

Abstract
Hepcidin, a peptide hormone produced in the liver, decreases intestinal iron absorption and macrophage iron release via effects on ferroportin. Bone morphogenic protein and Stat3 signaling regulate Hepcidin's transcription. Hepcidin is a potential drug target for patients with iron overload syndromes because its levels are inappropriately low in these individuals. To generate a tool for identifying small molecules that modulate Hepcidin expression, we stably transfected human hepatocytes (HepG2) cells with a reporter construct containing 2.7 kilobases of the human Hepcidin promoter upstream of a firefly reporter gene. We used high throughput methods to screen 10,169 chemicals in duplicate for their effect on Hepcidin expression and cell viability. Regulators were identified as chemicals that caused a change >3 standard deviations above or >1.5 standard deviations below the mean of the other chemicals (z-score >3 or <-1.5), while not adversely affecting cell viability, quantified by fluorescence assay. Following validation assays, we identified 16 chemicals in a broad range of functional classes that promote Hepcidin expression. All of the chemicals identified increased expression of bone morphogenic protein-dependent and/or Stat3-dependent genes, however none of them strongly increased phosphorylation of Smad1,5,8 or Stat3.
Keywords: hemochromatosis, thalassemia, Stat3, bone morphogenic protein, Interleukin-6
Introduction
Hepcidin is a cysteine-rich peptide hormone that regulates the absorption and distribution of iron in humans and other animals[1]. Hepcidin production is transcriptionally regulated in the liver in response to body iron stores and inflammation [2]. Increases in plasma iron levels result in enhanced signaling via bone morphogenic proteins [3] and phosphorylation of Smad1,5, and 8, which facilitates Smad4 binding to the Hepcidin promoter and greater Hepcidin transcription [4]. The inflammatory cytokine, interleukin-6, IL-6, can also upregulate Hepcidin by activating Stat3 and enhancing Stat3 binding to the Hepcidin promoter [5]. Hepcidin binds ferroportin1, the only known vertebrate iron exporter, resulting in internalization and degradation of both proteins [6]. Degradation of ferroportin1 decreases intestinal iron absorption [6] and prevents the release of iron from macrophage iron stores to developing erythrocytes in the bone marrow [7].
Clinical studies have demonstrated that Hepcidin levels are inappropriately low in patients with hereditary diseases associated with iron overload, such as thalassemia, congenital dyserythropoietic anemia, and hereditary hemochromatosis [8]. Iron overload is the major cause of death in patients with thalassemia major [9] and an important cause of morbidity in transfusion-dependent patients, such as bone marrow transplant recipients [10]. Current therapies for iron overload are restricted to chelation or removing blood, phlebotomy [11]. These therapies are not well tolerated or completely effective in many patients [12]. Intriguingly, transgenic over-expression of Hepcidin in mouse models of hereditary hemochromatosis[13] or β-thalassemia [14] reduces iron overload. Thus, pharmacologically increasing Hepcidin levels may help patients with iron overload by decreasing intestinal iron absorption. Hepcidin agonists under development include Hepcidin mimics, such as rationally designed peptides (minihepcidins), and Hepcidin stimulators, such as anti-sense oligonucleotides directed against inihibitors of Hepcidin expression, bone morphogenic protein 6 (BMP6) and small molecules therapies that activate the Stat and/or Smad pathways.[12].
Chemical screens are unbiased approaches to identifying small molecules that affect biological processes. They have been useful in identifying antagonists of specific pathways. For instance the bone morphogenic protein receptor 1 antagonist, dorsomorphin, was identified in a chemical screen for small molecules that affect zebrafish embryonic development [15]. Chemical screens identifying small molecules that impact specific biological processes have improved our understanding of these processes and led to clinical trials. For instance, prostaglandin E2, was shown to be important in hematopoietic stem cell proliferation [16] and is now being evaluated in human trials to improve the efficiency of umbilical cord hematopoietic stem cell transplants[17].
In a preliminary chemical screen evaluating the effect of isoflavones and related compounds in zebrafish embryos and human hepatocytes, we identified the small molecule genistein, a phytoestrogen that is one of the major components of soybeans, as a stimulator of Hepcidin expression that activated Stat3 and Smad signaling [18]. In order to identify additional small molecules that act via different mechanisms and may have greater potency, we undertook a high throughput chemical screen for small molecules that increase Hepcidin expression in human hepatocytes. To achieve this, we generated a line of human hepatoma cells, HepG2 Hepcidin-luciferase, that express 2.7 kb of the human Hepcidin promoter upstream of a firefly luciferase reporter. We screened a total of 10,169 small molecules in duplicate for their ability to increase or decrease Hepcidin expression without impairing cell viability. We validated our hits with quantitative realtime RT-PCR assays for Hepcidin expression and characterized them by their effects on genes regulated by BMP's or Stat3, as well as Western blots to detect phosphorylation of Smad1,5,8 or Stat3. We confirmed 16 small molecule Hepcidin stimulating agents in a broad range of functional classes. All of the chemicals identified increased expression of bone morphogenic protein-dependent and/or Stat3-dependent genes, however none of them strongly increased phosphorylation of Smad1,5,8 or Stat3. Several of the Hepcidin stimulatory chemicals inhibit growth factor receptor dependent signaling (AG1296, GTP 14564, AS252424, 10058-F, SU6668, and pterostilbene), decrease inflammation (leflunomide, amlexanox), or impair DNA repair and promote apoptosis (daunorubicin, 9-aminocridine, ethacridine), while the small molecules, vorinostat and SB 204741, inhibit histone deacetylase and serotonin receptor 2B, respectively. Two of the molecules, ipriflavone and vorinostat, were active at concentrations that were 10-fold below those required for genistein's effect and thus appear to be intriguing candidates for further development.
Materials and Methods
Cell culture and Reagents
The human hepatocarcinoma cell line, HepG2, (American Type Culture Collection, Manassas, VA) was maintained in α-Minimum Essential Medium (α-MEM)/10% certified endotoxin-free fetal bovine serum (FBS)/1% penicillin-streptomycin (Life Technologies, Grand Island, NY) at 37°C, 5% CO2. To generate a Hepcidin reporter cell line, HepG2 cells, were transfected using SuperFect (Qiagen, Valencia, CA) transfection reagent and a reporter construct including a 2.7 kb fragment of the human Hepcidin promoter upstream of a firefly luciferase promoter (gift of Drs. Ganz and Nemeth). Transfected clones were selected for resistance to G418 (Life Technologies) and subsequently maintained in the conditions described above with the addition of G418 1 mg/mL. Bone morphogenic protein 6 (BMP6) (R&D Systems, Minneapolis, MN), interleukin-6 (R&D Systems), and genistein (Sigma-Aldrich, St. Louis, MO) were used as positive controls for Hepcidin-luciferase activity, while dorsomorphin (#171260, Calbiochem, Billerica, MA) was used as a negative control. WP1066 (#573097, Calbiochem) was used as an inhibitor of Stat3 signaling. Interleukin-6, EGF, FGF, PDGF, and VEGFA were obtained from R&D Systems.
Chemical screen
The Institute of Chemistry and Cell Biology (ICCB) Screening Facility at Harvard Medical School provided drug libraries. The complete list of chemicals screened and the screening data are provided in Supplementary Table 1. The day before the addition of compounds, HepG2-Hepcidin luciferase cells were plated at 5000 cells in 30 μl per well of a 384-well microtiter plate (Nunc 142762) in α-MEM/1% penicillin/streptomycin using a WellMate MicroPlate Dispenser (Thermo Scientific, Rockford, IL). Twenty-four hours later, 100 nL aliquot of chemical was pin-transferred from each well on the chemical library plate to a corresponding well on the screening plate. BMP6 (50 ng/ml) was used as a positive control while vehicle only, DMSO (0.3%), was used as a negative control. After 24 hours of treatment, the cell viability and Hepcidin promoter activity were measured with the OneGlo+Tox Cell Viability and Luciferase Reporter assay (E7120, Promega, Madison, WI) according to the manufacturer's instructions using an EnVision 2102 Plate Reader (PerkinElmer, Waltham, MA). Fluorescence was measured using an excitation wavelength of 380-400 nm and emission wavelength of 505 nm. The entire screen was performed in duplicate.
