enhanced green fluorescent protein homomultimers. Anal Biochem. 2007;368:95–99. doi: 10.1016/j.ab.2007.05.025. [DOI] [PubMed] [Google Scholar]
- Sun H, Gong S, Carmody RJ, Hilliard A, Li L, Sun J, Kong L, Xu L, Hilliard B, Hu S, et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133:415–426. doi: 10.1016/j.cell.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature reviews Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Lucast L, Gong LW, Liu L, Sasaki J, Sasaki T, Abrams CS, Kanaho Y, De Camilli P. Phosphatidylinositol-4-phosphate 5-kinases and phosphatidylinositol 4,5-bisphosphate synthesis in the brain. The Journal of biological chemistry. 2010;285:28708–28714. doi: 10.1074/jbc.M110.132191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen X, Lian L, Tang T, Stalker TJ, Sasaki T, Kanaho Y, Brass LF, Choi JK, Hartwig JH, et al. Loss of PIP5KIbeta demonstrates that PIP5KI isoform-specific PIP2 synthesis is required for IP3 formation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14064–14069. doi: 10.1073/pnas.0804139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fayngerts S, Wang P, Sun H, Johnson DS, Ruan Q, Guo W, Chen YH. TIPE2 protein serves as a negative regulator of phagocytosis and oxidative burst during infection. Proc Natl Acad Sci U S A. 2012;109:15413–15418. doi: 10.1073/pnas.1204525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang J, Fan C, Li H, Sun H, Gong S, Chen YH, Shi Y. Crystal structure of TIPE2 provides insights into immune homeostasis. Nat Struct Mol Biol. 2009;16:89–90. doi: 10.1038/nsmb.1522. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang MY, He J, Wang JC, Yang YJ, Jin L, Chen ZY, Ma XJ, Sun MH, Xia KQ, et al. Tumor necrosis factor-alpha induced protein 8 polymorphism and risk of non-Hodgkin's lymphoma in a Chinese population: a case-control study. PloS one. 2012;7:e37846. doi: 10.1371/journal.pone.0037846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.