

Abstract
Cardiovascular disease (CV) remains the leading cause of death worldwide. Low levels of vitamin D are associated with high risk of myocardial infarction, even after controlling for factors associated with coronary artery disease. A growing body of evidence suggests that vitamin D plays an important role in CV related signaling pathways. However, little is known about the molecular mechanism by which vitamin D modulates cardiac development. The Wnt signaling pathway plays a pivotal role in tissue development by controlling stem cell renewal, lineage selection and even more importantly heart development. In this study, we examined the role of 1,25-D3 (active form of vitamin D) on cardiomyocyte proliferation, apoptosis, cell phenotype, cell cycle progression and differentiation into cardiomyotubes. We determined that the addition of 1,25-D3 to cardiomyocytes cells: 1) Inhibits cell proliferation without promoting apoptosis; 2) Decreases expression of genes related to the regulation of the cell cycle; 3) Promotes cardiomyotubes formation; 4) Induces the expression of Casein kinase-1-α1, a negative regulator of the canonical Wnt signaling pathway; and 5) Increases the expression of the noncanonical Wnt11, which it has been demonstrated to induce cardiac differentiation during embryonic development in adult cells. In conclusion, we postulate that vitamin D promotes cardiac differentiation through a negative modulation of the canonical Wnt signaling pathway and by up-regulating the expression of Wnt11. These results suggest that vitamin D repletion to prevent and/or improve cardiovascular disorders that are linked to abnormal cardiac differentiation, such as post infarction cardiac remodeling, deserve further study.
Keywords: VDR, Wnt11, Csnk1α1, β-catenin, Gsk3β, APC
1. Introduction
Hypovitaminosis D as well as other chronic medical conditions is increasingly recognized as an independent predictor for primary cardiovascular events and related diseases (CVD) (Martins et al 2007, Wang et al 2008, Giovannucci et al 2008, van Holten et al 2013). In addition, vitamin D deficiency is highly prevalent in heart failure patients being a significant predictor of reduced survival, whereas supplementation was associated with an improved outcome (Gotsman et al 2012).
The National Health and Nutritional Examination Surveys (NHANES) (1988–1994, 2000–2004) conducted in the US have provided a means to explore cross-sectional associations between vitamin D status and CVD. Kendrick et al reported that individuals surveyed in NHANES 1988–1994 with vitamin D deficiency (25-D <20 ng/mL) had higher prevalence of self-reported angina, myocardial infarction, and heart failure compared with individuals with higher levels of vitamin D (Kendrick et al 2009). In the NHANES 2000–2004 survey, vitamin D deficiency was associated with increased prevalence of self-reported coronary heart disease, heart failure, and peripheral vascular disease (Kim et al 2008, Melamed et al 2008). Moreover, several cardiovascular risk factors have been associated with lower vitamin D status, including hypertension, diabetes, elevated body mass index (>30), elevated triglyceride level, and microalbuminuria in NHANES 1988–1994 (Martins et al 2007). Prevalence of peripheral arterial disease was also increased comparing lowest quartile of 25-D to highest quartile of 25-D (Artaza et al, 2009). It has been also shown that in non-hypertensive individuals from NHANES 1988–1994, optimal vitamin D status (>32 ng/mL) was associated with a 20% reduction in the rate of blood pressure rise with age (Scragg et al 2007, Judd et al 2008).
A few in vitro and in vivo studies have also evaluated the role of vitamin D acting directly on cardiac tissue, especially in response to injury. It has been demonstrated that matrix metalloproteinases (MMP) proteins, which contribute to aberrant cardiomyocyte remodeling in response to injury and atherosclerosis, were upregulated in vitamin D receptor (VDR) knockout mice (Rahman et al 2007). It has been also shown that VDR knockout mice have impaired cardiac relaxation and contractility and develop left ventricular hypertrophy (Simpson et al 2007, Tishkoff et al 2008). A study in HL-1 murine cardiac myocytes showed that 1,25-D3 significantly: decreased cell proliferation, increased cell size, leading to hypoplasia, slight hypertrophy, and altered morphology of dividing cardiomyocytes, demonstrating that 1,25-D3 is involved in maintaining heart cell structure and function at the cellular level (Nibbelink et al 2007). In addition, our group demonstrated that 1,25-D3 inhibited profibrotic markers in vitro in mesenchymal multipotent cells, suggesting that 1,25-D3 may also have a direct effect on the vasculature fibrotic response to injury (Artaza et al 2009).
Even though most evidence indicates an adverse effect of low vitamin D on CVD, the role of 1,25-D3 on cardiac cell differentiation and repair remains poorly understood. In this study, we employed a cardiac myoblast cell line H9c2, derived from embryonic rat heart, which it has been extensively used as an in vitro model for cardiac muscle function (Kimes & Brandt 1976, Artaza et al 2007). The aim of the present study was to test the hypothesis that 1,25-D3 promotes myocardiac cell differentiation through inhibition of Wnt signaling pathway and to determine the associated molecular mechanism(s) in a well known and widely used heart-derived cell model.
2. Materials and Methods
2.1. Cell Culture
H9c2 rat embryonic myocardium cells (ATCC, Manassas, VA, USA) grown in DMEM and supplemented with 10% dialyzed fetal bovine serum at 37°C and 5% CO2 were seeded at 60-70% confluence in T75 flasks, eight-well chamber slides or six-well plates. The next day, cells were incubated for 4 and 7 days with or without 100 nM of 1,25-D3 (1α, 25(OH) 2 Vitamin D3) also known as calcitriol (Sigma-Aldrich, St. Louis, MO, USA) dissolved in less of 0.1% ethanol as vehicle in DMEM-10% dialyzed fetal bovine serum. For proliferation studies, cells were incubated with 1,25-D3 (10-500nM) for 4 days. Control groups were incubated in parallel with 0.1% ethanol in DMEM-10% dialyzed fetal bovine serum (Invitrogen, Carlsbad, CA, USA). The 100nM supra-physiological concentration of 1,25-D3 applied in the experimental design was based on the present and previous dose-response studies and it is in alignment with a commonly used dose on different cell lines or in primary cell culture studies (Barbosa et al 2004, Cardus et al 2006, Artaza & Norris 2009, Artaza et al 2010, Khanna-jain et al 2010, Ramirez et al 2010). Because of the short half-life of 1,25-D3, the cell culture media was replaced every 24h (Garcia et al 2011, 2013).
2.2. Cell proliferation assay
Cell proliferation was determined in 96-well plates by the Formazan dye assay (Promega Corp., Madison, WI, USA). Cells were grown at an initial density of 4,000 cells/well; and then treated for 4 days with 1,25-D3 in a concentration range from 10 to 500 nM. At the end of the incubation time, 100 μl of Formazan substrate buffer was added to the cultures for 3 h at 37 °C in 5% CO2, and the absorbance at 490 nm was read. For cell counting, cells were removed by trypsinization and the number of viable cells was counted in a hemocytometer with Trypan blue staining (Artaza et al 2007).
In parallel experiments, cell proliferation was also evaluated by changes in gene expression of related cell proliferation markers after 4 days incubation time with or without 1,25-D3. The cell proliferation markers applied were: a) proliferating cell nuclear antigen (PCNA), a protein essential for DNA replication, repair and cell proliferation (Essers et al 2005), and b) Gene expression of MKi-67 by real time PCR, a cellular marker strictly associated with cell proliferation (Gerdes et al 1983).
2.3. Cell Morphology/Phenotype
Changes in cell morphology/phenotype were determined in living cells in culture by the PKH2 Green Fluorescent Cell Linker assay (Sigma-Aldrich, Saint Louis, MO, USA) (Horan & Slezak 1989). After an initial and stable incorporation of the PKH2-GL (2× 10-6 M) into the lipid regions of the cell membrane, cells were counterstained with DAPI 0.02% (Invitrogen, Carlsbad, CA, USA). Cells were grown at an initial density of 8,000 cells/well in 8-wells chamber slides and were treated for 7 days with or without 1,25-D3 (10-500 nM). Serial pictures at 200× magnification were taken every 24h with a Leica DMLB fluorescence microscope coupled to Spot RT digital camera. The areas occupied by cells per field were analyzed using ImagePro-Plus 7.1 software (Media Cybernetics, Silver Spring, MD, USA). At least 10 pictures were taken per treatment group per wells done in duplicate. All the experiments were repeated at least three times.
2.4. Apoptosis
The apoptotic index was determined by the TdT-mediated dUTP nick end labeling (TUNEL) method, based on the ability of terminal TdT to catalyze addition of digoxenin-dUTP and dATP to 3′-OH ends of cleaved DNA. Cells were seeded on 8-well chamber slides and were incubated with or without 1,25-D3 (100 nM) for 4 days and then fixed in 2% p-formaldehyde in 1× PBS. Cells were treated with 2% H2O2 for quenching endogenous peroxidase activity, digoxigenin-conjugated nucleotides and TdT, and finally anti-digoxigenin-peroxidase. Slides were stained with 0.5% diaminobenzidine/0.01% H2O2, and counterstained with haematoxylin. As a negative control, PBS (1×) buffer was substituted for the TdT enzyme. Quantification of the apoptotic index was done at 400 ×. In all cases, 20 fields were randomly selected and the apoptotic index of each field was calculated as the percent of TUNEL-positive cells (Essers 2005, Artaza et al 2007).
