

Abstract
The aggressiveness of pancreatic cancer is associated with the acquisition of mesenchymal characteristics by a subset of pancreatic cancer cells. The factors driving the development of this subset are not well understood. In this study, we tested the hypothesis that acquisition of a mesenchymal phenotype occurs selectively in tumor cells that harbor specific enabling genetic alterations. We obtained whole‐genome comparative genomic hybridization (CGH) measurements on pancreatic cancer cell lines that have either an epithelial‐like (17 cell lines) or a mesenchymal‐like (9 cell lines) phenotype in vitro. The total amounts of amplifications and deletions were equivalent between the epithelial and mesenchymal groups, but 20 genes showed a major difference between the groups in prevalence of alterations. All 20 alterations (18 deletions and 2 amplifications) were more prevalent in the mesenchymal group, confirming the advanced nature of this cellular subtype. CDKN2A was altered in more than 50% of both groups, but co‐deletions in neighboring genes, and concomitant loss of gene expression, were more prevalent in the mesenchymal group, suggesting that the size of the loss around CDKN2A affects cell phenotype. Whole‐genome CGH on 11 primary cancer tissues revealed that the 20 genes were altered at a higher prevalence (up to 55% of the cases for certain genes) than randomly selected sets of 20 genes, with the same direction of alteration as in the cell lines. These findings support the concept that specific genetic alterations enable phenotype plasticity and provide promising candidate genes for further research.
Keywords: Comparative genomic hybridization, Epithelial–mesenchymal transition, Metastasis, Pancreatic cancer aggressiveness, Cellular plasticity
Highlights
We present a novel approach to identify genes possibly involved with a phenotype.
We identified specific genes frequently altered in mesenchymal‐like cancer cells.
We confirmed a corresponding high rate of alteration in primary tissues.
Most of the genes previously were not implicated in cancer progression.
Alterations near the tumor suppressor p16 might be more than passenger mutations.
1. Introduction
The standard treatments for pancreatic cancer are frustratingly ineffective for long term benefit. Surgical removal of the tumor typically extends survival, and chemotherapeutic treatment can reduce the bulk of tumors, but neither method completely eliminates cancer cells or their ability to spread and grow. As a result, disease recurrence is the norm. The failure of surgical removal indicates that, by the time a cancerous mass in the pancreas is recognized, some of the cancer cells already have disseminated to other parts of the body and established metastatic sites. Likewise, the failure of drugs to kill all cancer cells suggests that certain cells within the tumor have increased resistance to the treatment. Thus the cause of the failure of these treatments potentially can be traced to a subpopulation of cancer cells within each tumor.
Previous research provides more information on the nature of the cancer cells that could be particularly aggressive. Pancreatic cancer cells that express some combination of CD44, CD24, ESA, ALDH, or MET are more competent than the general population of cancer cells in forming tumors in immune‐compromised mice (Herreros‐Villanueva et al., 2012; Kim et al., 2011; Li et al., 2007). Studies on other cancers also demonstrated the existence of a subpopulation tumor‐initiating cells, or cancer stem cells (Al‐Hajj et al., 2003; Ginestier et al., 2007; Haraguchi et al., 2013; Patrawala et al., 2007). Such cells can acquire their stem‐cell properties by transitioning from an epithelial phenotype to a mesenchymal phenotype (Mani et al., 2008; Morel et al., 2008; Rhim et al., 2012). Clinical and experimental observations support the role of a mesenchymal subpopulation in pancreatic cancer progression: patients with cancer cells showing increased levels of mesenchymal markers have particularly poor outcomes (Javle et al., 2007; Nakajima et al., 2004; Oida et al., 2006; Yin et al., 2007), and cultured pancreatic cancer cells with mesenchymal properties are more invasive and resistant to drugs that those with epithelial properties (Arumugam et al., 2009). The epithelial–mesenchymal transition (EMT) therefore could generate the subset of cancer cells responsible for many aspects of pancreatic cancer progression (Yang and Weinberg, 2008).
The conditions required for the induction of EMT typically are present in the inflammatory environment of most pancreatic tumors, such as the activation of transcription factors like TGF‐β, reorganization and expression of particular cytoskeletal proteins, production of extracellular matrix‐degrading enzymes, and changes in the expression of specific microRNAs (Kalluri and Weinberg, 2009). At the same time, several molecules can counter the process of EMT. BMP7 can reverse TGF‐β1‐induced EMT by expression of E‐cadherin, and the expression of miR‐200 family can prevent TGF‐β‐induced EMT (Gregory et al., 2008; Zeisberg et al., 2003). Not all cancer cells undergo EMT, as most of the cancer cells in established pancreatic tumors have an epithelial morphology. Given the myriad routes of initiating EMT and the abundance of EMT‐inducing factors in most pancreatic tumors, the question arises why only a subset of cancer cells undergo this process. One possibility is that only certain cells are predisposed to respond to the external factors that can induce EMT. EMT could occur selectively in cells that harbor specific, enabling genomic alterations.
The most common mutations in pancreatic cancers are in KRAS, TP53, SMAD4, and CDKN2A (Caldas et al., 1994; Goggins et al., 1996; Jones et al., 2008; Moskaluk et al., 1997; Su et al., 1998), and other regions of genetic amplification and deletion frequently appear at multiple loci (Aguirre et al., 2004; Bashyam et al., 2005; Gysin et al., 2005; Harada et al., 2009; Heidenblad et al., 2004; Holzmann et al., 2004; Mahlamaki et al., 2004). Such mutations likely contribute to the initiation of cancer and may not necessarily be involved in the progression of a subset of cells to an invasive and resistant phenotype. A study finding specific amplifications and deletions enriched in patients with venous invasion and shortened survival (Loukopoulos et al., 2007) points to genetic hits that affect cancer cell progression, similar to findings that amplifications at the 7q21‐q22 locus involving the SMURF1 and ARPCIA genes promote cellular invasiveness (Kwei et al., 2011; Laurila et al., 2009). However, most of these studies identified genes that were altered in a very small subset of the cell lines/tissues being studied.
Here we examined the hypothesis that specific genomic alterations are more prevalent in pancreatic cancer cells with a mesenchymal phenotype than those with an epithelial phenotype. Cultured pancreatic cancer cells provided a good model system to test this hypothesis, since they tend to take on either a mesenchymal or an epithelial phenotype. Previously we showed that mesenchymal‐like cancer cells have distinct features of glycosylation in comparison to epithelial‐like cancer cells (Maupin et al., 2010; Wu et al., 2009), and in this study we examined differences in gene copy number gains and losses using comparative genomic hybridization (CGH). Unlike previous studies, we aimed to identify only those genes that were altered in a significant number of the cell lines being examined. We found that the mesenchymal‐like cells harbored specific deletions and amplifications not found in the epithelial‐like cells, whereas the average total number of alterations was not different between the groups. We found that the same genetic alterations were present in primary tumors at a significantly higher prevalence than other genetic alterations.
2. Materials and methods
2.1. Cell culture
The pancreatic cell lines were obtained from the American Type Culture Collection (Manassas, VA) and passaged for fewer than 6 months after resuscitation. The cells were maintained in RPMI medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (Invitrogen). The three‐dimensional cell cultures were performed in a 75:25 mixture of Matrigel (BD Biosciences, Bedford, MA) and DMEM media (Invitrogen) with a protocol similar to the ‘thin gel method’ advised by BD. The cells were recovered from the Matrigel using the Cell Recovery Solution (BD Biosciences) following the protocol supplied.
2.2. Flow cytometry
Pancreatic ductal adenocarcinoma samples were obtained under a WIRB protocol (20040832) for an NIH funded biospecimen repository (NCI P01 Grant CA109552). Biopsies were minced in the presence of NST buffer and DAPI according to published protocols. Briefly, nuclei were disaggregated then filtered through a 40 μm mesh prior to flow sorting with an Influx cytometer (Becton–Dickinson, San Jose, CA) with ultraviolet excitation. DAPI emission was collected at >450 nm. DNA content and cell cycle were analyzed using the software program MultiCycle (Phoenix Flow Systems, San Diego, CA).
2.3. Comparative genomic hybridization (CGH)
DNAs were extracted using Qiagen micro kits (Qiagen, Valencia, CA). For each hybridization, 1 μg of genomic DNA from each sample and 1 μg of pooled commercial 46XX reference DNA (Promega Corp., Madison, WI) were digested with DNaseI and labeled with Cy‐5 dUTP and Cy‐3 dUTP, respectively, using a BioPrime labeling kit (Invitrogen). All labeling reactions were assessed using a Nanodrop assay (Nanodrop, Wilmington, DE). We used Agilent 244K chips to obtain copy number data from the cell lines and patient samples as previously described (Ruiz et al., 2011).