The primary readout was the crosstalk-corrected Hepcidin luminescence for each well. The secondary readout was cell viability fluorescence for each well. For each readout and each well, a z-score was calculated using the formula: z-score [z = (x - mean of samples on the plate)/standard deviation of samples on the plate] where x=the fluorescence or luminescence intensity for the particular well. The positive and negative controls were excluded from calculation of the mean and standard deviation for the plate. An agonist compound was considered a hit if the luciferase z-scores for both replicates were >3. An antagonist compound was considered a hit if the luciferase z-score for each replicate was <-1. Any agonist or antagonist with a cell viability fluorescence z-score <-1 on either replicate was excluded from being considered a hit.
After identifying hits in the screening, we re-screened selected regulators at the original and two additional dilutions using the same luciferase and fluorescence assays. We considered a hit to be validated if it increased Hepcidin promoter activity at least 2-fold above the vehicle-only control (1% DMSO) at one of the concentrations. Negative regulators were identified as those that produced at least a 50% reduction in Hepcidin promoter activity. Supplementary Table 2 provides the sources, functional categories, and chemical structures for the candidate regulators that were characterized further by quantitative realtime RT-PCR and Western blots.
Quantitative realtime RT-PCR
In order to evaluate whether or not candidate regulators upregulate Hepcidin via the Stat3 pathway and/or the Smad4 pathway, we plated 400,000 wild type HepG2 cells per well of a 12-well tissue culture plate. After 8 hours of serum starvation in α-MEM/1% FBS, we added each candidate regulator. After 24 hours of treatment, we extracted RNA, and generated cDNA according to the method [18]. We measured the transcript levels of Hepcidin and key genes in each of these pathways in quantitative realtime RT-PCR using primers and probes as described (Supplementary Table 3).
Western Blots
To test for the effects of the Hepcidin regulators on proteins involved with the Smad4 or Stat3 signaling pathways, we plated 400,000 cells in a 12-well tissue culture plate and changed the media to α-MEM/1% FBS for 16 hours prior to treating the cells with chemicals for 1 hour. The cells were lysed in radio-immunoprecipitation assay buffer (RIPA buffer) with 1x Protease Inhibitor/1x Phosphatase Inhibitor Cocktail (both from Roche). The Pierce BCA Assay (Thermo Scientific) was used to measure the protein concentrations. Twenty micrograms of protein lysate was loaded with 0.5x TruSep SDS Sample Buffer (NuSep Inc., Bogart, GA) in each lane of a Tris-Glycine 4-10% SDS polyacrylamide gel (NuSep Inc.). After the gel was run and transferred to a polyvinylidene difluoride (PVDF) membrane, the membrane was blocked with TBS/0.05% Tween20/5% bovine serum albumin for antibodies against phosphorylated proteins or Pierce Protein-Free TBS Blocking Buffer (Thermo Scientific) for all other antibodies. The primary antibodies, all rabbit anti-human, were used at the following dilutions: phospho-Smad1, 5, and 8 (#9511S, Cell Signaling Technology, Inc., Danvers, MA) 1:200, phospho-Stat3 (SC-8001-R, Santa Cruz Biotechnology Inc., Dallas, TX) 1:100, Smad1 (#9743S, Cell Signaling Technology Inc.) 1:500, Stat3 (#SC-482, Santa Cruz Biotechnology, Inc.) 1:200, or β-actin (#4967S, Cell Signaling Technology Inc.) 1:1000. The blots were developed with secondary antibody, mouse anti-rabbit IgG-horseradish peroxidase (#SC-2357, Santa Cruz Biotechnology Inc.) 1:5000, followed by addition of Pierce ECL Western Blotting Substrate (Thermo Scientific) according to the manufacturer's instructions. The blots were exposed to Kodak Biomax light film (Sigma-Aldrich) for 5-30 min at room temperature.
Statistical analysis
Graphs were created and statistical analyses were performed using Prism 6.0c (Graphpad, San Diego, CA). We used the Kruskal-Wallis method to generate a global p-value for each experiment. Where the global p-value was <0.05, Student's t-tests were performed. P<0.05 was considered a significant result on the Student's t-test.
Structural analysis of small molecules
To assess patterns of structural similarity, the structures of all the compounds producing an average crosstalk corrected Hepcidin-luciferase z-score >3 or <-1, regardless of effects of viability were analyzed. The 405 compound structures were imported into Vortex (Dotmatics, Inc., version 2012.07.15406) and a 1024-bit Dotmatics hex-packed fingerprint was generated. Compounds were clustered on the basis of this fingerprint using Rogers-Tanimoto similarity, leading to 57 structural clusters (378 compounds) plus 27 singleton compounds that were not included in any of the clusters.
Results
In order to evaluate the effects of a broad range of small molecules on Hepcidin expression, we screened 10,169 chemicals in a dual Hepcidin luciferase assay and viability assay. The screening assays were performed in HepG2 cells stably transfected with a human Hepcidin promoter fragment (2.7kb) upstream of a firefly luciferase reporter. Hepcidin-luciferase activity in treated cells was measured as fold-change over controls treated with vehicle only (DMSO ≤1%). We confirmed that treatment for 24 hours with positive controls, BMP6 50 ng/ml, IL-6 100 ng/ml, or genistein 10 μM, significantly increased Hepcidin-luciferase activity in the stably transfected cells (2378±185.4, 3.48±0.24, 2.64±0.28, respectively), while dorsomorphin 40 μM decreased it (0.59±0.35) (Figure 1A).
Figure 1.
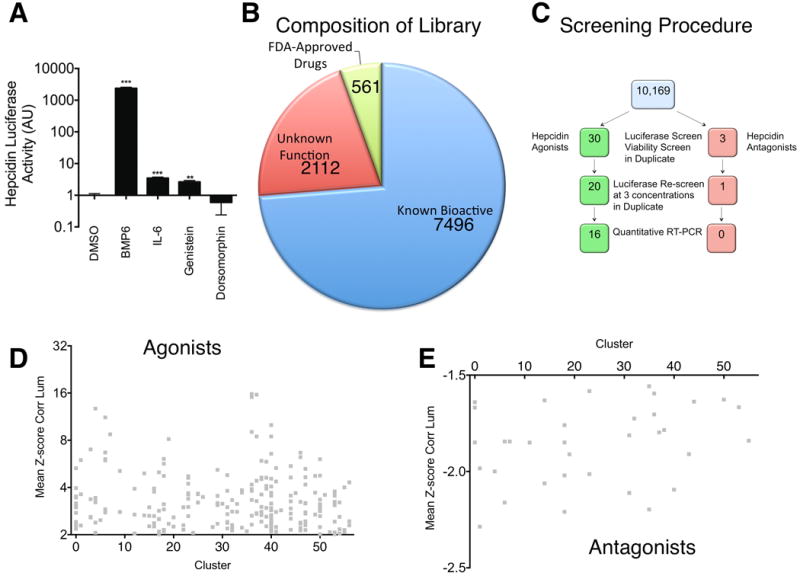
A. Effect of positive and negative controls on Hepcidin-luciferase activity in stably transfected HepG2 cells. After 16 hours of serum starvation in α-MEM/0% FBS, HepG2 cells stably transfected with Hepcidin-luciferase were treated for 24 hours with DMSO 1%, BMP6 50 ng/ml, IL-6 100 ng/ml, Genistein 10 μM, or Dorsomorphin 40 μM. Hepcidin-luciferase activity was measured using the OneGlo Assay (Promega) and is shown as mean fold-change over DMSO-treated control. The global P-value generated using the Kruskal-Wallis test was <0.0001. Unpaired Student's t-tests were performed compared to DMSO alone. ***denotes 0.0001≤P<0.0009 and **denotes 0.0009≤P<0.009. N=3 biological replicates per condition. B. Library Composition. The screening library included known bioactive molecules, molecules of unknown function, and FDA-approved drugs. C. Screening Method. 10,169 chemicals were evaluated for Hepcidin-luciferase activity and viability in HepG2 cells stably transfected with a Hepcidin-luciferase promoter construct. The hits were then re-evaluated in the same assay at three concentrations and in a quantitative realtime RT-PCR assay. D,E. Scatter-plot of structural cluster vs. mean z-score for Hepcidin-luciferase activity for 343 molecules found to increase (D) or 62 molecules found to decrease (E) Hepcidin-luciferase activity.
