

Abstract
Manganese (Mn) is an essential metal, but elevated cellular levels are toxic and may lead to the development of an irreversible parkinsonian-like syndrome that has no treatment. Mn-induced parkinsonism generally occurs as a result of exposure to elevated Mn levels in occupational or environmental settings. Additionally, patients with compromised liver function attributable to diseases, such as cirrhosis, fail to excrete Mn and may develop Mn-induced parkinsonism in the absence of exposure to elevated Mn. Recently, a new form of familial parkinsonism was reported to occur as a result of mutations in SLC30A10. The cellular function of SLC30A10 and the mechanisms by which mutations in this protein cause parkinsonism are unclear. Here, using a combination of mechanistic and functional studies in cell culture, Caenorhabditis elegans, and primary midbrain neurons, we show that SLC30A10 is a cell surface-localized Mn efflux transporter that reduces cellular Mn levels and protects against Mn-induced toxicity. Importantly, mutations in SLC30A10 that cause familial parkinsonism blocked the ability of the transporter to traffic to the cell surface and to mediate Mn efflux. Although expression of disease-causing SLC30A10 mutations were not deleterious by themselves, neurons and worms expressing these mutants exhibited enhanced sensitivity to Mn toxicity. Our results provide novel insights into the mechanisms involved in the onset of a familial form of parkinsonism and highlight the possibility of using enhanced Mn efflux as a therapeutic strategy for the potential management of Mn-induced parkinsonism, including that occurring as a result of mutations in SLC30A10.
Keywords: efflux, GPP130, manganese, parkinsonism, SLC30A10, trafficking
Introduction
Manganese (Mn) is an essential metal that functions as a cofactor required for the activity of numerous cellular enzymes (Aschner et al., 2009). However, at elevated cellular levels, Mn is toxic because it enhances oxidative stress, compromises mitochondrial function, and induces cell death (Milatovic et al., 2009; Stanwood et al., 2009). In humans, Mn is primarily excreted by the liver via bile (Butterworth, 2013). Excess Mn that is not excreted attributable to either hepatic dysfunction or increased systemic Mn levels secondary to elevated exposure preferentially accumulates in the basal ganglia, and the toxic effects of Mn lead to the development of an irreversible and incurable parkinsonian-like syndrome (Olanow, 2004; Perl and Olanow, 2007; Aschner et al., 2009; Butterworth, 2013). Historically, Mn-induced parkinsonism has been reported under certain occupational settings (e.g., welding, manufacture of dry batteries and steel, and mining) in which workers are chronically exposed to elevated Mn (Aschner et al., 2009). Recent studies indicate that Mn-induced parkinsonism may also occur as a result of elevated exposure from environmental sources (Lucchini et al., 2012) or in patients who receive total parenteral nutrition, which contains high Mn levels (Nagatomo et al., 1999). Furthermore, individuals with defective liver function attributable to diseases, such as cirrhosis, fail to adequately excrete Mn and may develop Mn-induced parkinsonism in the absence of exposure to elevated Mn levels (Butterworth, 2013). Indeed, in one study, the prevalence of Mn-induced parkinsonism in cirrhotic patients was >20% (Burkhard et al., 2003). The nature of the injury to basal ganglia neurons in Mn toxicity is different from that in Parkinson's disease, and the two diseases can be distinguished by clinical and pathological features (Olanow, 2004; Perl and Olanow, 2007). However, chronic exposure to elevated Mn (>20 years) was reported to increase the risk of developing Parkinson's disease (Gorell et al., 1999), suggesting that, in addition to inducing a parkinsonian-like syndrome, Mn may also represent an environmental risk factor for developing Parkinson's disease.
Recently, a new form of familial parkinsonism was reported attributable to mutations in SLC30A10 (Tuschl et al., 2008, 2012; Quadri et al., 2012). The disease exhibited autosomal recessive inheritance. Affected patients were born to consanguineous parents and were homozygous for mutations in SLC30A10 (Quadri et al., 2012; Tuschl et al., 2012). On clinical analyses, the major findings were >10-fold increase in serum Mn with evidence of Mn deposition in the basal ganglia (Quadri et al., 2012; Tuschl et al., 2012). Importantly, these patients had no history of exposure to elevated Mn levels, suggesting that Mn homeostasis was perturbed. The function of the SLC30A10 protein and the reasons why mutations in this gene cause parkinsonism are unclear.
Here, we show that SLC30A10 is a cell surface-localized Mn efflux transporter that reduces cellular Mn levels and protects against Mn toxicity. Furthermore, we report that parkinsonism-causing mutations in SLC30A10 block the trafficking and efflux activity of SLC30A10, and neurons and worms expressing these mutants exhibit enhanced sensitivity to Mn toxicity. These results provide novel, mechanistic insights into a new and as yet poorly understood familial parkinsonian syndrome.
Materials and Methods
Cell and neuronal culture experiments
Cell culture, DNA transfections, and Mn treatments in HeLa cells.
These were performed essentially as described previously (Mukhopadhyay et al., 2010, 2013; Mukhopadhyay and Linstedt, 2011, 2012). To summarize here, HeLa cells were grown in minimum essential media (MEM) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin-G, and 100 μg/ml streptomycin. DNA transfections were performed with the JetPEI reagent (VWR) using the recommendations of the manufacturer. Cultures were generally transfected 24 h after plating and used 48 h after transfection. For Mn treatments, unless otherwise specified, freshly prepared MnCl2 was added to the media at a final concentration of 500 μm for the indicated times. Chemicals were from Thermo Fisher Scientific or Sigma-Aldrich.
Viability assays in HeLa cells.
Cell viability was assessed using the methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay as described previously (Mukhopadhyay et al., 2010). Briefly, for Mn toxicity assays, cells were exposed to 0, 1, or 2 mm Mn for 16 h. For zinc (Zn) and copper (Cu) toxicity assays, cells were exposed to 0 or 2 mm Zn (in the form of ZnSO4) or 0 or 2 mm Cu (in the form of CuSO4) for 16 h. After the metal treatments, cells were washed with PBS, incubated in HBSS containing 0.05% MTT (w/v; EMD Millipore) for 2 h at 37°C, and lysed using 0.1N hydrochloric acid in isopropanol and 1% Triton X-100. Absorption at 570 nm was subsequently measured using a plate reader.
Culture, DNA and siRNA transfections, Mn treatments, and viability assays in AF5 cells.
AF5 cells, a gift from Dr. Donald Smith (University of California at Santa Cruz, Santa Cruz, CA), were cultured essentially as described previously (Sanchez et al., 2006; Crooks et al., 2007). For maintenance, cells were grown in DMEM/Ham's F-12 (Life Technologies) with 10% fetal bovine serum, 2 mm l-glutamine, 100 IU/ml penicillin-G, and 100 μg/ml streptomycin (maintenance media). For differentiation, cells were transferred to Neurobasal media supplemented with serum-free human B27 (differentiation media; Life Technologies). Depending on the experiment, differentiation was performed for 2 or 3 d. Previous studies have shown that culturing AF5 cells in differentiation media for 2–4 d induces neural differentiation (Sanchez et al., 2006; Crooks et al., 2007). In our studies, 2 d after transfer to differentiation medium, cells began generating long neurite-like processes. These processes persisted at days 3 and 4 after transfer to differentiation medium. The presence of these processes in >80% of cells in the culture was used as a sign of differentiation of the culture.
For detection of SLC30A10 localization, cells were plated on glass coverslips and cultured for 24 h in maintenance media. Cells were then transfected with FLAG-tagged SLC30A10–wild-type (WT) or Δ105-107 constructs using JetPEI (VWR) and grown in maintenance media for an additional 24 h. After this, cells were washed with PBS and transferred to differentiation media for 48 h. At the end of the differentiation, cells were fixed and processed for immunofluorescence microscopy as described below.
