

Abstract
The neuromuscular junction (NMJ) is a synapse between motor neurons and skeletal muscle fibers, and is critical for control of muscle contraction. Its formation requires neuronal agrin that acts by binding to LRP4 to stimulate MuSK. Mutations have been identified in agrin, MuSK, and LRP4 in patients with congenital myasthenic syndrome, and patients with myasthenia gravis develop antibodies against agrin, LRP4, and MuSK. However, it remains unclear whether the agrin signaling pathway is critical for NMJ maintenance because null mutation of any of the three genes is perinatal lethal. In this study, we generated imKO mice, a mutant strain whose LRP4 gene can be deleted in muscles by doxycycline (Dox) treatment. Ablation of the LRP4 gene in adult muscle enabled studies of its role in NMJ maintenance. We demonstrate that Dox treatment of P30 mice reduced muscle strength and compound muscle action potentials. AChR clusters became fragmented with diminished junctional folds and synaptic vesicles. The amplitude and frequency of miniature endplate potentials were reduced, indicating impaired neuromuscular transmission and providing cellular mechanisms of adult LRP4 deficiency. We showed that LRP4 ablation led to the loss of synaptic agrin and the 90 kDa fragments, which occurred ahead of other prejunctional and postjunctional components, suggesting that LRP4 may regulate the stability of synaptic agrin. These observations demonstrate that LRP4 is essential for maintaining the structural and functional integrity of the NMJ and that loss of muscle LRP4 in adulthood alone is sufficient to cause myasthenic symptoms.
Keywords: AChRs, agrin, congenital myasthenic syndrome, LRP4, NMJ, synaptic basal lamina
Introduction
Neuromuscular junction (NMJ) formation requires intricate interaction between motor neurons (MNs), muscle fibers, and Schwann cells that wrap up the synapse (Sohal, 1995; Sanes and Lichtman, 2001; Feng and Ko, 2008; Wu et al., 2010). Agrin is a heparan sulfate proteoglycan used by MNs to instruct postjunctional differentiation, including AChR clustering and synapse-specific transcription (McMahan, 1990; Brenner et al., 1992; Jo and Burden, 1992; Herczeg et al., 1995; Meier et al., 1997). Mice lacking agrin do not form NMJ, but form primitive aneural AChR clusters that are thought to be involved in nerve-induced AChR clusters (Gautam et al., 1996; Lin et al., 2001). Agrin is enriched at the synaptic basal lamina (SBL) between presynaptic and postsynaptic membranes (Ruegg et al., 1992; Jones et al., 1997; Burgess et al., 1999, 2000; Eusebio et al., 2003; Bolliger et al., 2010) and remains at the NMJ for weeks after nerve degeneration. MuSK is a receptor tyrosine kinase that is essential for agrin-induced clustering and for NMJ formation in vivo (DeChiara et al., 1996; Glass et al., 1996; Lin et al., 2001; Kim et al., 2008; Zhang et al., 2008). Agrin and MuSK do not directly interact (Glass et al., 1996); rather, MuSK activation by agrin requires LRP4, a member of the low-density lipoprotein (LDL) receptor (LDLR) family (Kim et al., 2008; Zhang et al., 2008). Evidence indicates that LRP4 is a receptor of agrin (Weatherbee et al., 2006; Kim et al., 2008; Zong et al., 2012). It is necessary for NMJ formation and agrin-induced activation of MuSK and AChR clustering in muscle cells (Weatherbee et al., 2006; Kim et al., 2008; Zhang et al., 2008). In addition, muscle LRP4 may also regulate presynaptic differentiation (Wu et al., 2012b; Yumoto et al., 2012).
The NMJ is a target of various disorders, including myasthenia gravis (MG), congenital myasthenic syndrome (CMS), and amyotropic lateral sclerosis (ALS). CMS patients could possess mutations in genes in agrin signaling, including agrin itself, LRP4, MuSK, or rapsyn, a cytoskeletal protein that interacts with AChR (Müller et al., 2004, 2006; Beeson et al., 2006; Hamuro et al., 2008; Maselli et al., 2010, 2012; Ben Ammar et al., 2013; Ohkawara et al., 2014). On the other hand, MG patients could develop antibodies against agrin, MuSK, and/or LRP4 that are pathogenic (Hoch et al., 2001; Punga et al., 2011; Pevzner et al., 2012; Huijbers et al., 2013; Shen et al., 2013). These observations suggest that NMJ maintenance or function in adulthood may require agrin-LRP4-MuSK signaling. The hypothesis is supported by somatic mutation of agrin (Bogdanik and Burgess, 2011; Samuel et al., 2012) or MuSK (Chevessier et al., 2008), or neonatal mutation of MuSK in muscle cells (Hesser et al., 2006). However, little is known about the effect of disrupting agrin signaling after NMJ development. Due to the embryonic lethality of null mutation of LRP4 or MuSK, it remains unclear whether they are involved in NMJ maintenance. Here, we generated inducible LRP4 mutant mice, which enabled us to eliminate LRP4 expression in adult mice, to investigate the role of LRP4 in NMJ maintenance in adult mice. A combination of biochemical, morphological, and functional approaches was used to characterize the NMJ. Results indicate a critical role of LRP4 in maintaining NMJ presynaptic and postsynaptic structure and function. Experiments designed toward deciphering underlining mechanisms showed specific, concomitant loss of synaptic agrin from the SBL. This suggests that LRP4 may be necessary for agrin stability in the SBL, identifying a novel function of LRP4.
Materials and Methods
Mouse strains.
Mice with LRP4 floxed alleles (LRP4f/f) were described previously (Wu et al., 2012b). B6;C3-Tg(ACTA1-rtTA; tetO-Cre) mice were from The Jackson Laboratory (stock #012433), which carry two transgenes: tetO-Cre, which expresses Cre under the control of the tetracycline-responsive regulatory element tetO; and ACTA1-rtTA, which expresses reverse tetracycline-controlled transactivator rtTA under the control of the promoter of the human α 1-actin gene ACTA1 or HSA (Rao and Monks, 2009). LRP4f/f mice were backcrossed with C57BL/5J mice before being crossed with B6; C3-Tg (tetO-Cre; ACTA1-rtTA) mice. Resulting tetO-Cre; ACTA1-rtTA; LRP4f/f mice are referred as inducible muscle-specific LRP4 knock-out (imKO) mice. Genotyping of LRP4f/f allele was performed as described previously (Wu et al., 2012b). tetO-Cre; ACTA1-rtTA was genotyped with primers: tetO-Cre with forward ACT GAG AGG TGG GAA GCT CA and reverse GGC GAG TTT ACG GGT TGT TA and for ACTA1-rtTA with primers: forward AGG TGT AGA GAA GGC ACT TA and reverse CTA ATC GCC ATC TTC CAG CA. Crosses generated the expected Mendelian numbers of each genotype. To induce Cre expression, imKO mice were treated with doxycycline in drinking water (2 mg/ml in 5% sucrose, in light proof bottles) ad libitum at 30 d of age for 3 weeks when doxycycline and glucose were omitted from the drinking water. Mice were housed in a room with a 12 h light/dark cycle with ad libitum access to water and rodent chow diet (Diet 7097, Harlan Teklad). Unless otherwise indicated, male mice were characterized in the current study. The Institutional Animal Care and Use Committee of the Georgia Regents University approved experimental procedures.
Measurement of muscle strength and weight.
Limb muscle strength was measured using an SR-1 hanging scale (American Weigh Scales) as described previously (Shen et al., 2013). Briefly, forelimbs were allowed to grip a square metal grid that was connected to a hanging scale. With hind limbs suspended, mice were gently pulled horizontally by tail until grip was released. Mice body weight was taken using a Mettler Toledo tabletop portable weighing scale.
Western blot analysis.
Muscles were dissected from TA muscles and lysed in modified RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% NP-40, 2% SDS, 2% DOC, 1 mm PMSF, 1 mm EDTA, 5 mm sodium fluoride, 2 mm sodium orthovanadate, and protease inhibitors). After centrifuging at 10,000 rpm at 4°C, supernatant was collected and used as sample lysates. Lysate protein concentrations were measured using Pierce BCA kit. Samples (50 μg of protein, unless otherwise indicated) were resolved by SDS-PAGE and transferred to nitrocellulose membrane. The membrane was first incubated in 5% milk in PBS-0.3% Tween 20 overnight at 4°C and then incubated with primary antibodies in 2% milk in TBS-Tween buffer: anti-MuSK (1:1000) (Luo et al., 2008; Zhu et al., 2008; Wu et al., 2012b); anti-α-tubulin (1:2000, sc-23948, Santa Cruz Biotechnology); anti-rapsyn (Luo et al., 2008); anti-α-dystroglycan (1:1000, ab106110, Abcam); anti-agrin clone-R132 (1:1000, kindly provided by Peter Sonderegger), anti-agrin C-95 (1:500, kindly provided by Markus Ruegg); and anti-LRP4 against the extracellular domain (ECD) (1:1000, clone N207/27, UC Davis/NIH NeuroMab Facility). After washing, the membrane was incubated with TBST buffer containing HRP-conjugated goat anti-mouse and rabbit IgG from Pierce (1:5000, PI-31430, anti-mouse; PI-31460, anti-rabbit). Immunoreactive bands were visualized by using enhanced chemiluminescence (Pierce). Quantitative densitometric analysis of the captured images was analyzed with ImageJ (National Institutes of Health), as described previously (Wu et al., 2012b).
Electron microscopy analysis.
Electron microscopy was performed as described previously (Wu et al., 2012a, b). TA muscles were dissected in PBS and were lightly stained with R-BTX (1:1000, in ice-cold PBS at 4°C) to mark the central region where NMJs are enriched. The regions were dissected with a microscalpel (Harvard Apparatus, #PY2 56-5673) under a Leica fluorescent dissection scope and fixed (blocks of ∼4 mm × 4 mm) in 2% glutaraldehyde and 2% PFA in 0.1 m PBS for overnight at 4°C. Tissues were further fixed in sodium cacodylate-buffered, pH 7.3, 1% osmium tetroxide for 1 h at 25°C. Fixed tissues were washed three times with PBS and subjected to dehydration through a series of ethanol: 30%, 50%, 70%, 80%, 90%, and 100%. After three rinses with 100% propylene oxide, samples were embedded in plastic resin (EM-bed 812, EMSciences). Serial sections (1–2 μm) were stained with 1% Toluidine Blue to identify motor nerves and were cut into ultrathin sections. Alternate longitudinal sections were not chosen to avoid duplicity of obtaining images from same terminals. They were mounted on 200-mesh unsupported copper grids and stained with a solution containing 3% uranyl acetate, 50% methanol, 2.6% lead nitrate, and 3.5% sodium citrate, pH 12.0. Electron micrographs were taken using a JEOL 100CXII operated at 80 KeV.