The composition of the library screened (Figure 1B) included a diverse range of chemicals with the majority known bioactives (7496), followed by molecules of unknown function (2112), and FDA-approved drugs (561). To each well, 100 nL of a single small molecule was transferred prior to incubation of the cells at 37°C for 24 hours. The entire screen was performed in duplicate. Of the 10,169 chemicals originally screened, 343 agonists and 62 antagonists were initially identified by producing a z-score >3 or <-1 for Hepcidin expression, respectively (Figure 1C). Analysis of these chemicals with the Vortex program separated these chemicals into 57 structural groups. Agonists (Figure 1D) and antagonists (Figure 1E) were scattered across the structural groups without a dominant structure. When toxic chemicals were excluded by eliminating compounds that produced a z-score for viability <-1, i.e. <1 standard deviation reduction in cell viability, 30 agonists and 3 antagonists remained. We re-screened these molecules at the original concentration and at 2 dilutions in duplicate. Of these chemicals, 22 agonists and 1 antagonist were confirmed on re-screening (Table 1). We did not evaluate acrisorcin further, because it is a salt of 9-aminoacridine with 4-hexylresorcinol [19], that produced a similar effect to 9-aminoacridine, one of the other Hepcidin stimulating agents (Table 1). We also did not evaluate #532270 further because it was only moderately active (5.03±0.21) at 66 μM and weakly active (1.42±0.04) at 12 μM.
Table 1. Re-screening data on potential agonists or antagonists identified in the initial chemical screen.
Small molecules that were identified as potential regulators in the screen were retested in duplicate at the original concentration screened (Concentration #1) and two dilutions (Concentrations #2 and #3) for their effects on Hepcidin-luciferase activity, measured as fold-change over the activity from cells treated with DMSO only. Candidate regulators that increased mean Hepcidin-luciferase activity at least 2-fold at the original concentration were considered agonists (shaded in green), while candidate regulators that decreased mean Hepcidin-luciferase activity at least 2-fold were considered antagonists (shaded in orange).
| Regulator Name | Concentration #1 (μM) | Hepcidin-Luciferase Activity at Conc. #1 Mean±SE (AU) N=2 | Concentration #2 (μM) | Hepcidin-Luciferase Activity at Conc. #2 Mean±SE (AU) N=2 | Concentration #3 (μM) | Hepcidin-Luciferase Activity at Conc. #3 Mean±SE (AU) N=2 |
|---|---|---|---|---|---|---|
| POTENTIAL AGONISTS | ||||||
| Vorinostat | 33 | 56.08±35.27 | 6.6 | 48.14±6.70 | 1.3 | 14.60±4.68 |
| 9-aminoacridine | 33 | 49.76±12.27 | 6.6 | 1.14±0.40 | 1.3 | 0.68±0.07 |
| Acrisorcin | 33 | 29.85±1.50 | 6.6 | 0.99±0.06 | 1.3 | 0.77±0.00 |
| Ethacridine lactate | 33 | 15.18±1.13 | 6.6 | 1.35±0.24 | 1.3 | 1.12±0.18 |
| Doxorubicin | 12 | 11.39±0.49 | 2.4 | 0.68±0.06 | 0.5 | 0.77±0.02 |
| Daunorubicin | 12 | 11.33±0.07 | 2.4 | 0.99±0.05 | 0.5 | 0.67±0.05 |
| Leflunomide | 33 | 5.50±0.29 | 6.6 | 3.10±0.23 | 1.3 | 1.12±0.18 |
| #5322770 | 66 | 5.03±0.21 | 12.2 | 1.42±0.04 | 2.6 | 0.84±0.06 |
| Ipriflavone | 33 | 4.57±0.30 | 6.6 | 3.41±0.08 | 1.3 | 2.56±0.07 |
| Lansoprazole | 33 | 4.09±0.44 | 6.6 | 0.98±0.01 | 1.3 | 0.58±0.01 |
| Pterostilbene | 33 | 4.06±0.59 | 16.5 | 3.91±0.28 | 8.25 | 2.21±0.19 |
| Amlexanox | 33 | 3.84±0.95 | 6.6 | 2.65±0.06 | 1.3 | 0.67±0.02 |
| Nabumetone | 33 | 3.74±0.19 | 16.5 | 3.16±0.30 | 8.25 | 3.61±0.95 |
| Topotecan | 33 | 3.63±1.39 | 6.6 | 0.57±0.05 | 1.3 | 0.73±0.04 |
| Camptothecin | 33 | 3.62±0.62 | 6.6 | 1.55±0.34 | 1.3 | 0.60±0.05 |
| Chrysin | 26 | 3.53±0.60 | 5.2 | 1.09±0.11 | 1.0 | 0.76±0.01 |
| GTP-14564 | 11 | 3.07±0.60 | 2.2 | 1.35±0.07 | 0.4 | 1.17±0.15 |
| 10058-F4 | 33 | 2.92±0.23 | 6.6 | 2.17±0.01 | 1.3 | 1.25±0.13 |
| SB 204741 | 33 | 2.87±0.22 | 6.6 | 1.96±0.18 | 1.3 | 1.09±0.04 |
| Phenazopyridine | 26 | 2.85±0.04 | 5.2 | 1.51±0.08 | 1.0 | 0.95±0.12 |
| AS-252424 | 33 | 2.84±0.02 | 6.6 | 1.40±0.03 | 1.3 | 0.73±0.02 |
| AG 1296 | 11 | 2.41±0.14 | 2.2 | 1.03±0.11 | 0.4 | 0.90±0.07 |
| SP 600125 | 33 | 1.86±0.00 | 6.6 | 1.11±0.02 | 1.3 | 0.93±0.09 |
| Fenbendazole | 22 | 1.75±0.08 | 4.4 | 1.03±0.20 | 0.9 | 0.96±0.12 |
| #5105276 | 66 | 1.73±0.04 | 12.2 | 0.75±0.13 | 2.6 | 0.74±0.02 |
| GW9662 | 33 | 1.66±0.08 | 6.6 | 1.12±0.05 | 1.3 | 1.09±0.01 |
| #5102420 | 66 | 1.66±0.06 | 12.2 | 0.80±0.11 | 2.6 | 0.83±0.16 |
| Imatinib | 33 | 1.24±0.13 | 16.5 | 1.18±0.06 | 8.25 | 1.31±0.06 |
| Nocodazole | 22 | 1.13±0.02 | 4.4 | 0.84±0.11 | 0.9 | 0.84±0.01 |
| #5193825 | 66 | 1.13±0.14 | 12.2 | 0.67±0.01 | 2.6 | 0.69±0.06 |
| POTENTIAL ANTAGONISTS | ||||||
| #5215082 | 66 | 1.09±0.08 | 12.2 | 0.87±0.04 | 2.6 | 0.79±0.02 |
| #5146972 | 66 | 1.08±0.00 | 12.2 | 0.72±0.05 | 2.6 | 0.66±0.03 |
| SU-6668 | 33 | 0.31±0.02 | 6.6 | 0.67±0.09 | 1.3 | 0.88±0.01 |
The remaining twenty potential Hepcidin agonists and one antagonist were subsequently evaluated by quantitative realtime RT-PCR for Hepcidin expression at the same concentrations that were effective in the Hepcidin-luciferase assay. BMP6 and dorsomorphin, used as positive and negative controls, respectively, produced the expected effects on Hepcidin expression (Figure 2A). Sixteen of the 20 putative agonists significantly increased Hepcidin transcript levels, however, the putative agonists, topotecan, campthothecin, nabumetone, and chrysin, failed to increase Hepcidin transcript levels, despite increasing Hepcidin-luciferase activity, while the putative antagonist, SU6668, increased Hepcidin transcript levels, despite decreasing Hepcidin-luciferase activity.
Figure 2.