The anti-SLC30A10 siRNA targeted the sequence AAC GCA GTC TTC CTC ACC GCG in rat SLC30A10. The sequence of the control siRNA (sense strand) was GAC CAG CCA UCG U AG UAC UTT. This siRNA was not predicted to deplete any rat or human gene. We have used this siRNA as a control in previous work (Mukhopadhyay and Linstedt, 2011). The siRNA transfections were performed essentially as described previously for HeLa cells (Mukhopadhyay and Linstedt, 2011). Briefly, AF5 cells were plated on glass coverslips and grown in maintenance media for 24 h. After this, cells were washed with PBS and replenished with fresh maintenance media, and siRNAs were added at a final concentration of 80 nm using the Oligofectamine reagent (Life Technologies). One day after transfection, cultures were washed with PBS, transferred to differentiation medium, and, depending on the experiment, cultured for an additional 2–3 d (details provided in specific figure legends).
For viability assays after DNA transfections, cells that had been transfected in maintenance media were transferred to differentiation media 24 h after transfection. Depending on the experiment, differentiation was performed for 48–72 h. Viability was then assessed using the MTT reagent. Similarly, for assessment of viability after knockdown, 1 d after transfection with the control or anti-SLC30A10 siRNA, cells were transferred to differentiation medium for 48–72 h, and, after this, the MTT assay was used to assess viability.
Culture, transfections, and Mn treatments in primary neuronal cells.
Primary neuronal cultures were generated as described previously (Diaz et al., 2004) with some modifications. Cultures were obtained from WT C57BL/6 mice, aged postnatal days 2–3, of either sex. Breeding procedures were approved by the Institutional Animal Care and Use Committee. The brain was dissected and placed in ice-cold MEM (10 ml/brain) on ice. Remaining steps were performed under sterile conditions. The midbrain was dissected and minced with a razor blade. This tissue was added to a 15 ml conical tube that contained 4 ml of trypsin (0.05%; VWR). The tube was incubated for 30 min in a 37°C water bath with periodic mixing by inversion. Cells were then collected by centrifugation at 200 × g for 2 min. The trypsin was removed, and 2 ml of MEM was added to the pelleted tissue. This tissue was triturated, very carefully, by repeatedly passing through a 1 ml pipette tip connected to a P1000 pipettor. The diameter of the pipette tip was 0.8–0.9 mm. Tissue from each midbrain had to be pipetted ∼40–50 times to obtain a homogenous mixture. Care was taken to ensure that no air bubbles were introduced during this process. The tube was then centrifuged as described above, the supernatant was discarded, and cells were resuspended in 2 ml of MEM that contained 10% fetal bovine serum. Viability was assessed using Trypan Blue (Sigma) and was routinely >90%. Cells were then plated on polylysine (Sigma)-coated glass coverslips placed within wells of 24-well plates. Approximately 80–85,000 cells were plated per well. Cells were allowed to attach for 24 h, and the media was then changed to Neurobasal/B27 (Life Technologies), which allows neurons to grow but inhibits growth of glial cells (Brewer et al., 1993). Cells were grown in Neurobasal/B27 media for 4–7 d and then transfected with SLC30A10 constructs. Transfections were performed using the NeuroFect transfection reagent (Gelantis) exactly as described by the manufacturer. For localization studies, cultures were fixed 48 h after transfection and used for immunofluorescence microscopy. For Mn toxicity studies, 24 h after transfection, cultures were treated with 0 or 200 μm MnCl2 for 16 h. After the Mn treatment, cultures were processed for immunofluorescence as described below.
Antibodies.
Custom polyclonal and monoclonal antibodies against Golgi phosphoprotein of 130 kDa (GPP130) have been described previously (Mukhopadhyay et al., 2010). Polyclonal antibody against calnexin was from Abcam. Polyclonal and monoclonal antibodies against the FLAG epitope and monoclonal antibodies against tubulin and the HA epitope were from Sigma. The polyclonal antibody against glutamate decarboxylase 65 and 67 (GAD 65/67) was from EMD Millipore.
Constructs.
FLAG-tagged SLC30A10–WT was a gift from Dr. Ruth Valentine (Newcastle University, Newcastle, UK). Point mutations and deletions were introduced into this construct using the QuikChange mutagenesis kit (Agilent Technologies). Green fluorescent protein (GFP)-tagged Rab5 construct used as a control and HA-tagged dipeptidyl protease 4 (DPP4) were described previously (Bachert et al., 2001; Mukhopadhyay et al., 2010).
Immunofluorescence and microscopy.
HeLa and AF5 cells and primary neuronal cultures were all fixed using 3% paraformaldehyde, and immunofluorescence stainings were performed exactly as described previously (Mukhopadhyay et al., 2010, 2013; Mukhopadhyay and Linstedt, 2011, 2012). Images were captured using a Nikon swept field confocal equipped with a four-line high-power laser launch and 100×, 1.45 numerical aperture oil-immersion objective (Nikon). An iXon3 X3 DU897 EM-CCD camera (Andor Technology) was used for image capture. All images were captured as Z-stacks with 0.5 μm spacing between individual frames. Images depicted in the figures are maximum intensity projections of the stacks.
Image analysis.
Within each experiment, all images were captured using identical settings. Quantification was performed as described below.
For GPP130 fluorescence measurements, average intensity projections of images were generated from individual Z-stacks using the functions ND Processing and ND Image Average (average all frames) of NIS Elements (Nikon) software. Additional analysis was performed using NIS Elements. An outline was drawn around the cell using the Simple ROI Editor tool. Temporary image adjustments were used to ensure accuracy of the outlining, but all measurements were made after reverting to original values. Background was then separately subtracted for each image, and total GPP130 fluorescence per cell was obtained using the ROI Statistics tool.
For calculating Pearson's correlation coefficient, average intensity projections of images being analyzed were generated as described above using NIS Elements (Nikon). An outline was drawn around each cell separately. The Pearson's coefficient was then obtained using the Colocalization function of NIS Elements.
For analyses of neurite morphology in primary neurons, maximum intensity projections of images were generated on NIS Elements. Additional processing was performed using NIH ImageJ. The images were converted to 8 bit, and the NeuronJ plugin was used for quantification. Using this plugin, primary, secondary, and tertiary neurites were traced from each neuron, and the following parameters were computed per neuron: (1) length of longest primary neurite; (2) total length of the neurite tree; (3) total number of primary neurites; and (4) total number of neurites.
Concentration-dependent Mn uptake in HeLa cells.
HeLa cells were seeded in 35 mm dishes and transfected the next day as indicated in specific figure legends. Two days after transfection, cells were treated with indicated concentrations of Mn in serum-containing growth media for 16 h. After the Mn treatment, cells were washed with PBS and collected by incubating with 500 μl of trypsin–EDTA at 37°C for 5 min. After this, the trypsin was neutralized by adding 1 ml of serum-containing growth media, and cells were collected by spinning at 0.5 × g for 3 min in a microfuge. Supernatants were discarded, and cell pellets were washed two times by adding 1 ml of PBS that contained 10 mm EDTA and collecting cells by centrifugation as described above. A final wash with PBS (without EDTA) was performed, and the cell pellets obtained were resuspended in 20 μl of PBS. From this pellet, 1–2 μl was used to assay for total protein, and the remaining was transferred to acid-washed vials and digested by addition of 200 μl of 70% metal-free HNO3, followed by incubation at 85°C for 1 d and then at room temperature for an additional day. Acid-washed vials were prepared by incubating the vials with 10% HNO3 for 2 d, followed by extensive washing in MilliQ water. Digested cell pellet samples were then diluted to 2% HNO3 with ultrapure water (Life Technologies). Measurements of intracellular Mn were performed using an Agilent 7500ce Quadrapole inductively coupled plasma mass spectrometer.
Mn pulse-chase assay.
HeLa cells were transfected with plasmids coding for Rab5, SLC30A10–WT, or SLC30A10–Δ105-107. Two days after transfection, cells were washed three times with PBS, replenished with fresh serum-containing growth media, and treated with 500 μm Mn for 16 h at 37°C (pulse phase). At the end of the pulse, cultures were washed five times with PBS, transferred to HBSS without Mn, and incubated at 37°C for 1 h (chase phase). At the end of the chase, media was collected to assess Mn secreted from cells, and cells were digested as described in the section on Mn measurements above to assess intracellular Mn retained. Mn measurements were performed by inductively coupled plasma mass spectrometry (ICP-MS).
Uptake of Zn and Cu in HeLa cells.