Synaptic vesicles were distinguished from other intracellular vesicles by uniform diameters (∼40 nm) and subcellular localization. Active zones were determined by electron-dense regions on presynaptic membranes, sometimes with fused vesicles. Synaptic cleft width was measured by calculating the distance between the presynaptic and postsynaptic membranes. Synaptic vesicle polarization was calculated as the number of vesicles in the half-terminal facing the muscle divided by the number in the other half terminal (Patton et al., 2001). Four animals of each group (treated or untreated imKO) were subjected to EM analysis. Quantitative analysis was done with 15 or more electron micrographs by FIJI (National Institutes of Health).
Electromyography and electrophysiological recording.
Recording was performed as described previously (Wu et al., 2012a, b; Shen et al., 2013). Briefly, mice were anesthetized with ketamine and xylazine (80 mg/kg and 20 mg/kg i.p., respectively). For electromyography, the stimulation electrode was inserted into the left leg thigh close to the path of sciatic nerves. A reference electrode was inserted near the Achille's tendon while the recording needle electrode was inserted into the middle of the gastrocnemius. Supramaximal stimulation was applied to the sciatic nerve with trains of 10 stimuli at 2, 5, 10, 20, and 40 Hz. Compound muscle action potentials (CMAPs) were collected by the reference and recording electrodes via axopatch 200B amplifier and Digidata 1322A (Molecular Devices). Peak-to-peak amplitudes were analyzed in Clampfit 9.2 (Molecular Devices). During the experiment, mice were maintained at 37°C on a heating pad.
For the analysis of neuromuscular transmission, mouse left hemi-diaphragms with ribs and phrenic nerve distal endings were dissected and pinned on Sylgard gel in oxygenated (95% O2/5% CO2), 26°C–28°C Ringer's solution (136.8 mm NaCl, 5 mm KCl, 12 mm NaHCO3, 1 mm NaH2PO4, 1 mm MgCl2, 2 mm CaCl2 and 11 mm d-glucose, pH 7.3). Microelectrodes, 20–40 MW when filled with 3 m KCl, were pierced into the center of muscle fibers (Wu et al., 2012a, b; Shen et al., 2013). Resting membrane potentials (−65 to −75 mV) remained stable during the experiment. Five or more muscle fibers were recorded from each hemidiaphragm over a 3 min period. To record endplate potentials (EPPs) and pair-pulse facilitations, the phrenic nerve was held with sucking and stimulated by a platinum electrode. Trigger signals (1 ms duration) were programmed in Clampex 9.2 (Molecular Devices) and elicited from Digidata 1322A digital output channel to stimulus isolator (AMPI, ISO-Flex). The intensity of stimulation was kept ∼≥130% of action potential threshold. Muscle contraction was blocked by 2.5 mm m-Conotoxin GIIIB (Bachem Americas) when phrenic nerves were stimulated. Data were collected with Axopatch 200B amplifier, digitized (10-kHz low-pass filtered) with Digidata 1322A, and analyzed in Clampfit 9.2. Four animals of each group (treated or untreated imKO) were subjected to electrophysiological analysis.
Light microscopic analysis of AChR clusters.
TA muscle fibers were fixed in 4% PFA overnight and permeabilized for 2 h with 0.5% Triton X-100 in 3% BSA and 3% goat serum. They were then incubated with a mixture of rhodamine-conjugated bungarotoxin (R-BTX, 1:2000, Invitrogen), Alexa488-conjugated Fasciculin II (488-Fasciculin II), and/or antibodies against neurofilament (NF) (1:1000, ab7795, Millipore) and synaptophysin (1:500, DAK-SYNAP, DAKO) at 4°C overnight. After washing with PBS, three times for 1 h each, muscle fibers were incubated with goat anti-mouse/rabbit IgG conjugated with AlexaFluor-488 or AlexaFluor-633 (1:500, Invitrogen) overnight at 4°C. Fasciculin II conjugated with the Alexa-484 dye was generously provided by Dr. Richard Rotundo (University of Miami). Images were collected with a Zeiss 510 upright confocal microscope with 40×, 63× oil-immersion objectives. Three-dimensional reconstruction of confocal z-stacks and image analysis was conducted using FIJI (National Institutes of Health).
Statistical analysis.
Statistically significant difference between two groups was determined by Student's t test. Unless otherwise indicated, data were expressed as mean ± SEM, and statistical significance was considered when p < 0.05.
Results
Reduced muscle strength and body weight in Dox-treated imKO mice
LRP4 is a receptor of agrin and critical for NMJ formation (Weatherbee et al., 2006; Kim et al., 2008; Zhang et al., 2008; Zhang et al., 2011; Wu et al., 2012b; Zong et al., 2012; Ahn et al., 2013; Shen et al., 2013). LRP4 mutant mice die prematurely (Weatherbee et al., 2006), which prevents investigating the role of LRP4 in NMJ stability. To restrict deletion of LPR4 to adult muscles, we have used a conditional and inducible tetracycline-controlled expression system (Tet-On) in which LRP4 can be deleted in a time- and muscle-specific manner (Rao and Monks, 2009). Tg(tetO-Cre; ACTA1-rtTA) mice were bred with LRP4 floxed alleles (LRP4f/f), to obtain Tg(tetO-Cre; ACTA1-rtTA; LRP4f/f), which we referred to as inducible muscle-specific knockof LRP4 or imKO for short (Fig. 1A). Unless otherwise stated, imKO mice at 30 d of age (P30) were given Dox ad libitum in drinking water for 3 weeks. To determine whether LRP4 in imKO mice was altered by Dox treatment, muscles were isolated from Dox-treated imKO mice and subjected to Western blotting. As control, muscles were isolated from untreated imKO mice and Dox-treated LRP4f/f mice. As shown in Figure 1B, LRP4 levels were similar between wild-type and imKO, suggesting that expression of the two tet-regulatory transgenes or Dox treatment alone had no effect on LRP4 expression. In contrast, LRP4 was reduced in Dox-treated imKO mice, compared with wild-type (Fig. 1B). This effect required the two transgenes because Dox had no effect on LRP4 level in LRP4f/f mice. These results validated successful ablation of the LRP4 gene and reduction of LRP4 in muscles of imKO mice. Twenty days after Dox treatment, imKO mice started losing weight and muscle strength, compared with untreated imKO mice (Fig. 1C,D). The body weight and muscle strength of LRP4f/f mice did not change in response to Dox treatment (Fig. 1C,D). By 30 d after treatment, many Dox-treated imKO mice developed scoliosis (Fig. 1E). Dox-treated imKO mice began to die ∼80 d after Dox administration; and by ∼130 d, none survived (Fig. 1F). These observations indicate that muscle ablation of LRP4 in adult mice led to muscle weakness, body weight loss, and eventual death, suggesting that LRP4 is required for NMJ maintenance. Unless otherwise indicated, we focused on imKO mice that had been treated with Dox for 30 d, which effectively ablated LRP4 in muscle cells.
Figure 1.

LRP4 is depleted from NMJs following conditional deletion in adults. A, Genotypes and LRP4 deletion in skeletal muscle. In ACTA1-rtTA; tetO-cre transgenic mice (Rao and Monks, 2009), cre is expressed under control of the ACTA1 (human actin α1, skeletal muscle) promoter, whereas cre expression is controlled by tetracycline inducible element (tetO). The bitransgenic mice do not express Cre until doxycycline is administered. ACTA1-rtTA; tetO-cre mice were bred with LRP4f/f mice (Wu et al., 2012b) to generate ACTA1-rtTA; tetO-cre; LRP4f/f (imKO). Dox-treated imKO mice did not express LRP4. Unless otherwise specified, LRP4f/f mice were used as control. B, Reduction of LRP4 in Dox-treated imKO mice. Tibialis anterior muscle was isolated from mice of indicated genotypes and treatment and blotted for LRP4, MuSK, and β-actin. C, Dox-treated imKO mice progressively lost weight, compared with imKO or LRP4f/f+DOX mice; n = 3 per genotype (two-way ANOVA). *p < 0.05 (Student's t test). D, Decreased grip strength in Dox-treated imKO mice; n = 3 per genotype (two-way ANOVA). *p < 0.05 (Student's t test). E, Dox-treated imKO mice were considerably smaller in size, compared with imKO mice, and developed scoliosis after 30 d of Dox treatment. F, Kaplan–Meier survival curves of control and Dox-treated imKO mice. χ2 = 22.67, p < 0.0001 (Log-Rank; Mantel–Cox test).
Fragmented AChR clusters in Dox-treated imKO mice
In adult mice, AChR clusters are organized in a “pretzel”-like structure with complex and continuous branches on the muscle membrane (Fig. 2A). To investigate the mechanisms of muscle weakness in Dox-treated imKO mice, we stained TA muscles with R-BTX, which labels AChRs irreversibly. As shown in Figure 2A, B, the NMJs in Dox-treated imKO mice were not continuous, but fragmented. Quantitative analysis revealed that total R-BTX-labeled area per pretzel was reduced from 373 ± 34.2 μm2 in imKO mice to 245 ± 54.3 μm2 in Dox-treated imKO mice (p < 0.01, n = 10) (Fig. 2A,B). The numbers of AChR cluster fragments were increased from 3.21 ± 0.8 to 9.11 ± 1.7 (p < 0.01, n = 10) (Fig. 2A,B). The area of each cluster or fragment was also reduced from 143 ± 21.5 μm2 in imKO mice to 90 ± 46.7 μm2 in Dox-treated imKO mice (p < 0.01, n = 10) (Fig. 2A,B). The fluorescent intensity of the clusters, measured by mean pixel value, was reduced to 52.03 ± 19.48 in Dox-treated imKO mice, compared with 85.4 ± 8.9 in imKO mice (p < 0.01, n = 10). No difference was observed between wild-type, imKO, or Dox-treated LRP4 floxed mice (Fig. 2A; data not shown). AChR clusters in Dox-treated LRP4f/fmice appeared similar to those in wild-type and untreated imKO mice (Fig. 2A,B), in agreement with the observations that these mice exhibit normal muscle strength and body weight. Together, these experiments demonstrate that NMJs became fragmented when LRP4 was ablated in adult mice.
Figure 2.