2.4. Identification of genes with differential alterations
We averaged the log10 ratios of each probe within each group (X avg) and then smoothed the values using a sliding window of 5 probes using the formula (X smooth at probe N = 0.1 × X avg probe (N − 2) + 0.2 × X avg probe (N − 1) + 0.4 × X avg probe N + 0.2 × X avg probe (N + 1) + 0.1 × X avg probe (N + 2)), where probes N − 2 to N + 2 are adjacent on a chromosome. We filtered out probes showing low average levels of amplification or deletion by retaining probes with an absolute value of X smooth > 0.3, indicating a two‐fold change relative to the reference DNA. For subsequent analyses, we selected the probe from each gene with the highest fold change, using the non‐smoothed values. We next calculated, for each probe, the percentage of cell lines in each group for which the probe had an absolute value >0.3. We selected the probes that exceeded the 0.3 threshold in at least 50% of the cell lines in both group and that had at least a two‐fold difference between the groups in the percentage of cell lines altered. For example, a probe that was altered (defined as greater than two‐fold change) in 60% of the cell lines in one group and in 25% of the other group passed this criterion. We separately searched for probes altered in at least 50% of both groups.
2.5. Quantitative PCR validation of genomic amplifications and deletions
We extracted genomic DNA using the PrepEase genomic DNA isolation kit (Affymetrix, Santa Clara, CA) following the kit protocol, except using pipetting to homogenize cells instead of a homogenizer. We determined genomic DNA concentrations using UV absorption (Nanodrop, Thermo Scientific) with semi‐quantitative confirmation by gel electrophoresis. Neuroblastoma Amplified Gene (NAG) protein from chromosome 2 was used as a loading control, as according to the CGH data it was in a region where none of the cell lines showed any major alteration. The primers were from Integrated DNA Technologies (Coralville, IA).
We used the Taq PCR Master Mix Kit (QIAGEN, Germantown, MD) to confirm the primer fidelity and performed gradient PCR to identify the optimum annealing temperature of 64 °C. Power SYBR Green PCR master mix (Life Technologies, Carlsbad, CA) was used for qPCR which was measured using the StepOne Plus RT‐PCR system (Applied Biosystems, Carlsbad, CA). A standard curve was made for NAG using template DNA from the Su86.86 cell line to calibrate the data. The qPCR was performed twice to confirm reproducibility.
2.6. Correlation with gene expression
We used gene expression measurements from the Cancer Cell Line Encyclopedia (CCLE) project (Barretina et al., 2012). Gene expression data were available for 25 out of the 26 cell lines that were used in the study. In case of multiple probes (3 or more) for a particular gene, the correlation between the different probes was obtained. If correlation was above 0.75, the values were averaged. If a single probe did the correlate well with the other ones, the outlier was removed while averaging. In case of only two probes and a poor correlation, their correlation with copy numbers was calculated separately as in the case of M34428. We also performed reverse transcription PCR for five genes for which probes were not present in the chip. mRNA was extracted using RNeasy Mini Kit (Qiagen) and treated with DNase I to remove genomic DNA contamination (Thermo Scientific, Pittsburgh PA). Conversion to cDNA was done using the iScript select cDNA synthesis kit (BIO‐RAD, Hercules, CA). A standard curve based on GAPDH expression was used to calibrate the expression levels of the genes. We calculated the significance of the correlation between gene expression and relative gene copy number using Microsoft Excel, with the formula t = r √ ((n − 2)/(1 − r 2)), where t is the value from Student's t distribution. Using the formula, it was found that r values greater than 0.34 were significant at p < 0.05.
2.7. Hierarchical clustering
To account for bias, hierarchical clustering was performed on the gene expression data from the Cancer Cell Line Encyclopedia (CCLE) Project. The gene expression data was logged to the base 2 and then mean centered before clustering using average linkage and Euclidean distance.
3. Results
3.1. Genomic differences between epithelial‐like and mesenchymal‐like cell lines
In order to test the hypothesis that specific genomic changes are more prevalent in pancreatic cancer cells with a mesenchymal phenotype, we classified 26 different pancreatic cancer cell lines into either an epithelial‐like group (n = 17) or a mesenchymal‐like (n = 9) group (Table 1). We observed molecular and morphological diversity between the cell lines and could easily classify most of the cell lines into one of these broad categories, as in a previous study (Maupin et al., 2010). We based the classification on morphology and expression levels of E‐cadherin, vimentin, and ZEB1, which we obtained from gene expression experiments or published information (Table 1). In cases where we did not have gene expression measurements for the molecular markers, we based the classification on the state of apical–basolateral organization, judging that mesenchymal cells typically lose such organization (Schock and Perrimon, 2002). We found that the cell lines showed correspondence between 2D and 3D cultures in morphology and E‐cadherin expression (Fig. S1), a result that agrees with previous findings using xenografts (Neureiter et al., 2005) and that supports the use of phenotypes in 2‐D cell cultures as a means of classification. We also asked whether whole‐genome expression profiling would reveal major differences between the cell lines that might represent a more accurate or natural way to group the cells. Un‐supervised clustering resulted in no clear grouping among the cells (Fig. S2), indicating that the cell lines do not have fundamental differences that result in genome‐wide transcriptional differences. But when we clustered the cells using only 20 genes involved in EMT (Zeisberg and Neilson, 2009), the cell lines segregated generally according to our classification (Fig. S2), confirming a transcriptional difference relating to their mesenchymal and epithelial phenotypes.
Table 1.
Classification of the cell lines. The column called “Classification basis” gives the basis used to classify the cells. The epithelial cells generally had higher E‐cadherin, lower vimentin, lower zeb‐1, and epithelial‐like morphology. In some cases the classification was based on other traits as indicated.
| Cell line | Classification basis | References | Origin of tissue | Classification |
|---|---|---|---|---|
| BxPC‐3 | E‐cadherin (E‐cad), Vimentin (Vim), Zeb1, Morphology | (Maupin et al., 2010) | Primary | Epithelial |
| Capan1 | E‐cad, Vim, Zeb1 | (ATCC; Buck et al., 2007) | Metastasis (liver) | Epithelial |
| Capan‐2 | E‐cad, Vim, Zeb1 | (Buck et al., 2007; Maupin et al., 2010) | Primary | Epithelial |
| DAN‐G | Morphology | (CLS; Frixen et al., 1991) | Primary | Epithelial |
| HPAC | E‐cad, Vim, Zeb1 | (ATCC; Buck et al., 2007) | Primary | Epithelial |
| HPAF‐II | E‐cad, Vim, zeb1 | (ATCC; Buck et al., 2007) | Metastasis (ascites) | Epithelial |
| HUP‐T4 | Morphology | (Nishimura et al., 1993) | Metastasis (ascites) | Epithelial |
| KC1‐MOH1 | Morphology, susceptibility to drugs | (BioInformationWeb; Mohammad et al., 1998; Mohammad et al., 1999) | Primary | Epithelial |
| Panc02.03 | Vim, E‐cad, Zeb1 | (ATCC; Maupin et al., 2010) | Primary | Epithelial |
| Panc03.27 | Vim, E‐cad, Zeb1 | (ATCC; Maupin et al., 2010) | Primary | Epithelial |
| Panc04.03 | E‐cad, vim, Zeb1 | (ATCC; Oncomine) | Primary | Epithelial |
| Panc05.04 | Vim, E‐cad, Zeb1 | (ATCC; Maupin et al., 2010) | Primary | Epithelial |
| Patu8902 | E‐cad, Morphology | (Elsasser et al., 1993; Jesse et al., 2010) | Primary | Epithelial |
| Patu8988T | Polar cells with apical–basolateral organization | (Elsasser et al., 1992) | Metastasis (Liver) | Epithelial |
| Su86.86 | Vim, E‐cad, Zeb1 | (ATCC; Maupin et al., 2010) | Metastasis (liver) | Epithelial |
| SW1990 | Morphology | (Kyriazis et al., 1983) | Primary/metastasis (spleen) | Epithelial |
| YAPC | Morphology, moderate‐poorly differentiated tumors formed in vivo | (Yamada et al., 1998) | Metastasis (ascites) | Epithelial |
| Aspc1 | E‐cad, Zeb1, Vim | (Maupin et al., 2010) | Metastasis (ascites) | Mesenchymal |
| Hs700T | Poorly differentiated tumors formed in vivo, Morphology | (ATCC; Owens, 1976) | Metastasis (pelvis) | Mesenchymal |
| Hs766T | E‐cad, Zeb1, Vim | (Maupin et al., 2010) | Metastasis (Lymph node) | Mesenchymal |
| Hup‐T3 | Poorly differentiated tumor formed in vivo | (Nishimura et al., 1993) | Metastasis (ascites) | Mesenchymal |
| Miapaca‐2 | E‐cad, Zeb1, Vim | (Maupin et al., 2010) | Primary | Mesenchymal |
| Panc‐1 | E‐cad, Zeb1, Vim | (Maupin et al., 2010) | Primary | Mesenchymal |
| Panc8.13 | E‐cad, Zeb1, Vim | (Maupin et al., 2010) | Primary | Mesenchymal |
| Patu8988s | No apical–basolateral organization | (Elsasser et al., 1992) | Metastasis (Liver) | Mesenchymal |
| PSN‐1 | E‐cad, Zeb1, Vim | (Oncomine) | Primary | Mesenchymal |
We obtained measurements of relative genomic amplifications and deletions for each cell line using a 244K‐probe microarray with an average spatial resolution of approximately 8.9 kb. We then tested whether the mesenchymal‐like cells harbored particular genomic alterations more frequently than the epithelial‐like cells. We initially usedStudent's t‐test to identify regions with significantly different levels between the groups but found the method insufficient to distinguish meaningful from irrelevant information, mainly because the test does not include a threshold for minimum level of amplification or deletion. For example, the TTN gene on chromosome 2 was significantly different between the groups (p < 0.0001), but none of the individual cell lines showed major amplification or deletion for that probe (defined as a two‐fold change relative to the reference DNA) (Fig. S3).