For uptake of Zn and Cu, HeLa cells were transfected with the Rab5 control construct or with SLC30A10–WT. Two days after transfection, cultures were treated with 100 μm Zn (in the form of ZnSO4) or 100 μm Cu (in the form of CuSO4) for 16 h. Cells were then processed for ICP-MS exactly as described for the measurement of intracellular Mn above. Zn and Cu amounts were measured by ICP-MS.
Surface biotinylation assay.
All reagents for the biotinylation assay were from Thermo Fisher Scientific. We used the EZ-link sulfo-NHS-SS-biotin, which is cell impermeable and water soluble. This form of biotin includes a cleavable disulfide bond for reversible labeling of proteins and cell surface primary amines. A stock solution of 125 μm biotin in water was prepared. For each transfection condition, cells from one confluent 35 mm dish were used. Before labeling, cells were washed three times in ice-cold PBS and then scraped and resuspended in 200 μl of PBS. Two microliters of the biotin stock solution was added to the cell suspension to obtain a final biotin concentration of 1.25 μm. Cells were incubated for 30 min at room temperature with end-over-end shaking. The reaction was then washed two times using 800 μl of quenching solution (50 mm Tris, pH 7.4, and 150 mm NaCl). After each wash with the quenching solution, the cell pellet was collected by spinning in a microfuge at 500 × g for 3 min at room temperature. Cells were then lysed by adding 100 μl of lysis buffer (25 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1.0% NP-40, 5% glycerol, 10 μg/ml leupeptin, 10 μg /ml pepstatin, and 1 mm phenylmethlysulfonyl fluoride) and incubating on ice for 30 min. During this 30 min incubation, cells were vortexed every 5–10 min. The lysate was spun at 10,000 × g for 2 min at 4°C. The clarified lysate was transferred into a new tube and used for the subsequent steps. For isolation of biotinylated proteins, a 50 μl slurry of neutravidin agarose was added to a spin column. The slurry was centrifuged at 1000 × g for 1 min, and the flow-through was discarded. The column was then washed with 500 μl of wash buffer (50 mm Tris, pH 7.4, 50 mm NaCl, and 0.5% Triton X-100). Centrifugation was as described above. The clarified cell lysate was added to the column, the bottom was capped, and the lysate was incubated in the column for 60 min at room temperature. At the end of this incubation, the column was spun at 1000 × g for 1 min and the flow-through was collected. This flow-through contained the unbiotinylated fraction of cellular proteins. The column was then washed three times with 500 μl of wash buffer. Finally, to collect biotinylated proteins, 60 μl of 2× reducing sample buffer (50 mm Tris, pH 6.8, 3.33% SDS, 5.3 m urea, 16.6% glycerol, 0.03% bromophenol blue, 5% β-mercaptoethanol, and 16.6 mm DTT) was added to the column, and the column was incubated at 95°C for 5 min. After this, the column was centrifuged at 1000 × g for 2 min at room temperature. The flow-through was collected and contained the biotinylated fraction of cellular proteins.
Quantitative real-time reverse-transcription PCR.
For real-time PCR assays, total RNA was isolated using the PureLink RNA minikit (Life Technologies). RNA was reverse transcribed to cDNA using multiscribe reverse transcriptase, GeneAmp PCR Buffer II, MgCl2, RNase inhibitor, random hexamers (Life Technologies), and deoxynucleotide solution mix (New England Biolabs). Real-time PCR was performed on the resulting cDNAs using iTaq Universal SYBR Green Supermix (Bio-Rad). The PCR conditions were as follows: 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. Primers used to amplify SLC30A10 were as follows: forward, 5′ CGC GAG CTA GTG GCA TAA A 3′ and reverse, 5′ ACA GGT CTT GCC CGA ATA AC 3′. Primers used to amplify GAPDH were as follows: forward, 5′ ATG ATT CTA CCC ACG GCA AG 3′ and reverse, 5′ CTG GAA GAT GGT GAT GGG TT 3′. For both primer sets, efficiency of amplification was calculated by generating standard curves using serial dilutions of cDNA. The calculated efficiencies were 100 and 94% for SLC30A10 and GAPDH, respectively. Additionally, dissociation curves were generated to guarantee amplification of the correct genes. SLC30A10 transcript levels were then quantified with the ΔΔCT method using GAPDH as an internal control (Livak and Schmittgen, 2001).
Immunoblot analyses.
Cells were lysed using Tris-SDS lysis buffer (25 mm Tris-Cl, pH 6.8, 1.5% SDS, 0.8% β-mercaptoethanol, 1 mm dithiothreitol, and 1 mm phenylmethylsulfonyl fluoride). Immunoblotting was performed as described previously (Mukhopadhyay et al., 2010, 2013; Mukhopadhyay and Linstedt, 2011, 2012). Immunoblots were quantitated using NIH ImageJ.
Statistical analyses.
All findings presented were replicated in at least three independent experiments. We used the Prism 6 software (GraphPad Software) for all statistical analyses. Comparisons between two groups were performed using two-tailed Student's t test assuming equal variances. Multiple group comparison at the same time was done using single-factor ANOVA with Tukey–Kramer or Dunnett's post hoc tests. In general, p < 0.05 was considered significant. Asterisks in graphs, wherever present, denote statistically significant differences.
Caenorhabditis elegans experiments
Plasmid constructs.
Full-length SLC30A10–WT and L89P variants were PCR amplified using primers 5′- GGGGACAAGTTTGTACAAAAAAGCAGGCTACATGGGCCGCTACTCTGG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTTAAAAATGCGTTCTGTTGAC-3′ from mRNAs provided by M. Diana Neely (Vanderbilt University, Nashville, TN) and cDNAs provided by Karin Tuschl (University College London Institute of Child Health, London, UK). The GFP tag was fused to both WT and L89P SLC30A10 by fusion PCR using three primer sets: 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTACATGGGCCGCTACTCTGG-3′ and 5′-CTTTACTCATGGCACCGGCTCCAGCGCCTGCACCAGCTCCAAAATGCGTTCTGTTGACATAAC-3′; 5′- ACGCAT TTTGGAGCTGGTGCAGGCGCTGGAGCCGGTGCCATGAGTAAAGGAGAAGAACTTTTC-3′ and 5′- GGGGACCACTTTGTACAAGAAAGCTGGGTTCTATTTGTATA GTTCATCCATGC-3′; and 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTACATGGGCCGCTACTCTGG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCTATTTGTATAGTTCATCCATGC-3′. Plasmids were created with Gateway recombinational cloning (Invitrogen). The above PCR products were initially recombined with the pDONR221 vector to create pENTRY clones. For whole-worm expression, SLC30A10 pENTRY constructs were then recombined into pDEST–sur-5 vector, under the promoter from the acetoacetyl-coenzyme A synthetase (sur-5) gene. Briefly, the sur-5 promoter region was cut out from plasmid pMLH280 (sur-5::mCherry; a gift from Dr. David Miller, Vanderbilt University, Nashville, TN) and replaced the unc-54 promoter region in plasmid pDEST–unc-54 (Chen et al., 2010) to create the pDEST–sur-5 vector. For expression in dopaminergic (DAergic) neurons, SLC30A10 pENTRY constructs were then recombined into pDEST–dat-1 vector, under the promoter from the DA transporter (dat-1) gene. For body wall muscle expression, SLC30A10–GFP pENTRY constructs were then recombined into pDEST–unc-54 vector, under the promoter from a muscle myosin class II heavy chain (unc-54) gene. These plasmids were then used to create transgenic worms.
C. elegans strains and protocols.
Nematodes were grown and maintained using standard procedures (Brenner, 1974). Transgenic worms were created using microinjection as described previously (Mello et al., 1991). Plasmids were injected at a concentration of 50 mg/ml, except pBCN43-R4R3 at 30 mg/ml. BY200 [dat-1::GFP(vtIs1) V] worms were crossed into VC171 [smf-2(gk133) X] worms to create MAB300 [dat-1::GFP(vtIs1) V, smf-2(gk133) X] strain. For whole-worm expression, sur-5::SLC30A10 WT or L89P alone were coinjected into the MAB300 strain with pBCN43–R4R3 (rpl-28::PuroR, myo-2::mCherry; Addgene), which were used as the selective markers for transformation. For expression in DAergic neurons, dat-1::SLC30A10 WT or L89P alone were coinjected into the MAB300 strain with pMHL280 (sur-5::mCherry; a gift from Dr. David Miller) and pBCN27–R4R3 (rpl-28::PuroR; Addgene). For GFP fusion protein expression in DAergic neurons, dat-1::SLC30A10–GFP WT or L89P alone were coinjected into VC171 worms with rol-6 and pBCN27–R4R3. For GFP fusion protein expression in the body wall muscle cells, unc-54::SLC30A10–GFP WT and L89P were coinjected into VC171 worms with rol-6, pBCN27–R4R3, and pBCN27–Pmyo-2::GFP (rpl-28::PuroR, myo-2::GFP; Addgene), respectively. For each injection mixture, at least three stable lines were generated and analyzed. Representative lines were selectively integrated by using UV irradiation using a Spectroline UV crosslinker with an energy setting of 500 mJ/cm2.