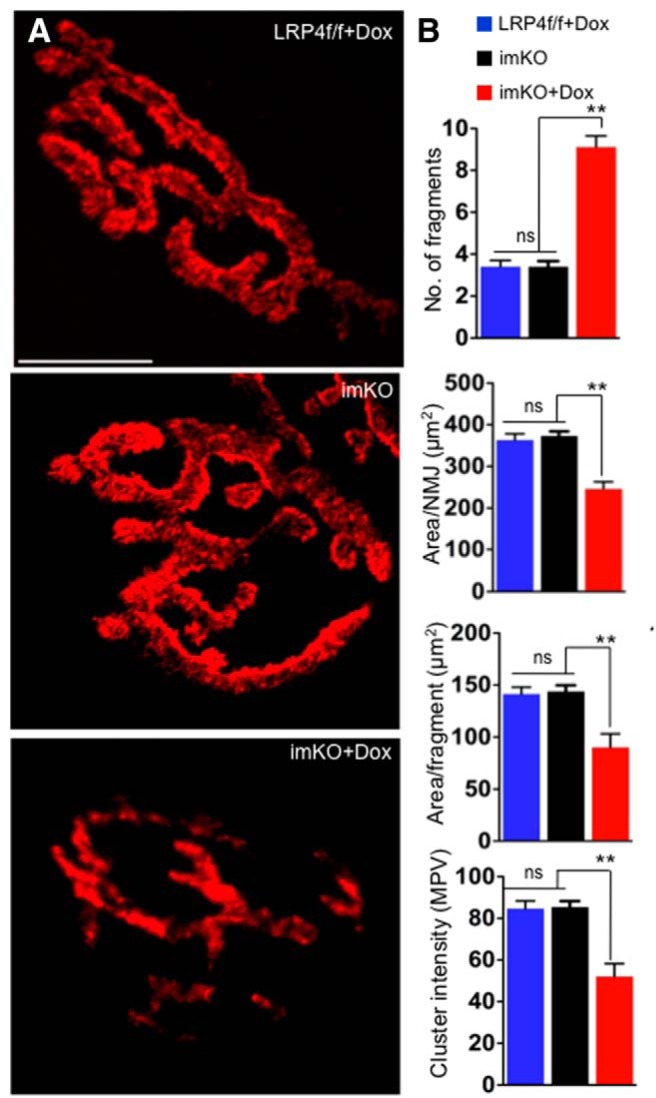
Fragmented NMJs with reduced AChR in Dox-treated imKO mice. A, TA muscles were stained whole mount with R-BTX to label AChR (red). In Dox-treated imKO mice, the endplates were smaller in size, fragmented, and displayed reduced R-BTX fluorescent intensity, compared with controls. Scale bar, 10 μm. B, Quantitative analysis revealed more fragmented AChR clusters, AChR-rich endplate area per synapse (area/NMJ), area per fragment, and AChR intensity in clusters (measured by mean pixel value). **p < 0.01 (Student's t test). n = 10. ns, Not significant. Scale bar, 10 μm.
Dox treatment reduced CMAPs and impaired neuromuscular transmission in imKO mice
To determine whether neuromuscular transmission is altered in Dox-treated imKO mice, we measured the CMAPs in gastrocnemius muscle in response to needle electrode stimulation (Shen et al., 2013). In control imKO mice, CMAPs did not significantly differ over 10 consecutive stimuli delivered at 40 Hz (Fig. 3A–C). In contrast, there was a significant reduction in CMAPs over 10 consecutive stimuli in Dox-treated imKO mice (Fig. 3C). The reduction was significant even after the second stimulation, and the 10th CMAP was reduced by 40%, compared with the first CMAP (Fig. 3C). The reduction of CMAPs was frequency-dependent (Fig. 3D), suggesting that there was a progressive loss of successful neuromuscular transmission after repeated stimulations. Together, these observations indicate that NMJ transmission was impaired in mice whose LRP4 was ablated in adulthood.
Figure 3.
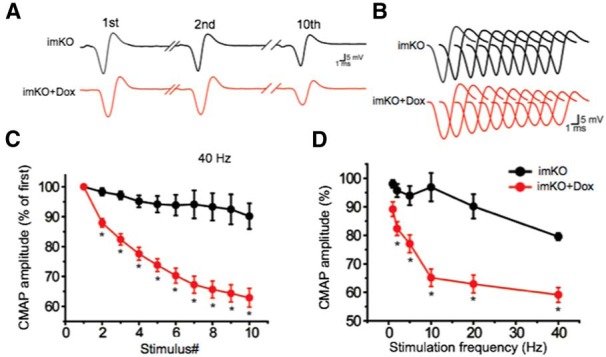
CMAP reduction in Dox-treated imKO mice. CMAPs were recorded in gastrocnemius in response to a train of 10 submaximal stimuli at different frequencies. The first stimulus response in control mice was assigned as 100%. A, Representative CMAP traces in response to the first, second, and 10th stimuli. B, All 10 CMAP traces, shown stacked in succession for better comparison. C, Reduced CMAP amplitudes at 40 Hz. D, CMAP amplitudes of the 10th stimulation at different stimulation frequencies. n = 4 mice per group. *p < 0.05 (Student's t test).
To investigate whether CMAP deficits result from presynaptic and/or postsynaptic impairment, we measured miniature EPPs (mEPPs), muscle membrane potentials due to spontaneous ACh release (Meier et al., 1996; Wu et al., 2012a, b; Shen et al., 2013). Whereas changes in frequency are suggestive of presynaptic defects, alteration in amplitude is suggestive of postsynaptic defects. As shown (Fig. 4A,B), mEPP amplitude was reduced by 30% in Dox-treated imKO mice (0.78 ± 0.04 mV, compared with 0.51 ± 0.03 mV in imKO mice; p < 0.05, n = 11). The reduction in amplitude suggests that the density of AChR clusters on the postsynaptic side was reduced in the mutants. Similarly, mEPP frequency was also decreased (by 90%) in Dox-treated imKO mice compared with controls (imKO, 1.18 ± 0.05 Hz; Dox-treated imKO, 0.17 ± 0.01; p < 0.05, n = 11) (Fig. 4A,C). This was suggestive of a presynaptic defect most likely stemming from a reduction in spontaneous ACh release from motor nerve terminals. Together, these results suggest that loss of LRP4 in muscles of adult mice affected both presynaptic and postsynaptic components of the NMJ.
Figure 4.

Impaired neuromuscular transmission in Dox-treated imKO mice. A, Representative mEPP traces. Underlined regions in the top were enlarged in the bottom. B, C, Reduced mEPP amplitudes (B) and frequencies (C); n = 6 in each group. D, Reduced EPP amplitude and representative traces; n = 5 in each group. E, F, Increased EPP rising time (E) and half-width (F); n = 5 NMJs in each group. G, Increased pair-pulse facilitation; n = 7 NMJs in each group. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test).
Moreover, LRP4 loss also caused reduction in end plate potentials (EPPs), which are an indicator of neurotransmission elicited by motor nerve stimulation. The quantal content, the ratio of EPP amplitude/mEPP amplitude, is a measure of the number of synaptic vesicles released. Dox treatment reduced the quantal content in imKO mice, suggesting that evoked release is impaired in mutants (Fig. 4D). Intriguingly, the half-width and the rising time of the EPP in Dox-treated imKO mice were higher than the imKO mice, indicating synaptic vesicle release deficits at the motoneuron terminals (Fig. 4E,F). Further, we examined paired-pulse facilitation (PPF) at different interstimulus intervals. When presynaptic neurons are stimulated by two consecutive stimuli at rapid succession, the second endplate potential is usually larger than the first one due to increased intracellular calcium. At 10 ms interval, PPF was significantly higher at the NMJs of Dox-treated imKO mice, compared with controls (Fig. 4G), indicating diminished readily releasable pools and/or reduced probability of release. The difference in PPF between groups reduced with an increase in the duration of interpulse interval; and at 100 ms interval, there was no significant difference between the groups. This suggests normal ability of endoplasmic reticulum to buffer calcium in motoneurons of Dox-treated imKO mice, although calcium-dependent vesicle release may be impaired. The results described above indicate that loss of muscle LRP4 in adulthood is sufficient to cause both presynaptic and postsynaptic neuromuscular defects and suggest that LRP4 is critical for maintaining functional NMJs. These results provide pathophysiological mechanisms of LRP4 mutations in CMS patients and LRP4 antibodies in MG patients.
Reduced synaptic vesicles and junctional folds in Dox-treated imKO mice
Under the electron microscope, muscle membrane at mature NMJ is invaginated with deep folds at regularly spaced intervals (Porter and Barnard, 1976). AChRs are concentrated at the crests of junctional folds (Fertuck and Salpeter, 1976), whereas active zones of motoneuron terminals are opposed to the troughs of junctional folds (Matthews-Bellinger and Salpeter, 1983). In imKO mice, postjunctional folds were evident at the NMJ (Fig. 5A, arrowheads). They appeared to be electron dense, presumably reflecting high concentration of AChRs (Fig. 5A) (Fertuck and Salpeter, 1976; Marques et al., 2000). However, the NMJs of Dox-treated imKO mice were almost devoid of junctional folds, with the number of junctional folds per terminal reduced to 1 ± 0.44 (compared with 6 ± 0.27 in imKO; p < 0.01; n = 15) (Fig. 5A, bottom, stars). Because of the diminished folds, it was difficult to identify typical junctional membranes. We identified them as the ones closely opposed to the active zones. As shown in Figure 5A, the electron density of the junctional membrane was reduced in Dox-treated imKO mice. The synaptic cleft width did not change by LRP4 loss (Fig. 5A,B). However, the electron density of synaptic basal lamina in the synaptic cleft was reduced. These observations suggest that muscle LRP4 is critical for maintenance of synaptic basal lamina, junctional folds as well as postjunctional density.
Figure 5.
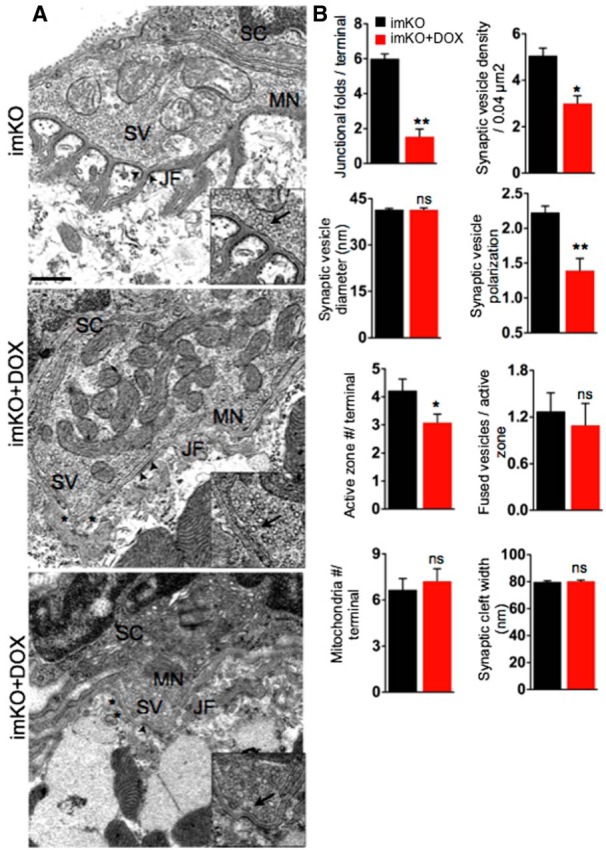
Abnormal NMJ ultrastructure in Dox-treated imKO mice. A, Representative EM NMJ images of control and Dox-treated imKO mice. NMJs in control mice displayed typical NMJ structures, where the nerve terminals were covered by terminal Schwann cells (SC) and enriched with synaptic vesicles (SV). Occasionally, fused SVs were present at the active zones (inset, arrows). Postsynaptic membranes invaginated to form junctional folds (JF), whose crests were electron dense and contained AChR-rich regions (arrows). In Dox-treated imKO mice, the numbers of SVs, JFs (asterisks), and active zones were reduced. B, Quantification of various synaptic attributes (n = 18, for each genotype), *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ns, Not significant. Scale bar, 500 nm.