Therefore, we based our search on the percentage of cell lines in each group with significant amplifications or deletions, searching for regions that were altered frequently in one group but not the other. First we selected the probes with high average levels of amplifications or deletions, and we then searched for probes with differences between the epithelial and mesenchymal groups in frequency of amplification or deletion. For the first step we chose probes with at least an average two‐fold amplification or deletion in either group, resulting in 783 probes spanning 72 genes. For each gene, we found the probe with the highest average alteration (Table S1) and calculated the difference between the groups in the percentage of cell lines showing either amplification or deletion of that probe. We searched for probes with at least a two‐fold difference between the groups in frequency of alteration and at least 50% alteration in either of the groups. We also required the immediately adjacent probes on either side to be affected in the same direction as the probe being tested. We used these stringent criteria in order to identify the loci that have the highest likelihood of functional contributions to the mesenchymal phenotype.
The search returned 20 genes (Figure 1A, Table S2). All of the 20 alterations were more prevalent in the mesenchymal group. This highly non‐random, preferential enrichment suggested either that the mesenchymal pancreatic cancer cells have evolved beyond the epithelial pancreatic cancer cells or that they have acquired more total genomic alterations. The total number of altered probes was not significantly different between the two groups (Figure 1B), supporting the former proposition. Furthermore, 18 of the 20 alterations were deletions. The total number of amplifications and deletions were similar to each other (Figure 1B), suggesting a higher selection for deletions in the mesenchymal‐like cells (Figure 1C).
Figure 1.
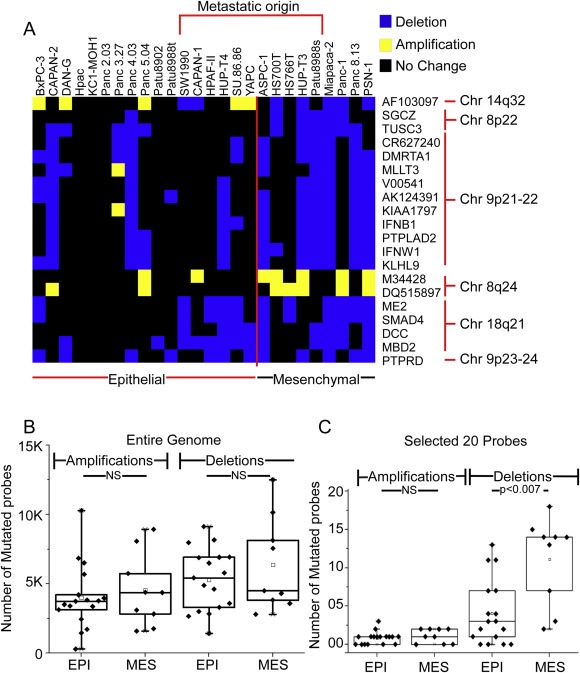
Enriched deletions and amplifications in mesenchymal‐like cancer cells. (A) Patterns of amplification and deletion among the most‐altered 20 genes. For each gene (given in the row labels) and each cell line (given in the column labels), a blue square indicates deletion, a yellow square indicates amplification, and a black square indicates no change. A clustering algorithm grouped the rows by similarity in pattern. (B) Total numbers of amplified or deleted probes in each group for the entire genome. (C) Total numbers of amplified or deleted probes in each group for the 17 hits. Each point represents a cell line. EPI, epithelial cell lines; MES, mesenchymal cell lines.
The alterations were confined to chromosomes 9 (11 deletions), 18 (4 deletions), 8 (2 amplifications and 2 deletions), and 14 (1 gene showing amplification in the mesenchymal group and deletion in the epithelial group). Some cell lines were exceptions to the general trends, like Hs766t, which we classified as mesenchymal but had an alteration in only three of the 20 genes (Figure 1A). Such exceptions might result from imperfect classification or diversity in genetic alterations resulting in similar phenotypes. The Hs766t cell line in particular was harder to classify because morphologically it appears somewhat epithelial but has certain molecular characteristics that are mesenchymal. Some of the epithelial‐like cells had been derived from a metastatic site and therefore may previously have displayed mesenchymal characteristics but returned to the epithelial state via the process of mesenchymal–epithelial transition (Thiery, 2002). Two of the five epithelial cell lines derived from metastatic lesions showed alterations that were akin to the mesenchymal cells as opposed to the epithelial cells (Figure 1A). None of the 20 genes, nor their chromosomal regions, was among those previously associated with adaptation to in vitro culture conditions (Baharvand et al., 2012; Baker et al., 2007; Enver et al., 2005; Imreh et al., 2006), suggesting that the mutations did not occur after the cells were cultured.
We asked whether the identified mutations were focal or broad when averaged over the cell lines, which may give information about which loci are important functionally. Chromosomes 8, 14, and 9 showed divergent patterns (Figure 2). The deletions of chromosome 8 were clustered in the p arm, whereas the amplifications were clustered in the q arm. In contrast, chromosome 14 showed no clustering but a narrow alteration at the putative gene AF103097, unusual because it was both deleted in the mesenchymal cells and amplified in the epithelial cells. However, not all the probes spanning AF103097 were amplified, so the implication of this change is not clear. The deletions of chromosome 9 were clustered on the p arm. The rest of the chromosomes show varying sizes of amplifications and deletions of in the epithelial and mesenchymal cells (Figs. S4 and S5), and in the differences between the mesenchymal and epithelial cells (Fig. S6), but no regions as significant as those noted above.
Figure 2.
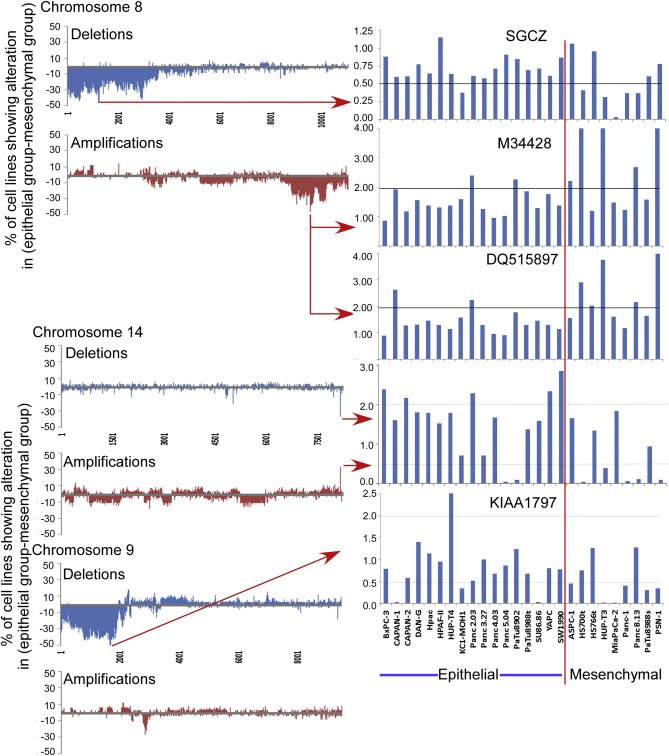
Rates of alterations of selected genes. For each probe, we calculated the difference between the epithelial group and the mesenchymal group in the percentage of cell lines showing a deletion or amplification. We smoothed the resulting numbers by a sliding window of 5 probes, using the formula (0.1 × probe X − 2) + (0.2 × probe X − 1) + (0.4 × probe X) + (0.2 × probe X + 1) + (0.1 × probe X + 2), prior to plotting data from all probes for chromosomes 8, 14, and 9. The column graphs at right show the raw CGH values for selected genes across all cell lines.