Lethality analysis.
Synchronized larva 1 (L1) stage animals were grown on nematode growth medium (NGM) plates to L4 stage. For each tested strain, 2500 L4 worms were exposed to 0, 100, and 200 mm Mn (in the form of MnCl2 as in the cell culture assays) in glass vials for 1 h. Worms were then transferred to siliconized tubes and washed three times by centrifugation at 1000 rpm for 1 min in 85 mm NaCl with 1% Tween 20. After acute Mn treatments, ∼30 worms from each treatment were transferred to individual 35 mm NGM plates seeded with E. coli OP50. Twenty-four hours later, live worms were scored for each plate, and the survival rate was calculated as the percentage of live worms in the worms placed on plates a day before. In SLC30A10-overexpressing worms, each survival rate was normalized to the survival rate of control worms in the absence of Mn exposure. Experiments were performed in three independent replicates.
Analysis of DAergic neurodegeneration.
Acute Mn treatments were performed as described above, except that synchronized L1 stage animals were treated with 0, 10, or 25 mm of MnCl2. Worms were washed and then transferred to NGM plates and allowed to recover for 4 h. Later, worms were transferred to 2% agarose pads and anesthetized with 3 mm levamisole for microscopy. Fluorescence microscopy was performed as described previously (Benedetto et al., 2010). If one or more of the six-head DAergic neurons showed morphological changes, such as puncta, blebs, neuronal absence or shrinkage, presence of vacuoles, dorsal or ventral cord gaps, loss of cell bodies, or strand breaks in neuronal processes, the worm was scored as neurodegenerative; only worms with six intact-head DAergic neurons were counted as “intact” (Cao et al., 2005; Benedetto et al., 2010). For each strain and condition, at least 20 animals were quantitated in three independent replicates.
Confocal microscopy.
Confocal microscopy was performed as described previously (Benedetto et al., 2010).
Basal slowing response.
The basal slowing response was quantified as the locomotor rate of worms moving on NGM plates with or without bacteria, by scoring the number of body bends in the anterior region in a 20 s interval. This assay was performed as described previously (Sawin et al., 2000), except that the worms were exposed to Mn at L1 stage and assayed 1 d later.
Semiquantitative RT-PCR.
Semiquantitative RT-PCR was performed on worms to validate the expression of SLC30A10. RNA isolation was performed as described previously (Chen et al., 2010), except that glucogen (Ambion) instead of glycoblue was used to visualize RNAs. The following primers were used to detect SLC30A10: forward, 5′-CTTGGGCTCTTGAGATGTTC-3′; and reverse, 5′-TTTGAATCAGCAGTGGGAAC-3′. For the ama-1 (RNA polymerase II large subunit) loading control, the forward primer was 5′-GCTACTCTGGCAAGACGTG-3′, and the reverse primer was 5′-CGAGCGCATCGATGACCC-3′.
Results
Localization of SLC30A10–WT and mutants
To gain insight into the function of SLC30A10 and determine why mutations in the protein cause parkinsonism, we first examined the localization of SLC30A10–WT and disease-causing mutants. Immunofluorescence analyses in cell culture revealed that FLAG-tagged SLC30A10–WT trafficked to the cell periphery and outlined the cell boundary, suggesting that the WT protein trafficked to the cell surface (Fig. 1A,B). We then tested the localization of five separate SLC30A10 mutants that cause familial parkinsonism: L89P, Δ98-134, Δ105-107, T196P, and Q308Stop (Tuschl et al., 2012). Remarkably, all mutant proteins failed to traffic to the cell periphery and outline the cell boundary and were instead retained in a compartment that overlapped with calnexin, a marker of the endoplasmic reticulum (Fig. 1A,B). To quantitatively assess the overlap between SLC30A10–WT or mutants and calnexin, we performed colocalization analyses using Pearson's correlation coefficient (Mukhopadhyay et al., 2010; Dunn et al., 2011). Values for Pearson's coefficient for two fluorophores imaged in two channels can range from +1 to −1 (Dunn et al., 2011). Values close to +1 indicate a high degree of colocalization or overlap, whereas values close to 0 and negative values imply lack of colocalization (Dunn et al., 2011). In our assays, the Pearson's coefficient for the colocalization between the SLC30A10 mutants and calnexin was >0.93, whereas that for SLC30A10–WT and calnexin was −0.24 ± 0.055 (Fig. 1C). The above results indicate that the mutants were trapped in the endoplasmic reticulum, whereas the WT protein trafficked to a compartment distinct from the endoplasmic reticulum, likely the cell surface. To validate that SLC30A10–WT indeed trafficked to the cell surface, we analyzed the colocalization between SLC30A10–WT and DPP4, an integral plasma membrane protein (Bachert et al., 2001). SLC30A10–WT strongly overlapped with DPP4 with a Pearson's coefficient of 0.9 ± 0.01 (Fig. 1D–F). As a control, we verified that SLC30A10–Δ105-107 did not overlap with DPP4 and had a significantly lower Pearson's coefficient (Fig. 1D–F). We then confirmed the microscopy results with a biochemical approach, using a surface biotinylation assay. Using a cell-impermeable biotin that covalently modifies surface-exposed lysine residues, we established that SLC30A10–WT, but not the Δ105-107 mutant, was biotinylated (Fig. 1G,H), indicating that only the WT protein trafficked to the cell surface. The recovery of SLC30A10–WT in the surface biotinylated elute was lower than that expected from the microscopy assays, likely because SLC30A10 is predicted to have only one surface-exposed lysine. Importantly, as a specificity control, we verified that GPP130, an endogenous protein localized to the Golgi apparatus that does not traffic to the cell surface but has 41 lysine residues, was not biotinylated (Fig. 1G,H). Overall, the microscopy and surface biotinylation results indicate that SLC30A10–WT traffics to the cell surface, whereas the disease-causing mutants are trapped in the endoplasmic reticulum.
Figure 1.

Localization of SLC30A10–WT and disease-causing mutants in HeLa cells and C. elegans. A, HeLa cells were transfected with various FLAG-tagged SLC30A10 constructs. Two days after transfection, cultures were processed for immunofluorescence. SLC30A10 was detected using a monoclonal antibody against the FLAG epitope, and a polyclonal antibody against calnexin was used to demarcate the endoplasmic reticulum. Scale bar, 10 μm. B, Quantification of percentage cells with surface-localized SLC30A10 from A (mean ± SE; n = 3 experiments with >50 cells per experiment; *p < 0.05 for the difference between SLC30A10–WT and other mutants by one-way ANOVA, followed by Tukey–Kramer post hoc test). C, Quantification of the Pearson's coefficient for colocalization between SLC30A10 and calnexin from A (mean ± SE; n = 15–20 cells per SLC30A10 construct; *p < 0.05 for the difference between SLC30A10–WT and other mutants by one-way ANOVA, followed by Tukey–Kramer post hoc test). D, HeLa cells were cotransfected with FLAG-tagged SLC30A10–WT or SLC30A10–Δ105-107 and HA-tagged DPP4. Two days after transfection, SLC30A10 was detected using an antibody against FLAG and DPP4 with an antibody against HA. Scale bar, 10 μm. E, Quantification of percentage cells from D in which signals for SLC30A10 and DPP4 overlapped (mean ± SE; n = 3 experiments with >50 cells per experiment; *p < 0.05 for the difference between SLC30A10–WT and Δ105-107 by Student's t test). F, Quantification of the Pearson's coefficient for colocalization between SLC30A10 and DPP4 from D (mean ± SE; n = 17 cells per SLC30A10 construct; *p < 0.05 for the difference between SLC30A10–WT and the Δ105-107 mutant by Student's t test). G, HeLa cells were transfected with SLC30A10–WT or Δ105-107. Two days after transfection, cell surface proteins were biotinylated as described in Materials and Methods. For each transfection condition, 17% of the biotinylated elute and 10% of the unbiotinylated flow-through was loaded on a SDS-PAGE gel. Immunoblot assays were then performed to detect SLC30A10, using an anti-FLAG antibody, and GPP130. H, Quantification of the recovery of different proteins in the surface biotinylated elute from G. The signal intensity for SLC30A10–WT, SLC30A10–Δ105-107, and GPP130 in the surface biotinylated elute was calculated using NIH ImageJ. The sum of the signals was adjusted to 100, and the signal for each probe (SLC30A10–WT, SLC30A10–Δ105-107, or GPP130) was expressed as a percentage of the sum (mean ± SE; n = 3 experiments; *p < 0.05 for the difference between SLC30A10–WT and other probes by one-way ANOVA, followed by Tukey–Kramer post hoc test). I, GFP-tagged SLC30A10–WT or L89P were expressed in C. elegans under control of the unc-54 promoter. Body wall muscle cells were then imaged as described in Materials and Methods. Scale bar, 10 μm. J, GFP-tagged SLC30A10–WT or L89P were expressed in C. elegans under control of the dat-1 promoter. Localization of SLC30A10 in DAergic neurons was then assessed by microscopy. Arrowheads in the inset show surface localization of SLC30A10–WT. Scale bars: 10 μm; insets, 5 μm.
