

Abstract
Alzheimer's disease is characterized by the accumulation and deposition of plaques of β-amyloid (Aβ) peptide in the brain. Given its pivotal role, new therapies targeting Aβ are in demand. We rationally designed liposomes targeting the brain and promoting the disaggregation of Aβ assemblies and evaluated their efficiency in reducing the Aβ burden in Alzheimer's disease mouse models. Liposomes were bifunctionalized with a peptide derived from the apolipoprotein-E receptor-binding domain for blood–brain barrier targeting and with phosphatidic acid for Aβ binding. Bifunctionalized liposomes display the unique ability to hinder the formation of, and disaggregate, Aβ assemblies in vitro (EM experiments). Administration of bifunctionalized liposomes to APP/presenilin 1 transgenic mice (aged 10 months) for 3 weeks (three injections per week) decreased total brain-insoluble Aβ1–42 (−33%), assessed by ELISA, and the number and total area of plaques (−34%) detected histologically. Also, brain Aβ oligomers were reduced (−70.5%), as assessed by SDS-PAGE. Plaque reduction was confirmed in APP23 transgenic mice (aged 15 months) either histologically or by PET imaging with [11C]Pittsburgh compound B (PIB). The reduction of brain Aβ was associated with its increase in liver (+18%) and spleen (+20%). Notably, the novel-object recognition test showed that the treatment ameliorated mouse impaired memory. Finally, liposomes reached the brain in an intact form, as determined by confocal microscopy experiments with fluorescently labeled liposomes. These data suggest that bifunctionalized liposomes destabilize brain Aβ aggregates and promote peptide removal across the blood–brain barrier and its peripheral clearance. This all-in-one multitask therapeutic device can be considered as a candidate for the treatment of Alzheimer's disease.
Keywords: Abeta, Alzheimer, cognitive impairment, liposomes, nanomedicine, oligomers
Introduction
Alzheimer's disease (AD), the most common form of dementia afflicting ∼36 million people worldwide, is a neurodegenerative disease characterized by synaptic dysfunction, memory loss, and neuronal cell death (Selkoe et al., 2012). It is mainly diffused in its sporadic form, although in a minor population, it is of the familial type, which is caused by mutations in the amyloid precursor protein (APP) or presenilin 1 (PS1) or 2 (PS2) genes (Bertram and Tanzi, 2012). AD brains are characterized by extracellular plaques, mainly composed of β-amyloid (Aβ), a 40–42 aa (Aβ1–40; Aβ1–42) proteolytic fragment of the membrane-associated APP (Verbeek et al., 1997). Aβ undergoes an aggregation process leading to the formation of small, soluble oligomeric species and large, insoluble fibrillar species (Bruggink et al., 2012) and ending with the deposition of plaques. Elevated levels of Aβ and its neurotoxic aggregates, oligomers in particular, in the brain are believed to be associated with perturbations of synaptic function and neural network activity, leading to cognitive deficits and neurodegeneration (Palop and Mucke, 2010).
Based on this knowledge, different Aβ-directed therapeutic strategies attempting to reduce brain Aβ burden are currently under investigation, including possibly drawing the Aβ excess out of the brain by peripheral administration of Aβ-binding agents: the so-called “sink effect” (Matsuoka et al., 2003; Biscaro et al., 2009; Sutcliffe et al., 2011). Nanotechnological devices, and in particular nanoparticles, have been suggested as potential tools for the therapy of CNS diseases (Re et al., 2012). Liposomes (LIPs), the best known nanoparticles (Chang and Yeh, 2012), are currently used in the clinic as drug vehicles. However, the possibility of multifunctionalization may confer on them the ability to perform multiple tasks at the same time. Following this view, within the present investigation, we have designed multifunctional LIPs for AD therapy.
The objectives for their construction were to confer on them the abilities to (1) cross the blood–brain barrier (BBB), (2) hinder the formation of and enhance the disruption of brain Aβ aggregates into smaller soluble assemblies, and (3) enhance their clearance from the brain. To reach this goal, relying on previous observations obtained in vitro, we have bifunctionalized LIPs composed of sphingomyelin (Sm) and cholesterol (Chol) with phosphatidic acid (PA) with the task of binding Aβ (Gobbi et al., 2010) and with a peptide (mApoE) derived from the receptor-binding domain of apolipoprotein E, with the task of targeting and crossing the BBB (Re et al., 2010, 2011; Bana et al., 2013). The present study reports the therapeutic effectiveness of bifunctionalized LIPs (mApoE–PA–LIP) in transgenic (Tg) AD mouse models, demonstrating their effects on both the reduction of amyloid burden and memory improvement.
Materials and Methods
Preparation and characterization of LIPs.
Bifunctionalized LIPs (mApoE–PA–LIP) were prepared as described previously (Re et al., 2010; Bana et al., 2013) by an extrusion procedure using polycarbonate filters (100 nm pore size diameter) and were composed of a matrix of bovine brain Sm and Chol at 1:1 molar ratio, mixed with 5% molar of dimyristoyl-PA and further surface functionalized with 1.25% molar of mApoE peptide. mApoE peptide, carrying the amino acid sequence CWG-LRKLRKRLLR corresponding to residues 141–150 of human ApoE, modified with the addition of a tryptophan, glycine, and cysteine residue at the C-terminal, was synthesized and purified as described previously (Re et al., 2010, 2011). As controls, monofunctionalized LIPs with PA (PA–LIP) or mApoE (mApoE–LIP) were also prepared as described previously (Gobbi et al., 2010; Re et al., 2011).
For pharmacokinetic and biodistribution experiments, LIPs contained 6 × 105 dpm of either [14C]PA or [3H]Sm (∼0.001–0.002 molar percentage of total lipids) added as tracers to follow lipid distribution by radioactivity counting.
For confocal microscopy experiments, fluorescent LIPs were used carrying BODIPY-FL C12-sphingomyelin in the lipid bilayer and Rhodamine B encapsulated in the aqueous core. BODIPY-FL C12-sphingomyelin (Invitrogen) was added to the lipid mixture during the preparation of LIPs, and the lipid film was rehydrated with a solution of 20 mm Rhodamine B (Sigma-Aldrich) and submitted to six cycles of freezing and thawing before being extruded. To remove any un-encapsulated material, LIPs were subjected to three cycles of diafiltration through 30,000 molecular weight (MW) cutoff membranes.