This deletion on chromosome 9p includes the tumor suppressor CDKN2A, suggesting that the alterations neighboring CDKN2A might be passenger mutations. Indeed, in no cases were the identified genes on chromosome 9 deleted when CDKN2A was not (Fig. S7). However, neighboring deletions were more frequent in the mesenchymal cells, whereas deletions specific to CDKN2A were evenly spread among the epithelial and mesenchymal cells (Fig. S7). This relationship suggests that the size of the deletion of the CDKN2A locus may affect cell phenotype; loss of CDKN2A promotes tumor formation, but the co‐deletion of certain neighboring genes may promote cancer plasticity and invasiveness.
3.2. Genomic alterations common to both groups
To further understand the relationships between all the cell lines in their genomic alterations, we also searched for genes that were altered across both groups with high frequency (at least 50% in each group). Only CDKN2A and CDKN2B were altered in both groups (Table S3), and both were deletions. CDKN2A, also known as P16, is a well‐known tumor suppressor and one of the most frequently deleted genes in pancreatic tumors (Bardeesy and DePinho, 2002; Caldas et al., 1994; Moskaluk et al., 1997; Oshima et al., 2013). CDKN2B encodes a cyclin‐dependent kinase inhibitor and is a potential effector of TGF‐β induced cell cycle arrest (UniProt). The identification of these two genes is consistent with previous research showing that the most common genetic hits are in tumor suppressors or oncogenes.
Well‐known alterations associated with pancreatic cancer, such as MYC amplification, were present in the initial selection of 72 genes (Table S1) but were not included in the final list of 20 genes due to a prevalence of less than 50% in both groups. Other well‐known pancreatic cancer associated genes such as KRAS and TP53 did not show up in the final lists because they typically are affected by point mutations and low copy number aberrations.
3.3. Validation of selected copy number changes
We selected at least one gene from each chromosome for validation by qPCR of the alterations found by CGH. We confirmed the integrity of the genomic DNA from the cell lines by gel electrophoresis and normalized the qPCR measurements to the NAG gene, which was within a region of minimum genomic alteration. The qPCR data correlated well with the CGH data for the genes KIAA1797, TUSC3, AF103097, M34428 and SMAD4 (Fig. S8). To further test the validity of our findings, we compared the alterations found here to the ones identified in a previous CGH analysis of pancreatic cancer cell lines (Bashyam et al., 2005), which shared 14 cell lines in common with our study. Of the 34 genes that were altered in the previous study, all but one was altered in the matched cell lines of this study. We did not find an amplification of STK19 in the SW1990 cell line, for which our analysis only had one probe.
Another test of validity is to randomly permute the grouping and repeat the analysis. We asked, would we achieve similar results with just any grouping of cell lines? We segregated the 26 cell lines into random groups of 17 (group A) and 9 (group B) and performed the exact same analysis as before, searching for genes that were consistently altered in one of the groups, and we then repeated the random grouping and analysis five separate times. For each of the random groupings, we did not see major differences between the groups. For example, on chromosome 8, only one grouping showed a single difference between any of the groups. We saw similar results with chromosomes 9, 17, and 18 (not shown). This result supports the concept of consistent genetic differences between the mesenchymal and epithelial cells.
3.4. Relationship of copy number to gene expression
Genomic amplifications and deletions could contribute to cancer progression by affecting the expression levels of certain genes (Kwei et al., 2011; Laurila et al., 2009; Legoffic et al., 2009). We obtained gene expression measurements from the CCLE Project (Barretina et al., 2012) for 21 of the 22 genes (including CDKN2A and CDKN2B) identified in our analysis and for 25 of the 26 cell lines. The gene copy numbers correlated significantly with the mRNA expression levels for 9 of 14 genes that had average expression levels above background (Table S4). The strong correlation for SMAD4 was expected, and we also observed strong correlations in genes neighboring CDKN2A, including CDKN2B, KLHL9, and KIAA1797 (Figure 3A), supporting the potential cancer‐promoting functions of losses near CDKN2A. The amplification of M34428 DNA copy number levels correlated with higher gene expression (Figure 3B), supporting a functional component of the DNA alteration. On the other hand, M34428 and DQ515897 are located on either side of the MYC gene, an oncogene that also is amplified in 6 of the cell lines, so they could be co‐altered as passengers with MYC. Although we were not able to detect expression of the AF103097 putative gene—interesting due to its gain in epithelial cells and loss in mesenchymal cells—genetic alterations to this locus nevertheless could contribute to cancer progression, perhaps through affecting the expression of other genes. Overall, a functional role for gene expression loss caused by DNA deletion is supported mainly for the chromosome 9 loci.
Figure 3.
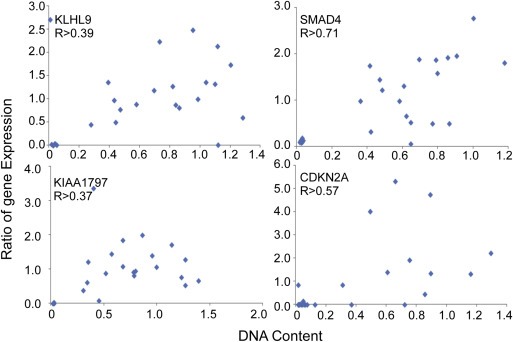
DNA–mRNA correlation. Correlation between the DNA copy numbers (unlogged raw ratios) and the mRNA expression data obtained using U133 Affymetrix mRNA chips.
3.5. Gains and losses of the selected genes in primary tumors
If any of the identified genes is a driver of a cancer‐promoting phenotype, we would expect increased prevalence of the mutation across primary tumors. To test this proposition, we obtained CGH data for 11 primary pancreatic adenocarcinoma samples using the same platform as used for the cell lines. Because a tumor specimen contains a mixture of both normal and neoplastic cells, we flow‐sorted the cells based on DNA ploidy and used only the aneuploid cells (the cancer cell population) for the CGH. This system gives a purer look at the DNA of the cancer cells (Ruiz et al., 2011) and previously provided information on the clonal origins of metastases (Yachida et al., 2010).
Among all the probes, the rate of alteration was similar between the cell lines and tissue, but the prevalence of deletions in the tissue samples was much higher for the 20 probes (Figure 4, and see Fig. S9 for rates of alteration over all chromosomes). For example, 8 (40%) of the 20 genes were deleted in 4 or more tissue samples, compared to less than 2% of all the probes. When we randomly selected 20 different probes 10 separate times and averaged the results, the rate of deletion was similar to that from all probes. Furthermore, the directions of the alterations of the 20 genes were the same between the tissue and cell lines: M34428 and DQ515897 showed amplifications, AF103097 was both amplified and deleted, and the rest showed deletions (Figure 4C). SGCZ, AF103097, KLHL9 and TUSC3 also were aberrant in a high proportion of the samples. CDKN2A, one of the common aberrations, was detected in 7 out of the 11 tumors (not shown), consistent with its major role in pancreatic cancer pathogenesis. The high rate of alteration of the 20 probes, relative to randomly chosen probes, and the agreement in direction of change between cell lines and tissue, support the involvement of these alterations in cancer progression.
Figure 4.