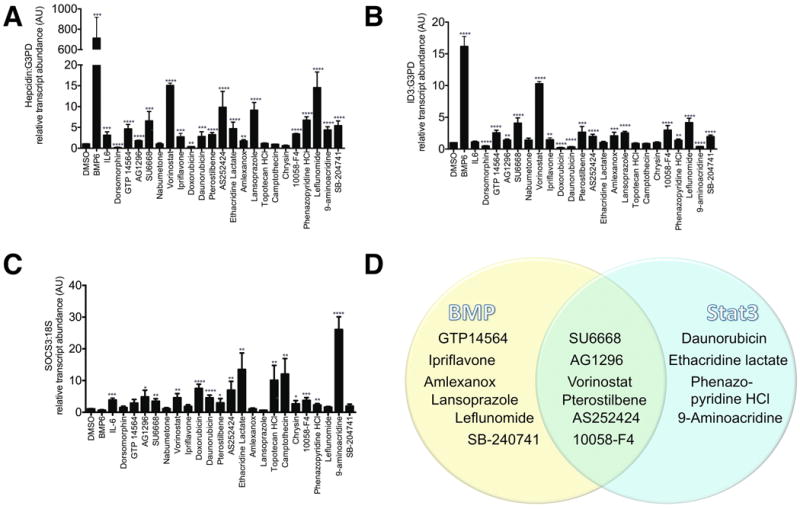
Quantitative realtime RT-PCR for A. Hepcidin, B. ID3, C. SOCS3. Following 8 hours of serum starvation in α-MEM/1%FBS, HepG2 cells were treated for 24 hours in α-MEM/1%FBS with DMSO 1%, BMP6 100 ng/ml, IL-6 100 ng/ml, dorsomorphin (an inhibitor of BMP signaling) 10 μM, GTP 14564 5 μM, AG1296 5 μM, SU6668 10 μM, nabumetone 5 μM, vorinostat 1 μM, ipriflavone 1 μM, doxorubicin 10 μM, daunorubicin 10 μM, pterostilbene 10 μM, AS252424 33 μM, ethacridine lactate 33 μM, amlexanox 33 μM, lansoprazole 33 μM, topotecan HCl 33 μM, camptothecin 33 μM, chrysin 33 μM, 10058-F4 33 μM, phenazopyridine 33 μM, leflunomide 33 μM, 9-aminoacridine 33 μM, SB-204741 33 μM. RNA was then extracted for quantitative realtime RT-PCR. Data shown are means±SE's. N=3-6 biological replicates per treatment. The global P-value generated using the Kruskal-Wallis test was <0.0001 for each experiment. Unpaired Student's t-tests were performed compared to DMSO alone. * denotes 0.009≤P<0.05, **denotes 0.0009≤P<0.009, ***denotes 0.0001≤P<0.0009, ****P<0.0001. D. Venn diagram illustrating which chemicals appear to increase RNA transcript levels of Hepcidin and the BMP-dependent transcript, ID3, and/or the Stat3-dependent transcript, SOCS3.
In previous RNA sequencing and quantitative RT-PCR experiments [18], we had identified the BMP-regulated transcript, ID3 [20-22], and the Stat3-regulated transcript SOCS3 [23], as genes whose expression increased significantly in HepG2 cells following treatment with BMP6 or IL-6, respectively. Thus, we evaluated the effects of the chemicals on ID3 (Figure 2B) and SOCS3 (Figure 2C) transcript levels, as readouts for bone morphogenic protein signaling and Stat3 signaling [18]. As expected, BMP6 produced a significant increase in ID3 expression over DMSO alone (16.17±1.57, p<0.0001) that was not observed with IL-6 treatment, while IL-6 increased SOCS3 expression (3.88±0.59, p=0.0002), but BMP6 did not. The BMP receptor antagonist, dorsomorphin, used as a negative control, inhibited ID3 expression (0.48±0.16, p<0.0001).
The HDAC inhibitor, vorinostat, which was one of the most potent Hepcidin stimulating chemicals identified in the screen, produced a particularly strong increase in Hepcidin (15.09±0.55, p<0.0001) and ID3 expression (10.3±0.33, p<0.0001). The majority of chemicals that significantly upregulated Hepcidin transcript levels significantly upregulated ID3, with the exception of daunorubicin, ethacridine, and 9-aminoacridine, which either decreased or did not affect ID3 expression (Figure 2B). Interestingly, the Hepcidin agonists that did not upregulate ID3, did upregulate SOCS3, consistent with Stat3 pathway activation (Figure 2C). Thus the Hepcidin agonists can be divided into three classes: (1) upregulators of BMP signaling only, (2) upregulators of Stat3 signaling only, (3) upregulators of both pathways (Figure 2D).
We were particularly interested in the kinase inhibitors, GTP 14564, AG1296, and SU6668, since they each affect growth factor dependent signaling, which has previously been shown to affect Hepcidin expression [24]. GTP 14564 inhibits FLT3, c-Fms, c-Kit, and PDGFRβ[25], while AG1296 impairs signaling by both PDGF-α and β receptors and by c-Kit [26]. SU6668 has broad effects against receptor tyrosine kinases and inhibits VEGF, FGF, and PDGF receptors [27]. To validate the initial classification of these compounds by ID3 and SOCS3 expression (Figure 2B-D), we evaluated these chemicals for their effects on transcription of an additional BMP-dependent gene, ID1 [20-22], and additional Stat3-dependent genes [23][18], IL6 receptor (IL6R) and VEGFA. We found that GTP 14564 and SU6668 each significantly upregulated expression of ID1, as well as IL6R and VEGFA (Figure 3A-C). Although AG1296 did not significantly increase expression of ID1, it did significantly increase transcript levels of BMP and Stat3-dependent genes, including ID3 (Figure 2B), SOCS3 (Figure 2C), and VEGFA (Figure 3C). Thus it appears that these growth factor receptor tyrosine kinase inhibitors upregulate both the BMP and Stat3 signaling pathways.
Figure 3.
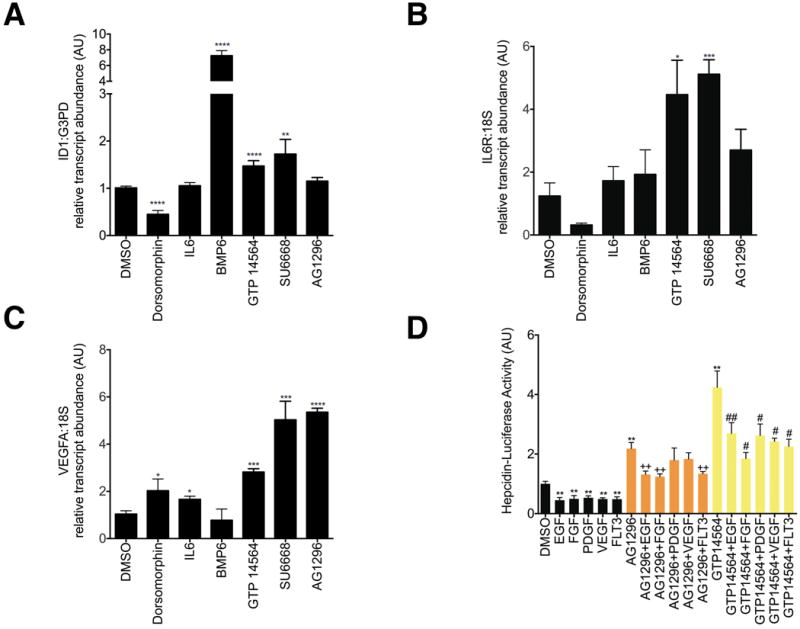
A-C. Effect of AG1296 and GTP 14564 on transcript levels of ID1(A), IL6 receptor (B), and VEGFA (C). HepG2 cells were treated with chemicals at the same doses and with the same conditions as described in Figure 2. Data shown are means±SE's. N=3-6 biological replicates per treatment. The global P-value generated using the Kruskal-Wallis test was <0.0001 for ID1 and =0.01 for IL6R and VEGFA. Unpaired Student's t-tests were performed compared to DMSO alone. * denotes 0.009≤P<0.05, **denotes 0.0009≤P<0.009, ***denotes 0.0001≤P<0.0009, ****P<0.0001. D. Effect of AG1296 or GTP 14564 on Hepcidin-Luciferase Activity in the presence or absence of growth factors or FLT3. After 16 hours of serum starvation in α-MEM/0% FBS, HepG2 cells stably transfected with Hepcidin-luciferase were treated for 24 hours with AG1296 (5 μM) or GTP 14564 (5 μM) in the presence or absence of EGF 150 ng/ml, FGF 200 ng/ml, PDGF 50 ng/ml, VEGF 150 ng/ml, or FLT3 150 ng/ml. Hepcidin-luciferase activity was measured using the OneGlo Assay (Promega) and is shown as mean fold-change over DMSO-treated control. The global P-value generated using the Kruskal-Wallis test was <0.0001. Unpaired Student's t-tests were performed. ** denotes 0.0009≤P<0.009, compared to DMSO-treated control. ++ denotes 0.0009≤P<0.009, compared to AG1296-treated cells. # denotes 0.009≤P<0.05, compared to GTP 14564-treated cells. ## denotes 0.0009≤P<0.009, compared to GTP 14564-treated cells. N=5-9 biological replicates for each condition.