LIP size and ζ-potential were characterized as described previously (Re et al., 2010; Bana et al., 2013) and were stable for at least 5 d, as reported (Bana et al., 2013). However, LIPs for animal treatment were freshly prepared on the same day of each injection.
Electron microscopy.
Electron microscopy (EM) was used to characterize bifunctionalized LIPs and to investigate their ability to either hinder the formation of fibrils or disrupt preformed Aβ1–42 aggregates. To verify the shape and size, 400 μm mApoE–PA–LIP in PBS were dropped onto nickel Formvar–carbon-coated 300 mesh EM grids (Electron Microscopy Science) for 3 min, successively stained for 5 min with a saturated solution of uranyl acetate, washed to eliminate excess uranyl acetate, and allowed to air dry. To study the ability of LIPs to hinder the formation of or to disaggregate preformed fibrils of Aβ1–42, the following procedure was used: in the former case, a solution of 25 μm Aβ1–42 was incubated alone or in the presence of a 1 mm solution of LIPs for 5 d at 37°C in PBS, and, in the latter case, after peptide aggregation, fibril suspension was incubated for 5 d with 1 mm LIP solution. At the end of the incubation in either case, all samples were diluted to a final concentration of 5 μm Aβ1–42 and dropped onto nickel Formvar–carbon grids. EM analyses was done with a Libra 120 transmission electron microscope operating at 120 kV equipped with a Proscan Slow Scan CCD camera (Carl Zeiss SMT).
Pharmacokinetic and biodistribution experiments.
Six- to 8-week-old BALB/c mice weighting 22–25 g were used for these studies. One hundred microliters of 40 mm (total lipid concentration) PA–LIP or mApoE–PA–LIP, containing 130 μg of PA and 6 × 105 dpm of [14C]PA and [3H]Sm (in 1:1 ratio), were administered by three intraperitoneal injections (48 h apart). Mice were killed 24 h after the last injection (three mice per experimental group). Blood, liver, spleen, kidneys, lungs, and brain were collected and solubilized by digestion as described previously (Wan et al., 2007). Radioactivity was measured by a Packard Tricarb 2200CA liquid scintillation counter (PerkinElmer Life and Analytical Sciences).
Animals.
Forty APPswe/PS1Δe9 (APP/PS1) 10-month-old Tg male mice [B6C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax mice; The Jackson Laboratory], mean weight of 33–34 g, and 20 non-Tg (WT) age-matched littermates were used. For some confirmatory positron emission tomography (PET) experiments, three APPswe single Tg (APP23) mice, 15 months old (Novartis Pharma), and three non-Tg (WT) C57/6N mice, 18 months old, were used. All animals were specific pathogen free (SPF) and were housed in an SPF facility in groups of four in standard mouse cages containing sawdust with food (2018S Harlan diet) and water ad libitum, under conventional laboratory conditions (room temperature, 20 ± 2°C; humidity, 60%) and a 12 h light/dark cycle (7:00 A.M. to 7:00 P.M.). No environmental enrichment was used because it notably improves AD pathology in mouse models of AD (Lazarov et al., 2005; Valero et al., 2011). Mice were all drug and behavioral test naive, and the experiments were all conducted during the light cycle. All procedures involving animals and their care were conducted according to European Union (EEC Council Directive 86/609, OJ L 358,1; December 12, 1987) and Italian (Decreto legislativo 116, Gazzetta Ufficiale s40, February 18, 1992) laws and policies and in accordance with the United States Department of Agriculture Animal Welfare Act and the National Institutes of Health policy on Human Care and Use of Laboratory Animals. They were reviewed and approved by the Mario Negri Institute Animal Care and Use Committee, which includes ad hoc members for ethical issues (1/04-D).
Animal treatment.
All animals (Tg or WT) were intraperitoneally injected with mApoE–PA–LIP (100 μl, 73.5 mg of total lipids/kg) or with PBS as a vehicle (100 μl) once every other day for 3 weeks. The weight of the animals was recorded before each treatment. Two experimental groups were treated with mApoE–PA–LIP (APP/PS1 and WT mice, n = 10 for each), two control groups were treated with PBS (APP/PS1 and WT, n = 19 for each), and two more Tg groups received monofunctionalized PA–LIP or mApoE–LIP (n = 10 for each). To minimize the effect of subjective bias, animals were allocated to treatment by an operator not involved in the study, and animal groups were named with numbers. Drug treatments were performed in a blind manner by naming them with alphabetic letters. Mice were treated always at the same time of the day (9:00–10:00 A.M.) in a specific room inside the animal facility, following a randomized order. Each single mouse was our experimental unit.
Blood and tissue collection.
Animals were deeply anesthetized with an overdose of ketamine/medetomidine (1.5 and 1.0 mg/kg, respectively), and the blood was collected from the heart for plasma separation. Afterward, liver, spleen, and brain were dissected and weighed. One brain hemisphere was fixed and processed for immunohistochemistry; the other hemisphere, liver, spleen, and plasma were snap frozen in dry ice and stored at −80°C (Cramer et al., 2012) until Aβ dosage by ELISA.
Brain immunohistochemistry.
APP/PS1 plaque deposition was examined using the 6E10 monoclonal anti-Aβ antibody (Covance), microglia with anti-ionized calcium binding adaptor molecule 1 (Iba1; DBA), and astrocytes with anti-glial fibrillary acidic protein (GFAP; Millipore) antibodies. Brain coronal cryostat sections (30 μm; three slices per mouse) were incubated for 1 h at room temperature with blocking solutions [6E10: 10% normal goat serum (NGS); Iba1: 0.3% Triton X-100 plus 10% NGS; GFAP: 0.4% Triton X-100 plus 3% NGS] and then overnight at 4°C with the primary antibodies (6E10, 1:500; Iba1, 1:1000; GFAP, 1:3500). After incubation with the anti-mouse biotinylated secondary antibody (1:200; 1 h at room temperature; Vector Laboratories) immunostaining was developed using the avidin–biotin kit (Vector Laboratories) and diaminobenzidine (Sigma). Tissue analysis and image acquisition were done using an Olympus image analyzer and the Cell-R software. Plaques were quantified by an operator blind to genotype and treatment using Fiji software, through the application of a homemade macro. Plaque deposition was also examined on APP23 mice using either the 6E10 monoclonal anti-Aβ antibody as described above or Thioflavin-S as described previously (Snellman et al., 2013).