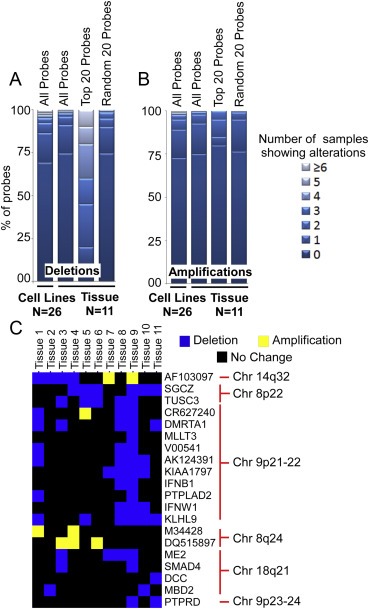
Genomic alterations in primary tumors. The columns indicate the percentage of probes altered in no sample, one sample, two samples and so on. The percentages were calculated for all the probes in the cell lines and primary tissues, the 20 probes selected from the cell lines in the primary tissue as well as random 20 probes in the primary tissue (averaged over 10 times). (A) Deletions. (B) Amplifications. (C) Amplifications and deletions to the 20 selected probes in individual tissue samples. For each gene (given in the row labels) and each tissue sample (columns), a blue square indicates deletion, a yellow square indicates amplification, and a black square indicates no change.
4. Discussion
Cancer cells with a mesenchymal phenotype may be responsible for the early dissemination, treatment resistance, and rapid recurrence that are common in pancreatic cancer. Our over‐arching model was that specific genetic alterations could enable a switch to such a phenotype. The switch might be induced by some external factor, like a cytokine from the tumor environment, but the switch acts only in the population containing the right genetic alteration. This model does not preclude conversion back to an epithelial phenotype under the right circumstance, as in the process of mesenchymal–epithelial transition, since the alteration might enable plasticity rather than a permanent change. We found that specific deletions and amplifications are more prevalent in cancer cells with a mesenchymal phenotype relative to those with an epithelial phenotype. Several features of the results support the validity of the strategy and findings: the enrichment of specific alterations only in the mesenchymal cell lines, despite equivalence between the groups in total alterations; the consistency between the primary tissue and the cell lines in the direction of alterations among the 20 selected genes; and the high prevalence of alterations of the 20 selected genes in primary cancer tissue, relative to all other probes. These findings suggest that some of the identified alterations are cancer promoting rather than initiating, since initiating alterations—such as CDKN2A loss—would appear at equal rates in the epithelial and mesenchymal phenotypes.
The design of this study was different from previous studies examining DNA copy number alterations in pancreatic cancer, but comparisons of results between the studies could give useful insights. As noted above, our study identified nearly all the alterations previously found in matched cell lines (Bashyam et al., 2005). We did not find some of the alterations that were previously observed in small subsets of cases, such as amplifications in 7q21‐q22 (Mahlamaki et al., 2004), most likely because of our more stringent search criteria. Our study is the first to find that some previously identified alterations, including the losses at 8p22 (Bashyam et al., 2005; Griffin et al., 2007), 9p21‐p22 (Aguirre et al., 2004; Gysin et al., 2005; Heidenblad et al., 2004), and 18q21 (Aguirre et al., 2004; Griffin et al., 2007; Gysin et al., 2005) and the amplification at 8q24 (Griffin et al., 2007, 2005, 2004, 2004), may be associated with the development of a mesenchymal phenotype affecting aggressiveness. The deletion of SMAD4 at 18q21 is one of the most frequent events in pancreatic cancer (Herreros‐Villanueva et al., 2013; Kowalski et al., 2007). SMAD4 is also involved in the EMT pathway but its functional significance is debatable. Intact SMAD4 can lead to EMT through TGF‐β signaling which has been shown to increase migration of cells, but SMAD4 inactivation is associated with poor prognosis, presumably through the loss of TGF‐β tumor‐suppressive capabilities (Blackford et al., 2009; Derynck et al., 2001; Herreros‐Villanueva et al., 2013), and with widespread metastases (Iacobuzio‐Donahue et al., 2009; Maitra et al., 2000). Our data support a dedifferentiation promoted by SMAD4 loss.
Two features of the present study may be particularly useful in future work. The system of analyzing only the aneuploid population, using flow‐sorting by DNA content (Mahlamaki et al., 1997), likely enabled the observation of concordance between the cell lines and the tissue samples. Previous studies using primary cancer samples may have been partially confounded by the presence of normal stromal cells within the tumors. The analysis of only the aneuploid population may have given us a cleaner, more accurate view of the cancer‐associated alterations. Another potentially useful feature of this work is the study design. Instead of searching for abnormalities that are commonly found among cancer cases, we grouped the cancers by phenotype and searched for alterations selectively enriched in one of the groups. A search for genes that were common to both groups, as in a typical study design, turned up only known oncogenes and tumor suppressors, whereas the division of cells by phenotype showed revealed alterations in the mesenchymal‐like cell lines that also were present at high prevalence in the primary tissue samples.
Several of the genes identified here are promising subjects for further study. Clearly the losses clustered on chromosome 9 were co‐deleted with the important tumor suppressor CDKN2A, as none of the genes were lost independently from CDKN2A. However, the higher prevalence of certain co‐deletions in the mesenchymal cells and the concomitant loss of gene expression argue for the functional importance of the co‐deleted genes. One of those genes, KIAA1797, was shown to accelerate colony formation, cell migration, and cell invasion (Brockschmidt et al., 2012). Another neighboring gene, KLHL9, is a substrate specific adapter of BCR E3 ligase required for mitotic expression and cytokinesis. This gene is involved in the correct alignment of chromosomes during mitosis, the completion of cytokinesis, and the removal of Aurora B from the complex (Sumara et al., 2007; UniProt). The loss of KLHL9 potentially could disrupt regulation of mitosis.
Losses to the p arm of chromosome 8, affecting SGCZ and TUSC3, also were highly prevalent in the mesenchymal cells. SGCZ (sarcoglycan zeta) is part of the sarcoglycan complex involved in forming a connection between the cytoskeleton and the extracellular matrix (Arco et al., 2012). The loss of sarcoglycans therefore could give ductal epithelial cells increased invasive capability (Arco et al., 2012). The downregulation of TUSC3, either through epigenetic silencing or DNA deletion, is prevalent in prostate cancer (Bova et al., 1993). It is also associated with poor prognosis in ovarian cancer (Pils et al., 2005, 2013). TUSC3 shares high sequence homology with OST3P, a subunit of the oligosaccharyltransferase complex involved in protein N‐glycosylation. Changes to protein glycosylation influence cancer progression in a variety of ways (Dennis et al., 1999), but the effects of TUSC3 loss on glycosylation are not yet known.
In summary, this study supports the hypothesis that specific genetic alterations enable phenotypic switches or cell plasticity that contributes to cancer progression. The mesenchymal cells tend to show larger losses of the region around CDKN2A on chromosome 9, marked by co‐deletion of KLHL9 and KIAA1797 and the associated loss of gene expression. The mesenchymal cells also show losses in the p arm of chromosome 8 involving SGCZ and TUSC3, and highly focal amplifications and deletions to the AF103097 putative gene on chromosome 14. Future studies of human tissue should further explore the relationship of the alterations identified here with the phenotype of the cancer cell. Potentially one could use fluorescence in‐situ hybridization (FISH) to examine gains or losses at the single‐cell level in tissue and then look at correspondence with histology or specific markers. Alternatively, one could flow‐sort the cancer cells by a molecular marker and then examine differences between the groups in genetic gains or losses. A confirmation of the contribution of particular genes to an aggressive phenotype could lead to better strategies to eliminate highly lethal cancer cells and to assess patient prognosis.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
We thank the NCI (R03CA139225, 5P01CA109552‐04, 1R21CA137687), the Van Andel Research Institute, and the Translational Genomics Institute for support of this work.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.04.007.
Sinha Arkadeep, Cherba David, Bartlam Heather, Lenkiewicz Elizabeth, Evers Lisa, Barrett Michael T., Haab Brian B., (2014), Mesenchymal-like pancreatic cancer cells harbor specific genomic alterations more frequently than their epithelial-like counterparts, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.04.007.
References
- Aguirre, A.J. , Brennan, C. , Bailey, G. , Sinha, R. , Feng, B. , Leo, C. , Zhang, Y. , Zhang, J. , Gans, J.D. , Bardeesy, N. , Cauwels, C. , Cordon-Cardo, C. , Redston, M.S. , DePinho, R.A. , Chin, L. , 2004. High-resolution characterization of the pancreatic adenocarcinoma genome. Proc. Natl. Acad. Sci. U.S.A. 101, 9067–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj, M. , Wicha, M.S. , Benito-Hernandez, A. , Morrison, S.J. , Clarke, M.F. , 2003. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arco, A. , Favaloro, A. , Gioffre, M. , Santoro, G. , Speciale, F. , Vermiglio, G. , Cutroneo, G. , 2012. Sarcoglycans in the normal and pathological breast tissue of humans: an immunohistochemical and molecular study. Cells Tissues Organs. 195, 550–562. [DOI] [PubMed] [Google Scholar]
- Arumugam, T. , Ramachandran, V. , Fournier, K.F. , Wang, H. , Marquis, L. , Abbruzzese, J.L. , Gallick, G.E. , Logsdon, C.D. , McConkey, D.J. , Choi, W. , 2009. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 69, 5820–5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATCC, American Type Culture Collection (ATCC). http://www.atcc.org/.