2.5. Detection of VDR by immunofluorescence
Cells plated on eight-well chamber slides were incubated with or without 1,25-D3 (100nM; based on results of dose response analyses) in a time-course dependent manner from 0-4 days. At the end of the corresponding incubation times, cells were washed thrice with PBS (1×) and fixed in 2% p-formaldehyde. Cells were blocked with goat serum, and incubated with rabbit polyclonal antibody against VDR at a dilution of 1:50 (Santa Cruz Biothecnology, Santa Cruz, CA). The detection was followed by a 1/200 dilution of anti-rabbit biotinylated secondary antibody (Calbiochem, La Jolla, CA), followed by Streptavidine-Texas Red (10 μg/ml) (Vector Labs, Burlinghame, CA). After several washes, cells were counterstained with DAPI. In negative controls, we either omitted the first antibody or used a rabbit nonspecific IgG (Artaza et al. 2005, 2008). To ensure specific staining, an additional negative control was included by pre-absorbing the primary VDR antibody with a VDR specific peptide (1μM), SC1008P (Santa Cruz Biotechnology, Santa Cruz, CA), after 1h incubation at RT the mixture was centrifuged at full speed for 15 minutes and the supernatant was used as primary antibody at the same dilution applied for the experiment. No positive staining was observed in the cells by immunofluorescence in this negative control. At the end, the slides were detached and mounted in “prolong anti fade” (Molecular Probes, Eugene, OR) and were examined under a fluorescence microscope Leica DMLB, coupled to a Spot RT digital camera.
2.6. RT2 Profiler PCR Array Analysis of Cell Cycle and Wnt related Target Genes
Total cellular RNA was isolated with Trizol-Reagent (Invitrogen, Carlsbad, CA, USA) from H9c2 cells treated with or without 1,25-D3 (100nM) for 4 days. Isolated RNA was subjected to reverse transcription, and the resulting cDNA was analyzed by RT2 profiler Rat Cell Cycle PCR array (PARN-020) and by RT2 profiler Rat Wnt Signaling Pathway PCR Array (PARN-043A) (SABiosciences Corp., Frederik, MD, USA). Analysis of cell cycle and Wnt related target genes were done in triplicates. The Rat Cell Cycle RT² Profiler™ PCR Array contains genes that regulate the cell cycle, the transitions between each of the phases, DNA replication, checkpoints and arrest. The Rat Wnt RT² Profiler™ PCR Array profiles the expression of genes related to Wnt-mediated signal transduction. Each array contains a panel of 84 primer sets related to the cell cycle and to Wnt signaling pathway plus 5 housekeeping genes and 2 negative controls. Real time PCRs were performed as follows: melting for 10 min at 95°C, 40 cycles of two-step PCR including melting for 15 sec at 95°C, annealing for 1 min at 60°C. The raw data were analyzed using the ΔΔCt method following the manufacturer's instructions (SABiosciences Corp., Frederick, MD, USA) (Garcia et al 2011, 2013).
2.7. Real-Time Quantitative PCR
Total RNA was extracted using Trizol-Reagent (Invitrogen, Carlsbad, CA) and equal amounts (1μg) of RNA were reverse transcribed using a RNA PCR kit (Applied Biosystems, Foster City, CA, USA). Rat gene PCR primer sets (RT2) for Mki67, PCNA, Casp3, Bcl-2, Casein kinase 1 α-1 and Wnt11 were obtained from SABiosciences Corp (Frederick, MD, USA). The QIAGEN Sybr Green PCR kit with HotStar Taq DNA polymerase (QIAGEN, Valencia, CA, USA) was used with i-Cycler PCR thermocycler and fluorescent detector lid (Bio-Rad, Hercules, CA, USA) (Garcia et al 2011, 2013).
The protocol included melting for 15 min at 95°C, 40 cycles of three-step PCR including melting for 15 sec at 95°C, annealing for 30 sec at 58°C, elongation for 30 sec at 72°C with an additional detection step of 15 sec at 81°C, followed by a melting curve from 55–95°C at the rate of 0.5°C per 10 sec. Samples of 25 ng cDNA were analyzed in quadruplicate in parallel with GAPDH controls; standard curves (threshold cycle vs. log pg cDNA) were generated by log dilutions of from 0.1 pg to 100 ng standard cDNA (reverse-transcribed mRNA from H9c2 cells in AM). Experimental mRNA starting quantities were then calculated from the standard curves and averaged using i-Cycler, iQ software as described previously (Garcia et al, 2013; 2011). The ratios of marker experimental gene (e.g. Mki67, PCNA, Casp3, Bcl-2, casein kinase 1 α-1 and Wnt11 mRNA) to GAPDH mRNA were computed and normalized to control (untreated) samples as 100%.
2.8. Immunocytochemical analyses of PCNA, c Troponin 3, casein kinase 1 α-1 and Wnt11 antigens
After incubating the cardiac cells for 4 days with or without 1,25-D3 (100nM), cells were washed five times with PBS (1×) and fixed in 2% p-formaldehyde, quenched with H2O2, blocked with horse or rabbit serum and incubated with: anti-proliferating cell nuclear antigen (PCNA) MoAb (1:400) (Millipore, Temecula, CA, USA), anti-cardiac muscle cTroponin 3 MoAb (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Csnk1α1 rabbit polyclonal antibody (1/100) and anti-Wnt11 rabbit polyclonal antibody (1/100) (Abcam Inc., Cambridge, MA, USA). Detection was based on a secondary biotinylated antibody (1:200), followed by the addition of the streptavidin-horseradish peroxidase ABC complex (1:100), Vectastain (Elite ABC System, Vector Laboratories, Burlingame, CA, USA) and 3,3′-diaminobenzidine and H2O2 mixture (Sigma, St. Louis, MO). Cells were counterstained with Mayer's hematoxylin solution (Sigma-Aldrich, St. Louis, MO, USA). In negative controls, we either omitted the first antibody or used a rabbit non-specific IgG (Garcia et al 2011).
2.9. Western Blots and Densitometry Analysis
Cell lysates (25-40μg of protein) were subjected to western blot analyses by 4-15% Tris-HCl polyacrylamide gel electrophoresis (PAGE) (Bio-Rad, Hercules, CA, USA) in running buffer (Tris/Glycine/SDS). Proteins were transferred overnight at 4°C, to nitrocellulose membranes in transfer buffer (Tris/Glycine/Methanol). Next day the non-specific binding was blocked by immersing the membranes into 5% non-fat dried milk, 0.1% (v/v) Tween 20 in PBS for 2 h at RT. After several washes with washing buffer (PBS Tween 0.1%), membranes were incubated with the primary antibodies for 3 hours at RT or overnight at 4°C, MoAb were as follow: a) PCNA (1/500) (Millipore, Temecula, CA, USA) b) GAPDH (1/10,000) (Chemicon International, Temecula, CA, USA), c) Bcl-2 (1/500), d) GSK-3β (1/500) (BD Biosciences, San Jose, CA, USA), e) Cyclin D1 (1/500), f) Cyclin D3 (1/500) and g) CDk4 (1/500) (Cell Signaling Technology, Inc., Danvers, MA, USA). Polyclonal antibodies were used for a) VDR (1/500), b) CDk2 (1/500), c) p21 (1/500), d) p27 (1/500), e) APC (1/500) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), f) Wnt11 (1/200) and g) Csnk1α1 (1/200) (Abcam Inc., Cambridge, MA, USA), h) anti-cardiac muscle Troponin 3 monoclonal antibody (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA). After several washes with buffer, the membranes were incubated for 1 hour at RT with 1/3,000 dilution (anti-mouse) or 1:2,000 dilution (anti-rabbit) of secondary antibody linked to horseradish peroxidase, respectively (Cell Signaling Technology, Inc., Danvers, MA, USA). After several washes, the immunoreactive bands were visualized using the SuperSignal western blotting chemiluminescence detection system (Thermo Fisher Scientific, Inc., Rockford, IL, USA). The densitometry analysis of the bands was done with the Scion Image software beta 4.02 (Scion Corp., Frederick, MD, USA) (Garcia et al 2011, 2013).
2.10. Qualitative and Quantitative Immunocytochemical Analyses
In all cases, the immunoreactivity was quantified by image analysis using ImagePro-Plus 7.1 software (Media Cybernetics, Silver Spring, MD, USA). Two independent observers count fields blindly and each experiment was repeated at least three times. For PCNA and TUNEL determinations, the number of positive cells at 400× was counted in a computerized grid and results were expressed as a ratio of the percentage of positive cells/total cells per field. In all cases, 10 fields at 400× were analyzed per well. Cell size was determined by outlining and measuring the area of each cell per field. Approximately 100 cells were measured for each experiment and the results were plotted as cardiomyotubes area expressed in μm2 per 1,25-D3 concentration. For cTroponine 3, Csnk1α1 and Wnt11 immunocytochemistry determinations, the images were first calibrated for background lighting and the Integrated Optical density (IOD) was determined. The IOD results are proportional to the mean optical density per area and determine the amount of immunoreactive antigen present in each cell. The IOD values expressed in arbitrary units were determined in at least 20 pictures per treatment and time points (Artaza et al, 2010, Garcia et al, 2011).
To test the precision of the QIA, we evaluated both inter- and intra-assay variability. Standard deviation was 0.38 for assays in triplicates, CV=1.7% for intra-assay determinations and CV 5% for inter-assay. The results expressed as Mean ± S.E.M. represent the average of three independent experiments.
2.11. Statistical Analysis
All data are presented as mean ± S.E.M, and between-group differences were analyzed using ANOVA. If the overall ANOVA revealed significant differences, then pair-wise comparisons between groups were performed by Tukey's multiple comparison test. All comparisons were two-tailed, and a p value < 0.05 was considered statistically significant. The in vitro experiments were repeated thrice, and data from representative experiments are shown. Specifically, the RT2 Profiler PCR arrays were done in triplicate and in some cases further confirmed by qRT-PCR done in triplicate.