To assess the effects of growth factors on Hepcidin promoter activity, we treated the Hepcidin-Luciferase HepG2 cells with EGF 150 ng/ml, FGF 200 ng/ml, PDGF 50 ng/ml, VEGF 150 ng/ml, or FLT3 150 ng/ml for 24 hours in the presence or absence of the tyrosine kinase inhibitors, AG1296 (5 μM) or GTP 14564 (5 μM) (Figure 3D). We found that each of these proteins significantly reduced baseline Hepcidin promoter activity. In combination with GTP 14564 each of the tested proteins significantly impaired GTP 14564's stimulation of Hepcidin promoter activity, while in combination with AG1296, only EGF, FGF, or FLT3 significantly blocked AG1296's induction of Hepcidin promoter activity. Thus growth factor- or FLT3-dependent signaling appears to inhibit Hepcidin promoter activity and to impair the stimulatory effects of AG1296 and GTP 14564, but we did not observe a phenomenon that was limited to one particular growth factor or ligand.
We had hypothesized that the Hepcidin stimulatory molecules identified in the screen would increase phosphorylation of Smad1,5, and 8 and/or phosphorylation of Stat3. To evaluate this hypothesis, we performed Western blots to evaluate the ratio of P-Smad1,5,8 to Smad1 (Figure 4A) and P-Stat3 to Stat3 (Figure 4B). As expected, BMP6 treatment increased the intensity of P-Smad1,5,8 relative to Smad1 after 1 hour of treatment, however, none of the small molecules significantly increased the intensity of P-Smad1,5,8 relative to Smad1, as assessed by densitometry. Furthermore, in the one hour time frame, neither IL-6 nor any of the small molecules tested increased the intensity of P-Stat3 relative to Stat3. WP1066, a known inhibitor of Jak2 and Stat3 phosphorylation [28] for Jak/Stat signaling, did not decrease P-Stat3 to Stat3, however WP1066 is reported to be more effective after 24-48 hours of incubation.[28] After 24 hours of treatment, we observed a significant increase in Stat3 protein levels relative to DMSO-treated controls in the hepatocytes treated with lansoprazole or vorinostat (2.34+0.96, p=0.047 and 1.88+0.43, p=0.03, respectively, Supplementary Figure 1), but no significant change in phosphorylation of Stat3 relative to Stat3 levels.
Figure 4. Western blots.
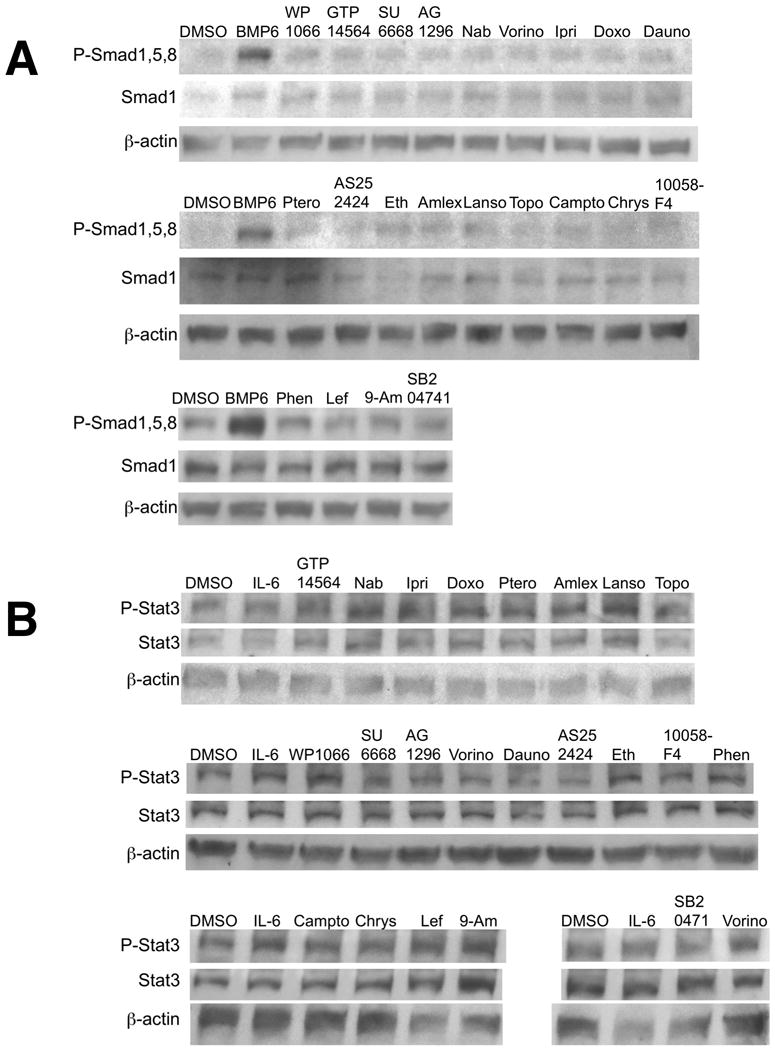
Following 16 hours of serum starvation in α-MEM/1%FBS, HepG2 cells were treated for 1 hour in α-MEM/1%FBS with the chemicals at the same concentrations as given in Figure 2. Protein was extracted from the cells, separated by SDS-PAGE, and blotted for incubation with antibodies against anti-P-Smad1,5,8 (A) or P-Stat3 (B). Following immunoblotting, the membranes were stripped and re-probed with either antibody against anti-Smad1,5,8 (A) or Stat3 (B). The blots were then stripped again and probed for β-actin as a loading control. Each chemical was evaluated in two or three biological replicates.
Discussion
In this study, we have demonstrated a high throughput screening method to identify small molecules that regulate Hepcidin gene expression using a Hepcidin-luciferase reporter cell line. Our study was the first large-scale screen for small molecules upregulating Hepcidin transcript levels. Using a screening approach that includes toxicity evaluation, we have identified the largest number of non-toxic small molecules that stimulate Hepcidin, which will facilitate future preclinical studies in iron overload syndromes. Several of the Hepcidin stimulating agents that we identified are drugs that are orally bioavailable or have been approved by the United States Food and Drug Administration (FDA) for other indications. These factors will facilitate their testing in preclinical models. The FDA-approved drugs that we identified include amlexanox, lansoprazole, leflunomide, vorinostat, and phenazopyridine, while pterostilbene and isoflavone are already commercially available as nutritional supplements. Small scale screening efforts previously identified genistein [18] and three kinase inhibitors [24] as small molecules that stimulate Hepcidin expression. Peptide analogs of hepcidin, minihepcidins, have also been injected into Hepcidin-deficient mice to prevent iron overload[29], but are not orally available. High throughput screening has previously been used to identify small molecules that function as Hepcidin antagonists, but not agonists [30]. Other antagonists to Hepcidin that have been developed include an antibody to Hepcidin [31], soluble hemojuvelin[32], and the bone morphogenic protein receptor antagonists, dorsomorphin and LDN-193189 [32].
Having screened 10,169 molecules, we identified 33 potential hits, which were reduced to 21 after re-screening with the same assay. Further characterization with quantitative realtime RT-PCR for Hepcidin transcript level reduced the number of hits to 16 agonists and no antagonists. Of the publically available small molecule screens in PubChem, 20% rely on bioluminescent assays, such as ours [33]. A recent study of 360,864 compounds in the NIH Molecular Libraries Small Molecule Repository revealed that 12% of the library inhibits firefly luciferase [34]. Interestingly, some of these inhibitors can prolong the half-life of the firefly luciferase enzyme causing an increase in bioluminescence, which can be misinterpreted as increased transcriptional activation of the gene [35-37]. Another possibility, is that the discrepancies between findings in the Hepcidin luciferase assay and the Hepcidin quantitative realtime RT-PCR assay are caused by the absence of distal elements in the 2.7 kb fragment of the Hepcidin promoter-Luciferase construct that are present in the endogenous Hepcidin promoter. For these reasons, we believe that it is not surprising that 24% of the 21 hits that we identified did not produce the anticipated effect on Hepcidin transcript levels in the quantitative RT-PCR assay.
In our previous work, we identified genistein as a small molecule that increases Hepcidin expression in human hepatocytes and zebrafish embryos by activating both bone morphogenic protein and Stat3 signaling pathways [18]. Genistein strongly upregulated transcript levels of ID3 and SOCS3[18], BMP- and Stat3-dependent genes, respectively, thus we assayed for effects on expression of these genes as a short-hand for BMP and Stat3-dependent gene expression associated with treatment by the hits identified in the screen. We found that all the hits that increased Hepcidin expression in the screen upregulated one or both of these genes (Figure 2A-C). Thus we were able to classify the hits by their association with BMP or Stat3 signaling pathways (Figure 2D).