Aβ plaque imaging by PET in APP23 mice.
APP23 mice were used for PET experiments because it has been shown that the probe does not sufficiently bind to the plaques in the APP/PS1 mouse brain (Snellman et al., 2013). [11C]Pittsburgh compound B (PIB) was synthesized as published previously (Snellman et al., 2013). Mean specific radioactivity of the batches was 536 ± 112 GBq/μmol at the end of synthesis. [11C]PIB (injected dose, 10.4 ± 0.7 MBq) was administered via the tail vain. PET/computed tomography (CT) scans were performed with Inveon Multimodality PET/CT device (Siemens), and dynamic 60 min scans (timeframes, 30 × 10, 15 × 60, 4 × 300, and 2 × 600 s) in 3-D list mode were initiated simultaneously with the injection. Images were reconstructed with a 2-D filtered backprojection algorithm. Animals were first imaged during the week before treatment (scan 1 at 15 and 18 months of age), then during the week after the treatment (scan 2 at 16 and 19 months of age), and finally 3 months after the treatment (scan 3 at 19 and 22 months of age). From the dynamic PET images, time–radioactivity curves for brain, frontal cortex, and cerebellum were obtained from regions of interest manually drawn to the CT image and projected to the PET image. Bound-to-free ratios (B/F40–60) for the frontal cortex were determined from the late phase (40–60 min) of the time–radioactivity curves as described recently using cerebellum as a reference region (Snellman et al., 2013).
Novel-object recognition test.
The novel-object recognition (NOR) test is a memory test that relies on spontaneous animal behavior without the need of stressful elements, such as food or water deprivation or electric footshock (Antunes and Biala, 2012). In the NOR test, mice are introduced into an arena containing two identical objects that they can explore freely. Twenty-four hours later, mice are reintroduced into the arena containing two different objects, one of which was presented previously (familiar) and a new completely different one (novel). At the end of treatment, mice were tested in an open-square gray arena (40 × 40 cm), 30 cm high, with the floor divided into 25 squares by black lines, placed in a specific room dedicated to behavioral analysis and separated from the operator's room. The following objects were used: a black plastic cylinder (4 × 5 cm), a glass vial with a white cup (3 × 6 cm), and a metal cube (3 × 5 cm). The task started with a habituation trial during which the animals were placed in the empty arena for 5 min, and their movements were recorded as the number of line crossings, which provide an indication of both WT and Tg mice motor activity. Mice were tested following a predefined scheme (five mice for each treatment group and the remaining mice by following the same scheme) so to precisely maintain the 24 h of retest for each mouse. The next day, mice were again placed in the same arena containing two identical objects (familiarization phase). Exploration was recorded in a 10 min trial by an investigator blinded to the genotype and treatment. Sniffing, touching, and stretching the head toward the object at a distance of no more than 2 cm were scored as object investigation. Twenty-four hours later (test phase), mice were again placed in the arena containing two objects, one of the objects presented during the familiarization phase (familiar object) and a new different one (novel object), and the time spent exploring the two objects was recorded for 10 min. Results were expressed as percentage time of investigation on objects per 10 min or as discrimination index (DI), i.e., (seconds spent on novel − seconds spent on familiar)/(total time spent on objects). Animals with no memory impairment spent a longer time investigating the novel object, giving a higher DI.
Aβ quantification in animal organs.
Mouse brains were treated as described previously (Steinerman et al., 2008) with some modifications. Mouse brain hemispheres were homogenized in a Tris buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 50 mm EDTA, 1% Triton X-100, and 2% protease inhibitor. After centrifugation (15,000 rpm, 21,000 × g, 4°C for 25 min), the supernatant was retained as the Triton-soluble fraction (soluble Aβ). The pellet was homogenized a second time in the presence of 70% formic acid (FA) (10% v/w) and ultracentrifuged (55,000 rpm, 100,000 × g, 4°C, 1 h), and the resulting FA-extracted supernatant was neutralized with 1 m Tris buffer, pH 11, representing the FA-extracted insoluble fraction. Levels of Aβ1–40 and Aβ1–42 in each fraction were quantified by sandwich ELISA (ELISA kit; IBL).
Liver and spleen were homogenized, and their Aβ levels were quantified as for the brain. Levels of plasma Aβ1–40 and Aβ1–42 were quantified by ELISA (Wako Chemicals). Each sample was assayed in triplicate.
Brain Aβ oligomer analysis.
Aliquots of the Triton-soluble fractions (soluble Aβ), containing 45 μg of total protein, were run on a precast NuPAGE 4–12% bis-Tris gel (Invitrogen), transferred to a nitrocellulose membrane, probed with the 6E10 anti-Aβ antibody (1:1000 dilution), and visualized with enhanced chemiluminescence (ECL) by ImageQuant LAS4000. The protein load was controlled either by Ponceau S staining or β-actin immunoblotting using rabbit anti-β-actin antibody (1:1500 dilution; Invitrogen). The content of soluble Aβ assemblies was quantified by the intensity of the chemiluminescent bands using NIH ImageJ Software and normalized with respect to the β-actin content of the same sample.
Confocal microscopy.
To investigate whether mApoE–PA–LIP entered the brain in an intact form, APP/PS1 mice (100 μl, 73.5 mg of total lipids/kg) were intraperitoneally injected with fluorescently labeled mApoE–PA–LIP or with PBS as vehicle (100 μl), once a day for 3 consecutive days. Three hours after the last injection, animals were killed, and the brains were fixed in 4% paraformaldehyde for 24 h, transferred to 30% sucrose until the tissue sank, and frozen at −80°C. Brain coronal cryostat sections (30 μm) were washed three times in PBS, and the nuclei were stained with DAPI (1:500 in PBS) for 10 min and then washed again four times in PBS. The sections were finally mounted with Fluorsave (Calbiochem) and viewed under a laser-scan confocal microscope (Zeiss LSM 710) using a 20× objective in fluorescence and bright-field combined acquisition mode.