Our electrophysiological studies suggested that the number of ACh-laden synaptic vesicles could be reduced in mutants. To test this hypothesis, we examined the presynaptic structure in mutants. In control or imKO mice, axon terminals were filled with synaptic vesicles; some of them were docked at active zones (Fig. 5A; inset, arrows, active zones) (Shen et al., 2013). However, electron micrographs of Dox-treated imKO NMJs revealed significant reduction in synaptic vesicles (from imKO, 5 ± 0.32 to 3 ± 0.33 vesicles per 0.04 μm2 in control and treated imKO mice, respectively; p < 0.01; n = 17) and in active zones per terminal (from 4 ± 0.41 μm to 3 ± 0.33 μm in control and treated imKO mice, respectively; p < 0.05; n = 13) (Fig. 5A,B). In control mice, synaptic vesicle distribution is polarized: being more in regions close to active zones, compared with regions facing away from the muscle (Patton et al., 2001; Shen et al., 2013). Interestingly, the ratios of vesicles in the synaptic half (i.e., area close to active zones) over those in distal half (i.e., area facing away from the muscle) was 1.3 ± 0.17 in imKO mice and 2.2 ± 0.09 in Dox-treated imKO mice (p < 0.01, n = 10) (Fig. 5A,B). This suggests that the loss of synaptic vesicles in LRP4 mutant mice was not the same between the two regions; rather, the loss was greater in the synaptic region than in nonsynaptic region. This also suggests that the reduction in overall vesicle number in the mutants is not because of impaired production of vesicles.
Mitochondria at axon terminals generate ATP and sequester calcium to maintain cytosolic calcium concentration that is conducive for normal neurotransmission (Thayer and Miller, 1990; García-Chacón et al., 2006). The number of mitochondria per terminal was similar between control (6.6 ± 0.72) and Dox-treated imKO mice (7.2 ± 0.81) (p < 0.05, n = 10) (Fig. 5A,B), in agreement with electrophysiological data that calcium buffering was normal in Dox-treated imKO NMJ (Fig. 4E). Schwann cells covered both Dox-treated imKO mice NMJs as wells as control (Fig. 5A). We did not notice any Schwann cell processes in the synaptic cleft separating the nerve terminal and muscles. In summary, LRP4 deletion in skeletal muscles during adulthood alone is sufficient to cause a loss of synaptic vesicles from the MN terminals, reduction of active zones, and retraction of synaptic folds. These ultrastructural defects are commensurate with the electrophysiological deficits as well as changes observed by light microscopy.
Nerve terminal retraction and sprouting with Schwann cell extension at Dox-treated imKO NMJs
Having observed fragmented AChR clusters in imKO mice, we investigated whether nerve terminals and Schwann cells were altered subsequent to postsynaptic deficits. To this end, TA muscles were stained whole mount with antibodies against neurofilament/synaptophysin (NF/Syn) for motor nerve terminals, S100β for differentiated Schwann cells, and R-BTX for AChR clusters. In control adult NMJs, motor neuron terminals were in perfect registry with AChR clusters under light microscope (Fig. 6A). Moreover, terminal Schwann cell processes covered the synapse, and sprouting or denervation was negligible (Fig. 6A,B). In Dox-treated imKO mice (at P60), AChR clusters were fragmented (Figs. 2 and 6A). Some R-BTX-labeled AChR clusters were interestingly not covered by motor nerve terminals but Schwann cell processes (Fig. 6A, bottom, pink arrows). This suggests the fragmented AChR clusters may have lost innervation. There were areas where Schwann cell processes were present, but not nerve terminals or AChR clusters (Fig. 6B, bottom, yellow arrows). This could suggest that during NMJ disintegration, Schwann cell processes may be more stable than AChR clusters or nerve terminals as observed in NMJ elimination, and thus remain at the site where the NMJ used to be (Culican et al., 1998). Alternatively, Schwann cells may sprout as a prelude to nerve terminal sprouting as compensatory mechanism for reduced NMJ transmission. Schwann cell sprouting has been shown to be critical for reinnervation after injury (Son and Thompson, 1995a, b). As shown in Figure 6A (bottom, stars), Schwann cell sprouting was observed in NMJs in Dox-treated imKO mice, with (bottom, star) or without (top, star) nerve terminals. Quantitatively, the percentage of NMJs with nerve retraction (including those with Schwann cell processes) was 45.4 ± 2.37% in Dox-treated imKO mice and 1.76 ± 0.43% in control mice (n = 36, p < 0.01). Nerve terminal sprouts were observed in 36.5 ± 3.17% of NMJs in Dox-treated imKO mice and only in 5.47 ± 1.12% of NMJs in control mice (Fig. 6B) (n = 36, p < 0.01).
Figure 6.

Partial denervation and extensive sprouting at Dox-treated imKO NMJs. In control NMJs, AChR clusters were completely innervated by nerve terminals labeled by neurofilament (NF) and synaptophysin (syn) (green). The NMJs were covered almost entirely by terminal Schwann cells and their processes (labeled by S100β in cyan). None or negligible sprouting was observed for nerve terminals and Schwann cell processes (A, B). In Dox-treated imKO mice, NMJs became fragmented and lost innervation (pink arrow); yellow represents lost innervation and AChR clusters. Stars indicate sprouting of nerve terminals and Schwann cells; n = 30 for each genotype. **p < 0.01 (Student's t test). Scale bar, 10 μm.
Reduction of synaptophysin in Dox-treated imKO mice
To identify how loss of LRP4 from adult muscle could lead to synaptic disruption, we sought to understand which components were changed at the NMJ. To this end, TA muscles were stained whole mount with antibodies against synaptophysin (syn), a protein expressed in synaptic vesicles; α-dystroglycan, a protein enriched in SBL; or Fasciculin II, which binds to AChE irreversibly (Sugiyama et al., 1994; Peng et al., 1999; Li et al., 2008; Rotundo et al., 2008). Muscle sections were stained for the following antibodies because they do not work in whole-mount samples: antibodies against MuSK; rapsyn, an adaptor protein that interacts with AChR (Luo et al., 2008); and utrophin, a cytoskeletal protein is also known to colocalize with AChRs at the junctional folds (Blake et al., 1994; Weatherbee et al., 2006; Friese et al., 2007).
We analyzed the distribution of three postsynaptic proteins, including MuSK, rapsyn, and utrophin (Fig. 7E–I). The fluorescence intensities of MuSK, rapsyn, and utrophin were not altered in Dox-treated imKO mice at P60. Additionally, AChR clusters were reduced in number and became fragmented in Dox-treated imKO mice; there appeared to be a good registry between R-BTX staining and that of these proteins (Fig. 7E–I). These results suggest that NMJ disruption in the absence of adult LRP4 was not due to alteration of MuSK, rapsyn, or utrophin; rather, their loss in Dox-treated imKO mice may be secondary to NMJ disruption.
Figure 7.

Loss of agrin and synaptophysin from Dox-treated imKO NMJs. A–G, Muscles were stained whole mount with antibodies against syn, α-dystroglycan or agrin, or with Alexa488-conjugated Fasciculin II. Muscle sections were stained with antibodies against MuSK, rapsyn, or utrophin. Dox-treated imKO NMJs showed reduced syn or agrin, but no change in Fasciculin II, α-dystroglycan, MuSK, rapsyn, or utrophin. H, Quantification of fluorescent intensity; n = 15. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). I, R-BTX area covered by individual synaptic proteins in in Dox-treated imKO NMJs; n = 15. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ns., Not significant. Scale bar, 10 μm.
SBL is composed of extracellular matrix proteins secreted by motoneurons, muscles, and Schwann cells and is enriched in factors critical for formation and maintenance of presynaptic and postsynaptic structures at the NMJ. α-Dystroglycan is a glycoprotein that is abundantly expressed at the NMJ. It can bind agrin and has been implicated in aggregation and stabilization of AChR clusters (Sugiyama et al., 1994; Côté et al., 1999). Loss of α-dystroglycan in mice results in defective NMJ (Côté et al., 1999; Samuel et al., 2012). As shown in Fig. 7C, H, the intensity of α-dystroglycan staining was not altered in Dox-treated imKO mice, compared with controls. On the contrary, the area of α-dystroglycan staining appeared to be higher in Dox-treated imKO mice, compared with that in controls. This increase was most likely due to reduced areas of AChR clusters in Dox-treated imKO mice. These results indicate that, despite reduced fragmented AChR clusters and reduced R-BTX staining intensity, the SBL scaffold remained intact at P60. This notion was supported by the observations that no change occurred in staining intensity of Alexa488-conjugated Fasciculin II that labeled AChE, another SBL component. These suggest that fragmentation and instability of AChR clusters may not be due to changes of α-dystroglycan or AChE in the SBL (Fig. 7B–D,H,I).
As shown in Figure 7A, staining with anti-syn antibody alone exhibited reduced fluorescence intensity in Dox-treated imKO mice, compared with that in controls (Fig. 7A,H). Moreover, in control mice, syn staining covered almost 100% AChR cluster area, but only 73 ± 0.03% of AChR cluster area was covered by syn (n = 11; p < 0.01) in Dox-treated imKO mice. These results are indicative of reduction in number of synaptic vesicles at nerve terminals in the absence of muscle LRP4, in agreement with results of EM analysis (Fig. 5).
Agrin reduction in Dox-treated imKO mice
Knowing that LRP4 serves as a receptor of agrin, we determined whether loss of muscle LRP4 in adulthood changes the amount of agrin. To this end, muscles were stained with a well-characterized anti-agrin antibody (R132). This antibody was generated against the C-terminal fragment of agrin (Reif et al., 2007). Agrin staining was present in AChR clusters, but also in extrasynaptic areas in the pretzel-like structure in control mice (Fig. 7D). Intriguingly, agrin staining intensity in Dox-treated imKO mice was significantly reduced, compared with that in control mice (Fig. 7D,H,I) (43 ± 1.35 and 119 ± 3.02, respectively) (n = 10, p < 0.05). Moreover, 70% of AChR cluster area lacked agrin (1.2 ± 0.03 in control and 0.3 ± 0.49 in Dox-treated imKO mice; n = 10, p < 0.01). These results suggest that ablation of LRP4 in adult muscle reduces agrin that is critical for NMJ formation.