Next, we validated our cell culture results at the whole-organism level in C. elegans. A GFP tag was fused to the C terminus of both SLC30A10–WT and L89P, and the fusion proteins were overexpressed in the worm's body wall muscle cells under the control of the unc-54 promoter. SLC30A10–WT was localized at the cell surface in body wall muscle cells (Fig. 1I), whereas the L89P mutant was distributed throughout the cytoplasm in a reticular pattern consistent with retention in the endoplasmic reticulum (Fig. 1I). Because SLC30A10 is highly expressed in the basal ganglia of human brains (Quadri et al., 2012), we posited that muscle cells might not reflect the “real” localization of this protein. Accordingly, we also expressed the fusion proteins in DAergic neurons of C. elegans under the control of the dopamine transporter dat-1 promoter. In DAergic neurons, a similar expression pattern was observed, with SLC30A10–WT trafficking to the plasma membrane (Fig. 1J) and the L89P mutant trapped in the cytoplasm (Fig. 1J). These results were subsequently verified in a mammalian neuronal cell line and in primary midbrain neurons (see Figs. 10–12).
Figure 10.

SLC30A10 mediates Mn detoxification in GABAergic AF5 cells. A, AF5 cells were transfected with FLAG-tagged SLC30A10–WT or Δ105-107 constructs. One day after transfection, cultures were differentiated as described in Materials and Methods. Cells were then imaged to detect FLAG and calnexin. Scale bar, 10 μm. B, Quantification of percentage cells with surface localized SLC30A10 from A (mean ± SE; n = 3 experiments with >50 cells per experiment; *p < 0.05 for the difference between SLC30A10–WT and Δ105-107 by Student's t test). C, AF5 cells were transfected with control (Rab5; Control), SLC30A10–WT, or Δ105-107 constructs. One day after transfection, cultures were transferred to differentiation medium for 48 h. Viability was then assessed using the MTT assay. Viability of cells expressing the control construct was set to 100 and used for normalization of viability of other transfection conditions (mean ± SE; n = 3 experiments; p > 0.05 by one-way ANOVA). D, AF5 cells were transfected with the control Rab5, SLC30A10–WT, or Δ105-107 constructs. One day after transfection, cultures were shifted to differentiation medium for 32 h. After this, cultures were treated with 0 or 500 μm Mn for 16 h in differentiation media. At the end of the Mn treatment, cell viability was assessed using the MTT assay. Percentage viability of the transfection control cells that were treated with Mn was set to 50 and used for normalization of the viability of Mn-treated cells from SLC30A10–WT and SLC30A10–Δ105-107 groups (mean ± SE; n = 3 experiments; *p < 0.05 for the difference between SLC30A10–WT and other transfection conditions by one-way ANOVA, followed by Tukey–Kramer post hoc test). E, AF5 cells were transfected with control or anti-SLC30A10 siRNAs. One day after transfection, cultures were transferred to differentiation medium. After 48 h of differentiation (i.e., 72 h after knockdown), cells were harvested for real-time analyses. RNA isolation and generation of cDNA was performed as described in Materials and Methods. mRNA levels of SLC30A10 and a housekeeping gene, GAPDH, were then quantified using real-time RT-PCR. For each transfection condition, level of SLC30A10 mRNA was normalized to that of GAPDH to correct for generalized changes in gene expression. Normalized levels of SLC30A10 mRNA of the control siRNA group was then expressed as 1 (mean ± SE; n = 3 experiments; *p < 0.05 by Student's t test). F, AF5 cells were transfected with the control or anti-SLC30A10 siRNAs and, 1 d after transfection, shifted to differentiation media for 48 h as above. After this, cell viability was assessed using the MTT assay (mean ± SE; n = 3 experiments; p > 0.05 by Student's t test). G, AF5 cells, transfected with the control or anti-SLC30A10 siRNA, were shifted to differentiation media 1 d after knockdown. Cells were kept in differentiation medium for 32 h. After this, cells were treated with 0 or 500 μm Mn for an additional 16 h in differentiation media. Percentage viability of the Mn-treated transfection control cells was expressed as 50 and used for normalization of the viability of Mn-treated cells from the anti-SLC30A10 siRNA group (mean ± SE; n = 3 experiments; *p < 0.05 by Student's t test).
Figure 11.
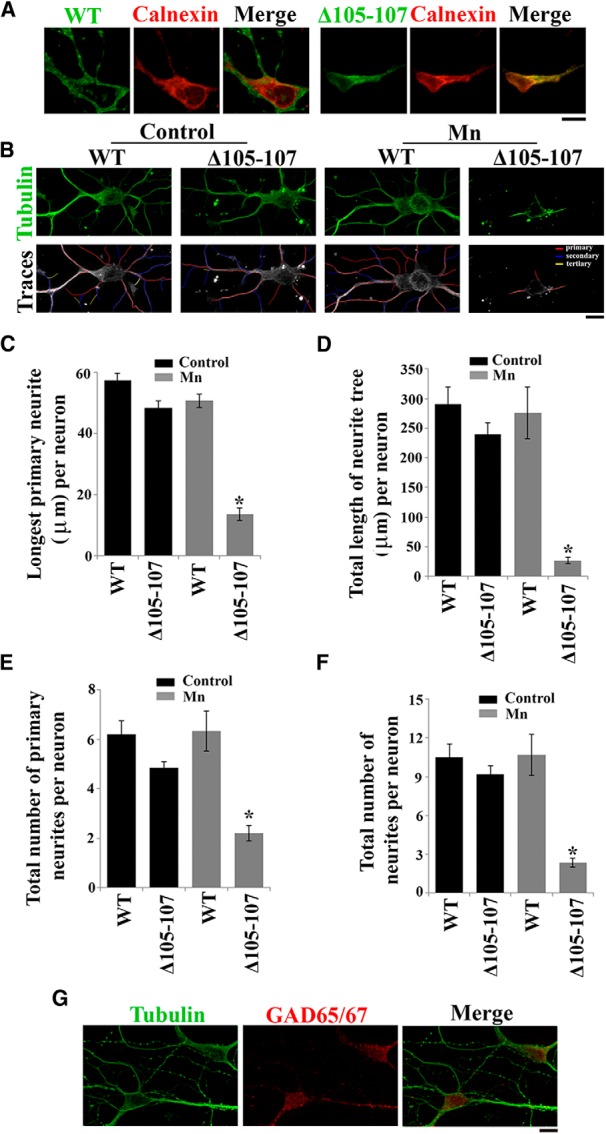
SLC30A10 protects primary midbrain neurons from Mn-induced neurodegeneration. A, Primary midbrain neurons were generated as described in Materials and Methods. Four days after plating, cultures were transfected with FLAG-tagged SLC30A10–WT or Δ105-107. Two days after transfection, cultures were fixed and stained to detect FLAG using a monoclonal antibody and calnexin using a polyclonal antibody. FLAG expression in the cell body appeared to be stronger than in neurites. Scale bar, 10 μm. B, Primary midbrain neurons were generated and transfected with FLAG-tagged SLC30A10 constructs as described above. One day after transfection, cultures were treated with 0 (Control) or 200 μm Mn for 16 h. Cultures were then fixed and stained using a monoclonal antibody against tubulin to visualize neurites and a polyclonal antibody against FLAG to detect transfection. Expression of the FLAG-tagged SLC30A10 constructs in the cells depicted in this panel is shown in Figure 12. For the bottom row, the tubulin images were converted to grayscale, and primary (red), secondary (blue), and tertiary (yellow) neurites that emanated from the neurons were drawn using the NeuronJ plugin of NIH ImageJ. Scale bar, 10 μm. C–F, Quantification of the morphology of neurites from B above. For each neuron, the length of the longest primary neurite, total length of the neuritic tree, total number of primary neurites, and total number of neurites were calculated using the NeuronJ plugin of NIH ImageJ (for each parameter measured, n = 15–25 neurons for each transfection and Mn treatment condition; *p < 0.05 for the difference between Mn-treated cells expressing SLC30A10–Δ105-107 and the other transfection and Mn treatment conditions using one-way ANOVA, followed by Tukey–Kramer post hoc test). G, Primary midbrain neurons were generated as described above and stained to detect tubulin using a monoclonal antibody and GAD 65/67 using a polyclonal antibody. Scale bar, 10 μm.