- Baharvand, H. , Aghdami, N. , Harrison, N. , Baker, D. , Andrews, P. , 2012. The Significance of Culture Adaptation of Embryonic Stem Cells for Regenerative Medicine, Advances in Stem Cell Research Humana Press; 17–27. [Google Scholar]
- Baker, D.E. , Harrison, N.J. , Maltby, E. , Smith, K. , Moore, H.D. , Shaw, P.J. , Heath, P.R. , Holden, H. , Andrews, P.W. , 2007. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 25, 207–215. [DOI] [PubMed] [Google Scholar]
- Bardeesy, N. , DePinho, R.A. , 2002. Pancreatic cancer biology and genetics. Nat. Rev. Cancer. 2, 897–909. [DOI] [PubMed] [Google Scholar]
- Barretina, J. , Caponigro, G. , Stransky, N. , Venkatesan, K. , Margolin, A.A. , Kim, S. , Wilson, C.J. , Lehar, J. , Kryukov, G.V. , Sonkin, D. , Reddy, A. , Liu, M. , Murray, L. , Berger, M.F. , Monahan, J.E. , Morais, P. , Meltzer, J. , Korejwa, A. , Jane-Valbuena, J. , Mapa, F.A. , Thibault, J. , Bric-Furlong, E. , Raman, P. , Shipway, A. , Engels, I.H. , Cheng, J. , Yu, G.K. , Yu, J. , Aspesi, P. , de Silva, M. , Jagtap, K. , Jones, M.D. , Wang, L. , Hatton, C. , Palescandolo, E. , Gupta, S. , Mahan, S. , Sougnez, C. , Onofrio, R.C. , Liefeld, T. , MacConaill, L. , Winckler, W. , Reich, M. , Li, N. , Mesirov, J.P. , Gabriel, S.B. , Getz, G. , Ardlie, K. , Chan, V. , Myer, V.E. , Weber, B.L. , Porter, J. , Warmuth, M. , Finan, P. , Harris, J.L. , Meyerson, M. , Golub, T.R. , Morrissey, M.P. , Sellers, W.R. , Schlegel, R. , Garraway, L.A. , 2012. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashyam, M.D. , Bair, R. , Kim, Y.H. , Wang, P. , Hernandez-Boussard, T. , Karikari, C.A. , Tibshirani, R. , Maitra, A. , Pollack, J.R. , 2005. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia (New York, NY). 7, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BioInformationWeb, BioInformationWeb. http://bioinfoweb.com/.
- Blackford, A. , Serrano, O.K. , Wolfgang, C.L. , Parmigiani, G. , Jones, S. , Zhang, X. , Parsons, D.W. , Lin, J.C. , Leary, R.J. , Eshleman, J.R. , Goggins, M. , Jaffee, E.M. , Iacobuzio-Donahue, C.A. , Maitra, A. , Cameron, J.L. , Olino, K. , Schulick, R. , Winter, J. , Herman, J.M. , Laheru, D. , Klein, A.P. , Vogelstein, B. , Kinzler, K.W. , Velculescu, V.E. , Hruban, R.H. , 2009. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 15, 4674–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bova, G.S. , Carter, B.S. , Bussemakers, M.J. , Emi, M. , Fujiwara, Y. , Kyprianou, N. , Jacobs, S.C. , Robinson, J.C. , Epstein, J.I. , Walsh, P.C. , 1993. Homozygous deletion and frequent allelic loss of chromosome 8p22 loci in human prostate cancer. Cancer Res. 53, 3869–3873. [PubMed] [Google Scholar]
- Brockschmidt, A. , Trost, D. , Peterziel, H. , Zimmermann, K. , Ehrler, M. , Grassmann, H. , Pfenning, P.N. , Waha, A. , Wohlleber, D. , Brockschmidt, F.F. , Jugold, M. , Hoischen, A. , Kalla, C. , Seifert, G. , Knolle, P.A. , Latz, E. , Hans, V.H. , Wick, W. , Pfeifer, A. , Angel, P. , Weber, R.G. , 2012. KIAA1797/FOCAD encodes a novel focal adhesion protein with tumour suppressor function in gliomas. Brain. 135, 1027–1041. [DOI] [PubMed] [Google Scholar]
- Buck, E. , Eyzaguirre, A. , Barr, S. , Thompson, S. , Sennello, R. , Young, D. , Iwata, K.K. , Gibson, N.W. , Cagnoni, P. , Haley, J.D. , 2007. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol. Cancer Ther. 6, 532–541. [DOI] [PubMed] [Google Scholar]
- Caldas, C. , Hahn, S.A. , da Costa, L.T. , Redston, M.S. , Schutte, M. , Seymour, A.B. , Weinstein, C.L. , Hruban, R.H. , Yeo, C.J. , Kern, S.E. , 1994. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 8, 27–32. [DOI] [PubMed] [Google Scholar]
- CLS, Cell Line Service. http://www.cell-lines-service.de/content/index_eng.html.
- Dennis, J.W. , Granovsky, M. , Warren, C.E. , 1999. Glycoprotein glycosylation and cancer progression. Biochim. Biophys. Acta. 1473, 21–34. [DOI] [PubMed] [Google Scholar]
- Derynck, R. , Akhurst, R.J. , Balmain, A. , 2001. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29, 117–129. [DOI] [PubMed] [Google Scholar]
- Elsasser, H.P. , Lehr, U. , Agricola, B. , Kern, H.F. , 1992. Establishment and characterisation of two cell lines with different grade of differentiation derived from one primary human pancreatic adenocarcinoma. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 61, 295–306. [DOI] [PubMed] [Google Scholar]
- Elsasser, H.P. , Lehr, U. , Agricola, B. , Kern, H.F. , 1993. Structural analysis of a new highly metastatic cell line PaTu 8902 from a primary human pancreatic adenocarcinoma. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 64, 201–207. [DOI] [PubMed] [Google Scholar]
- Enver, T. , Soneji, S. , Joshi, C. , Brown, J. , Iborra, F. , Orntoft, T. , Thykjaer, T. , Maltby, E. , Smith, K. , Abu Dawud, R. , Jones, M. , Matin, M. , Gokhale, P. , Draper, J. , Andrews, P.W. , 2005. Cellular differentiation hierarchies in normal and culture-adapted human embryonic stem cells. Hum. Mol. Genet. 14, 3129–3140. [DOI] [PubMed] [Google Scholar]
- Frixen, U.H. , Behrens, J. , Sachs, M. , Eberle, G. , Voss, B. , Warda, A. , Lochner, D. , Birchmeier, W. , 1991. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 113, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier, C. , Hur, M.H. , Charafe-Jauffret, E. , Monville, F. , Dutcher, J. , Brown, M. , Jacquemier, J. , Viens, P. , Kleer, C.G. , Liu, S. , Schott, A. , Hayes, D. , Birnbaum, D. , Wicha, M.S. , Dontu, G. , 2007. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 1, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggins, M. , Schutte, M. , Lu, J. , Moskaluk, C.A. , Weinstein, C.L. , Petersen, G.M. , Yeo, C.J. , Jackson, C.E. , Lynch, H.T. , Hruban, R.H. , Kern, S.E. , 1996. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 56, 5360–5364. [PubMed] [Google Scholar]
- Gregory, P.A. , Bert, A.G. , Paterson, E.L. , Barry, S.C. , Tsykin, A. , Farshid, G. , Vadas, M.A. , Khew-Goodall, Y. , Goodall, G.J. , 2008. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell. Biol. 10, 593–601. [DOI] [PubMed] [Google Scholar]
- Griffin, C.A. , Morsberger, L. , Hawkins, A.L. , Haddadin, M. , Patel, A. , Ried, T. , Schrock, E. , Perlman, E.J. , Jaffee, E. , 2007. Molecular cytogenetic characterization of pancreas cancer cell lines reveals high complexity chromosomal alterations. Cytogenet. Genome Res. 118, 148–156. [DOI] [PubMed] [Google Scholar]
- Gysin, S. , Rickert, P. , Kastury, K. , McMahon, M. , 2005. Analysis of genomic DNA alterations and mRNA expression patterns in a panel of human pancreatic cancer cell lines. Genes Chromosomes Cancer. 44, 37–51. [DOI] [PubMed] [Google Scholar]
- Harada, T. , Chelala, C. , Crnogorac-Jurcevic, T. , Lemoine, N.R. , 2009. Genome-wide analysis of pancreatic cancer using microarray-based techniques. Pancreatology. 9, 13–24. [DOI] [PubMed] [Google Scholar]