Next, we determined the time course of agrin reduction in Dox-treated imKO mice. To exclude the possibility that agrin reduction was due to or secondary of AChR cluster disruption, we normalized agrin signals by two denominators, including AChR clusters, whose level was reduced, and AChE, whose level did not change, in Dox-treated imKO mice at P60 (Fig. 8A,B,G,H). As shown in Figure 8C–H, the ratio of agrin/AChR was reduced from P30 to P60, which indicates that agrin reduction occurred ahead of AChR reduction. However, the ratio of Syn/AChR remained similar during the same period, suggesting that syn reduction was in parallel to that of AChR in Dox-treated imKO mice. Further, the ratio of agrin/Fasciculin II staining showed a leftward shift of the time course, indicating that agrin reduction was independent of SBL scaffold (Fig. 8H). Similar reduction time was observed in syn/Fasciculin II ratio (Fig. 8H). These results suggest that loss of agrin preceded the loss of AChR or synaptophysin.
Figure 8.

Time course depletion of synaptic agrin and synaptophysin from the NMJs in Dox-treated imKO mice. Whole-mount staining was performed as in Figure 7. A–F, Confocal images of NMJs at indicated times. A gradual reduction of agrin and syn was observed in Dox-treated imKO NMJs. Fasciculin II levels remained unchanged. G, Quantitative analysis of loss of agrin and Syn, relative to R-BTX (n = 6). H, Quantitative analysis of loss of Agrin and Syn, relative to Fasciculin II (n = 6). Scale bar, 10 μm.
Finally, we determined whether reduced agrin signal in staining was due to agrin loss or dispersion from the NMJ; we performed Western blot analysis of entire TA muscles at different times after Dox treatment of imKO mice. Blotting for LRP4 exhibited time-dependent reduction of LRP4, which became barely detectable at P60 (Fig. 9A,B). Blotting with antibodies against α-dystroglycan, MuSK, and rapsyn did not reveal a reduction in total protein level (Fig. 9A,B), although immunostaining signal of each protein was reduced, suggesting that the reduction in staining signal was due to dispersal of these proteins into nonsynaptic areas. The agrin gene encodes a protein of 2000 amino acids with predicted molecular weight of 225 kDa but can display as 600 kDa protein due to extensive N-terminal glycosylation (Bezakova and Ruegg, 2003). Full-length agrin (220 kDa) is cleaved in the C-terminal region by a serine-protease to yield a 95 kDa fragment. This fragment contains the domain that binds to LRP4 and a sequence for α-dystroglycan (Ruegg et al., 1992; Fallon and Hall, 1994; Neumann et al., 2001; Bezakova and Ruegg, 2003; Bolliger et al., 2010). Interestingly, this 95 kDa fragment revealed by the R132 antibody, was reduced progressively after Dox treatment in imKO mice. Similar reduction of the 95 kDa fragment was observed when blotted with the C95 antibody that was independently generated and recognized agrin (Eusebio et al., 2003). Full-length agrin ranging from 200 to 600 kDa remained unchanged. These observations suggest that the reduction of agrin was 95 kDa-specific. Because reduction of agrin was observed in both immunostaining and Western blot analysis, we interpret these results as evidence for a role of muscle LRP4 in controlling the stability of agrin.
Figure 9.

Loss of the 95 kDa agrin fragment in Dox-treated imKO muscle. A, Time course of alteration of various synaptic proteins. Muscle homogenates of indicated mice were subjected to Western blotting with respective antibodies. B, Quantitative analysis of data in A.
Discussion
This study provides evidence that loss of LRP4 in adult muscles results in severe structural and functional defects of both presynaptic and postsynaptic components (Figs. 1, 2, 3, 4, and 5). First, Dox treatment of imKO reduced LRP4 levels to undetectable 30 d after treatment. The mice displayed weight loss, muscle weakness, scoliosis, and premature death, which resemble myasthenic symptoms of CMS and MG patients. Second, morphological analysis revealed that NMJs lost characteristic “pretzel”-like morphology and reduced AChR intensity in fragmented AChR clusters. The clusters were partially innervated, and the numbers of active zones and synaptic vesicles were reduced in motoneuron terminals. Third, functionally, CMAPs were reduced, indicative of reduced excitability of postsynaptic muscle. The amplitude and frequency of mEPPs were diminished, suggesting impairment of both presynaptic and postsynaptic compartments. These observations demonstrate that LRP4 is essential for maintaining the structural and functional integrity of the NMJ and that loss of muscle LRP4 in adulthood alone is sufficient to cause myasthenic symptoms.
LRP4 belongs to the LDL receptor family, which consists of many evolutionarily conserved, transmembrane proteins (Nykjaer and Willnow, 2002). Members of this family are known to serve as coreceptor of various ligands, including ApoE and Wnts (Pinson et al., 2000; Tamai et al., 2000; Wehrli et al., 2000). The ECD of LRP4 has 8 LDLa (LDL Class A) repeats at the N terminus, followed by four homologous YWTD motif-containing β-propeller domains that are separated by EGF-like modules. Several extracellular binding partners have been identified for LRP4, including ApoE (that binds to LDLa), agrin (that binds to the first β-propeller domain), MuSK and Wnt modulators (SOST and Dkk) (that bind to the third β-propeller domain), BMP signaling regulators (Wise and GREM1), and APP (whose interacting motif in LRP4 was unclear) (Kim et al., 2008; Zhang et al., 2008, 2012a; Choi et al., 2009; Zhang et al., 2011; Zong et al., 2012; Ahn et al., 2013; Choi et al., 2013; Barik et al., 2014). Mutations in the LRP4 ECD were observed in patients with various diseases, including Cenani–Lenz syndrome, Potocki–Shaffer syndrome, and bone disorders, including osteoporosis, Richter syndrome, and Cenani–Lenz syndactyly syndrome (Wakui et al., 2005; Li et al., 2010; Rasi et al., 2011; Boudin et al., 2013; Ohkawara et al., 2014). Which signaling pathway(s) is altered by these mutations remains to be unclear, although Wnt signaling is thought to be a strong suspect for bone-related deficits (Johnson et al., 2005; Choi et al., 2009; Li et al., 2010; Ohazama et al., 2010).
Neither is it clear how LRP4 regulate NMJ maintenance. We offer the following hypotheses. First, neuromuscular disorders, such as CMS and MG often involve structural defects, including fewer synaptic vesicles, reduced AChR density, diminished junctional folds, and abnormal NMJ transmission, such as reduced CMAPs. In CMS patients, mutations have been identified in NMJ structural and functional proteins, such as ChAT, collagen Q, laminin, AChR, sodium channels. In addition, proteins in agrin signaling are often mutated, including agrin, LRP4, MuSK, rapsyn, and Dok7 (Maselli et al., 2010, 2012; Ben Ammar et al., 2013; Ohkawara et al., 2014). In mice, a missense mutation of MuSK that reduces its kinase activity causes pronounced myasthenic deficits (Chevessier et al., 2008). Agrin deletion in a subset of motoneurons (by Thy1-CreERT2 or SLICK-creERT2) in adult mice results in AChR loss and loss of other components in the postjunctional membrane and synaptic cleft. However, mutant mice survived because agrin was still expressed in majority of motoneurons (Samuel et al., 2012). Similarly, neonatal, mosaic mutation of MuSK by muscle creatine kinase (MCK)-Cre leads to loss of AChRs and disassembly of postjunctional organization (Hesser et al., 2006). On the other hand, MG is caused by autoantibodies against AChR as well as agrin signaling proteins, including agrin, LRP4, and MuSK (Culican et al., 1998; Kraner et al., 2003; Sanders et al., 2003; Reif et al., 2007; Rotundo et al., 2008; Zhang et al., 2012b, 2014; Zisimopoulou et al., 2014). Antibodies against agrin, LRP4, and MuSK may act by disrupting agrin signaling (Hoch et al., 2001; Shigemoto et al., 2006, 2008; Zhang et al., 2012b, 2014; Shen et al., 2013). These observations suggest a role of the agrin signaling pathway in NMJ maintenance. At present, two heteroallelic mutations in LRP4 have been identified in CMS patients: p.Glu1233Lys (c.3697G>A) and p.Arg1277His (c.3830G>A). They are located at the border region of the third β-propeller domain (Ohkawara et al., 2014), a domain that interacts with MuSK (Zhang et al., 2011). These mutations were shown to diminish agrin signaling, but not Wnt signaling (Ohkawara et al., 2014). Therefore, diminished agrin signaling may be a pathophysiological mechanism underlying LRP4 loss in adult mice.
The time-dependent loss of synaptic agrin and the 90 kDa fragments, which occurred ahead of other prejunctional and postjunctional components, after LRP4 ablation in adult mouse muscles, suggests that LRP4 may regulate the stability of synaptic agrin, in addition to transducing agrin signaling. Agrin is expressed in transmembrane and secreted isoforms (Burgess et al., 2000; Neumann et al., 2001). The secreted isoforms contain a signal peptide and an N-terminal fragment that binds to laminin (SS-NtA-agrin), whereas the transmembrane (TM)-agrin has a TM domain instead of the SS-Nt fragment (Burgess et al., 1999, 2000; Bezakova and Ruegg, 2003). TM-agrin is expressed in the brain and may be involved in dendrite development and synaptogenesis (Annies et al., 2006; Ramseger et al., 2009). SS-NtA-agrin is enriched at the NMJ (Denzer et al., 1997; Burgess et al., 1999, 2000). Agrin undergoes various post-translational modifications, including glycosylation and proteolytic cleavage (Ruegg et al., 1992; Gesemann et al., 1995, 1996; Burgess et al., 1999, 2000; Bezakova and Ruegg, 2003; Bolliger et al., 2010; Bogdanik and Burgess, 2011). How these processes are regulated was not well understood. Mutation of the glycosylation site in agrin reduces agrin secretion and causes CMS-like phenotypes in mutant mice (Bogdanik and Burgess, 2011). Loss of agrin from the SBL following LRP4 ablation would reduce local agrin signaling strength and consequently reduced AChR expression, membrane insertion, and anchoring (Herczeg et al., 1995; Jones et al., 1997; Bezakova et al., 2001; Moransard et al., 2003; Lin et al., 2008; Brenner and Akaaboune, 2014). In support of this hypothesis was the finding that overexpression of muscle agrin was able to rescue dystrophic phenotypes in lamininα2 mutant mice (Moll et al., 2001).