3. Results
3.1. Time course of expression and nuclear translocation of VDR in H9c2 cardiac derived cells incubated with 1,25-D3
To determine whether the H9c2 cardiac cells expressed the VDR at basal level and whether its expression and nuclear translocation is induced upon incubation with 1,25-D3, experiments using immunofluorescence and western blots were carried out at different time points. Cells were continuously incubated or without 1,25-D3 (100 nM) for 30min, 1h, 5h, 24h and 4 days. By immunofluorescence (Fig 1, A) and under basal conditions (no 1,25-D3 added), VDR immunofluorescence (red) was barely detected in the cells. After 30 min incubation with 1,25-D3, VDR expression was clearly increased in the cytoplasm and the nuclear compartment. A similar observation was made at 1 and 5h (Fig 1, A, upper panel). In contrast, after 24h and 4 days of continuous 1,25-D3 incubation, most of the red-fluorescence staining was observed in the nucleus (Fig 1, A, lower panel) indicating nuclear translocation of the VDR upon continuous 1,25-D3 incubation for 24h. The counterstaining with DAPI (blue) confirms the nuclear localization of VDR after continuous incubation with 1,25 D3.
Figure 1. Expression, nuclear translocation and protein up-regulation levels of VDR upon incubation of H9c2 cardiac cells with 1,25-D3.
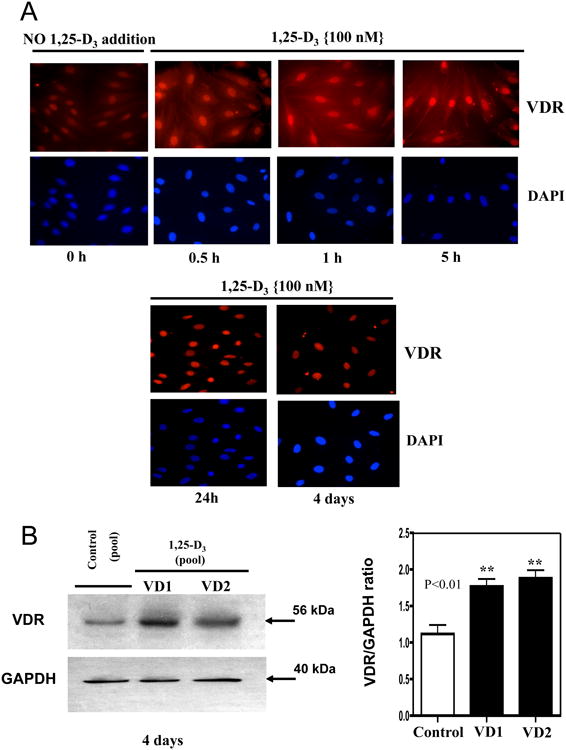
Cultures of H9c2 cardiac cells were incubated or not with 1,25-D3 (100nM) in a time course manner (0-4 days) on 8-well removable chambers slide and subjected to immunofluorescence using a polyclonal antibody for VDR followed by a Texas Red-conjugated secondary antibody (red). Cells were counterstained with DAPI (blue) to indicate nuclear localization. Magnification ×400 (A). Protein extracts isolated at 4 days were subjected to western blots with the corresponding image analysis (B). Mean ± SEM corresponds to experiments done in triplicates, **, P< 0.01. Control, VD1 and VD2 are different pools of two samples each. Samples and controls were normalized with GAPDH housekeeping gene.
The increased expression of VDR upon 1,25-D3 exposure was further confirmed by Western blot analysis in whole cell culture homogenates after 4 days of continuous incubation with 1,25-D3 (100nM) (Fig 1, B). The densitometry analysis shows a 2.0 fold increase in VDR expression after incubation with 1,25-D3 at 4 days in comparison with the control.
3.2. 1,25-D3 inhibits cell proliferation of H9c2 cardiomyocytes
To assess whether 1,25-D3 inhibits H9c2 cardiomyocytes cells proliferation and stimulates cell differentiation by exiting cells from the cell cycle, H9C2 cells were seeded in a low confluence (4,000 cells/well) and incubated in triplicate with increasing concentrations of 1,25-D3, for 4 days. At the end of the incubation time, cell proliferation was evaluated by the Formazan assay (Fig 2, A). Starting at 50 nM, 1,25-D3 induced a statistically significant reduction in cell number reaching a plateau at 100nM with no statistically significant difference at 500 nM compared with 100nM in agreement with our previous published data on multipotent cells (Artaza et al 2009). Based on these results, 100nM was the selected dose used throughout the study to evaluate the effects of 1,25-D3 on H9c2 cardiomyocytes cells.
Figure 2. Increasing concentrations of 1,25-D3 reduce Cardiomyocytes cell proliferation.

H9c2 cells were incubated for 4 days with increasing concentrations of 1,25-D (0-500nM), at the end of the incubation time, cell proliferation was evaluated by the formazan assay (A). In a parallel experiment, inhibition of cell proliferation was also demonstrated by the steady-state mRNA down-regulation of Mki67, a well-known proliferation antigen (B). Mean ± SEM corresponds to experiments done in triplicates, ***, P< 0.001. Samples and controls were normalized with GAPDH housekeeping gene.
Inhibition of cell proliferation was also demonstrated by real time PCR by showing a decrease in the expression of MKi67 a well-known proliferation antigen, which is induced in G1, S, G2 and M phases of the cell cycle (Fig 2, B). Quiescent or resting cells in the G0 phase of the cell cycle do not express the MKi67 antigen. (Endl et al 1997, Preusser et al 2008).
To further corroborate the inhibition of cardiac embryonic cell proliferation promoted by continuous incubation with 100nM 1,25-D3 the expression of the proliferating cell nuclear antigen (PCNA) was studied by immunocytochemistry (ICC). Fig 3A, upper panel shows representative pictures at 200× magnification of the decreased nuclear expression of PCNA after incubating the cardiac cells with 100 nM of 1,25-D3 for 4 days. The visual inspection was confirmed by quantitative image analysis (Fig 3 A, lower panel) where PCNA expression was decreased 2.7-fold with respect to control (p<0.001). The decreased expression of PCNA was even further confirmed by real time PCR (PCNA/GAPDH ratio: -2.35) (Fig 3B) and at the protein level by western blot (Fig 3C, left) with the corresponding densitometry analysis (Fig 3C, right).
Figure 3. 1,25-D3 down-regulates the expression of PCNA.
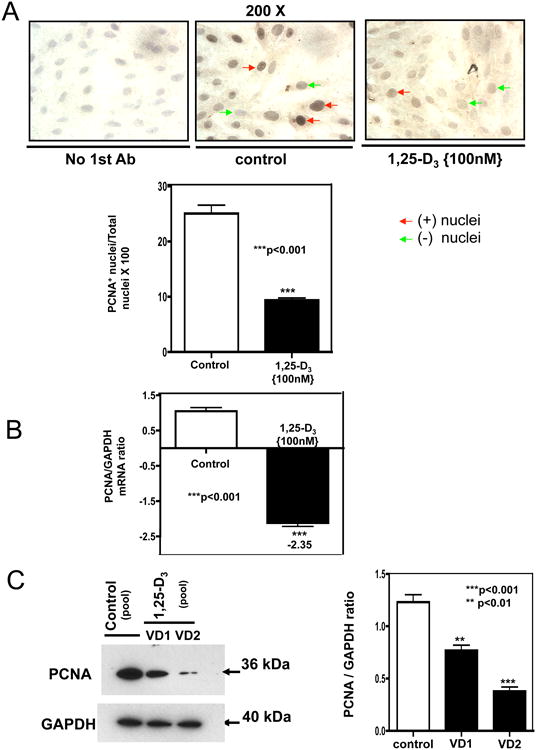
Cultures of H9c2 cells were treated as in Figure 2 for 4 days. ICC (immunocytochemistry) reactions, real time PCR and western blottings were performed at the end of the incubation time. (A) Representative ICC pictures with the corresponding image analysis expressing percentage of positive cells (brown nuclear staining) for experiments done in triplicates. ***, P< 0.001. Magnification 200×. (B) Steady-state mRNA down-regulation of PCNA. Mean ± SEM corresponds to experiments done in triplicates, ***, P< 0.001. (C) Protein extracts were subjected to western blots with the corresponding image analysis. Mean ± SEM corresponds to experiments done in triplicates, **, P< 0.01 and ***, P< 0.001. Control, VD1 and VD2 are different pools of two samples each. In both cases, real time PCR and western blots, samples and controls were normalized with GAPDH housekeeping gene.
3.3. 1,25-D3 enhances cardiomyotube area and promotes the expression of cardiac troponin
In order to determine whether 1,25-D3 promotes cardiac cell differentiation and increases cell size, the cells were pre-labeled with the cell membrane marker PKH2 Green Fluorescence Cell Linker and counterstained with DAPI, to visualize cell boundaries and cell nuclei respectively. Fig 4, A, upper panel shows representative fluorescence pictures of 1,25-D3 non-treated and treated cardiac cells at different concentrations and Fig 4, A, lower panel shows the respective image analysis indicating a statistically significant increase in cardiomyotube area upon incubating the cardiac cells with increasing doses of 1,25-D3 starting at 50 nM for seven days.
Figure 4. 1,25-D3 increases cardiomyotubes area and the expression of cardiac troponin.