Images were acquired focusing on the hippocampus using a 63× oil-immersion objective, generating a 3-D reconstruction using an appropriate optical sectioning (Z-stack). The images shown have been obtained as the sum of several focal planes with a thickness of 0.63 μm each, giving information related to the entire volume of the analyzed sections. This procedure ensures a “full-volume” data collection. Acquisition parameters were set to select the specific wavelength of the fluorescent LIPs injected, reducing any possible interference from autofluorescence, and were maintained constant for all the experiments. In detail, DAPI signals were recorded in the 406–475 nm emission range using a 405 nm laser wavelength, and mApoE–PA–LIP signals were detected in the 493–542 nm emission range using a 488 nm laser wavelength for BODIPY-FL and in the 562–626 nm emission range using a 561 nm laser wavelength for Rhodamine B.
Statistical analysis.
Data were expressed as mean ± SEM. For ELISA assay, Western blot and plaque quantification data were analyzed by Student's t test. For the NOR test, data were analyzed by a two-way ANOVA. In the presence of a significant interaction between the factors Tg × treatment, the Tukey's post hoc test was applied. p < 0.05 was considered significant.
Results
The physicochemical features of mApoE–PA–LIP are reported in Table 1. An EM image of the LIP preparation is shown in Figure 1A. EM experiments confirmed their ability to inhibit, in vitro, the formation of amyloid aggregates and to disrupt preformed fibrils, as reported previously using other techniques (Bana et al., 2013). In fact, as shown in Figure 1B–D, the meshwork of the amyloid fibrils formed by Aβ1–42 was significantly reduced when the peptide aggregation was performed in the presence, or after incubation, of mApoE–PA–LIP with preformed fibrils.
Table 1.
Physicochemical features of LIPs used in the present investigation
| Liposome | Size (nm) | PDI | ζ potential (mV) |
|---|---|---|---|
| PA–LIP | 109 ± 8 | 0.10 | −23.6 ± 4 |
| mApoE–LIP | 119 ± 7 | 0.12 | −15.3 ± 3 |
| mApoE–PA–LIP | 121 ± 7 | 0.15 | −18.7 ± 4 |
Size, polydispersity index (PDI), and ζ-potential of LIPs were measured by the dynamic light scattering technique and interferometric Doppler velocimetry.
Figure 1.

EM characterization of mApoE–PA–LIP and their ability to hinder the formation and disaggregate Aβ assemblies in vitro. A, Electron micrograph of mApoE–PA–LIP used in the present investigation. B, Fibrillary assemblies of 25 μm Aβ1–42 formed after 5 d incubation at 37°C. C, Fibrillary assemblies of 25 μm Aβ1–42 formed after 5 d incubation with 1 mm mApoE–PA–LIP at 37°C. D, Fibrillary assemblies of Aβ1–42 obtained as in B and then incubated with 1 mm mApoE–PA–LIP.
Pharmacokinetic experiments, reported in Figure 2, were performed by intraperitoneal administration of dually radiolabeled ([14C]PA and [3H]Sm) PA–LIP or mApoE–PA–LIP in BALB/c mice to assess the radioactivity distribution in blood, liver, spleen, kidneys, lungs, and brain. The results show that the amount of radioactivity reaching the brain in vivo is higher for mApoE–PA–LIP than for monofunctionalized PA–LIP. Furthermore, the data show that the ratio between 14C and 3H detected in the brain is comparable with the ratio between the two isotopes (∼1:1) of the mApoE–PA–LIP injected.
Figure 2.

The additional functionalization with mApoE of radiolabeled PA–LIP administered to healthy mice increases the amount of brain-associated radioactivity. Dually radiolabeled ([3H]Sm and [14C]PA) mApoE–PA–LIP or PA–LIP (100 μl, 73.5 mg of total lipids/kg) were administered intraperitoneally to BALB/c mice (n = 3; 6–8 weeks old), three injections (1 injection every 48 h). Mice were killed 3 h after the injections, blood, liver, spleen, kidneys, lungs, and brain were collected, and radioactivity was measured. A, Amount of [14C]PA (expressed as percentage of injected dose) in mouse brain after one, two, or three injections of PA–LIP or mApoE–PA–LIP. B, Amount of [3H]Sm (expressed as percentage of injected dose) in mouse brain after one, two, or three injections of PA–LIP or mApoE–PA–LIP. C, [14C]PA biodistribution (expressed as radioactivity percentage of injected dose) in blood, liver, spleen, kidney, lung, and brain. *p < 0.05 by Student's t test.
APP/PS1 Tg mice were treated with either bifunctionalized mApoE–PA–LIP or monofunctionalized LIP (PA–LIP or mApoE–LIP) or with PBS, as vehicle, for 3 weeks and submitted to an NOR test. Figure 3 shows that, although PBS-treated APP/PS1 mice were unable to discriminate between the familiar and the novel object (percentage time of investigation per 10 min: familiar, 47.2 ± 2.2; novel, 52.8 ± 2.2; DI, 0.02 ± 0.04; n = 19), after treatment, only mice receiving mApoE–PA–LIP significantly recovered their long-term recognition memory (percentage time of investigation per 10 min: familiar, 36.2 ± 5.1; novel, 63.8 ± 5.1; DI, 0.28 ± 0.1; n = 10), close to the values of PBS-treated WT mice (percentage time of investigation per 10 min: familiar, 37.0 ± 2.4; novel, 63.0 ± 2.4; DI, 0.28 ± 0.04; n = 19). One-way ANOVA for the DI found a significant effect of treatment (F(5,72) = 3.5, p = 0.006). Interestingly, in contrast to bifunctionalized LIPs, only a slight, albeit nonstatistically, significant memory improvement was observed after treatment with monofunctionalized LIP (percentage time of investigation per 10 min: PA–LIP, familiar, 41.3 ± 4.2; novel, 59.0 ± 4.2; DI, 0.18 ± 0.08; n = 10; mApoE-LIP, familiar, 43.7 ± 2.8; novel, 56.3 ± 2.8; DI, 0.13 ± 0.06; n = 10).
Figure 3.