Second, LRP4 may regulate proteins that interact with agrin. For example, APP is enriched at the NMJ and can bind to both agrin and LRP4 (Akaaboune et al., 2000; Choi et al., 2013). APP–LRP4 interaction is enhanced by agrin and is thought to be involved in NMJ formation (Akaaboune et al., 2000; Choi et al., 2013). β1 integrin binds to various basal lamina components, including agrin, and muscle-specific β1 integrin mutant mouse displays NMJ defects similar to those in agrin mutants (Gautam et al., 1996; Martin and Sanes, 1997; Schwander et al., 2004). Of particular interest is agrin interaction with α-dystroglycan, a central component of the dystrophin–glycoprotein complex (including utrophin, dystrophin, dystrobrevin, and syntrophin) (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Côté et al., 1999). Together with dystrophin, dystroglycan levels are significantly reduced in the sarcolemma of duchenne muscular dystrophy patients and dystrophic mdx mice (Ervasti et al., 1990; Ohlendieck and Campbell, 1991; Ibraghimov-Beskrovnaya et al., 1992; Durbeej et al., 1998). This complex is thought to link AChRs to the SBL and the intracellular cytoskeleton (Banks et al., 2003; Shi et al., 2012). Mutations of α-dystrobrevin and α-syntrophin led to postjunctional destruction and AChR cluster fragmentation (Grady et al., 2003; Martinez-Pena y Valenzuela et al., 2011). In zebrafish, dystroglycan was shown to cooperate with MuSK in determining the pattern innervation of AChR clusters (Lefebvre et al., 2007). Whether LRP4 interacts with the dystrophin–glycoprotein complex remains unclear. When LRP4 is ablated in muscles, levels of α-dystroglycan were not changed in Dox-treated imKO mice. Therefore, myasthenic deficit here is caused by a dystroglycan-independent mechanism.
In addition to agrin signaling, reduced levels of LRP4 may impair Wnt signaling. In vitro studies showed that Wnts could stimulate AChR clusters in cultured muscle cells in the absence of agrin, although some Wnts may inhibit agrin-induced clustering (Barik, 2012; Strochlic et al., 2012; Zhang et al., 2012a; Barik et al., 2014). Wnt4 mutant mice showed NMJ deficits (Strochlic et al., 2012). Wnt signaling proteins, such as dishevelled (dvl) and β-catenin, interact with MuSK and rapsyn, respectively, and are thought to regulate AChR clustering (Luo et al., 2002; Zhang et al., 2007). APC, a protein critical for controlling β-catenin stability, interacts with AChR (Wang et al., 2003). Disruption of Dvl function alters AChR clustering in zebrafish and in mice (Henriquez et al., 2008; Jing et al., 2009; Jing et al., 2010; Gordon et al., 2012). Finally, muscle β-catenin, a critical effector of Wnt signaling in regulating transcription, may direct a retrograde signal for presynaptic differentiation (Li et al., 2008, Li et al., 2012; Wu et al., 2012a). These observations support the notion that Wnt signaling may contribute to NMJ formation. As described above, LRP4 could associate with Wnt signaling modulators (such as DKK and SOST) (Choi et al., 2009). It could also interact, at least indirectly, with Wnt (Zhang et al., 2012a). It would be interesting to know whether Wnt signaling is necessary for NMJ maintenance, and if so, what the underlying mechanism is. An answer to these questions would require future studies to identify LRP4 domains necessary for agrin, agrin-binding partners, Wnt, and Wnt signaling modulators. Interestingly, the mutations identified in patients with the Cenani–Lenz syndactyly syndrome are located in the center of the third β-propeller domain (Li et al., 2010), the region that interacts with MuSK. However, these mutations had no effect on agrin-MuSK signaling but rather were thought to alter Wnt signaling (Ohkawara et al., 2014).
Footnotes
This work was supported in part by the National Institutes of Health to W.-c.X. and L.M.
The authors declare no competing financial interests.
References
- Ahn Y, Sims C, Logue JM, Weatherbee SD, Krumlauf R. Lrp4 and Wise interplay controls the formation and patterning of mammary and other skin appendage placodes by modulating Wnt signaling. Development. 2013;140:583–593. doi: 10.1242/dev.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaaboune M, Allinquant B, Farza H, Roy K, Magoul R, Fiszman M, Festoff BW, Hantaï D. Developmental regulation of amyloid precursor protein at the neuromuscular junction in mouse skeletal muscle. Mol Cell Neurosci. 2000;15:355–367. doi: 10.1006/mcne.2000.0834. [DOI] [PubMed] [Google Scholar]
- Annies M, Bittcher G, Ramseger R, Löschinger J, Wöll S, Porten E, Abraham C, Rüegg MA, Kröger S. Clustering transmembrane-agrin induces filopodia-like processes on axons and dendrites. Mol Cell Neurosci. 2006;31:515–524. doi: 10.1016/j.mcn.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Banks GB, Fuhrer C, Adams ME, Froehner SC. The postsynaptic submembrane machinery at the neuromuscular junction: requirement for rapsyn and the utrophin/dystrophin-associated complex. J Neurocytol. 2003;32:709–726. doi: 10.1023/B:NEUR.0000020619.24681.2b. [DOI] [PubMed] [Google Scholar]
- Barik A, Xiong Wc, Mei L. Mukai H, editor. MuSK: a kinase critical for the formation and maintenance of the neuromuscular junction. Protein kinase technologies. 2012;vol. 68:203–217. [Google Scholar]
- Barik A, Zhang B, Sohal GS, Xiong WC, Mei L. Crosstalk between Agrin and Wnt signaling pathways in development of vertebrate neuromuscular junction. Dev Neurobiol. 2014;74:828–838. doi: 10.1002/dneu.22190. [DOI] [PubMed] [Google Scholar]
- Beeson D, Higuchi O, Palace J, Cossins J, Spearman H, Maxwell S, Newsom-Davis J, Burke G, Fawcett P, Motomura M, Müller JS, Lochmüller H, Slater C, Vincent A, Yamanashi Y. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- Ben Ammar A, Soltanzadeh P, Bauché S, Richard P, Goillot E, Herbst R, Gaudon K, Huzé C, Schaeffer L, Yamanashi Y, Higuchi O, Taly A, Koenig J, Leroy JP, Hentati F, Najmabadi H, Kahrizi K, Ilkhani M, Fardeau M, Eymard B, et al. A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS One. 2013;8:e53826. doi: 10.1371/journal.pone.0053826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- Bezakova G, Rabben I, Sefland I, Fumagalli G, Lømo T. Neural agrin controls acetylcholine receptor stability in skeletal muscle fibers. Proc Natl Acad Sci U S A. 2001;98:9924–9929. doi: 10.1073/pnas.171539698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Tinsley JM, Davies KE. The emerging family of dystrophin-related proteins. Trends Cell Biol. 1994;4:19–23. doi: 10.1016/0962-8924(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Bogdanik LP, Burgess RW. A valid mouse model of AGRIN-associated congenital myasthenic syndrome. Hum Mol Genet. 2011;20:4617–4633. doi: 10.1093/hmg/ddr396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolliger MF, Zurlinden A, Lüscher D, Bütikofer L, Shakhova O, Francolini M, Kozlov SV, Cinelli P, Stephan A, Kistler AD, Rülicke T, Pelczar P, Ledermann B, Fumagalli G, Gloor SM, Kunz B, Sonderegger P. Specific proteolytic cleavage of agrin regulates maturation of the neuromuscular junction. J Cell Sci. 2010;123:3944–3955. doi: 10.1242/jcs.072090. [DOI] [PubMed] [Google Scholar]
- Boudin E, Steenackers E, de Freitas F, Nielsen TL, Andersen M, Brixen K, Van Hul W, Piters E. A common LRP4 haplotype is associated with bone mineral density and hip geometry in men: data from the Odense Androgen Study (OAS) Bone. 2013;53:414–420. doi: 10.1016/j.bone.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: a heteromeric complex related to the dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Brenner HR, Herczeg A, Slater CR. Synapse-specific expression of acetylcholine receptor genes and their products at original synaptic sites in rat soleus muscle fibres regenerating in the absence of innervation. Development. 1992;116:41–53. doi: 10.1242/dev.116.1.41. [DOI] [PubMed] [Google Scholar]
- Brenner HR, Akaaboune M. Recycling of acetylcholine receptors at ectopic postsynaptic clusters induced by exogenous agrin in living rats. Dev Biol. 2014 doi: 10.1016/j.ydbio.2014.07.018. doi: 10.1016/j.ydbio.2014.07.018. Advance online publication. Retrieved Aug. 4, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RW, Nguyen QT, Son YJ, Lichtman JW, Sanes JR. Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron. 1999;23:33–44. doi: 10.1016/S0896-6273(00)80751-5. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Skarnes WC, Sanes JR. Agrin isoforms with distinct amino termini: differential expression, localization, and function. J Cell Biol. 2000;151:41–52. doi: 10.1083/jcb.151.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Chevessier F, Girard E, Molgö J, Bartling S, Koenig J, Hantaï D, Witzemann V. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet. 2008;17:3577–3595. doi: 10.1093/hmg/ddn251. [DOI] [PubMed] [Google Scholar]
- Choi HY, Dieckmann M, Herz J, Niemeier A. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS One. 2009;4:e7930. doi: 10.1371/journal.pone.0007930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HY, Liu Y, Tennert C, Sugiura Y, Karakatsani A, Kröger S, Johnson EB, Hammer RE, Lin W, Herz J. APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife. 2013;2:e00220. doi: 10.7554/eLife.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté PD, Moukhles H, Lindenbaum M, Carbonetto S. Chimaeric mice deficient in dystroglycans develop muscular dystrophy and have disrupted myoneural synapses. Nat Genet. 1999;23:338–342. doi: 10.1038/15519. [DOI] [PubMed] [Google Scholar]
- Culican SM, Nelson CC, Lichtman JW. Axon withdrawal during synapse elimination at the neuromuscular junction is accompanied by disassembly of the postsynaptic specialization and withdrawal of Schwann cell processes. J Neurosci. 1998;18:4953–4965. doi: 10.1523/JNEUROSCI.18-13-04953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/S0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol. 1997;137:671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Henry MD, Campbell KP. Dystroglycan in development and disease. Curr Opin Cell Biol. 1998;10:594–601. doi: 10.1016/S0955-0674(98)80034-3. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- Eusebio A, Oliveri F, Barzaghi P, Ruegg MA. Expression of mouse agrin in normal, denervated and dystrophic muscle. Neuromuscul Disord. 2003;13:408–415. doi: 10.1016/S0960-8966(03)00036-1. [DOI] [PubMed] [Google Scholar]