Figure 12.
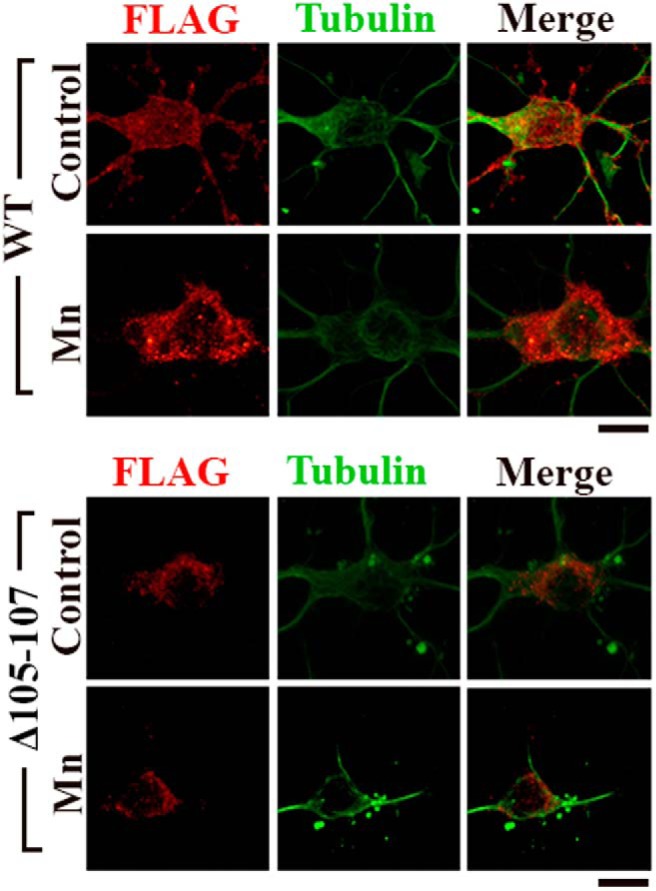
Presence of FLAG-tagged SLC30A10 constructs in neurons depicted in Figure 11. Cells shown in this figure are identical to those shown in Figure 11B. FLAG expression was stronger in the cell body compared with neurites. Note that the polyclonal anti-FLAG antibody used here was very specific and sensitive in identifying transfected cells but was not as effective as the monoclonal anti-FLAG antibody used in Figure 11A for determining intracellular FLAG localization. Use of the polyclonal FLAG antibody was essential because the monoclonal FLAG and tubulin antibodies were from the same species. Scale bars, 10 μm.
The fact that the disease-causing SLC30A10 mutants were trapped in the endoplasmic reticulum (Fig. 1A,B) suggested that these proteins were misfolded. Because misfolded proteins trapped in the endoplasmic reticulum are often unstable and degraded in the proteasome (Gardner et al., 2013), next we tested whether SLC30A10 mutants underwent a similar degradation in cultured cells. Although in immunofluorescence assays we could clearly detect cells in which expression of the SLC30A10 mutants was comparable with that of SLC30A10–WT, in a large number of transfected cells, expression of the mutants appeared to be less than that of the WT protein. To validate this observation, we performed immunoblot analyses and discovered that normalized protein levels of SLC30A10–L89P, SLC30A10–Δ98-134, and SLC30A10–Δ105-107 were significantly decreased compared with SLC30A10–WT (Fig. 2A,B). Transfection efficiency of SLC30A10–WT and the mutants were similar. Importantly, we then observed that treatment with the proteasome inhibitor MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal) increased expression of SLC30A10–L89P, SLC30A10–Δ98-134, and SLC30A10–Δ105-107 but did not affect expression of SLC30A10–WT (Fig. 2A–C), suggesting that the mutant proteins underwent proteasomal degradation. In summary, the results in Figures 1 and 2 show that, although SLC30A10–WT traffics to the plasma membrane, the disease causing mutants (L89P, Δ98-134, Δ105-107, T196P, and Q308Stop) exhibit a trafficking defect and cannot escape from the endoplasmic reticulum.
Figure 2.
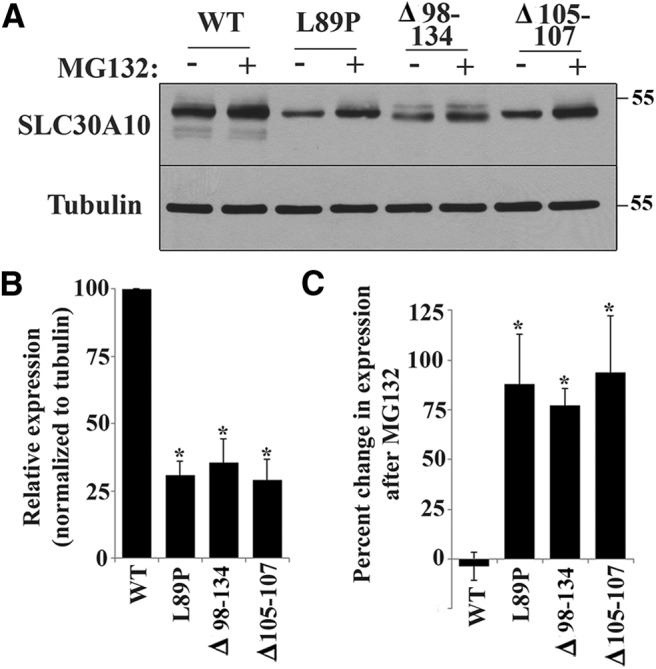
Rapid turnover of SLC30A10 mutants. A, HeLa cells were transfected with indicated FLAG-tagged SLC30A10 constructs. Two days after transfection, cultures were treated with or without 0.5 μm MG132 for 24 h. Cells were then lysed, and FLAG and tubulin were detected by immunoblot. B, Quantification of SLC30A10 levels from A. Expression of SLC30A10 from cultures that were not exposed to MG132 was quantified using NIH ImageJ, and the results were normalized to tubulin expression. Normalized expression of SLC30A10–WT was set to 100 (n = 3 experiments; *p < 0.05 for the difference in expression between each SLC30A10 mutant and WT using one-way ANOVA and Dunnett's post hoc test). C, Quantification of the MG132-induced increase in expression of SLC30A10 mutants from A. SLC30A10 levels after MG132 treatment were quantified using NIH ImageJ and normalized to tubulin. Percentage increase in SLC30A10 expression was then calculated using data in B (n = 3 experiments; *p < 0.05 for the difference between SLC30A10–WT and each mutant by one-way ANOVA and Dunnett's post hoc test. MG132 treatment did not induce a statistically significant change in SLC30A10–WT levels).