Treatment with bifunctionalized mApoE–PA–LIP significantly restores long-term recognition memory in APP/PS1 Tg mice. APP/PS1 Tg or WT mice were treated with mApoE–PA–LIP, PA–LIP, mApoE–LIP, or vehicle, and, at the end of treatment, their memory was tested with the NOR test. A, Histograms indicate the time percentage (mean ± SEM) of investigation of the familiar and novel objects of the experimental groups tested. B, Histograms are mean ± SEM of the corresponding DI. One-way ANOVA found a significant effect of treatment (F(5,72) = 3.5, p = 0.006). *p < 0.05, **p < 0.01 by Tukey's post hoc test.
In addition, we demonstrated that mApoE–PA–LIP treatment had no negative effect on the memory of WT mice (percentage time of investigation per 10 min: familiar, 37.3 ± 3.4; novel, 62.7 ± 3.4; DI, 0.25 ± 0.07; n = 10) and did not affect mouse weight and motor activity (data not shown).
At the end of behavioral investigation by the NOR test, APP/PS1 mice were killed, and half of the brain was postfixed and subsequently immunostained for plaque quantification. As expected at the age investigated (Balducci and Forloni, 2011), APP/PS1 mice treated with PBS displayed important deposits of plaques that were significantly reduced in APP/PS1 mice treated with mApoE–PA–LIP. As shown in Figure 4, we found that mApoE–PA–LIP reduced the number and total area of brain Aβ plaques by ∼34% in both the cortex and the hippocampus. Student's t test for the two treatment groups, mApoE–PA–LIP (n = 10) versus PBS (n = 10), found a significant reduction in the number of plaques (t(17) = −3.0, p = 0.008) and in the total plaque area (t(17) = −2.5, p = 0.02). The treatment of APP/PS1 Tg mice with PA–LIP or mApoE–LIP, which did not significantly recover from memory impairment, did not reduce brain plaques either (data not shown).
Figure 4.

mApoE–PA–LIP treatment significantly reduced Aβ plaque load in the brain of APP/PS1 Tg mice. APP/PS1 Tg mice were treated with mApoE–PA–LIP or vehicle, and, at the end of treatment, the brain Aβ burden was analyzed by immunohistochemistry. A, Representative cortical and hippocampal brain sections of mice treated with PBS. B, Representative cortical and hippocampal brain sections of mice treated with mApoE–PA–LIP. Brain sections were stained with the anti-Aβ 6E10 monoclonal antibody. C, Histograms report the percentage reduction (mean ± SEM) of the total number of plaques in mApoE–PA–LIP-treated mice. D, Histograms report the percentage reduction (mean ± SEM) of the total plaque area in mApoE–PA–LIP-treated mice. *p < 0.05, **p < 0.01 by Student's t test. Scale bar, 250 μm.
The plaque reduction induced by mApoE–PA–LIP treatment on APP/PS1 mice was also confirmed on APP23 mice by either Thioflavin-S or 6E10 anti-Aβ staining on brain sections (Fig. 5). The effect of decreasing the plaque load was also followed on APP23 mice by PET using [11C]PIB (Snellman et al., 2013) as the plaque-detecting probe (Fig. 5). Interestingly, PET imaging, performed 3 months after the completion of the treatment, suggested a scarce tendency to plaque reconstitution, because APP23 mice showed low B/F40–60 ratios (0.10; 0.13; −0.04) similar to WT mice (−0.09; −0.01).
Figure 5.

mApoE–PA–LIP treatment reduced Aβ plaque load, detected by [11C]PIB PET, in the brain of APP23 Tg mice. APP23 Tg or WT mice were treated with mApoE–PA–LIP, and, at the end of treatment, animals were imaged repeatedly with 60 min dynamic [11C]PIB PET scans. Three months after completion of the treatment, animals were imaged and subsequently killed. Brain Aβ deposition was evaluated on cortical cryosections. The figure displays summed (40–60 min after injection) PET images (PET) or cortical sections stained with Thioflavin-S (ThS) or 6E10 anti-Aβ antibody (6E10) of Tg and WT mice 3 months after the completion of the treatment with mApoE–PA–LIP (n = 3 for each group). Additional images of untreated Tg mouse present the expected Aβ deposition in this model at 18 months of age. Scale bar, 200 μm.
The effect of mApoE–PA–LIP-mediated plaque reduction in APP/PS1 mice was paralleled by a decrease in the total amount of brain Aβ levels, assayed by ELISA (Fig. 6). After treatment, the amount of insoluble and soluble brain Aβ1–40 (1249.7 ± 259.8 and 50.2 ± 12.1 pmol/g brain) was 33 and 32%, respectively, which is lower than in the brain of PBS-treated mice (p = 0.0000053, p = 0.00022 by Student's t test). The amount of insoluble and soluble brain Aβ1–42 (1001.7 ± 221.0 and 60.2 ± 10.7 pmol/g) was 26 and 11% lower (p = 0.007, p = 0.024 by Student's t test), respectively.
Figure 6.

Aβ levels were significantly reduced in the brain of mApoE–PA–LIP-treated APP/PS1 Tg mice. APP/PS1 Tg or WT mice were treated with mApoE–PA–LIP or vehicle, and, at the end of treatment, brains were homogenized and soluble or insoluble Aβ1–40 and Aβ1–42 amounts were measured by ELISA. A, Percentage change of insoluble brain Aβ1–40 and Aβ1–42 levels. B, Percentage change of soluble brain Aβ1–40 and Aβ1–42 levels. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01 by Student's t test.
To assess whether the decrease of brain Aβ burden was also involving oligomers, recognized as the best correlate of synaptic dysfunction and disease severity (Lue et al., 1999; McLean et al., 1999), their content was analyzed on brain homogenates of treated mice. It is noteworthy that the treatment strongly reduced (− 70.5%, p < 0.001) the levels of soluble Aβ species with MW up to 90 kDa (Fig. 7). This also holds true for the band with an MW of ∼100 kDa, likely corresponding to full-length APP (−33.9%, p = 0.055).
Figure 7.