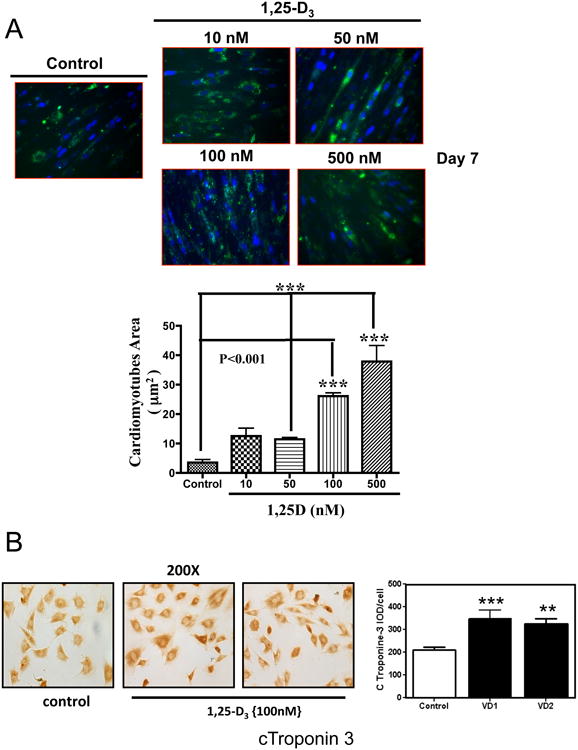
(A) Cultures of H9c2 cells pre-incubated with PKH2 Green Fluorescence Cell Linker and counterstained with DAPI were treated as in Figure 2 for 7 days. Representative pictures are presented for control (No 1,25-D3 addition) and for increasing concentrations (10-500 nM) of 1,25-D3 with the corresponding image analysis of the cardiomyotube area. ***, P< 0.001. Magnification. In a parallel experiment, cultures of H9c2 cells incubated on 8-well chamber slides were treated as in Figure 2 for 7 days. At the end of the incubation time, cells were fixed and subjected to ICC. (B) Representative ICC pictures of cardiac Troponin+ cells with the corresponding image analysis expressing percentage IOD (area × intensity) for experiments done in triplicates. **, P< 0.01 and ***, P< 0.001. Magnification, × 200.
To further corroborate the effect of 1,25-D3 on cardiac differentiation, the expression of cTroponin-3 a marker of cardiac tissue formation was evaluated. Fig 4, B shows an increased expression of the cardiac marker cTroponin-3 by immunocytochemistry (left) with the corresponding image analysis (right) upon incubating the cells with 1,25-D3 (100nM) for 7 days, indicating cardiac tissue formation.
3.4. 1,25-D3 induces cell cycle exit of H9c2 cardiac cells without inducing apoptosis
The effect of 1,25-D3 incubation on the expression of genes/proteins related to activation, promotion or exiting from the cell cycle were investigated by real time PCR arrays (Fig 5, A) and by western blots (Fig 5, B, C and D). Cell division relies on the activation of cyclins, which bind to cyclin-dependent kinases (CDKs) to induce cell-cycle progression towards S phase and later to initiate mitosis. Cyclins are a group of proteins that control the progression of cells through the cell cycle by activating cyclin-dependent kinase enzymes. Fig 5 shows a general decrease in the expression of Cyclins A1, D1, D3, C and D.
Figure 5. 1,25-D3 induces cell cycle exit of H9c2 cardiac cells.
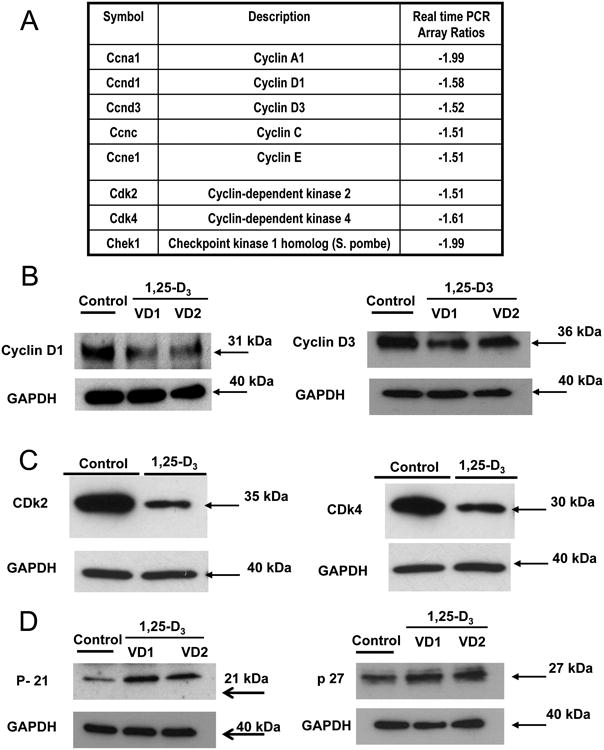
(A) Differential steady-state mRNA levels of cell cyclins, cyclin dependent kinases, DNA replication checkpoints and arrest factors between 1,25-D3 treated and untreated H9c2 cells. Total RNA from cells treated as in Fig. 1 was subjected to RT real-time PCR by the cell cycle PCR array, and the ratios between the 1,25-D3-treated and 1,25-D3-untreated cells corrected by GAPDH were calculated for assays performed in triplicate. (B) Confirmation of cyclin D1 and cyclin D3 PCR array results by western blots. (C) Confirmation of Cdk2 and CDk4 PCR array results by western blots. (D) Changes in p-21 and p-27 expression after 1,25-D incubation by western blots. VD1 and VD2 correspond to different pools of two samples each. In both cases, real-time PCR and Western blottings, samples and controls were normalized with GAPDH housekeeping gene.
Cyclin-dependent kinases (CDKs) are a group of protein kinases that are activated by the formation of a complex with cyclins and are involved in the regulation of the cell cycle. Fig 5, C shows a decrease expression of CDk2 and CDk4. 1,25-D3 also decreases the expression of Chek1, a checkpoint serine/threonine kinases that is involved in the control of the cell cycle. Even more interesting is the fact that p21 and p27 that bind to cyclin-CDK complexes to inhibit their catalytic activity and induce cell cycle are differentially expressed in cardiac cells incubated with 1,25-D3 (western blot; Fig 5, D). At least, in this cell model the exit from the cell cycle induced by incubation with 1,25-D3 appears to be solely due to an increased in p21 expression since the expression of p27 does not change by 1,25-D3 incubation (Fig 5, D).
Fig 6 demonstrates by different approaches that the reduction in cell number observed in the cell proliferation studies (Fig 2 and Fig 3) is not due to apoptosis. Fig 6, A shows that there was not a significant effect of 1,25-D3 on the number of apoptotic cells determined by TUNEL assay followed by quantitative image analysis. There was no change in the expression of the pro-apoptotic caspase-3 by realtime PCR after 1,25D3 incubation (Fig 6, B) or in the expression of the anti-apoptotic Bcl-2 evaluated by real time PCR and western blot (Fig 6, C).
Figure 6. 1,25-D3 induces no changes in apoptotic markers in H9c2 cardiac cells.
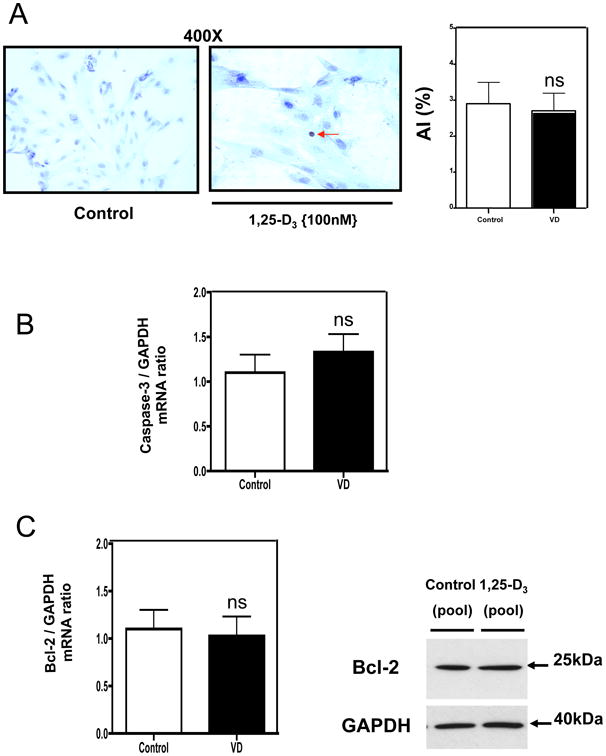
(A) Cells treated as in Fig 2, were seeded in 8-well chamber slides for TUNEL assay or in 6-well plates for RNA and protein isolation. At the end of the incubation period, samples were subjected to TUNEL assay where the apoptotic index (AI) was obtained for control and VD incubated samples. In another set of samples, total RNA isolation was followed by real time PCR for Caspase 3 and Bcl-2 normalized by GAPDH housekeeping gene (B) and (C). Mean ±SEM corresponds to experiments done in triplicates. (C) Western blot analysis was performed for extracts also obtained at 4 days incubation time for Bcl-2 normalized with GAPDH.
3.5. 1,25-D3 modulates the expression of key components of the canonical and non-canonical Wnt signaling pathway
The effects of 1,25-D3 incubation on the expression of key members of the canonical and non-canonical Wnt signaling pathway on cardiac cells was demonstrated by real time PCR arrays and confirmed by qPCR, western blots and immunocytochemistry analysis.
Fig 7 A, shows the differential expression of key Wnt signaling pathway members by real time PCR arrays after incubating the cardiac derived cells with 1,25-D3. No changes were noted in the expression of adenomatosis polyposis coli (APC), Axin 1 and 2, GsK3β, NKD1 and 2, and NLK after 1,25-D3 incubation. However, the expression of Csnk1α1, a negative regulator of the canonical Wnt signaling pathway, and Wnt11 a non-canonical Wnt signaling member, was remarkable increased. The analysis of the other components of the destruction complex responsible for degrading β-catenin and thus impairing the canonical Wnt signaling pathway such as APC, Axin 1, 2 and GsK3β showed no change in expression by real time PCR arrays. This observation was further confirmed by western blots with the corresponding densitometric analysis (Fig 7, B and C).