Interestingly, none of the chemicals tested caused enhanced phosphorylation of Smad1,5,8 or Stat3. While Western blots for P-Smad1,5,8 appeared to be highly sensitive, indicating a clear increase in P-Smad1,5,8 signal to Smad1 for hepatocytes treated with BMP6 (Figure 4A), Western blots for PStat3 to Stat3 (Figure 4B) were less sensitive and unable to detect the 3-fold increase in PStat3 to Stat3 that we had previously observed with an ELISA assay [18] performed on HepG2 cells treated with IL-6 for at the same concentration and conditions used in these experiments. Thus it is possible that the chemicals activated Stat3 at a level that was below the limit of detection for this assay. We did observe, however, that Stat3 protein levels significantly increased in hepatocytes after 24 hours of treatment with lansoprazole or vorinostat (Supplementary Figure 1). It appears likely that the chemicals either potentiated or stabilized Smad or Stat3 binding to the Hepcidin promoter without increasing phosphorylation of the proteins, caused phosphorylation at a later time point, which would most likely be an indirect effect after other signal transduction cascades were activated, or acted via other pathways.
Potent agonists
The two most potent agonists, ipriflavone and vorinostat, active at 1 μM concentrations, were 10-fold more potent than genistein [18]. Interestingly, ipriflavone, like genistein, is an isoflavone with estrogenic properties [38]. Ipriflavone is used to treat osteoporosis based on its ability to inhibit osteoclast activity, promote mineralization of osteoblasts [39], and increase bone mineral density in postmenopausal women[40]. However, our previous work indicated that estradiol does not increase Hepcidin expression and that blockade of the estrogen receptor fails to inhibit genistein's effect on Hepcidin expression [18], thus we think it is unlikely that ipriflavone is promoting Hepcidin expression in an estrogenic manner. Similar to our observation of genistein [18], ipriflavone increased expression of the BMP-dependent gene, ID3 (Figure 2B), however, unlike genistein, ipriflavone did not increase expression of the Stat3-dependent gene, SOCS3 (Figure 2C), or increase Stat3 phosphorylation (Figure 4B).
Tyrosine Kinase Inhibitors
Several of the hits that increased Hepcidin transcript levels were tyrosine kinase inhibitors affecting growth factor signaling (Figure 5), including SU6668, GTP 14564, and AG1296. SU6668 inhibits VEGF, FGF, and PDGF receptors [27]. We found that SU6668 exhibited the paradoxical effect of inhibiting Hepcidin-luciferase activity, but increasing Hepcidin transcript levels in the quantitative realtime RT-PCR experiments. GTP 14564 and AG1296, however, both increased Hepcidin-luciferase activity and Hepcidin transcript levels in quantitative realtime RT-PCR assays. GTP 14564 is a potent inhibitor of FLT3, c-Fms, c-Kit, and PDGFRβ[25], while AG1296 inhibits signaling by both PDGF-α and β receptors and by c-Kit, without affecting VEGF receptor signaling [26]. We demonstrated that AG1296 or GTP 14564's stimulatory effects on the Hepcidin promoter can be significantly impaired by co-treating with EGF, FGF, or FLT3 (for AG1296 or GTP 14564) or PDGF or VEGF (for GTP 14564). Both PDGF-α and β receptors signal via PI3 Kinase, among other pathways, and can activate Src leading to transcription of c-Myc [41]. Two of the other Hepcidin stimulating agents that we identified in the screen, AS252424 and 10058-F4, affect pathways that can act downstream of PDGF receptor. AS252424 inhibits PI3 Kinase isoform γ [42], while 10058-F4 blocks c-Myc's activity [43, 44]. Interestingly, pterostilbene, another Hepcidin agonist that we identified in the screen, is a naturally occurring polyphenolic anti-oxidant compound that has been shown to inhibit human growth factor signaling via PI3 Kinase in breast cancer cells and PDGF-driven proliferation in vascular smooth muscle cells [45].
Figure 5. Schematic illustrating the pathways implicated in transcriptional regulation of Hepcidin by the small molecules identified as Hepcidin stimulators in the screen.
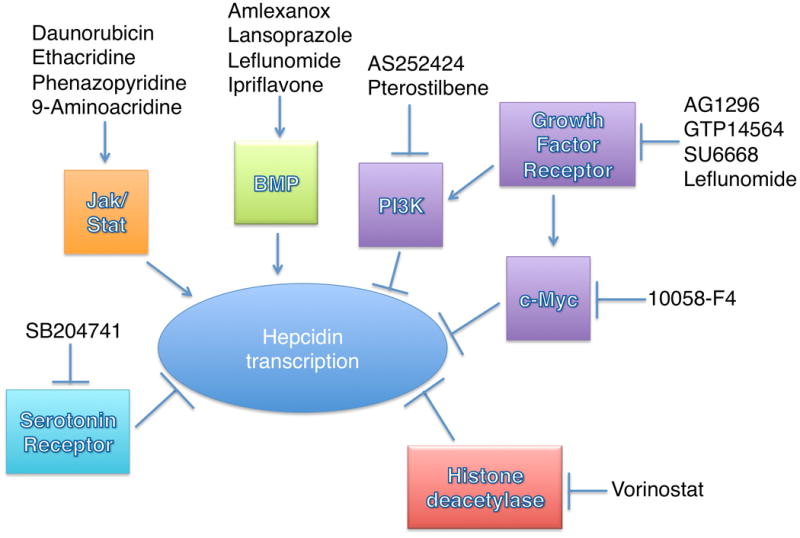
->Indicates a stimulatory effect, while -| indicates an inhibitory effect.
To facilitate high throughput screening, we performed our screen entirely in human hepatoma cells (HepG2). As hepatocellular carcinoma cells exhibit increased signaling by FGF, PDGF, and VEGF, this may have biased our results to identify antagonists of growth factor signaling. Others, however, have used small scale screening of kinase inhibitors to demonstrate that hepatocyte growth factor (HGF) and epidermal growth factor (EGF) reduce BMP-stimulated Hepcidin expression in a mitogen-activated ERK kinase/extracellular signal-related kinase (MEK/ERK) dependent manner in primary mouse hepatocytes [24]. HGF's inhibitory activity on Hepcidin expression can be suppressed by pre-treatment with small molecule inhibitors of Met (PHA665752), MEK1/2 (U0126), or PI3Kinase (LY2940021) [24]. Furthermore intraperitoneal injection of EGF in wild type mice reduces the induction of Hepcidin expression in response to iron loading[24]. Given these findings, we propose that SU6668, GTP 14564, AG1296, AS252424, 10058-F, and pterostilbene enhance Hepcidin transcript levels in HepG2 cells by inhibiting growth-factor dependent signaling.
HDAC inhibitors
One of the most interesting findings in the screen is that the nonselective histone deacetylase (HDAC) inhibitor, vorinostat, is a potent stimulant of Hepcidin expression. These data are consistent with the finding that histone acetylation increases Hepcidin expression [4, 46, 47]. In particular, post-translational modification of Histone H3, one of the core proteins of the nucleosome, regulates transcription and chromatin condensation[48]. Transfection of Smad4 into Smad4-null hepatocytes increases binding of Histone 3 acetylated at lysine 9 (H3K9) to the Hepcidin promoter [4]. Histone acetylation also appears to affect Stat3 binding to the Hepcidin promoter. Hepatitis C viral infection of cultured hepatoma cells causes hypoacetylation of histones and decreased Hepcidin expression, while treatment with the pan-HDAC inhibitor, trichostatin A, increases Stat3 binding to the Hepcidin promoter [46] and enhances Hepcidin expression [46, 47]. Vorinostat has been approved for the treatment of refractory cutaneous T-cell lymphoma[49] and thus may be amenable to clinical investigation in patients with iron overload syndromes who produce inappropriately low levels of Hepcidin.
Drugs that increase ID3 expression
The anti-inflammatory drugs, amlexanox, lansoprazole, and leflunomide each increased Hepcidin expression and ID3 expression in the screen. Amlexanox is an anti-allergic drug that binds the cytoskeletal protein S100A13 and inhibits heat shock-induced release of FGF1[50]. Lansoprazole, a drug commonly used to treat stomach ulcers, inhibits the H+/K+-adenosine triphosphatase in gastric parietal cells, but it also has been shown to have anti-inflammatory properties, in the esophagus, intestine, and lung and can stimulate heme oxygenase-1 expression [51]. Leflunomide, used for the treatment of rheumatoid arthritis, yields an active metabolite, A771726, which potently blocks pyrimidine synthesis by inhibiting dihydroorotate synthase. At higher concentrations, however, such as those used in this study, leflunomide inhibits phosphorylation of PDGF receptor or EGF receptor (IC50 30-55 and 150-200 μM, respectively) [52].