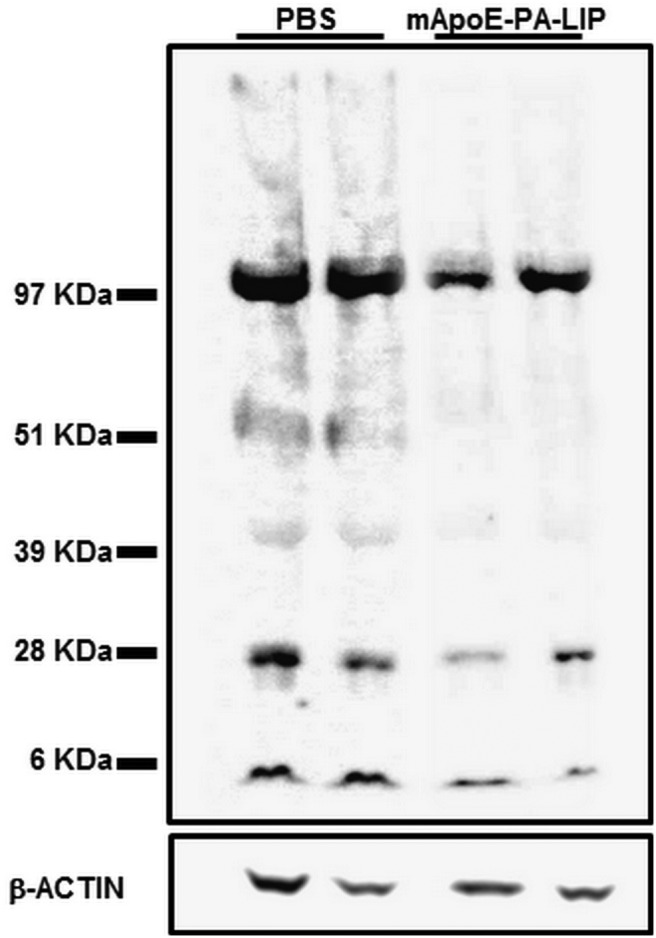
mApoE–PA–LIP significantly reduced Aβ oligomers in the brain of APP/PS1 Tg mice. APP/PS1 Tg or WT mice were treated with mApoE–PA–LIP or vehicle, and, at the end of treatment, brains were homogenized and soluble Aβ was submitted to SDS-PAGE or Western blot. Representative Western blot of brain-soluble Aβ probed with anti-Aβ 6E10 and visualized by ECL is shown.
Because of all this compelling evidence, highlighting the ability of the mApoE–PA–LIP to induce a significant brain Aβ decrease, we wondered what happened to the disappeared peptide. In an attempt to answer this question, we measured Aβ levels in organs and tissues of APP/PS1 mice treated with mApoE–PA–LIP. We found that the levels of Aβ in both the liver (Fig. 8A) and the spleen (Fig. 8B) had increased (liver, +18%, p = 0.038; spleen, +20%, p = 0.0057 by Student's t test), whereas Aβ levels in plasma, at the end of the treatment, did not significantly change with respect to PBS-treated mice (1.47 ± 0.69 versus 1.27 ± 0.79 pmol/ml Aβ).
Figure 8.

Aβ levels in the liver and spleen of APP/PS1 Tg mice are increased after treatment with mApoE–PA–LIP. APP/PS1 Tg or WT mice were treated with mApoE–PA–LIP or vehicle, and, at the end of treatment, liver and spleen were dissected and homogenized and total Aβ was extracted. Aβ1–40 and Aβ1–42 amounts were measured by ELISA. Data are expressed as a fold change of Aβ levels. A, Histogram of liver Aβ1–40 and Aβ1–42 levels. B, Histogram of spleen Aβ1–40 and Aβ1–42 levels. Data are expressed as mean ± SEM. *p < 0.05 by Student's t test.
The involvement of microglia and astrocytes in Aβ clearance (Agostinho et al., 2010) was ruled out through immunostaining experiments on APP/PS1 brains, which showed no increased activation in either the cortex or the hippocampus after mApoE–PA–LIP treatment (data not shown).
Finally, experiments with dually fluorescently labeled mApoE–PA–LIP were performed to assess their ability to cross the BBB in an intact form. After injection in APP/PS1 Tg mice, brain sections were imaged by confocal microscopy. Thanks to the incorporation of two different fluorophores into mApoE–PA–LIP, specific punctuated red and green signals with high level of colocalization were observed in the hippocampus (Fig. 9).
Figure 9.

Dually fluorescently labeled mApoE–PA–LIP administered to APP/PS1 mice are detected intact in the hippocampal regions. A, Bright-field and DAPI fluorescence of brain sections from APP/PS1 mice treated with PBS or LIPs, acquired to focus the additional analysis on the hippocampal area. B, Representative images of brain hippocampal regions of mice treated with PBS or dually fluorescently labeled mApoE–PA–LIP. The images shown have been obtained as the sum of several focal planes with a thickness of 0.63 μm each, giving information related to the entire volume of the analyzed sections. C, Magnification of the framed areas of B.
Discussion
Considering the demographic increase and the global trend in population aging, the influence of AD will be even more pronounced in the future: it is estimated that the number of AD-afflicted individuals will triple by the year 2050. Unfortunately, there are currently no effective means to prevent or cure this disease. Biotechnological devices, such as nanoparticles, represent a possible tool to reach this goal, relying on the possibility of conferring on them multitask features. Despite the fact that several nanoparticles have been synthesized and proposed for therapy and diagnosis of AD, evidence of their efficacy in vivo has not yet been reached (Brambilla et al., 2011). Therefore, in the present investigation, we designed multitask LIPs and tested their therapeutic efficacy in vivo in AD mouse models. Based on our previous in vitro studies (Gobbi et al., 2010; Re et al., 2010, 2011; Bana et al., 2013), these tasks were assigned to LIPs bifunctionalized with PA, which binds Aβ in different aggregation forms, and with mApoE, obtained by modification of a decapeptide from ApoE, targets the BBB and facilitates its crossing. This peptide sequence contains only the receptor-binding domain of ApoE and not the Aβ-binding sequence. The combination of the two ligands on the LIP surface confers to them the unforeseen feature of disaggregating Aβ assemblies, in vitro, as we have reported recently (Bana et al., 2013) and herein confirmed. This ability, not displayed by monofunctionalized LIPs (with either PA or mApoE), could arise from the synergic interaction of both the negatively charged PA phosphate group and the positively charged mApoE amino acids with oppositely charged residues present on Aβ peptide at physiological pH (Datta et al., 2000; Ahyayauch et al., 2012; Bana et al., 2013).