Figure 7. 1,25-D3 modulates the expression of key components of the canonical and non-canonical Wnt signaling pathway.
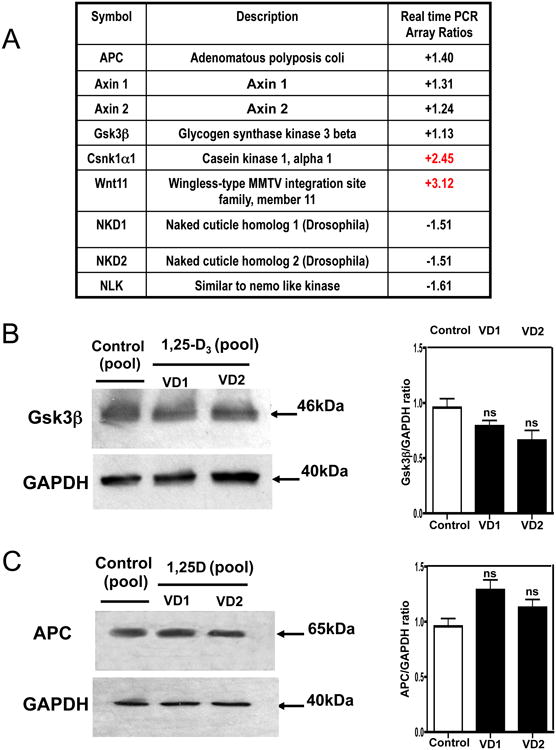
(A) Differential steady-state mRNA levels of Wnt signaling factors members between 1,25-D3 treated and untreated H9c2 cells. Total RNA from cells treated as in Fig. 1 was subjected to RT real-time PCR by the Wnt Signaling Pathway PCR Array, and the ratios between the 1,25-D3-treated and 1,25-D-untreated cells corrected by GAPDH were calculated for assays performed in triplicate. (B) Confirmation of Gsk3β PCR array results by western blots with the corresponding densitometric analysis. (C) Confirmation of APC PCR array results by western blots with the corresponding densitometric analysis. VD1 and VD2 correspond to different pools of two samples each. In both cases, real-time PCR arrays and Western blottings, samples and controls were normalized with GAPDH housekeeping gene.
In order to corroborate the finding obtained by Real time PCR arrays, the expression of Csnk1α1 was further evaluated at mRNA and protein levels. Fig 8 panel A shows a statistically significant increase of Csnk1α1 by real time PCR, panel B shows that Csnk1α1 is also up-regulated at the protein level as evidenced by an increased expression by western blot with the corresponding densitometric analysis; and panel C shows Csnk1α1 is localized in the nucleus and cytoplasm of the cardiac cell, and after incubation with 1,25-D3, its expression is highly up-regulated.
Figure 8. 1,25-D3 up-regulates the expression of Gsk3β.
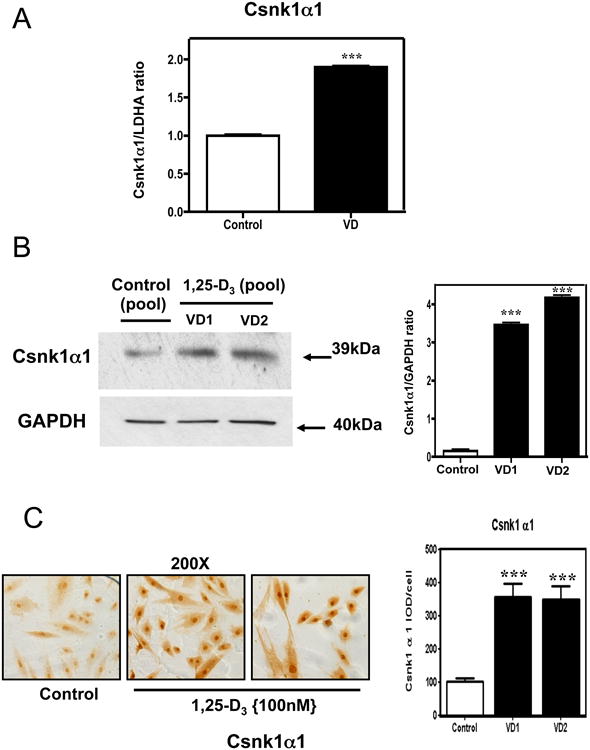
(A) Differential steady-state mRNA levels of Gsk3β between 1,25-D3 treated and untreated H9c2 cells. Total RNA from cells treated as in Fig. 1 was subjected to RT real-time PCR for Gsk3β, and the ratios between the 1,25-D3-treated and 1,25-D3-untreated cells corrected by GAPDH were calculated for assays performed in triplicate. (B) Confirmation of Gsk3β PCR array results by western blots with the corresponding densitometric analysis. (C) Representative immunocytochemistry pictures of Gsk3β+ cells with the corresponding image analysis expressing percentage IOD (area × intensity) for experiments done in triplicate. Magnification 200×. Mean ± SEM corresponds to experiments done in triplicates **, P< 0.01 and ***, P< 0.001.
The increased expression of Wnt 11 was also corroborated by different approaches: Fig 9. A shows a statistically significant increase of Wnt 11 by real time PCR, panel B shows an increase in Wnt 11 expression by western blots with the corresponding densitometric analysis; and panel C shows an increase in Wnt 11 expression by immunocytochemistry with the corresponding quantitative image analysis.
Figure 9. 1,25-D3 up-regulates the expression of Wnt 11.
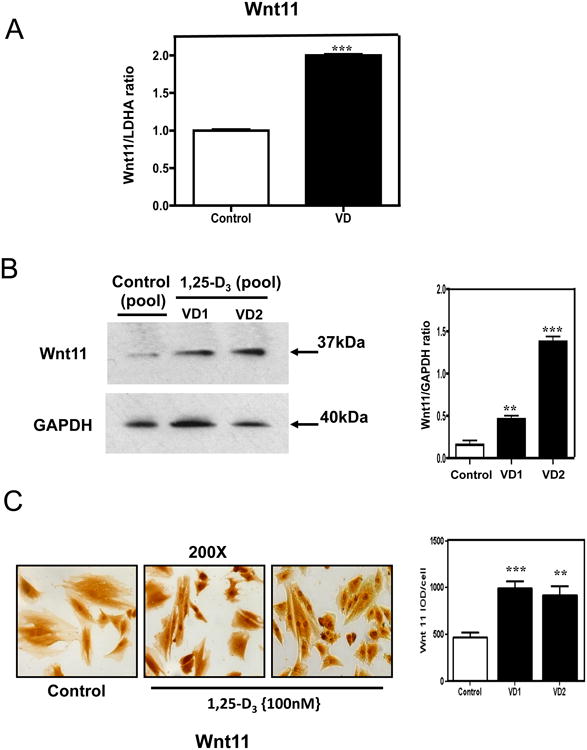
(A) Differential steady-state mRNA levels of Wnt 11 between 1,25-D3 treated and untreated H9c2 cells. Total RNA from cells treated as in Fig. 1 was subjected to RT real-time PCR for Wnt 11, and the ratios between the 1,25-D-treated and 1,25-D-untreated cells corrected by GAPDH were calculated for assays performed in triplicate. (B) Confirmation of Wnt 11 PCR array results by western blots with the corresponding densitometric analysis. (C) Representative immunocytochemistry pictures of Wnt 11 + cells with the corresponding image analysis expressing percentage IOD (area × intensity) for experiments done in triplicate. Magnification 200×. Mean ± SEM corresponds to experiments done in triplicates **, P< 0.01 and ***, P< 0.001.
4. Discussion
The discovery of adult eCSCs (endogenous cardiac stem cells) (Beltrami et al 2003) indicates that the heart is not a complete post-mitotic organ without any regenerative capability. The identification of eCSCs has provided an explanation for the hitherto unexplained existence of a subpopulation of immature cycling myocytes in the adult myocardium. Indeed, published evidence from a genetic fate-mapping study established that stem cells replenish adult mammalian cardiomyocytes lost by cardiac wear and tear and injury throughout the adult life (Hsieh et al 2007). Moreover, it is now accepted that myocyte death and myocyte renewal are the two sides of the proverbial coin of cardiac homeostasis in which the eCSCs play a central role (Nadal-Ginard et al 2003). These findings produced a paradigm shift in cardiac biology and opened new opportunities and approaches for future treatment of cardiac diseases by targeting eCSCs with growth factors and as showed in this manuscript vitamin D. There is recent publishing data demonstrates that activating differentiation may be critical to allow subsequent proliferation (Naqvi et al 2014).
The data presented in this manuscript demonstrate that the addition of 1,25-D3 to H9c2 cardiac cells inhibits cell proliferation without inducing apoptosis, and promotes cardiac differentiation through a mechanism mediated by an increase expression and nuclear translocation of the VDR. We cannot discard the possibility of the interaction of 1,25-D3 with a membrane-localized receptor that binds 1,25-D3 (Huhtakangas et al 2004) especially at the supra-physiological doses used for the in vitro cultures. It is speculated that the VDR normally associated with the cell nucleus and gene transcription (genomic response) can also be resident near to or associated with caveolae present in the plasma membrane to generate RR (rapid Response) (Razani et al 2002). The outcomes of these interactions would include opening of the voltage-gated calcium or chloride channels or generation of the indicated second messengers. Some of these second messengers, particularly RAF/MAPK, may engage in cross talk with the nucleus to modulate gene expression (Norman, 2006). It would be interesting to reproduce this experimental design in vivo applying vitamin D analogs or non-genomic vitamin D activators that may be able to induce the beneficial pro-differentiation and anti-proliferative effects without the adverse hyper-calcemic side effects.