The chemical screen described here also demonstrated a potential role for serotonin receptor 2B in regulating Hepcidin expression. We found that SB 204741, a serotonin receptor 2B (5-HT2B) antagonist, increased Hepcidin expression and ID3 expression. Serotonin stimulates proliferation of hepatocellular carcinoma cells [53], but represses liver regeneration via effects on hepatocyte stellate cells [54]. Animal studies indicate that the 5-HT2B inhibitor, SB 204741, confers the converse effect, decreasing growth of human hepatocellular carcinoma xenografts in mice [53], but enhancing liver regeneration following partial hepatectomy in animal models [54].
Drugs that increase SOCS3 expression
Daunorubicin, ethacridine lactate, phenazopyridine, and 9-aminoacridine each increased Hepcidin transcript levels and expression of the Stat3-dependent gene, SOCS3. As Stat3 is critically involved in liver injury and regeneration [55], it may be that these drugs stimulate Hepcidin expression by facilitating cell injury. Daunorubicin is an anti-cancer drug and DNA intercalator that inhibits Topoisomerase II resulting in breaks in double stranded DNA and increased apoptosis[56]. Daunorubicin has also been shown to increase expression of Stat3-dependent genes, such as SOCS3 [57]. Ethacridine lactate provokes uterine contractions and histamine release [58], but also inhibits poly(ADP-ribose) glycohydrolase [59], the major enzyme that catabolizes poly(ADP-ribose). Inhibition of poly(ADP-ribose) glycohydrolase has been shown to promote apoptosis and impair DNA repair in cells damaged by oxidative stress [60]. Phenazopyridine can cause liver injury [61, 62], while 9-aminoacridine is a DNA intercalator and experimental mutagen [63].
Conclusions
As a result of our screen, we have identified 16 small molecules that increase Hepcidin transcript levels in human HepG2 cells. Several of these chemicals affect growth factor receptor signaling, have anti-inflammatory properties, or impact DNA repair and apoptosis. The identification of multiple inhibitors of growth factor receptors and their downstream targets (Figure 5) indicates the importance of this pathway in regulating Hepcidin expression, while the discovery of inhibitors of histone deacetylase and serotonin receptor as Hepcidin stimulating agents indicates new avenues for further study. While each of these candidate molecules was associated with increases in transcript levels of other BMP and/or Stat3 associated genes, none of them exhibited a strong effect on Smad1,5,8 or Stat3 phosphorylation. Further studies will be needed to determine how each of these different molecules functions to increase Hepcidin transcript levels. We also plan experiments to determine if these chemicals are effective in raising Hepcidin levels in vivo. In the future, we would like to test these candidate Hepcidin stimulatory chemicals in animal models of iron overload to determine if they could be adapted into therapeutic agents for patients with iron overload syndromes.
Supplementary Material
Supplementary Table 1. Complete Screening Data
Supplementary Table 2. Sources of Hepcidin modulators identified in the screen that were characterized further in quantitative realtime RT-PCR and/or Western blot experiments.
Supplementary Table 3. Primers and probes used in realtime quantitative RT-PCR assays
Supplementary Figure 1. Effect of treatment with Hepcidin stimulating agents for 24 hours on P-Stat3 and Stat3 protein levels. A. Following 16 hours of serum starvation in α-MEM/1%FBS, HepG2 cells were treated for 24 hours in α-MEM/1%FBS with the chemicals at the same concentrations as given in Figure 2. Protein was extracted from the cells, separated by SDS-PAGE, and blotted for incubation with antibodies against P-Stat3. Following immunoblotting, the membranes were stripped and re-probed with antibody against Stat3. The blots were then stripped again and probed for β-actin as a loading control. Each chemical was evaluated in three biological replicates. B. Protein levels were measured as fold-change over DMSO-treated control by densitometry analysis using Image Studio Lite Version 3.1.4 (LiCor Biosciences, Lincoln, NE). The global P-value generated using the Kruskal-Wallis test was <0.05. Unpaired Student's t-tests were performed compared to DMSO alone. * denotes 0.009≤P<0.05, **denotes 0.0009≤P<0.009.
Acknowledgments
This work was supported by the National Institutes of Health (R01 DK085250-01A1 to P.G.F.), the Cooley's Anemia Foundation (to P.G.F.), the March of Dimes Research Foundation Basil O'Connor Starter Scholar Research Award (to P.G.F.), and the Harvard College Research Program (to J.V.). The funding sources played no role in the design of the research, writing of the report, or the decision to publish.
References
- 1.Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823(9):1434–43. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins JF, Wessling-Resnick M, Knutson MD. Hepcidin regulation of iron transport. J Nutr. 2008;138(11):2284–8. doi: 10.3945/jn.108.096347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corradini E, et al. Serum and liver iron differently regulate the bone morphogenetic protein 6 (BMP6)-SMAD signaling pathway in mice. Hepatology. 2011;54(1):273–84. doi: 10.1002/hep.24359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang RH, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2(6):399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108(9):3204–9. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 7.Fraenkel PG, et al. Ferroportin1 is required for normal iron cycling in zebrafish. J Clin Invest. 2005;115(6):1532–41. doi: 10.1172/JCI23780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papanikolaou G, et al. Hepcidin in iron overload disorders. Blood. 2005;105(10):4103–5. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lekawanvijit S, Chattipakorn N. Iron overload thalassemic cardiomyopathy: iron status assessment and mechanisms of mechanical and electrical disturbance due to iron toxicity. Can J Cardiol. 2009;25(4):213–8. doi: 10.1016/s0828-282x(09)70064-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angelucci E, et al. Phlebotomy to reduce iron overload in patients cured of thalassemia by bone marrow transplantation. Italian Cooperative Group for Phlebotomy Treatment of Transplanted Thalassemia Patients. Blood. 1997;90(3):994–8. [PubMed] [Google Scholar]
- 11.Barton JC. Chelation therapy for iron overload. Curr Gastroenterol Rep. 2007;9(1):74–82. doi: 10.1007/s11894-008-0024-9. [DOI] [PubMed] [Google Scholar]
- 12.Fung E, Nemeth E. Manipulation of the hepcidin pathway for therapeutic purposes. Haematologica. 2013;98(11):1667–76. doi: 10.3324/haematol.2013.084624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicolas G, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34(1):97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 14.Gardenghi S, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120(12):4466–77. doi: 10.1172/JCI41717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu PB, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4(1):33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.North TE, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447(7147):1007–11. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cutler C, et al. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood. 2013;122(17):3074–81. doi: 10.1182/blood-2013-05-503177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhen AW, et al. The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology. 2013;58(4):1315–25. doi: 10.1002/hep.26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.A new agent for the control of tinea versicolor. Acrisorcin (Akrinol) JAMA. 1966;196(11):1010. doi: 10.1001/jama.1966.03100240144035. [DOI] [PubMed] [Google Scholar]
- 20.Lopez-Rovira T, et al. Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J Biol Chem. 2002;277(5):3176–85. doi: 10.1074/jbc.M106826200. [DOI] [PubMed] [Google Scholar]
- 21.Conidi A, et al. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFbeta/BMP signaling in vivo. Cytokine Growth Factor Rev. 2011;22(5-6):287–300. doi: 10.1016/j.cytogfr.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Kersten C, et al. BMP-6 inhibits human bone marrow B lymphopoiesis--upregulation of Id1 and Id3. Exp Hematol. 2006;34(1):72–81. doi: 10.1016/j.exphem.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 23.Dauer DJ, et al. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24(21):3397–408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 24.Goodnough JB, et al. Inhibition of hepcidin transcription by growth factors. Hepatology. 2012;56(1):291–9. doi: 10.1002/hep.25615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murata K, et al. Selective cytotoxic mechanism of GTP-14564, a novel tyrosine kinase inhibitor in leukemia cells expressing a constitutively active Fms-like tyrosine kinase 3 (FLT3) J Biol Chem. 2003;278(35):32892–8. doi: 10.1074/jbc.M210405200. [DOI] [PubMed] [Google Scholar]