For the in vivo proof-of-principle, we used APP/PS1 as the AD mouse model. The APP/PS1 carry the human Swedish mutation and a deletion of the exon 9 on PS1 and show Aβ plaque deposition starting from 8–9 months of age (Lee et al., 1997; Jankowsky et al., 2001). Therefore, it represents a useful “early-onset” AD mouse model. We observed that the treatment with mApoE–PA–LIP induced an important reduction in the number of brain plaques. mApoE–PA–LIP-induced plaque reduction was also confirmed in some experiments on APP23 mice; APP23 mice were used also for PET imaging of plaques with [11C]PIB, because it has been shown that this probe does not sufficiently bind to the plaques in the APP/PS1 mouse brain (Snellman et al., 2013). Noticeably, PET experiments showed that the plaque reduction was still present 3 months after treatment. The comparison with monofunctionalized LIPs indicates that bifunctionalization is crucial to either affect the Aβ plaque load or enhance the amount of radioactivity crossing the BBB after injection of radiolabeled LIPs. Finally, an approach based on the use of dually fluorescent LIPs coupled to confocal microscopy imaging (Tanifum et al., 2012) showed colocalization of the two fluorescent probes in the hippocampus of treated mice, suggesting that mApoE–PA–LIP reached the brain in an intact form. Considering the results obtained, it is reasonable to speculate that mApoE–PA–LIP exert their activity by crossing the BBB interacting with, and destabilizing, brain Aβ aggregates. Successively, the generated lower MW Aβ species may be facilitated to move from brain to blood and are peripherally cleared in the liver and spleen through the so-called sink effect (Matsuoka et al., 2003; Biscaro et al., 2009; Sutcliffe et al., 2011), likely mediated by circulating mApoE–PA–LIP. To this regard, the peripheral increment of Aβ in the liver and spleen, detected at the end of the treatment, supports this hypothesis.
However, the increased Aβ recovery in peripheral organs is not sufficient to explain the total reduction of brain Aβ levels. Because we did not observe activation of microglia and astrocytes after treatment, we suggest that proteolytic degradation of brain Aβ might be integrated by the sink effect. The relevant role of proteolytic degradation in Aβ clearance, with respect to its elimination across the BBB, has been suggested recently (Ito et al., 2013).
In association with plaque reduction, the treatment with mApoE–PA–LIP also induced a significant recovery of Tg mouse impaired memory, at variance with monofunctionalized LIPs, which exerted a weaker effect that did not reach statistical significance. However, these findings suggest that monofunctionalized LIPs might exert, when administered together, an effect comparable with that of bifunctionalized LIPs. A deeper investigation with a larger number of data will clarify this issue.
This observation, together with the fact that the mApoE–PA–LIP also induced a significant reduction of Aβ oligomers, makes the treatment even more compelling. Aβ oligomers are indeed recognized as the main species responsible of the neuropathological process underlying the onset and progression of AD and the best correlates of synaptic dysfunction and disease severity (Lue et al., 1999; McLean et al., 1999; Wilcox et al., 2011; Scopes et al., 2012). In experimental models, oligomers have been shown to specifically inhibit long-term potentiation and mediate memory impairment, whereas Aβ monomer and fibrils were inactive (Walsh et al., 2002; Cleary et al., 2005; Balducci et al., 2010). Because it is more likely that different Aβ species, rather than a unique molecular assembly, affect neuronal functions at multiple levels, another relevant observation is that the Aβ oligomer reduction, mediated by the treatment, helps to reduce different MW species. It is also worth noting that full-length APP is seemingly reduced after treatment. This observation opens the possibility that additional mechanisms are underlying the effect of LIP. Of course, more focused studies are necessary to clarify these complex issues.
Together, our findings promote mApoE–PA–LIP as a well tolerated valuable new nanotechnological means for AD therapy. It is important to point out that the treatment with mApoE–PA–LIP does not eliminate the cause of Aβ overproduction but could slow down the neurodegeneration process. Therefore, it is conceivable that possible therapeutic protocols could require chronic treatments to minimize the progression of the disease.
Footnotes
The research leading to these results has received funding from the European Community's Seventh Framework Program (FP7/2007-2013) under Grant 212043 (NAD, Nanoparticles for therapy and diagnosis of Alzheimer disease).
The authors declare no competing financial interests.