Identification of SLC30A10 as a potential Mn efflux transporter using GPP130 as a Mn sensor
SLC30 family proteins, of which SLC30A10 is a member, belong to the cation diffusion facilitator superfamily of metal transporters (Huang and Tepaamorndech, 2013). There are 10 proteins in the SLC30 family, SLC30A1–SLC30A10 (Huang and Tepaamorndech, 2013). SLC30A1–SLC30A8 transport Zn from the cytosol to the cell exterior or the lumen of various intracellular organelles (the function of SLC30A9 is unclear; Huang and Tepaamorndech, 2013). Notably, in patients with parkinsonism attributable to mutations in SLC30A10, serum Mn levels are high (Tuschl et al., 2008, 2012; Quadri et al., 2012). This clinical picture raised the possibility that SLC30A10–WT-mediated efflux of Mn instead of Zn and the disease-causing mutants failed to do so. The above hypothesis was supported by our analyses of the SLC30A10 sequence, which revealed that the putative ion-binding site in SLC30A10 contained an asparagine residue not detected in Zn transporting cation diffusion facilitators, such as SLC30A5, a mammalian transporter, as well as YiiP, a bacterial transporter (Lu and Fu, 2007; Ohana et al., 2009). Although Mn can coordinate with asparagine, Zn does not show this preference (Dokmanić et al., 2008). To directly test whether SLC30A10 functions as a Mn efflux transporter, we used the Mn response of the Golgi protein GPP130. Our previous work demonstrated that, in response to a specific increase in intra-Golgi (but not simply cytosolic) Mn, GPP130 trafficked from the Golgi to late endosomes and lysosomes in which it was degraded (Mukhopadhyay et al., 2010, 2013; Mukhopadhyay and Linstedt, 2011, 2012). Activity of secretory pathway Ca2+-ATPase 1 (SPCA1), a Golgi-localized P-type ATPase, was required for the transport of Mn into the Golgi and subsequent degradation of GPP130 (Mukhopadhyay and Linstedt, 2011). For this study, because SLC30A10–WT was localized to the cell surface, we wondered whether its expression would block the Mn-induced degradation of GPP130. We posited that, if SLC30A10 is an efficient Mn efflux transporter, it might rapidly transport Mn from the cytosol to the cell exterior and thereby block or reduce the transport of Mn into the Golgi and inhibit GPP130 degradation. Immunofluorescence assays revealed that, in cells expressing a control construct, a 4 h treatment with Mn induced a profound loss of GPP130 (Fig. 3A). Expression of SLC30A10–WT blocked the Mn-induced loss of GPP130 (Fig. 3A–C), suggesting that expression of SLC30A10 blocks the transport of Mn into the Golgi. We confirmed that SPCA1 remained localized to the Golgi in SLC30A10–WT-expressing cells. Importantly, expression of the disease-causing SLC30A10 mutants did not exert an inhibitory effect on GPP130 degradation (Fig. 3B,C). For the mutants, the analysis was restricted to cells that expressed SLC30A10 at a level comparable with the WT protein. The results of the immunofluorescence assays were then validated with immunoblots (Fig. 3D,E). Note that expression of the control construct does not affect Mn homeostasis or the induced degradation of GPP130 (Mukhopadhyay et al., 2010; Mukhopadhyay and Linstedt, 2011; Figs. 4, 5).
Figure 3.

Expression of SLC30A10 blocks the Mn-induced degradation of GPP130. A, HeLa cells were transfected with plasmids coding for Rab5 (transfection control) or FLAG-tagged SLC30A10–WT. Two days after transfection, cultures were treated with or without 500 μm Mn for 4 h and imaged to detect Rab5 and GPP130 or SLC30A10 (using an anti-FLAG antibody) and GPP130. Scale bar, 10 μm. B, HeLa cells were transfected with various FLAG-tagged SLC30A10 constructs. Two days after transfection, cells were treated with Mn as described in A and imaged to detect FLAG and GPP130. Scale bar, 10 μm. C, Quantification of the amount of GPP130 remaining in cells after Mn treatment from B. Mock-transfected cells did not express any SLC30A10 construct. For each transfection condition, mean GPP130 fluorescence per cell with and without Mn treatment was quantified as described in Materials and Methods and used to calculate the percentage residual GPP130 after Mn (mean ± SE; n = 22 cells per Mn treatment condition per construct; *p < 0.05 for the difference between WT and other transfection conditions by one-way ANOVA and Tukey–Kramer post hoc test). D, HeLa cells were transfected with Rab5 (transfection control), SLC30A10–WT, or SLC30A10–Δ105-107. All constructs exhibited similar transfection efficiencies. Two days after transfection, cells were treated with or without 500 μm Mn for 4 h. GPP130 and tubulin were then detected by immunoblot. E, Quantification of the amount of GPP130 remaining (normalized to tubulin) after Mn treatment from D. For each transfection and Mn treatment condition, normalized GPP130 levels were quantified using NIH ImageJ and used to calculate the percentage residual GPP130 after Mn (mean ± SE; n = 3 experiments; *p < 0.05 for the difference between SLC30A10–WT and the other transfection conditions using one-way ANOVA, followed by Tukey–Kramer post hoc test).
Figure 4.

Expression of SLC30A10–WT increases Mn efflux. A, HeLa cells were transfected with a control construct (Rab5; denoted as Control) or SLC30A10–WT. Two days after transfection, cells were treated with the indicated amounts of Mn for 16 h. The absolute level of intracellular Mn was then measured using ICP-MS and normalized to total protein. Data shown are from one representative experiment; error bars depict SDs from three ICP-MS runs. Analysis from three independent experiments is presented in B. B, Quantification of the difference in intracellular Mn between cells expressing control (Rab5; Control) or SLC30A10–WT constructs from multiple experiments. HeLa cells were transfected as described in A and, 2 d after transfection, treated with 500 μm Mn for 16 h. Intracellular Mn normalized to total cellular protein was then measured as described in A. In each experiment, for cells expressing the control construct, the absolute value of intracellular Mn in parts per billion per microgram protein was adjusted to 100 (mean ± SE; n = 3 experiments; *p < 0.05 by Student's t test). C, HeLa cells were transfected with the control (Rab5; Control) construct or mock transfected. Two days later, cells were treated with 500 μm Mn for 16 h. Intracellular Mn in parts per billion per microgram protein was then calculated as described in A. Data are from one representative experiment; error bars depict SDs from three ICP-MS runs. Analysis from three independent experiments is presented in D below. D, Quantification of the difference in intracellular Mn between mock-transfected and control-transfected cells from multiple experiments. Cells were transfected and treated with Mn exactly as described in C. In each experiment, the value of the intracellular Mn in parts per billion per microgram protein obtained for mock-transfected cells was adjusted to 100 (mean ± SE; n = 3 experiments; p > 0.05 for the difference between the two groups by Student's t test). E, HeLa cells were transfected with control (Rab5; Control), SLC30A10–WT, or SLC30A10–Δ105-107 constructs. Two days after transfection, the Mn pulse-chase assay was performed as described in Materials and Methods. After the chase, intracellular Mn retained within the cells and released into the medium was measured using ICP-MS. For each transfection condition, the amount of Mn in each compartment (intracellular or extracellular) was expressed as a percentage of the total Mn (i.e., sum of the intracellular and extracellular Mn). As in A, data shown are from one representative experiment; error bars depict SDs from three ICP-MS runs. Analysis from three independent experiments is presented in F. F, Quantification of the difference in intracellular Mn after the pulse-chase assay from multiple experiments. The Mn pulse-chase assay was performed in HeLa cells expressing control (Rab5; Control), SLC30A10–WT, or SLC30A10–Δ105-107 constructs exactly as described in E. In each experiment, for control-transfected cells, the percentage intracellular Mn retained was adjusted to 50 (mean ± SE; n = 3 experiments; *p < 0.05 for the difference between WT and other transfection conditions by one-way ANOVA and Tukey–Kramer post hoc test). G, HeLa cells were transfected with the control Rab5 construct or SLC30A10–WT. Two days after transfection, cells were treated with 100 μm Zn for 16 h. The absolute level of intracellular Zn was then measured using ICP-MS and normalized to total protein. Data shown are from one representative experiment; error bars depict SDs from three ICP-MS runs. Analysis from three independent experiments is presented in H. H, Quantification of the difference in intracellular Zn between cells expressing control (Rab5) or SLC30A10–WT constructs from multiple experiments. Intracellular Zn levels were measured exactly as described in G. In each experiment, for cells expressing the control construct, the absolute value for intracellular Zn (parts per billion per microgram protein) was adjusted to 100 (mean ± SE; n = 3 experiments; p > 0.05 by Student's t test). In this panel, the SE is relatively high because, over multiple experiments, the difference in intracellular Zn levels between control and SLC30A10-transfected cells had a wide distribution. I, Two days after transfection, HeLa cells expressing the control Rab5 construct or SLC30A10–WT were treated with 100 μm Cu for 16 h. After this, intracellular Cu was measured using ICP-MS. Values were normalized to total protein. Data shown are from one representative experiment; error bars depict SDs from three ICP-MS runs. Analysis from three independent experiments is presented in J. J, Quantification of the difference in intracellular Cu between cells expressing control (Rab5) or SLC30A10–WT constructs from multiple experiments. Intracellular Cu was measured as described in I. In each experiment, for cells expressing the control construct, the absolute value for intracellular Cu (parts per billion per microgram protein) was adjusted to 100 (mean ± SE; n = 3 experiments; p > 0.05 by Student's t test).