- 26.Kovalenko M, et al. Selective platelet-derived growth factor receptor kinase blockers reverse sis-transformation. Cancer Res. 1994;54(23):6106–14. [PubMed] [Google Scholar]
- 27.Laird AD, et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000;60(15):4152–60. [PubMed] [Google Scholar]
- 28.Verstovsek S, et al. WP1066, a novel JAK2 inhibitor, suppresses proliferation and induces apoptosis in erythroid human cells carrying the JAK2 V617F mutation. Clin Cancer Res. 2008;14(3):788–96. doi: 10.1158/1078-0432.CCR-07-0524. [DOI] [PubMed] [Google Scholar]
- 29.Ramos E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120(18):3829–36. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fung E, et al. High-throughput screening of small molecules identifies hepcidin antagonists. Mol Pharmacol. 2013;83(3):681–90. doi: 10.1124/mol.112.083428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooke KS, et al. A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood. 2013;122(17):3054–61. doi: 10.1182/blood-2013-06-505792. [DOI] [PubMed] [Google Scholar]
- 32.Theurl I, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011;118(18):4977–84. doi: 10.1182/blood-2011-03-345066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thorne N, Inglese J, Auld DS. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol. 2010;17(6):646–57. doi: 10.1016/j.chembiol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorne N, et al. Firefly luciferase in chemical biology: a compendium of inhibitors, mechanistic evaluation of chemotypes, and suggested use as a reporter. Chem Biol. 2012;19(8):1060–72. doi: 10.1016/j.chembiol.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auld DS, et al. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008;51(8):2372–86. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- 36.Auld DS, et al. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A. 2009;106(9):3585–90. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson JF, Hayes LS, Lloyd DB. Modulation of firefly luciferase stability and impact on studies of gene regulation. Gene. 1991;103(2):171–7. doi: 10.1016/0378-1119(91)90270-l. [DOI] [PubMed] [Google Scholar]
- 38.Ise R, et al. Expression profiling of the estrogen responsive genes in response to phytoestrogens using a customized DNA microarray. FEBS Lett. 2005;579(7):1732–40. doi: 10.1016/j.febslet.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 39.Tseng CH, et al. Synthesis and anti-osteoporotic evaluation of certain 3-amino-2-hydroxypropoxyisoflavone derivatives. Eur J Med Chem. 2009;44(9):3621–6. doi: 10.1016/j.ejmech.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, et al. Effects of ipri fl avone on postmenopausal syndrome and osteoporosis. Gynecol Endocrinol. 2010;26(2):76–80. doi: 10.3109/09513590903184159. [DOI] [PubMed] [Google Scholar]
- 41.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22(10):1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shugg RP, et al. Effects of isoform-selective phosphatidylinositol 3-kinase inhibitors on osteoclasts: actions on cytoskeletal organization, survival, and resorption. J Biol Chem. 2013;288(49):35346–57. doi: 10.1074/jbc.M113.507525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomez-Curet I, et al. c-Myc inhibition negatively impacts lymphoma growth. J Pediatr Surg. 2006;41(1):207–11. doi: 10.1016/j.jpedsurg.2005.10.025. discussion 207-11. [DOI] [PubMed] [Google Scholar]
- 44.Huang MJ, et al. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol. 2006;34(11):1480–9. doi: 10.1016/j.exphem.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 45.McCormack D, McFadden D. A review of pterostilbene antioxidant activity and disease modification. Oxid Med Cell Longev. 2013;2013:575482. doi: 10.1155/2013/575482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miura K, et al. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48(5):1420–9. doi: 10.1002/hep.22486. [DOI] [PubMed] [Google Scholar]
- 47.Liu H, et al. Iron regulator hepcidin exhibits antiviral activity against hepatitis C virus. PLoS One. 2012;7(10):e46631. doi: 10.1371/journal.pone.0046631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He H, Lehming N. Global effects of histone modifications. Brief Funct Genomic Proteomic. 2003;2(3):234–43. doi: 10.1093/bfgp/2.3.234. [DOI] [PubMed] [Google Scholar]
- 49.Olsen EA, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 50.Mouta Carreira C, et al. S100A13 is involved in the regulation of fibroblast growth factor-1 and p40 synaptotagmin-1 release in vitro. J Biol Chem. 1998;273(35):22224–31. doi: 10.1074/jbc.273.35.22224. [DOI] [PubMed] [Google Scholar]
- 51.Takagi T, Naito Y, Yoshikawa T. The expression of heme oxygenase-1 induced by lansoprazole. J Clin Biochem Nutr. 2009;45(1):9–13. doi: 10.3164/jcbnSR09-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu X, et al. In vitro and in vivo antitumor activity of a novel immunomodulatory drug, leflunomide: mechanisms of action. Biochem Pharmacol. 1999;58(9):1405–13. doi: 10.1016/s0006-2952(99)00228-2. [DOI] [PubMed] [Google Scholar]
- 53.Soll C, et al. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology. 2010;51(4):1244–54. doi: 10.1002/hep.23441. [DOI] [PubMed] [Google Scholar]
- 54.Ebrahimkhani MR, et al. Stimulating healthy tissue regeneration by targeting the 5-HT(2)B receptor in chronic liver disease. Nat Med. 2011;17(12):1668–73. doi: 10.1038/nm.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawaratani H, et al. The effect of inflammatory cytokines in alcoholic liver disease. Mediators Inflamm. 2013;2013:495156. doi: 10.1155/2013/495156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9(5):338–50. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quotti Tubi L, et al. Inhibition of protein kinase CK2 with the clinical-grade small ATP-competitive compound CX-4945 or by RNA interference unveils its role in acute myeloid leukemia cell survival, p53-dependent apoptosis and daunorubicin-induced cytotoxicity. J Hematol Oncol. 2013;6:78. doi: 10.1186/1756-8722-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rudolph MI, et al. On the mechanism of action of ethodin in inducing myometrium contractions. Gen Pharmacol. 1997;28(3):381–5. doi: 10.1016/s0306-3623(96)00288-1. [DOI] [PubMed] [Google Scholar]
- 59.Bernardi R, et al. Analysis of poly(ADP-ribose) glycohydrolase activity in nuclear extracts from mammalian cells. Biochim Biophys Acta. 1997;1338(1):60–8. doi: 10.1016/s0167-4838(96)00188-4. [DOI] [PubMed] [Google Scholar]
- 60.Erdelyi K, et al. Dual role of poly(ADP-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed A549 cells. FASEB J. 2009;23(10):3553–63. doi: 10.1096/fj.09-133264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holahan ML, Littman MP, Hayes CL. Presumptive hepatotoxicity and rhabdomyolysis secondary to phenazopyridine toxicity in a dog. J Vet Emerg Crit Care (San Antonio) 2010;20(3):352–8. doi: 10.1111/j.1476-4431.2010.00541.x. [DOI] [PubMed] [Google Scholar]
- 62.Wander HJ, Pascoe DJ. Phenylazopyridine Hydrochloride Poisoning. Report of Case and Review of Literature. Am J Dis Child. 1965;110:105–7. [PubMed] [Google Scholar]
- 63.von Borstel RC, Higgins JA. Janus carcinogens and mutagens. Mutat Res. 1998;402(1-2):321–9. doi: 10.1016/s0027-5107(97)00312-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Complete Screening Data
Supplementary Table 2. Sources of Hepcidin modulators identified in the screen that were characterized further in quantitative realtime RT-PCR and/or Western blot experiments.
Supplementary Table 3. Primers and probes used in realtime quantitative RT-PCR assays
Supplementary Figure 1. Effect of treatment with Hepcidin stimulating agents for 24 hours on P-Stat3 and Stat3 protein levels. A. Following 16 hours of serum starvation in α-MEM/1%FBS, HepG2 cells were treated for 24 hours in α-MEM/1%FBS with the chemicals at the same concentrations as given in Figure 2. Protein was extracted from the cells, separated by SDS-PAGE, and blotted for incubation with antibodies against P-Stat3. Following immunoblotting, the membranes were stripped and re-probed with antibody against Stat3. The blots were then stripped again and probed for β-actin as a loading control. Each chemical was evaluated in three biological replicates. B. Protein levels were measured as fold-change over DMSO-treated control by densitometry analysis using Image Studio Lite Version 3.1.4 (LiCor Biosciences, Lincoln, NE). The global P-value generated using the Kruskal-Wallis test was <0.05. Unpaired Student's t-tests were performed compared to DMSO alone. * denotes 0.009≤P<0.05, **denotes 0.0009≤P<0.009.