We also demonstrated that the effects of 1,25-D3 to promote cardiac differentiation involve the modulation of members of the Wnt signaling pathway, including the increased expression of CK1α1 (Casein kinase-1, α1), a well-known inhibitor of the canonical Wnt signaling pathway (Thorne et al 2010). This observation agrees with the concept that canonical Wnt inhibition is required for heart development in vivo (Bergmann 2010). Remarkably, the inhibition of the canonical Wnt signaling pathway promoted by 1,25-D3 is accompanied by an increased expression of the noncanonical Wnt11, which has been reported per se to induce cardiac differentiation during embryonic development and in adult cells (Flaherty & Dawn 2008, Flaherty et al 2008, 2012).
We showed that VDR is minimally expressed in untreated H9c2 cardiomyocytes and incubation with 1,25-D3 stimulates de novo synthesis and nuclear translocation. This was expected since it is known that 1,25-D3 auto-regulates the expression of the VDR gene through intronic and upstream enhancers (Pike & Meyer 2010).
Our data show that the addition of 1,25-D3 to H9c2 cardiomyocytes inhibits cell proliferation in a dose dependent manner and down-regulates the expression of Mki67 and PCNA, two well-known and broadly used cell proliferation markers/indicators (Preusser et al 2008, Essers et al 2005). The inhibition of cell proliferation by 1,25-D3 is not accompanied by changes in apoptosis or the expression of the pro-apoptotic caspase-3 and/or the expression of the anti-apoptotic Bcl-2. These results are in agreement with our previous results in mesenchymal multipotent cells (Artaza et al 2010) and with the data presented by Assalin and coauthors where vitamin D deficiency is not associated with an increase in apoptosis in cardiac tissue (Assalin et al 2013).
The inhibition in cell proliferation induced by 1,25-D3 is accompanied by an increase in cell differentiation evidenced by an increase in cardiomyotubes cell area and the expression of c-troponin, a main cardiac differentiation marker (Perán et al 2010).
In agreement with the previous work of Eelen and collaborators (Eelen et al 2007), we found that the addition of 1,25-D3 to H9c2 cardiomyocytes blocks the transition from G1 to S1-phase of the cell cycle accumulating cells in G1 by decreasing the expression of cyclins such as: Cyclin A1, D1, D3, C and E; as well as cyclin dependent kinases (CDk) such as CDk2 and CDk4. Of particular interest is that in this cardiac cell model, 1,25-D3 G1/S-blocking effect was accompanied by an increased expression of p21, a CDk inhibitor. However, 1,25-D3 does not increase the expression of p27, making p21 a primary candidate for the antiproliferative effect in H9c2 cardiac cells. 1,25-D3 also decreases the expression of Chek1, a protein required for checkpoint mediated cell cycle arrest in response to DNA damage or the presence of unreplicated DNA.
Lastly, we demonstrated that the induction of cardiac differentiation promoted by 1,25-D3 in H9c2 cardiomyocytes involves the modulation of the expression of key members of the Wnt signaling pathway. The Wnt gene family consists of secreted lipid-modified and evolutionary conserved signaling glycoproteins (Cadigan & Nusse 1997). Wnt signals are transduced to the canonical pathway for cell fate determination, and to the noncanonical pathway for control of cell movement and tissue polarity (Katoh & Katoh 2007). Canonical Wnt signals are transduced through Frizzled family receptors and LRP5/LRP6 coreceptor to the β-catenin signaling cascade. Noncanonical Wnt signaling are transduced through Frizzled family receptors and ROR2/RYK coreceptors to the DVL-dependent or the Ca2+ dependent signaling cascades (Katoh & Katoh 2007). It has been demonstrated that canonical Wnt signaling inhibition is essential for cardiogenic activity and even the possibility that the pathway can be targeted for the design of drug-like cardiogenic molecules (Lanier et al 2012, Bergmann 2010). Even more, it has been shown that inhibitors of the canonical Wnt signaling pathway potently promote cardiomyocyte differentiation from human embryonic stem cell-derived mesoderm (Willems et al 2011).
Canonical Wnt signaling (or Wnt/β-catenin pathway) causes an accumulation of β-catenin in the cytoplasm and its eventual translocation into the nucleus to act as a transcriptional coactivator of transcription factors that belong to the TCF/LEF family. β-Catenin in the nucleus binds to TCF/LEF transcription factors to activate Wnt/β-catenin-responsive genes, such as CDK1 and cyclin D1, which are required for cell cycle progression. Inhibition of the canonical Wnt signaling pathway reverts this process. In other words, there is a synergistic effect prompted by inhibition of the canonical Wnt signaling pathway and the well-known antiproliferative effect and cell cycle arrest promoted by vitamin D as demonstrated by our group and others in different cell models (Wang et al, 1996, Verlinden et al 1998, Artaza et al 2010). The possibility of modulating the Wnt signaling pathway by vitamin D replenishment and/or supplementation will promote better outcomes in heart remodeling by inducing cardiac differentiation. This is very promising and possible translation to the clinic. Even more, several publications suggest that canonical Wnt signals play distinct roles during discrete developmental windows, first positively regulating pre-cardiac mesoderm commitmentand then playing a negative role in the initial induction of cardiac progenitors (Kwon et al 2007, Marvin et al 2001). Experiments performed by Naito et al on ES cells indicate that Wnt/β-catenin signaling, when activated after cells are committed to the cardiac lineage negatively regulates cardiomyocyte differentiation, and that inhibition of Wnt signaling at this stage decreases cell proliferation and even more important enhances cardiomyocyte differentiation (Naito et al 2006). Those results are in agreement with the data presented in this manuscript where the experiments were performed on a cell model, the H9c2 cardiomyocytes, already committed with the cardiac lineage. Without Wnt signaling, the β-catenin would not accumulate in the cytoplasm since a ‘destruction complex’ would normally degrade it. This destruction complex includes the following proteins: Axin, adenomatosis polyposis coli (APC), protein phosphatase 2A (PP2A), glycogen synthase kinase 3 (GSK3) and casein kinase 1α (CK1α). Our results show that the only member of the destruction complex that increases upon 1,25-D3 incubation is CK1α1, likely making CK1α1 the sole responsible for canonical Wnt inhibition in this system, thus promoting cardiac differentiation.
In this manuscript we show that 1,25-D3 incubation induces the increased expression of Wnt 11, a noncanonical Wnt signaling member that has been shown to induce cardiomyogenesis both during embryonic development and in adult cells (Flaherty & Dawn 2008, Flaherty et al 2012). It has also been reported that Wnt11 induces robust cardiomyogenic differentiation in bone marrow mononuclear cells (Flaherty et al 2008). The general consensus in the literature regarding cardiac differentiation indicates that an initial cardiac specification requires a balanced expression of both canonical and noncanonical Wnt signaling, but once cells are committed such as in the case of our cardiomyocytes, the balance must be tipped in the direction of canonical inhibition for cardiac specification to occur (Eisenberg & Eisenberg 2006, 2007, Ueno et al 2007).
There is increasing evidence that the Wnt signaling pathway is important in regulating cardiac function (Dawson et al 2013). It has been documented that the Wnt signaling pathway plays a pivotal role in heart development but also during adult cardiac hypertrophy and remodeling (Bergmann 2010). As Bergmann pointed out, inhibition of nuclear β-catenin signaling downstream of the canonical Wnt pathway significantly reduced post-infarct mortality and functional decline of LV (left ventricular) function following chronic left anterior descending coronary artery ligation. Cardiac hypertrophy is characterized by an increase in cell size, and is accompanied by protein synthesis, fibrosis and upregulation of a fetal-gene expression pattern (including atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and beta myosin heavy chain (β-MHC) (Rohini et al 2010). Initially an adaptive response, in later stages cardiac hypertrophy can lead to maladaptive remodeling and heart failure, especially if proliferation is occurring prior to appropriate cardiomyocyte differentiation. In normal adult cardiomyocytes, Wnt/Fzd signaling is quiescent (Cingolani, 2007). However, the pathway becomes reactivated in disease states, including hypertrophy. Inhibiting Wnt signaling with 1,25-D3 by activating Casein kinase 1, alpha 1 would attenuate the hypertrophic response. Even more, cardiac fibrosis can result from cardiac diseases such as congestive heart failure (HF) or acute myocardiac infarction (MI), but also from cardiac senescence, genetic predisposition and intense exercise (Benito et al 2011, Rohr 2012). A fibrotic process can impair cardiac relaxation, causing diastolic dysfunction and potentially HF. It also impedes electrical wave propagation, potentially causing arrhythmias. The Wnt pathway is well established to be involved in fibrosis of several organ systems (lung, kidney and liver) (He et al 2009, Henderson et al 2010, Akhmetshina et al 2012). Since canonical Wnt signaling is required for transforming growth factor-β (TGFβ)-mediated fibrosis (Akhmetshina et al 2012). The inhibition of the canonical Wnt signaling pathway promoted by 1,25-D3 could ameliorate the fibrotic process. Moreover, our group shown previously that 1,25-D3 is a potent anti-fibrotic factor promoting the decrease expression of TGF-β, PAI-1 and several collagen isoforms in mesenchymal multipotent cells (Artaza & Norris 2009) There is an in vivo study showing that vitamin D deficiency results in maladaptive cardiac remodeling attributable to progressive myocyte hypertrophy and interstitial fibrosis (Meems et al, 2012). A recent study by our group did show repletion of hypovitaminosis D in humans led to down-regulation of protein expression in abdomen fat biopsies for tumor necrosis factor alpha, IFN gamma, interleukin 6 and soluble intercellular adhesion molecule-1 (sICAM-1), a biomarker of inflammatory process associated with endothelial damage and platelet activation, supporting an in vivo role for vitamin D in related signaling pathways (Martins et al 2014).