- Fallon JR, Hall ZW. Building synapses: agrin and dystroglycan stick together. Trends Neurosci. 1994;17:469–473. doi: 10.1016/0166-2236(94)90135-X. [DOI] [PubMed] [Google Scholar]
- Feng Z, Ko CP. The role of glial cells in the formation and maintenance of the neuromuscular junction. Ann N Y Acad Sci. 2008;1132:19–28. doi: 10.1196/annals.1405.016. [DOI] [PubMed] [Google Scholar]
- Fertuck HC, Salpeter MM. Quantitation of junctional and extrajunctional acetylcholine receptors by electron microscope autoradiography after 125I-alpha-bungarotoxin binding at mouse neuromuscular junctions. J Cell Biol. 1976;69:144–158. doi: 10.1083/jcb.69.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MB, Blagden CS, Burden SJ. Synaptic differentiation is defective in mice lacking acetylcholine receptor beta-subunit tyrosine phosphorylation. Development. 2007;134:4167–4176. doi: 10.1242/dev.010702. [DOI] [PubMed] [Google Scholar]
- García-Chacón LE, Nguyen KT, David G, Barrett EF. Extrusion of Ca2+ from mouse motor terminal mitochondria via a Na+-Ca2+ exchanger increases post-tetanic evoked release. J Physiol. 2006;574:663–675. doi: 10.1113/jphysiol.2006.110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/S0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Gesemann M, Denzer AJ, Ruegg MA. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesemann M, Cavalli V, Denzer AJ, Brancaccio A, Schumacher B, Ruegg MA. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–767. doi: 10.1016/S0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/S0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Gordon LR, Gribble KD, Syrett CM, Granato M. Initiation of synapse formation by Wnt-induced MuSK endocytosis. Development. 2012;139:1023–1033. doi: 10.1242/dev.071555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Akaaboune M, Cohen AL, Maimone MM, Lichtman JW, Sanes JR. Tyrosine-phosphorylated and nonphosphorylated isoforms of α-dystrobrevin: roles in skeletal muscle and its neuromuscular and myotendinous junctions. J Cell Biol. 2003;160:741–752. doi: 10.1083/jcb.200209045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro J, Higuchi O, Okada K, Ueno M, Iemura S, Natsume T, Spearman H, Beeson D, Yamanashi Y. Mutations causing DOK7 congenital myasthenia ablate functional motifs in Dok-7. J Biol Chem. 2008;283:5518–5524. doi: 10.1074/jbc.M708607200. [DOI] [PubMed] [Google Scholar]
- Henriquez JP, Webb A, Bence M, Bildsoe H, Sahores M, Hughes SM, Salinas PC. Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc Natl Acad Sci U S A. 2008;105:18812–18817. doi: 10.1073/pnas.0806300105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herczeg A, Jones G, Brenner HR. Involvement of extracellular matrix in acetylcholine receptor ϵ-subunit gene expression at the rat neuromuscular junction. Neurosci Lett. 1995;193:33–36. doi: 10.1016/0304-3940(95)11661-F. [DOI] [PubMed] [Google Scholar]
- Hesser BA, Henschel O, Witzemann V. Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol Cell Neurosci. 2006;31:470–480. doi: 10.1016/j.mcn.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365–368. doi: 10.1038/85520. [DOI] [PubMed] [Google Scholar]
- Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, Thijssen PE, Vrolijk H, Plomp JJ, Vogels P, Losen M, Van der Maarel SM, Burden SJ, Verschuuren JJ. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci U S A. 2013;110:20783–20788. doi: 10.1073/pnas.1313944110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Jing L, Lefebvre JL, Gordon LR, Granato M. Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron. 2009;61:721–733. doi: 10.1016/j.neuron.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Gordon LR, Shtibin E, Granato M. Temporal and spatial requirements of unplugged/MuSK function during zebrafish neuromuscular development. PLoS One. 2010;5:e8843. doi: 10.1371/journal.pone.0008843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo SA, Burden SJ. Synaptic basal lamina contains a signal for synapse-specific transcription. Development. 1992;115:673–680. doi: 10.1242/dev.115.3.673. [DOI] [PubMed] [Google Scholar]
- Johnson EB, Hammer RE, Herz J. Abnormal development of the apical ectodermal ridge and polysyndactyly in Megf7-deficient mice. Hum Mol Genet. 2005;14:3523–3538. doi: 10.1093/hmg/ddi381. [DOI] [PubMed] [Google Scholar]
- Jones G, Meier T, Lichtsteiner M, Witzemann V, Sakmann B, Brenner HR. Induction by agrin of ectopic and functional postsynaptic-like membrane in innervated muscle. Proc Natl Acad Sci U S A. 1997;94:2654–2659. doi: 10.1073/pnas.94.6.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraner S, Laufenberg I, Strassburg HM, Sieb JP, Steinlein OK. Congenital myasthenic syndrome with episodic apnea in patients homozygous for a CHAT missense mutation. Arch Neurol. 2003;60:761–763. doi: 10.1001/archneur.60.5.761. [DOI] [PubMed] [Google Scholar]
- Lefebvre JL, Jing L, Becaficco S, Franzini-Armstrong C, Granato M. Differential requirement for MuSK and dystroglycan in generating patterns of neuromuscular innervation. Proc Natl Acad Sci U S A. 2007;104:2483–2488. doi: 10.1073/pnas.0610822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XM, Dong XP, Luo SW, Zhang B, Lee DH, Ting AK, Neiswender H, Kim CH, Carpenter-Hyland E, Gao TM, Xiong WC, Mei L. Retrograde regulation of motoneuron differentiation by muscle β-catenin. Nat Neurosci. 2008;11:262–268. doi: 10.1038/nn2053. [DOI] [PubMed] [Google Scholar]
- Li Y, Pawlik B, Elcioglu N, Aglan M, Kayserili H, Yigit G, Percin F, Goodman F, Nürnberg G, Cenani A, Urquhart J, Chung BD, Ismail S, Amr K, Aslanger AD, Becker C, Netzer C, Scambler P, Eyaid W, Hamamy H, et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani–Lenz syndrome. Am J Hum Genet. 2010;86:696–706. doi: 10.1016/j.ajhg.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Maj M, Bezakova G, Magyar JP, Brenner HR, Ruegg MA. Muscle-wide secretion of a miniaturized form of neural agrin rescues focal neuromuscular innervation in agrin mutant mice. Proc Natl Acad Sci U S A. 2008;105:11406–11411. doi: 10.1073/pnas.0801683105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sugiura Y, Wu F, Mi W, Taketo MM, Cannon S, Carroll T, Lin W. β-Catenin stabilization in skeletal muscles, but not in motor neurons, leads to aberrant motor innervation of the muscle during neuromuscular development in mice. Dev Biol. 2012;366:255–267. doi: 10.1016/j.ydbio.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Zhang B, Dong XP, Tao Y, Ting A, Zhou Z, Meixiong J, Luo J, Chiu FC, Xiong WC, Mei L. HSP90 beta regulates rapsyn turnover and subsequent AChR cluster formation and maintenance. Neuron. 2008;60:97–110. doi: 10.1016/j.neuron.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZG, Wang Q, Zhou JZ, Wang J, Luo Z, Liu M, He X, Wynshaw-Boris A, Xiong WC, Lu B, Mei L. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron. 2002;35:489–505. doi: 10.1016/S0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- Marques MJ, Conchello JA, Lichtman JW. From plaque to pretzel: fold formation and acetylcholine receptor loss at the developing neuromuscular junction. J Neurosci. 2000;20:3663–3675. doi: 10.1523/JNEUROSCI.20-10-03663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Sanes JR. Integrins mediate adhesion to agrin and modulate agrin signaling. Development. 1997;124:3909–3917. doi: 10.1242/dev.124.19.3909. [DOI] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela I, Mouslim C, Pires-Oliveira M, Adams ME, Froehner SC, Akaaboune M. Nicotinic acetylcholine receptor stability at the NMJ deficient in α-syntrophin in vivo. J Neurosci. 2011;31:15586–15596. doi: 10.1523/JNEUROSCI.4038-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maselli RA, Arredondo J, Cagney O, Ng JJ, Anderson JA, Williams C, Gerke BJ, Soliven B, Wollmann RL. Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum Mol Genet. 2010;19:2370–2379. doi: 10.1093/hmg/ddq110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maselli RA, Fernandez JM, Arredondo J, Navarro C, Ngo M, Beeson D, Cagney O, Williams DC, Wollmann RL, Yarov-Yarovoy V, Ferns MJ. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. 2012;131:1123–1135. doi: 10.1007/s00439-011-1132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews-Bellinger JA, Salpeter MM. Fine structural distribution of acetylcholine receptors at developing mouse neuromuscular junctions. J Neurosci. 1983;3:644–657. doi: 10.1523/JNEUROSCI.03-03-00644.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahan UJ. The agrin hypothesis. Cold Spring Harbor Symp Quant Biol. 1990;55:407–418. doi: 10.1101/SQB.1990.055.01.041. [DOI] [PubMed] [Google Scholar]
- Meier T, Gesemann M, Cavalli V, Ruegg MA, Wallace BG. AChR phosphorylation and aggregation induced by an agrin fragment that lacks the binding domain for α-dystroglycan. EMBO J. 1996;15:2625–2631. [PMC free article] [PubMed] [Google Scholar]
- Meier T, Hauser DM, Chiquet M, Landmann L, Ruegg MA, Brenner HR. Neural agrin induces ectopic postsynaptic specializations in innervated muscle fibers. J Neurosci. 1997;17:6534–6544. doi: 10.1523/JNEUROSCI.17-17-06534.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll J, Barzaghi P, Lin S, Bezakova G, Lochmüller H, Engvall E, Müller U, Ruegg MA. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- Moransard M, Borges LS, Willmann R, Marangi PA, Brenner HR, Ferns MJ, Fuhrer C. Agrin regulates rapsyn interaction with surface acetylcholine receptors, and this underlies cytoskeletal anchoring and clustering. J Biol Chem. 2003;27:7350–7359. doi: 10.1074/jbc.M210865200. [DOI] [PubMed] [Google Scholar]
- Müller JS, Abicht A, Christen HJ, Stucka R, Schara U, Mortier W, Huebner A, Lochmüller H. A newly identified chromosomal microdeletion of the rapsyn gene causes a congenital myasthenic syndrome. Neuromuscul Disord. 2004;14:744–749. doi: 10.1016/j.nmd.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Müller JS, Baumeister SK, Rasic VM, Krause S, Todorovic S, Kugler K, Müller-Felber W, Abicht A, Lochmüller H. Impaired receptor clustering in congenital myasthenic syndrome with novel RAPSN mutations. Neurology. 2006;67:1159–1164. doi: 10.1212/01.wnl.0000233837.79459.40. [DOI] [PubMed] [Google Scholar]