- Haraguchi, N. , Ishii, H. , Mimori, K. , Ohta, K. , Uemura, M. , Nishimura, J. , Hata, T. , Takemasa, I. , Mizushima, T. , Yamamoto, H. , Doki, Y. , Mori, M. , 2013. CD49f-positive cell population efficiently enriches colon cancer-initiating cells. Int. J. Oncol. 43, 425–430. [DOI] [PubMed] [Google Scholar]
- Heidenblad, M. , Lindgren, D. , Veltman, J.A. , Jonson, T. , Mahlamaki, E.H. , Gorunova, L. , van Kessel, A.G. , Schoenmakers, E.F. , Hoglund, M. , 2005. Microarray analyses reveal strong influence of DNA copy number alterations on the transcriptional patterns in pancreatic cancer: implications for the interpretation of genomic amplifications. Oncogene. 24, 1794–1801. [DOI] [PubMed] [Google Scholar]
- Heidenblad, M. , Schoenmakers, E.F. , Jonson, T. , Gorunova, L. , Veltman, J.A. , van Kessel, A.G. , Hoglund, M. , 2004. Genome-wide array-based comparative genomic hybridization reveals multiple amplification targets and novel homozygous deletions in pancreatic carcinoma cell lines. Cancer Res. 64, 3052–3059. [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva, M. , Gironella, M. , Castells, A. , Bujanda, L. , 2013. Molecular markers in pancreatic cancer diagnosis. Clin. Chim. Acta. 418, 22–29. [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva, M. , Zubia-Olascoaga, A. , Bujanda, L. , 2012. c-Met in pancreatic cancer stem cells: therapeutic implications. World J. Gastroenterol. 18, 5321–5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann, K. , Kohlhammer, H. , Schwaenen, C. , Wessendorf, S. , Kestler, H.A. , Schwoerer, A. , Rau, B. , Radlwimmer, B. , Dohner, H. , Lichter, P. , Gress, T. , Bentz, M. , 2004. Genomic DNA-chip hybridization reveals a higher incidence of genomic amplifications in pancreatic cancer than conventional comparative genomic hybridization and leads to the identification of novel candidate genes. Cancer Res. 64, 4428–4433. [DOI] [PubMed] [Google Scholar]
- Iacobuzio-Donahue, C.A. , Fu, B. , Yachida, S. , Luo, M. , Abe, H. , Henderson, C.M. , Vilardell, F. , Wang, Z. , Keller, J.W. , Banerjee, P. , Herman, J.M. , Cameron, J.L. , Yeo, C.J. , Halushka, M.K. , Eshleman, J.R. , Raben, M. , Klein, A.P. , Hruban, R.H. , Hidalgo, M. , Laheru, D. , 2009. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 27, 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imreh, M.P. , Gertow, K. , Cedervall, J. , Unger, C. , Holmberg, K. , Szoke, K. , Csoregh, L. , Fried, G. , Dilber, S. , Blennow, E. , Ahrlund-Richter, L. , 2006. In vitro culture conditions favoring selection of chromosomal abnormalities in human ES cells. J. Cell. Biochem. 99, 508–516. [DOI] [PubMed] [Google Scholar]
- Javle, M.M. , Gibbs, J.F. , Iwata, K.K. , Pak, Y. , Rutledge, P. , Yu, J. , Black, J.D. , Tan, D. , Khoury, T. , 2007. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann. Surg. Oncol. 14, 3527–3533. [DOI] [PubMed] [Google Scholar]
- Jesse, S. , Koenig, A. , Ellenrieder, V. , Menke, A. , 2010. Lef-1 isoforms regulate different target genes and reduce cellular adhesion. Int. J. Cancer. 126, 1109–1120. [DOI] [PubMed] [Google Scholar]
- Jones, S. , Zhang, X. , Parsons, D.W. , Lin, J.C. , Leary, R.J. , Angenendt, P. , Mankoo, P. , Carter, H. , Kamiyama, H. , Jimeno, A. , Hong, S.M. , Fu, B. , Lin, M.T. , Calhoun, E.S. , Kamiyama, M. , Walter, K. , Nikolskaya, T. , Nikolsky, Y. , Hartigan, J. , Smith, D.R. , Hidalgo, M. , Leach, S.D. , Klein, A.P. , Jaffee, E.M. , Goggins, M. , Maitra, A. , Iacobuzio-Donahue, C. , Eshleman, J.R. , Kern, S.E. , Hruban, R.H. , Karchin, R. , Papadopoulos, N. , Parmigiani, G. , Vogelstein, B. , Velculescu, V.E. , Kinzler, K.W. , 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. , Weinberg, R.A. , 2009. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M.P. , Fleming, J.B. , Wang, H. , Abbruzzese, J.L. , Choi, W. , Kopetz, S. , McConkey, D.J. , Evans, D.B. , Gallick, G.E. , 2011. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One. 6, e20636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski, J. , Morsberger, L.A. , Blackford, A. , Hawkins, A. , Yeo, C.J. , Hruban, R.H. , Griffin, C.A. , 2007. Chromosomal abnormalities of adenocarcinoma of the pancreas: identifying early and late changes. Cancer Genet. Cytogenet. 178, 26–35. [DOI] [PubMed] [Google Scholar]
- Kwei, K.A. , Shain, A.H. , Bair, R. , Montgomery, K. , Karikari, C.A. , van de Rijn, M. , Hidalgo, M. , Maitra, A. , Bashyam, M.D. , Pollack, J.R. , 2011. SMURF1 amplification promotes invasiveness in pancreatic cancer. PLoS One. 6, e23924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriazis, A.P. , McCombs, W.B. , Sandberg, A.A. , Kyriazis, A.A. , Sloane, N.H. , Lepera, R. , 1983. Establishment and characterization of human pancreatic adenocarcinoma cell line SW-1990 in tissue culture and the nude mouse. Cancer Res. 43, 4393–4401. [PubMed] [Google Scholar]
- Laurila, E. , Savinainen, K. , Kuuselo, R. , Karhu, R. , Kallioniemi, A. , 2009. Characterization of the 7q21-q22 amplicon identifies ARPC1A, a subunit of the Arp2/3 complex, as a regulator of cell migration and invasion in pancreatic cancer. Genes Chromosomes Cancer. 48, 330–339. [DOI] [PubMed] [Google Scholar]
- Legoffic, A. , Calvo, E.L. , Barthet, M. , Delpero, J.R. , Dagorn, J.C. , Iovanna, J.L. , 2009. Identification of genomic alterations associated with the aggressiveness of pancreatic cancer using an ultra-high-resolution CGH array. Pancreatology. 9, 267–272. [DOI] [PubMed] [Google Scholar]
- Li, C. , Heidt, D.G. , Dalerba, P. , Burant, C.F. , Zhang, L. , Adsay, V. , Wicha, M. , Clarke, M.F. , Simeone, D.M. , 2007. Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037. [DOI] [PubMed] [Google Scholar]
- Loukopoulos, P. , Shibata, T. , Katoh, H. , Kokubu, A. , Sakamoto, M. , Yamazaki, K. , Kosuge, T. , Kanai, Y. , Hosoda, F. , Imoto, I. , Ohki, M. , Inazawa, J. , Hirohashi, S. , 2007. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 98, 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlamaki, E.H. , Hoglund, M. , Gorunova, L. , Karhu, R. , Dawiskiba, S. , Andren-Sandberg, A. , Kallioniemi, O.P. , Johansson, B. , 1997. Comparative genomic hybridization reveals frequent gains of 20q, 8q, 11q, 12p, and 17q, and losses of 18q, 9p, and 15q in pancreatic cancer. Genes Chromosomes Cancer. 20, 383–391. [DOI] [PubMed] [Google Scholar]