The natural conclusion is that targeting or modulating the Wnt signaling pathway with vitamin D may be a suitable natural and cost effective therapeutic intervention that deserves further investigation.
In conclusion, the inhibition of the canonical Wnt signaling pathway through an increase expression of CK1α and at the same time the increase expression of Wnt 11 promoted in both cases by 1,25-D3 supplementation enhanced cardiomyocte differentiation and may prove useful for clinical strategies such as preprogramming stem cells before myocardial transplantation.
These results indicate that vitamin D repletion might enhance the outcomes of cardiac cell therapy or even cardiac cell remodeling, contributing to the prevention/treatment of cardiovascular disease and other related cardiac conditions by promoting cardiac differentiation.
The data presented in this manuscript establish a novel role of vitamin D in the modulation of Wnt signaling in cardiac derived cells promoting cardiac differentiation, which may ultimately be useful in directing differentiation of multipotent cells into cardiomyocytes for cardiac repair applications.
Acknowledgments
This work was supported in part by NIH-NIMHD grants 5U54MD007598-05 and U54MD008149, and by NIH-NIGMS grant SC1NS064611.
Abbreviations
- 1,25-D3
1α, 25-(OH)2D3 (calcitriol)
- VDR
vitamin D receptor
- Wnt11
Drosophila, wingless-related protein 11
- CK1α1
Casein kinase-1, α1
- Gsk3β
Glycogen synthase kinase 3 beta
- APC
adenomatosis polyposis coli
- GAPDH
glyceraldehyde-3-phosphate-dehydrogenase
Footnotes
Declaration of interest: The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the data reported.
References
- Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artaza JN, Bhasin S, Magee TR, Reisz-Porszasz S, Shen R, Groome NP, Meerasahib MF, Gonzalez-Cadavid NF. Myostatin inhibits myogenesis and promotes adipogenesis in C3H 10T(1/2) mesenchymal multipotent cells. Endocrinology. 2005;8:3547–57. doi: 10.1210/en.2005-0362. [DOI] [PubMed] [Google Scholar]
- Artaza JN, Mehrotra R, Norris KC. Vitamin D and the cardiovascular system. Clin J Am Soc Nephrol. 2009;4(9):1515–22. doi: 10.2215/CJN.02260409. [DOI] [PubMed] [Google Scholar]
- Artaza JN, Norris KC. Vitamin D reduces the expression of collagen and key profibrotic factors by inducing an antifibrotic phenotype in mesenchymal multipotent cells. J Endocrinol. 2009;200(2):207–21. doi: 10.1677/JOE-08-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artaza JN, Reisz-Porszasz S, Dow JS, Kloner RA, Tsao J, Bhasin S, Gonzalez-Cadavid NF. Alterations in myostatin expression are associated with changes in cardiac left ventricular mass but not ejection fraction in the mouse. J Endocrinol. 2007;194(1):63–76. doi: 10.1677/JOE-07-0072. [DOI] [PubMed] [Google Scholar]
- Artaza JN, Singh R, Ferrini MG, Braga M, Tsao J, Gonzalez-Cadavid NF. Myostatin promotes a fibrotic phenotypic switch in multipotent C3H 10T1/2 cells without affecting their differentiation into myofibroblasts. J Endocrinol. 2008;196(2):235–49. doi: 10.1677/JOE-07-0408. [DOI] [PubMed] [Google Scholar]
- Artaza JN, Sirad F, Ferrini MG, Norris KC. 1,25(OH)2vitamin D3 inhibits cell proliferation by promoting cell cycle arrest without inducing apoptosis and modifies cell morphology of mesenchymal multipotent cells. J Steroid Biochem Mol Biol. 2010;119(1-2):73–83. doi: 10.1016/j.jsbmb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assalin HB, Rafacho BP, dos Santos PP, Ardisson LP, Roscani MG, Chiuso-Minicucci F, Barbisan LF, Fernandes AA, Azevedo PS, Minicucci MF, et al. Impact of the length of vitamin D deficiency on cardiac remodeling. Circ Heart Fail. 2013;6(4):809–16. doi: 10.1161/CIRCHEARTFAILURE.112.000298. [DOI] [PubMed] [Google Scholar]
- Barbosa EM, Nonogaki S, Katayama ML, Folgueira MA, Alves VF, Brentani MM. Vitamin D3 modulation of plasminogen activator inhibitor type-1 in human breast carcinomas under organ culture. Virchows Arch. 2004;444(2):175–82. doi: 10.1007/s00428-003-0929-5. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114(6):763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- Benito B, Gay-Jordi G, Serrano-Mollar A, Guasch E, Shi Y, Tardif JC, Brugada J, Nattel S, Mont L. Cardiac arrhythmogenic remodeling in a rat model of long-term intensive exercise training. Circulation. 2011;123:13–22. doi: 10.1161/CIRCULATIONAHA.110.938282. [DOI] [PubMed] [Google Scholar]
- Bergmann MW. WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ Res. 2010;107(10):1198–208. doi: 10.1161/CIRCRESAHA.110.223768. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11(24):3286–305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Cardús A, Parisi E, Gallego C, Aldea M, Fernández E, Valdivielso JM. 1,25-Dihydroxyvitamin D3 stimulates vascular smooth muscle cell proliferation through a VEGF-mediated pathway. Kidney Int. 2006;69(8):1377–84. doi: 10.1038/sj.ki.5000304. [DOI] [PubMed] [Google Scholar]
- Cingolani OH. Cardiac hypertrophy and the Wnt/Frizzled pathway. Hypertension. 2007;49:427–428. doi: 10.1161/01.HYP.0000255947.79237.61. [DOI] [PubMed] [Google Scholar]
- Dawson K, Aflaki M, Nattel S. Role of the Wnt-Frizzled system in cardiac pathophysiology: a rapidly developing, poorly understood area with enormous potential. J Physiol. 2013;591(Pt 6):1409–32. doi: 10.1113/jphysiol.2012.235382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eelen G, Gysemans C, Verlinden L, Vanoirbeek E, De Clercq P, Van Haver D, Mathieu C, Bouillon R, Verstuyf A. Mechanism and potential of the growth-inhibitory actions of vitamin D and analogs. Curr Med Chem. 2007;14(17):1893–910. doi: 10.2174/092986707781058823. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Eisenberg CA. Wnt signal transduction and the formation of the myocardium. Dev Biol. 2006;293:305–315. doi: 10.1016/j.ydbio.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Eisenberg CA. Evaluating the role of Wnt signal transduction in promoting the development of the heart. Sci World J. 2007;7:161–176. doi: 10.1100/tsw.2007.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endl E, Steinbach P, Knuchel R, Hofstadter F. Analysis of cell cycle-related Ki-67 and p120 expression by flow cytometric BrdUrd-Hoechst/7AAD and immunolabeling technique. Cytometry. 1997;29:233–241. [PubMed] [Google Scholar]
- Essers J, Theil AF, Baldeyron C, van Cappellen WA, Houtsmuller AB, Kanaar R, Vermeulen W. Nuclear dynamics of PCNA in DNA replication and repair. Mol Cell Biol. 2005;25(21):9350–9. doi: 10.1128/MCB.25.21.9350-9359.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty MP, Abdel-Latif A, Li Q, Hunt G, Ranjan S, Ou Q, Tang XL, Johnson RK, Bolli R, Dawn B. 2008 Noncanonical Wnt11 signaling is sufficient to induce cardiomyogenic differentiation in unfractionated bone marrow mononuclear cells. Circulation. 2008;117(17):2241–52. doi: 10.1161/CIRCULATIONAHA.107.741066. [DOI] [PubMed] [Google Scholar]
- Flaherty MP, Dawn B. Noncanonical Wnt11 signaling and cardiomyogenic differentiation. Trends Cardiovasc Med. 2008;18(7):260–8. doi: 10.1016/j.tcm.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty MP, Kamerzell TJ, Dawn B. Wnt signaling and cardiac differentiation. Prog Mol Biol Transl Sci. 2012;111:153–74. doi: 10.1016/B978-0-12-398459-3.00007-1. [DOI] [PubMed] [Google Scholar]
- Garcia LA, Ferrini MG, Norris KC, Artaza JN. 1,25(OH)(2)vitamin D(3) enhances myogenic differentiation by modulating the expression of key angiogenic growth factors and angiogenic inhibitors in C(2)C(12) skeletal muscle cells. J Steroid Biochem Mol Biol. 2013;133:1–11. doi: 10.1016/j.jsbmb.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia LA, King KK, Ferrini MG, Norris KC, Artaza JN. 1,25(OH)2vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology. 2011;152(8):2976–86. doi: 10.1210/en.2011-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes J, Schwab U, Lemke H, Stein H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer. 1983;31(1):13–20. doi: 10.1002/ijc.2910310104. [DOI] [PubMed] [Google Scholar]
- Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med. 2008;168(11):1174–80. doi: 10.1001/archinte.168.11.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotsman I, Shauer A, Zwas DR, Hellman Y, Keren A, Lotan C, Admon D. Vitamin D deficiency is a predictor of reduced survival in patients with heart failure; vitamin D supplementation improves outcome. Eur J Heart Fail. 2012;14(4):357–66. doi: 10.1093/eurjhf/hfr175. [DOI] [PubMed] [Google Scholar]
- He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/β-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. 2009;20:765–776. doi: 10.1681/ASN.2008060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson WR, Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, Borok Z, Knight DA, Kahn M. Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010;107:14309–14314. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan PK, Slezak SE. Stable cell membrane labeling. Nature. 1989;340(6229):167–8. doi: 10.1038/340167a0. [DOI] [PubMed] [Google Scholar]