References
- Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer's disease. Curr Pharm Des. 2010;16:2766–2778. doi: 10.2174/138161210793176572. [DOI] [PubMed] [Google Scholar]
- Ahyayauch H, Raab M, Busto JV, Andraka N, Arrondo JL, Masserini M, Tvaroska I, Goñi FM. Binding of β-amyloid (1–42) peptide to negatively charged phospholipid membranes in the liquid-ordered state: modeling and experimental studies. Biophys J. 2012;103:453–463. doi: 10.1016/j.bpj.2012.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C, Forloni G. APP transgenic mice: their use and limitations. Neuromolecular Med. 2011;13:117–137. doi: 10.1007/s12017-010-8141-7. [DOI] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bana L, Minniti S, Salvati E, Sesana S, Zambelli V, Cagnotto A, Orlando A, Cazzaniga E, Zwart R, Scheper W, Masserini M, Re F. LIPosomes bi-functionalized with phosphatidic acid and an ApoE-derived peptide affect Aβ aggregation features and cross the blood-brain-barrier: implications for therapy of Alzheimer Disease. Nanomedicine. 2013 doi: 10.1016/j.nano.2013.12.001. doi: 10.1016/j.nano.2013.12.001. Advance online publication. Retrieved September 9, 2014. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. The genetics of Alzheimer's disease. Prog Mol Biol Transl Sci. 2012;107:79–100. doi: 10.1016/B978-0-12-385883-2.00008-4. [DOI] [PubMed] [Google Scholar]
- Biscaro B, Lindvall O, Hock C, Ekdahl CT, Nitsch RM. Abeta immunotherapy protects morphology and survival of adult-born neurons in doubly transgenic APP/PS1 mice. J Neurosci. 2009;29:14108–14119. doi: 10.1523/JNEUROSCI.2055-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla D, Le Droumaguet B, Nicolas J, Hashemi SH, Wu LP, Moghimi SM, Couvreur P, Andrieux K. Nanotechnologies for Alzheimer's disease: diagnosis, therapy, and safety issues. Nanomedicine. 2011;7:521–540. doi: 10.1016/j.nano.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Bruggink KA, Müller M, Kuiperij HB, Verbeek MM. Methods for analysis of amyloid-beta aggregates. J Alzheimers Dis. 2012;28:735–758. doi: 10.3233/JAD-2011-111421. [DOI] [PubMed] [Google Scholar]
- Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta G, Chaddha M, Garber DW, Chung BH, Tytler EM, Dashti N, Bradley WA, Gianturco SH, Anantharamaiah GM. The receptor binding domain of apolipoprotein E, linked to a model class A amphipathic helix, enhances internalization and degradation of LDL by fibroblasts. Biochemistry. 2000;39:213–220. doi: 10.1021/bi991209w. [DOI] [PubMed] [Google Scholar]
- Gobbi M, Re F, Canovi M, Beeg M, Gregori M, Sesana S, Sonnino S, Brogioli D, Musicanti C, Gasco P, Salmona M, Masserini ME. Lipid-based nanoparticles with high binding affinity for amyloid-beta1–42 peptide. Biomaterials. 2010;31:6519–6529. doi: 10.1016/j.biomaterials.2010.04.044. [DOI] [PubMed] [Google Scholar]
- Ito S, Matsumiya K, Ohtsuki S, Kamiie J, Terasaki T. Contributions of degradation and brain-to-blood elimination across the blood-brain barrier to cerebral clearance of human amyloid-beta peptide(1–40) in mouse brain. J Cereb Blood Flow Metab. 2013;33:1770–1777. doi: 10.1038/jcbfm.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, Jenkins N, Copeland N, Price DL, Sisodia SS. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med. 1997;3:756–760. doi: 10.1038/nm0797-756. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/S0002-9440(10)65184-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K. Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci. 2003;23:29–33. doi: 10.1523/JNEUROSCI.23-01-00029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re F, Cambianica I, Sesana S, Salvati E, Cagnotto A, Salmona M, Couraud PO, Moghimi SM, Masserini M, Sancini G. Functionalization with ApoE-derived peptides enhances the interaction with brain capillary endothelial cells of nanoliposomes binding amyloid-beta peptide. J Biotechnol. 2010;156:341–346. doi: 10.1016/j.jbiotec.2011.06.037. [DOI] [PubMed] [Google Scholar]
- Re F, Cambianica I, Zona C, Sesana S, Gregori M, Rigolio R, La Ferla B, Nicotra F, Forloni G, Cagnotto A, Salmona M, Masserini M, Sancini G. Functionalization of liposomes with ApoE-derived peptides at different density affects cellular uptake and drug transport across a blood-brain barrier model. Nanomedicine. 2011;7:551–559. doi: 10.1016/j.nano.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Re F, Gregori M, Masserini M. Nanotechnology for neurodegenerative disorders. Maturitas. 2012;73:45–51. doi: 10.1016/j.maturitas.2011.12.015. [DOI] [PubMed] [Google Scholar]
- Scopes DI, O'Hare E, Jeggo R, Whyment AD, Spanswick D, Kim EM, Gannon J, Amijee H, Treherne JM. Abeta oligomer toxicity inhibitor protects memory in models of synaptic toxicity. Br J Pharmacol. 2012;167:383–392. doi: 10.1111/j.1476-5381.2012.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D, Mandelkow E, Holtzman D. Deciphering Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a011460. doi: 10.1101/cshperspect.a011460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snellman A, López-Picón FR, Rokka J, Salmona M, Forloni G, Scheinin M, Solin O, Rinne JO, Haaparanta-Solin M. Longitudinal amyloid imaging in mouse brain with 11C-PIB: comparison of APP23, Tg2576, and APPswe-PS1dE9 mouse models of alzheimer disease. J Nucl Med. 2013;54:1434–1441. doi: 10.2967/jnumed.112.110163. [DOI] [PubMed] [Google Scholar]
- Steinerman JR, Irizarry M, Scarmeas N, Raju S, Brandt J, Albert M, Blacker D, Hyman B, Stern Y. Distinct pools of beta-amyloid in Alzheimer disease-affected brain: a clinicopathologic study. Arch Neurol. 2008;65:906–912. doi: 10.1001/archneur.65.7.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JG, Hedlund PB, Thomas EA, Bloom FE, Hilbush BS. Peripheral reduction of beta-amyloid is sufficient to reduce brain beta-amyloid: implications for Alzheimer's disease. J Neurosci Res. 2011;89:808–814. doi: 10.1002/jnr.22603. [DOI] [PubMed] [Google Scholar]
- Tanifum EA, Dasgupta I, Srivastava M, Bhavane RC, Sun L, Berridge J, Pourgarzham H, Kamath R, Espinosa G, Cook SC, Eriksen JL, Annapragada A. Intravenous delivery of targeted liposomes to amyloid-β pathology in APP/PSEN1 transgenic mice. PLoS One. 2012;7:e48515. doi: 10.1371/journal.pone.0048515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero J, España J, Parra-Damas A, Martín E, Rodríguez-Álvarez J, Saura CA. Short-term environmental enrichment rescues adult neurogenesis and memory deficits in APP(Sw, Ind) transgenic mice. PLoS One. 2011;6:e16832. doi: 10.1371/journal.pone.0016832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek MM, Ruiter DJ, de Waal RM. The role of amyloid in the pathogenesis of Alzheimer's disease. Biol Chem. 1997;378:937–950. doi: 10.1515/bchm.1997.378.9.937. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wan L, Pooyan S, Hu P, Leibowitz MJ, Stein S, Sinko PJ. Peritoneal macrophage uptake, pharmacokinetics and biodistribution of macrophage-targeted PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarriers for improving HIV drug delivery. Pharm Res. 2007;24:2110–2119. doi: 10.1007/s11095-007-9402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox KC, Lacor PN, Pitt J, Klein WL. Abeta oligomer-induced synapse degeneration in Alzheimer's disease. Cell Mol Neurobiol. 2011;31:939–948. doi: 10.1007/s10571-011-9691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]