- Mahlamaki, E.H. , Kauraniemi, P. , Monni, O. , Wolf, M. , Hautaniemi, S. , Kallioniemi, A. , 2004. High-resolution genomic and expression profiling reveals 105 putative amplification target genes in pancreatic cancer. Neoplasia (New York, N.Y.). 6, 432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra, A. , Molberg, K. , Albores-Saavedra, J. , Lindberg, G. , 2000. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. Am. J. Pathol. 157, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani, S.A. , Guo, W. , Liao, M.J. , Eaton, E.N. , Ayyanan, A. , Zhou, A.Y. , Brooks, M. , Reinhard, F. , Zhang, C.C. , Shipitsin, M. , Campbell, L.L. , Polyak, K. , Brisken, C. , Yang, J. , Weinberg, R.A. , 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupin, K.A. , Sinha, A. , Eugster, E. , Miller, J. , Ross, J. , Paulino, V. , Keshamouni, V.G. , Tran, N. , Berens, M. , Webb, C. , Haab, B.B. , 2010. Glycogene expression alterations associated with pancreatic cancer epithelial-mesenchymal transition in complementary model systems. PLoS One. 5, e13002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad, R.M. , Dugan, M.C. , Mohamed, A.N. , Almatchy, V.P. , Flake, T.M. , Dergham, S.T. , Shields, A.F. , Al-Katib, A.A. , Vaitkevicius, V.K. , Sarkar, F.H. , 1998. Establishment of a human pancreatic tumor xenograft model: potential application for preclinical evaluation of novel therapeutic agents. Pancreas. 16, 19–25. [DOI] [PubMed] [Google Scholar]
- Mohammad, R.M. , Li, Y. , Mohamed, A.N. , Pettit, G.R. , Adsay, V. , Vaitkevicius, V.K. , Al-Katib, A.M. , Sarkar, F.H. , 1999. Clonal preservation of human pancreatic cell line derived from primary pancreatic adenocarcinoma. Pancreas. 19, 353–361. [DOI] [PubMed] [Google Scholar]
- Morel, A.P. , Lievre, M. , Thomas, C. , Hinkal, G. , Ansieau, S. , Puisieux, A. , 2008. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 3, e2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskaluk, C.A. , Hruban, R.H. , Kern, S.E. , 1997. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 57, 2140–2143. [PubMed] [Google Scholar]
- Nakajima, S. , Doi, R. , Toyoda, E. , Tsuji, S. , Wada, M. , Koizumi, M. , Tulachan, S.S. , Ito, D. , Kami, K. , Mori, T. , Kawaguchi, Y. , Fujimoto, K. , Hosotani, R. , Imamura, M. , 2004. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 10, 4125–4133. [DOI] [PubMed] [Google Scholar]
- Neureiter, D. , Zopf, S. , Dimmler, A. , Stintzing, S. , Hahn, E.G. , Kirchner, T. , Herold, C. , Ocker, M. , 2005. Different capabilities of morphological pattern formation and its association with the expression of differentiation markers in a xenograft model of human pancreatic cancer cell lines. Pancreatology. 5, 387–397. [DOI] [PubMed] [Google Scholar]
- Nishimura, N. , Saito, S. , Kubota, Y. , Moto-o, N. , Taguchi, K. , Yamazaki, K. , Watanabe, A. , Sasaki, H. , 1993. Newly established human pancreatic carcinoma cell lines and their lectin binding properties. Int. J. Pancreatol. 13, 31–41. [DOI] [PubMed] [Google Scholar]
- Oida, Y. , Yamazaki, H. , Tobita, K. , Mukai, M. , Ohtani, Y. , Miyazaki, N. , Abe, Y. , Imaizumi, T. , Makuuchi, H. , Ueyama, Y. , Nakamura, M. , 2006. Increased S100A4 expression combined with decreased E-cadherin expression predicts a poor outcome of patients with pancreatic cancer. Oncol. Rep. 16, 457–463. [PubMed] [Google Scholar]
- Oncomine, Oncomine. https://www.oncomine.org/resource/login.html.
- Oshima, M. , Okano, K. , Muraki, S. , Haba, R. , Maeba, T. , Suzuki, Y. , Yachida, S. , 2013. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann. Surg. 258, 336–346. [DOI] [PubMed] [Google Scholar]
- Owens, R.B. , 1976. Selective Cultivation of Mammalian Epithelial Cells Academic Press Incorporated; [DOI] [PubMed] [Google Scholar]
- Patrawala, L. , Calhoun-Davis, T. , Schneider-Broussard, R. , Tang, D.G. , 2007. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 67, 6796–6805. [DOI] [PubMed] [Google Scholar]
- Pils, D. , Horak, P. , Gleiss, A. , Sax, C. , Fabjani, G. , Moebus, V.J. , Zielinski, C. , Reinthaller, A. , Zeillinger, R. , Krainer, M. , 2005. Five genes from chromosomal band 8p22 are significantly down-regulated in ovarian carcinoma: N33 and EFA6R have a potential impact on overall survival. Cancer. 104, 2417–2429. [DOI] [PubMed] [Google Scholar]
- Pils, D. , Horak, P. , Vanhara, P. , Anees, M. , Petz, M. , Alfanz, A. , Gugerell, A. , Wittinger, M. , Gleiss, A. , Auner, V. , Tong, D. , Zeillinger, R. , Braicu, E.I. , Sehouli, J. , Krainer, M. , 2013. Methylation status of TUSC3 is a prognostic factor in ovarian cancer. Cancer. 119, 946–954. [DOI] [PubMed] [Google Scholar]
- Rhim, A.D. , Mirek, E.T. , Aiello, N.M. , Maitra, A. , Bailey, J.M. , McAllister, F. , Reichert, M. , Beatty, G.L. , Rustgi, A.K. , Vonderheide, R.H. , Leach, S.D. , Stanger, B.Z. , 2012. EMT and dissemination precede pancreatic tumor formation. Cell. 148, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz, C. , Lenkiewicz, E. , Evers, L. , Holley, T. , Robeson, A. , Kiefer, J. , Demeure, M.J. , Hollingsworth, M.A. , Shen, M. , Prunkard, D. , Rabinovitch, P.S. , Zellweger, T. , Mousses, S. , Trent, J.M. , Carpten, J.D. , Bubendorf, L. , Von Hoff, D. , Barrett, M.T. , 2011. Advancing a clinically relevant perspective of the clonal nature of cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 12054–12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schock, F. , Perrimon, N. , 2002. Molecular mechanisms of epithelial morphogenesis. Annu. Rev. Cell Dev. Biol. 18, 463–493. [DOI] [PubMed] [Google Scholar]
- Su, G.H. , Hilgers, W. , Shekher, M.C. , Tang, D.J. , Yeo, C.J. , Hruban, R.H. , Kern, S.E. , 1998. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res. 58, 2339–2342. [PubMed] [Google Scholar]
- Sumara, I. , Quadroni, M. , Frei, C. , Olma, M.H. , Sumara, G. , Ricci, R. , Peter, M. , 2007. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev. Cell. 12, 887–900. [DOI] [PubMed] [Google Scholar]
- Thiery, J.P. , 2002. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2, 442–454. [DOI] [PubMed] [Google Scholar]
- UniProt, UniProt. http://www.uniprot.org/.
- Wu, Y.M. , Nowack, D.D. , Omenn, G.S. , Haab, B.B. , 2009. Mucin glycosylation is altered by pro-inflammatory signaling in pancreatic-cancer cells. J. Proteome Res. 8, 1876–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida, S. , Jones, S. , Bozic, I. , Antal, T. , Leary, R. , Fu, B. , Kamiyama, M. , Hruban, R.H. , Eshleman, J.R. , Nowak, M.A. , Velculescu, V.E. , Kinzler, K.W. , Vogelstein, B. , Iacobuzio-Donahue, C.A. , 2010. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 467, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, T. , Okajima, F. , Adachi, M. , Ohwada, S. , Kondo, Y. , 1998. Growth dependency of a new human pancreatic cancer cell line, YAPC, on autocrine interleukin-1alpha stimulation. Int. J. Cancer. 76, 141–147. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Weinberg, R.A. , 2008. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell. 14, 818–829. [DOI] [PubMed] [Google Scholar]
- Yin, T. , Wang, C. , Liu, T. , Zhao, G. , Zha, Y. , Yang, M. , 2007. Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J. Surg. Res. 141, 196–203. [DOI] [PubMed] [Google Scholar]
- Zeisberg, M. , Hanai, J. , Sugimoto, H. , Mammoto, T. , Charytan, D. , Strutz, F. , Kalluri, R. , 2003. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968. [DOI] [PubMed] [Google Scholar]
- Zeisberg, M. , Neilson, E.G. , 2009. Biomarkers for epithelial–mesenchymal transitions. J. Clin. Invest. 119, 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data