- Judd SE, Nanes MS, Ziegler TR, Wilson PW, Tangpricha V. Optimal vitamin D status attenuates the age-associated increase in systolic blood pressure in white Americans: results from the third National Health and Nutrition Examination Survey. Am J Clin Nutr. 2008;87(1):136–41. doi: 10.1093/ajcn/87.1.136. [DOI] [PubMed] [Google Scholar]
- Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13(8):970–4. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP, Norman AW. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1α,25(OH)2-vitamin D3 in vivo and in vitro. Mol Endocrinol. 2004;18:2660–2671. doi: 10.1210/me.2004-0116. [DOI] [PubMed] [Google Scholar]
- Khanna-Jain R, Vuorinen A, Sándor GK, Suuronen R, Miettinen S. Vitamin D(3) metabolites induce osteogenic differentiation in human dental pulp and human dental follicle cells. J Steroid Biochem Mol Biol. 2010;122(4):133–41. doi: 10.1016/j.jsbmb.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Katoh M, Katoh M. WNT signaling pathway and stem cell signaling network. Clin Cancer Res. 2007;1314:4042–5. doi: 10.1158/1078-0432.CCR-06-2316. Review. [DOI] [PubMed] [Google Scholar]
- Kendrick J, Targher G, Smits G, Chonchol M. 25-Hydroxyvitamin D deficiency is independently associated with cardiovascular disease in the Third National Health and Nutrition Examination Survey. Atherosclerosis. 2009;205(1):255–60. doi: 10.1016/j.atherosclerosis.2008.10.033. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004) Am J Cardiol. 2008;102(11):1540–4. doi: 10.1016/j.amjcard.2008.06.067. [DOI] [PubMed] [Google Scholar]
- Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Exp Cell Res. 1976;98:367–381. doi: 10.1016/0014-4827(76)90447-x. [DOI] [PubMed] [Google Scholar]
- Kwon C, Arnold J, Hsiao EC, Taketo MM, Conklin BR, Srivastava D. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc Natl Acad Sci U S A. 2007;104(26):10894–9. doi: 10.1073/pnas.0704044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier M, Schade D, Willems E, Tsuda M, Spiering S, Kalisiak J, Mercola M, Cashman JR. Wnt inhibition correlates with human embryonic stem cell cardiomyogenesis: a structure-activity relationship study based on inhibitors for the Wnt response. J Med Chem. 2012;55(2):697–708. doi: 10.1021/jm2010223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins D, Meng Y, Tareen N, Artaza J, Lee JE, Farodolu C, Gibbons G, Norris K. The Effect of Short Term Vitamin D Supplementation on the Inflammatory and Oxidative Mediators of Arterial Stiffness. Health. 2014;6:1503–1511. doi: 10.4236/health.2014.612185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins D, Wolf M, Pan D, Zadshir A, Tareen N, Thadhani R, Felsenfeld A, Levine B, Mehrotra R, Norris K. Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2007;167(11):1159–65. doi: 10.1001/archinte.167.11.1159. [DOI] [PubMed] [Google Scholar]
- Marvin MJ, Di Rocco G, Gardiner A, Bush SM, Lassar AB. Inhibition of Wnt activity induces heart formation from posterior mesoderm. Genes Dev. 2001;15(3):316–27. doi: 10.1101/gad.855501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meems LM, Cannon MV, Mahmud H, Voors AA, van Gilst WH, Silljé HH, Ruifrok WP, de Boer RA. The vitamin D receptor activator paricalcitol prevents fibrosis and diastolic dysfunction in a murine model of pressure overload. J Steroid Biochem Mol Biol. 2012;132(3-5):282–9. doi: 10.1016/j.jsbmb.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Melamed ML, Muntner P, Michos ED, Uribarri J, Weber C, Sharma J, Raggi P. Serum 25-hydroxyvitamin D levels and the prevalence of peripheral arterial disease: results from NHANES 2001 to 2004. Arterioscler Thromb Vasc Biol. 2008;28(6):1179–85. doi: 10.1161/ATVBAHA.108.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92:139–150. doi: 10.1161/01.res.0000053618.86362.df. [DOI] [PubMed] [Google Scholar]
- Naito T, Shiojima I, Akazawa H, Hidaka K, Morisaki T, Kikuchi A, Komuro I. 2006 Developmental stage-specific biphasic roles of Wnt/β-catenin signaling in cardiomyogenesis and hematopoiesis. Proc Natl Acad Sci U S A. 2006;103(52):19812–19817. doi: 10.1073/pnas.0605768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, et al. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell. 2014;157(4):795–807. doi: 10.1016/j.cell.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol. 2007;103(3-5):533–7. doi: 10.1016/j.jsbmb.2006.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman AW. Minireview: vitamin D receptor: new assignments for an already busy receptor. Endocrinology. 2006;147(12):5542–8. doi: 10.1210/en.2006-0946. [DOI] [PubMed] [Google Scholar]
- Perán M, Marchal JA, López E, Jiménez-Navarro M, Boulaiz H, Rodríguez-Serrano F, Carrillo E, Sánchez-Espin G, de Teresa E, Tosh D, et al. Human cardiac tissue induces transdifferentiation of adult stem cells towards cardiomyocytes. Cytotherapy. 2010;12(3):332–7. doi: 10.3109/14653240903548202. [DOI] [PubMed] [Google Scholar]
- Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3) Endocrinol Metab Clin North Am. 2010;39(2):255–69. doi: 10.1016/j.ecl.2010.02.007. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preusser M, Heinzl H, Gelpi E, Hoftberger R, Fischer I, Pipp I, Milenkovic I, Wohrer A, Popovici F, Wolfsberger S. Ki67 index in intracranial ependymoma: a promising histopathological candidate biomarker. Histopathology. 2008;53(1):39–47. doi: 10.1111/j.1365-2559.2008.03065.x. [DOI] [PubMed] [Google Scholar]
- Ramirez AM, Wongtrakool C, Welch T, Steinmeyer A, Zügel U, Roman J. Vitamin D inhibition of pro-fibrotic effects of transforming growth factor beta1 in lung fibroblasts and epithelial cells. J Steroid Biochem Mol Biol. 2010;118(3):142–50. doi: 10.1016/j.jsbmb.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Hershey S, Ahmed S, Nibbelink K, Simpson RU. 2007 Heart extracellular matrix gene expression profile in the vitamin D receptor knockout mice. J Steroid Biochem Mol Biol. 2007;103(3-5):416–9. doi: 10.1016/j.jsbmb.2006.12.081. [DOI] [PubMed] [Google Scholar]
- Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- Rohini A, Agrawal N, Koyani CN, Singh R. Molecular targets and regulators of cardiac hypertrophy. Pharmacol Res. 2010;61(4):269–80. doi: 10.1016/j.phrs.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Rohr S. Arrhythmogenic implications of fibroblast– myocyte interactions. Circ Arrhythm Electrophysiol. 2012;5:442–452. doi: 10.1161/CIRCEP.110.957647. [DOI] [PubMed] [Google Scholar]
- Scragg R, Sowers M, Bell C. Serum 25-hydroxyvitamin D, ethnicity, and blood pressure in the Third National Health and Nutrition Examination Survey. Am J Hypertens. 2007;20:713–9. doi: 10.1016/j.amjhyper.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Simpson RU, Hershey SH, Nibbelink KA. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J Steroid Biochem Mol Biol. 2007;103:521–4. doi: 10.1016/j.jsbmb.2006.12.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson AG, et al. 2010 Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat Chem Biol. 2010;6(11):829–36. doi: 10.1038/nchembio.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology. 2008;149:558–64. doi: 10.1210/en.2007-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104(23):9685–90. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holten TC, Waanders LF, de Groot PG, Vissers J, Hoefer IE, Pasterkamp G, Prins MW, Roest M. Circulating biomarkers for predicting cardiovascular disease risk; a systematic review and comprehensive overview of meta-analyses. PLoS. 2013;8(4):e62080. doi: 10.1371/journal.pone.0062080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlinden L, Verstuyf A, Convents R, Marcelis S, Van Camp M, Bouillon R. Action of 1,25(OH)2D3 on the cell cycle genes, cyclin D1, p21 and p27 in MCF-7 cells. Mol Cell Endocrinol. 1998;142(1-2):57–65. doi: 10.1016/s0303-7207(98)00117-8. [DOI] [PubMed] [Google Scholar]
- Wang QM, Jones JB, Studzinski GP. Cyclin-dependent kinase inhibitor p27 as a mediator of the G1-S phase block induced by 1,25-dihydroxyvitamin D3 in HL60 cells. Cancer Res. 1996;56(2):264–7. [PubMed] [Google Scholar]
- Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, Benjamin EJ, D'Agostino RB, Wolf M, Vasan RS. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117(4):503–11. doi: 10.1161/CIRCULATIONAHA.107.706127. 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems E, Spiering S, Davidovics H, Lanier M, Xia Z, Dawson M, Cashman J, Mercola M. Small-molecule inhibitors of the Wnt pathway potently promote cardiomyocytes from human embryonic stem cell-derived mesoderm. Circ Res. 2011;109(4):360–4. doi: 10.1161/CIRCRESAHA.111.249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Li H, Ma Z, Feng J, Gao P, Dong H, Zhang Z. Efficient cardiomyogenic differentiation of bone marrow mesenchymal stromal cells by combination of Wnt11 and bone morphogenetic protein 2. Exp Biol Med (Maywood) 2012;237(7):768–76. doi: 10.1258/ebm.2012.011291. [DOI] [PubMed] [Google Scholar]