- Neumann FR, Bittcher G, Annies M, Schumacher B, Kröger S, Ruegg MA. An alternative amino-terminus expressed in the central nervous system converts agrin to a type II transmembrane protein. Mol Cell Neurosci. 2001;17:208–225. doi: 10.1006/mcne.2000.0932. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Willnow TE. The low-density lipoprotein receptor gene family: a cellular Swiss army knife? Trends Cell Biol. 2002;12:273–280. doi: 10.1016/S0962-8924(02)02282-1. [DOI] [PubMed] [Google Scholar]
- Ohazama A, Porntaveetus T, Ota MS, Herz J, Sharpe PT. Lrp4: a novel modulator of extracellular signaling in craniofacial organogenesis. Am J Med Genet A. 2010;152:2974–2983. doi: 10.1002/ajmg.a.33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawara B, Cabrera-Serrano M, Nakata T, Milone M, Asai N, Ito K, Ito M, Masuda A, Ito Y, Engel AG, Ohno K. LRP4 third β-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet. 2014;23:1856–1868. doi: 10.1093/hmg/ddt578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton BL, Cunningham JM, Thyboll J, Kortesmaa J, Westerblad H, Edström L, Tryggvason K, Sanes JR. Properly formed but improperly localized synaptic specializations in the absence of laminin α4. Nat Neurosci. 2001;4:597–604. doi: 10.1038/88414. [DOI] [PubMed] [Google Scholar]
- Peng HB, Xie H, Rossi SG, Rotundo RL. Acetylcholinesterase clustering at the neuromuscular junction involves perlecan and dystroglycan. J Cell Biol. 1999;145:911–921. doi: 10.1083/jcb.145.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, Melms A, Kröger S. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol. 2012;259:427–435. doi: 10.1007/s00415-011-6194-7. [DOI] [PubMed] [Google Scholar]
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- Porter CW, Barnard EA. Ultrastructural studies on the acetylcholine receptor at motor end plates of normal and pathologic muscles. Ann N Y Acad Sci. 1976;274:85–107. doi: 10.1111/j.1749-6632.1976.tb47678.x. [DOI] [PubMed] [Google Scholar]
- Punga AR, Lin S, Oliveri F, Meinen S, Rüegg MA. Muscle-selective synaptic disassembly and reorganization in MuSK antibody positive MG mice. Exp Neurol. 2011;230:207–217. doi: 10.1016/j.expneurol.2011.04.018. [DOI] [PubMed] [Google Scholar]
- Ramseger R, White R, Kröger S. Transmembrane form agrin-induced process formation requires lipid rafts and the activation of Fyn and MAPK. J Biol Chem. 2009;284:7697–7705. doi: 10.1074/jbc.M806719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P, Monks DA. A tetracycline-inducible and skeletal muscle-specific Cre recombinase transgenic mouse. Dev Neurobiol. 2009;69:401–406. doi: 10.1002/dneu.20714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasi S, Spina V, Bruscaggin A, Vaisitti T, Tripodo C, Forconi F, De Paoli L, Fangazio M, Sozzi E, Cencini E, Laurenti L, Marasca R, Visco C, Xu-Monette ZY, Gattei V, Young KH, Malavasi F, Deaglio S, Gaidano G, Rossi D. A variant of the LRP4 gene affects the risk of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2011;152:284–294. doi: 10.1111/j.1365-2141.2010.08482.x. [DOI] [PubMed] [Google Scholar]
- Reif R, Sales S, Hettwer S, Dreier B, Gisler C, Wölfel J, Lüscher D, Zurlinden A, Stephan A, Ahmed S, Baici A, Ledermann B, Kunz B, Sonderegger P. Specific cleavage of agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J. 2007;21:3468–3478. doi: 10.1096/fj.07-8800com. [DOI] [PubMed] [Google Scholar]
- Rotundo RL, Ruiz CA, Marrero E, Kimbell LM, Rossi SG, Rosenberry T, Darr A, Tsoulfas P. Assembly and regulation of acetylcholinesterase at the vertebrate neuromuscular junction. Chem Biol Interact. 2008;175:26–29. doi: 10.1016/j.cbi.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg MA, Tsim KW, Horton SE, Kröger S, Escher G, Gensch EM, McMahan UJ. The agrin gene codes for a family of basal lamina proteins that differ in function and distribution. Neuron. 1992;8:691–699. doi: 10.1016/0896-6273(92)90090-Z. [DOI] [PubMed] [Google Scholar]
- Samuel MA, Valdez G, Tapia JC, Lichtman JW, Sanes JR. Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS One. 2012;7:e46663. doi: 10.1371/journal.pone.0046663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DB, El-Salem K, Massey JM, McConville J, Vincent A. Clinical aspects of MuSK antibody positive seronegative MG. Neurology. 2003;60:1978–1980. doi: 10.1212/01.WNL.0000065882.63904.53. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Schwander M, Shirasaki R, Pfaff SL, Müller U. Beta1 integrins in muscle, but not in motor neurons, are required for skeletal muscle innervation. J Neurosci. 2004;24:8181–8191. doi: 10.1523/JNEUROSCI.1345-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Lu Y, Zhang B, Figueiredo D, Bean J, Jung J, Wu H, Barik A, Yin DM, Xiong WC, Mei L. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest. 2013;123:5190–5202. doi: 10.1172/JCI66039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fu AK, Ip NY. Molecular mechanisms underlying maturation and maintenance of the vertebrate neuromuscular junction. Trends Neurosci. 2012;35:441–453. doi: 10.1016/j.tins.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Shigemoto K, Kubo S, Maruyama N, Hato N, Yamada H, Jie C, Kobayashi N, Mominoki K, Abe Y, Ueda N, Matsuda S. Induction of myasthenia by immunization against muscle-specific kinase. J Clin Invest. 2006;116:1016–1024. doi: 10.1172/JCI21545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto K, Kubo S, Jie C, Hato N, Abe Y, Ueda N, Kobayashi N, Kameda K, Mominoki K, Miyazawa A, Ishigami A, Matsude S, Maruyama N. Myasthenia gravis experimentally induced with muscle-specific kinase. Ann N Y Acad Sci. 2008;1132:93–98. doi: 10.1196/annals.1405.002. [DOI] [PubMed] [Google Scholar]
- Sohal GS. Sixth Annual Stuart Reiner Memorial Lecture: embryonic development of nerve and muscle. Muscle Nerve. 1995;18:2–14. doi: 10.1002/mus.880180103. [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron. 1995a;14:133–141. doi: 10.1016/0896-6273(95)90247-3. [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ. Schwann cell processes guide regeneration of peripheral axons. Neuron. 1995b;14:125–132. doi: 10.1016/0896-6273(95)90246-5. [DOI] [PubMed] [Google Scholar]
- Strochlic L, Falk J, Goillot E, Sigoillot S, Bourgeois F, Delers P, Rouvière J, Swain A, Castellani V, Schaeffer L, Legay C. Wnt4 participates in the formation of vertebrate neuromuscular junction. PLoS One. 2012;7:e29976. doi: 10.1371/journal.pone.0029976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama J, Bowen DC, Hall ZW. Dystroglycan binds nerve and muscle agrin. Neuron. 1994;13:103–115. doi: 10.1016/0896-6273(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407:530–535. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakui K, Gregato G, Ballif BC, Glotzbach CD, Bailey KA, Kuo PL, Sue WC, Sheffield LJ, Irons M, Gomez EG, Hecht JT, Potocki L, Shaffer LG. Construction of a natural panel of 11p11.2 deletions and further delineation of the critical region involved in Potocki-Shaffer syndrome. Eur J Hum Genet. 2005;13:528–540. doi: 10.1038/sj.ejhg.5201366. [DOI] [PubMed] [Google Scholar]
- Wang J, Jing Z, Zhang L, Zhou G, Braun J, Yao Y, Wang ZZ. Regulation of acetylcholine receptor clustering by the tumor suppressor APC. Nat Neurosci. 2003;6:1017–1018. doi: 10.1038/nn1128. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu Y, Barik A, Joseph A, Taketo MM, Xiong WC, Mei L. β-Catenin gain of function in muscles impairs neuromuscular junction formation. Development. 2012a;139:2392–2404. doi: 10.1242/dev.080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu Y, Shen C, Patel N, Gan L, Xiong WC, Mei L. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. 2012b;75:94–107. doi: 10.1016/j.neuron.2012.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumoto N, Kim N, Burden SJ. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. 2012;489:438–442. doi: 10.1038/nature11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Luo S, Dong XP, Zhang X, Liu C, Luo Z, Xiong WC, Mei L. Beta-catenin regulates acetylcholine receptor clustering in muscle cells through interaction with rapsyn. J Neurosci. 2007;27:3968–3973. doi: 10.1523/JNEUROSCI.4691-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Liang C, Bates R, Yin Y, Xiong WC, Mei L. Wnt proteins regulate acetylcholine receptor clustering in muscle cells. Mol Brain. 2012a;5:7. doi: 10.1186/1756-6606-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Tzartos JS, Belimezi M, Ragheb S, Bealmear B, Lewis RA, Xiong WC, Lisak RP, Tzartos SJ, Mei L. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol. 2012b;69:445–451. doi: 10.1001/archneurol.2011.2393. [DOI] [PubMed] [Google Scholar]
- Zhang B, Shen C, Bealmear B, Ragheb S, Xiong WC, Lewis RA, Lisak RP, Mei L. Autoantibodies to agrin in myasthenia gravis patients. PLoS One. 2014;9:e91816. doi: 10.1371/journal.pone.0091816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Coldefy AS, Hubbard SR, Burden SJ. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK) J Biol Chem. 2011;286:40624–40630. doi: 10.1074/jbc.M111.279307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Yang Z, Luo Z, Luo S, Xiong WC, Mei L. Muscle-specific receptor tyrosine kinase endocytosis in acetylcholine receptor clustering in response to agrin. J Neurosci. 2008;28:1688–1696. doi: 10.1523/JNEUROSCI.4130-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisimopoulou P, Evangelakou P, Tzartos J, Lazaridis K, Zouvelou V, Mantegazza R, Antozzi C, Andreetta F, Evoli A, Deymeer F, Saruhan-Direskeneli G, Durmus H, Brenner T, Vaknin A, Berrih-Aknin S, Frenkian Cuvelier M, Stojkovic T, DeBaets M, Losen M, Martinez-Martinez P, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun. 2014;52:139–145. doi: 10.1016/j.jaut.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Zong Y, Zhang B, Gu S, Lee K, Zhou J, Yao G, Figueiredo D, Perry K, Mei L, Jin R. Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 2012;26:247–258. doi: 10.1101/gad.180885.111. [DOI] [PMC free article] [PubMed] [Google Scholar]