Figure 5.

SLC30A10–WT protects against Mn toxicity. A, HeLa cells were transfected with a control construct (Rab5; Control) or SLC30A10–WT. Two days after transfection, cells were treated with 0, 1, or 2 mm Mn for 16 h. Cell viability was then assessed using the MTT assay as described in Materials and Methods. For each transfection condition, viability at 0 mm Mn was set to 100 and used to calculate the percentage viability after Mn treatment (mean ± SE; n = 3 experiments; *p < 0.05 for the difference between the viability of control and SLC30A10–WT-expressing cells at each Mn treatment condition; comparison between groups was performed at each Mn treatment condition separately using Student's t test). B, HeLa cells, transfected with the control (Rab5; Control), SLC30A10–WT, or SLC30A10–Δ105-107 constructs as described in A, were treated with 0 or 2 mm Mn for 16 h. The MTT assay was then performed to assess cell viability. For each experiment, percentage viability of the transfection control cells that were treated with Mn was set to 50 and used for normalization of the viability of Mn-treated cells from SLC30A10–WT and Δ105-107 groups (mean ± SE; n = 3 experiments; *p < 0.05 for the difference in viability between SLC30A10–WT and other transfection groups using one-way ANOVA, followed by Tukey–Kramer post hoc test). C, HeLa cells were transfected with the control Rab5 construct or mock transfected. Two days after transfection, cells were treated with 0 or 2 mm Mn for 16 h. For each experiment, percentage viability of the mock-transfected cells that were treated with Mn was set to 50 and used for normalization of the viability of Mn-treated SLC30A10–WT-expressing cells (mean ± SE; n = 3 experiments; p > 0.05 by Student's t test). D, E, HeLa cells were transfected with the control Rab5 construct or SLC30A10–WT. Two days later, cells were treated with 0 or 2 mm Zn (D) or 0 or 2 mm Cu (E). For each metal in each experiment, percentage viability for mock-transfected cells after metal treatment was adjusted to 50 and used for normalization of the viability of metal-treated SLC30A10–WT-expressing cells (for each panel, mean ± SE; n = 3 experiments; p > 0.05 by Student's t test).
Intracellular Mn measurements validate the Mn efflux activity of SLC30A10
To expand on the results obtained in the GPP130 degradation assay and directly determine whether SLC30A10 expression affected cellular Mn levels, we measured intracellular Mn in HeLa cells expressing SLC30A10–WT or a control construct. Mn measurements were performed using ICP-MS and after exposure of cells to 0–500 μm for 16 h. Previous studies have shown that the above levels of Mn are not toxic to HeLa cells (Mukhopadhyay et al., 2010; Mukhopadhyay and Linstedt, 2011). Intracellular Mn in SLC30A10–WT-expressing cells was lower than in control cells (Fig. 4A,B), showing that SLC30A10 expression reduces cellular Mn levels. There was no difference in intracellular Mn levels between cells expressing the control construct and mock-transfected cells, indicating that expression of the control construct did not alter Mn uptake or efflux (Fig. 4C,D). The decrease in intracellular Mn during SLC30A10 expression might reflect an increase in Mn efflux or a block in Mn uptake. To differentiate between these possibilities, we performed a pulse-chase assay. Cells expressing control or SLC30A10–WT constructs were treated with 500 μm Mn for 16 h (pulse phase) and then chased in Mn-free media for an additional 1 h. At the end of the chase, the amount of Mn retained in the cells and released into the medium was measured by ICP-MS. Compared with control cells, SLC30A10–WT-expressing cells retained less intracellular Mn within and secreted more Mn into the media (Fig. 4E,F), consistent with increased Mn efflux in SLC30A10–WT-expressing cells. To control for the possibility that the increase in Mn efflux seen on SLC30A10–WT expression was attributable to an indirect effect, we analyzed the efflux activity of cells expressing SLC30A10–Δ105-107 in the same experiment. Importantly, there was no difference in the amount of Mn retained within the intracellular compartment or released into the medium between control cells and those expressing the Δ105-107 mutant (Fig. 4E,F). SLC30A10 was initially considered to be a Zn transporter (Bosomworth et al., 2012), and a recent clinical study reported an increase in liver Zn (and Cu) in one patient who had homozygous mutations in SLC30A10 (Lechpammer et al., 2014). A mild increase in serum Cu was also reported in one other patient in a previous study (Quadri et al., 2012). Because changes in Cu and Zn levels were not seen in all patients with SLC30A10 mutations, it was unlikely that SLC30A10 was involved in regulating cellular levels of these metals (Tuschl et al., 2008, 2012; Quadri et al., 2012). However, to rule out this possibility, we measured intracellular Zn and Cu levels in control and SLC30A10–WT-expressing cells and discovered that there was no difference in the cellular levels of either metal between the two groups (Fig. 4G–J). The results of the metal measurement and GPP130 degradation assays combined with the fact that SLC30 family proteins are known to mediate metal efflux strongly suggest that SLC30A10 is a specific Mn efflux transporter and that disease-causing mutations in SLC30A10 block the efflux activity of the transporter.
SLC30A10 protects cells against Mn toxicity
To test the physiologic relevance of the efflux activity of SLC30A10, we sought to determine whether expression of SLC30A10 conferred protection against Mn-induced cell death. Assays were first performed using HeLa cells in culture. In previous work, we demonstrated that treatment with 1 mm Mn for 16 h induces 50% cell death (Mukhopadhyay and Linstedt, 2011). For this experiment, we treated cells expressing SLC30A10–WT or a control construct with 0, 1, or 2 mm Mn for 16 h and then measured viability using the MTT assay. In control cultures, viability of cells treated with 1 or 2 mm Mn was 48 ± 6.5 and 20 ± 2.5%, respectively, of that of cells not exposed to Mn (Fig. 5A). Expression of SLC30A10–WT strongly protected against Mn-induced cell death (Fig. 5A). We then compared the viability of cells expressing a control construct, SLC30A10–WT, or the Δ105-107 mutant and treated with 2 mm Mn for 16 h. Although expression of SLC30A10–WT continued to protect, the Δ105-107 mutant did not have any effect (Fig. 5B). There was no difference in the viability of cells expressing the control construct and those that were mock transfected after exposure to 2 mm Mn for 16 h (Fig. 5C), providing additional evidence in support of the fact that expression of the control construct did not perturb Mn homeostasis or detoxification. Furthermore, as expected, expression of SLC30A10–WT did not protect against Zn- or Cu-mediated cell death (Fig. 5D,E). In summary, results in Figures 3–5 show that expression of SLC30A10–WT reduces cellular Mn levels by increasing Mn efflux and thereby protects cells from Mn toxicity. In contrast, disease-causing mutants do not mediate efflux, and cells expressing these constructs exhibit enhanced sensitivity to Mn toxicity.
SLC30A10 protects C. elegans against Mn toxicity
To determine the relevance of SLC30A10 function in protecting against the onset of parkinsonism in vivo, we reverted to the C. elegans model system and used three separate assays: (1) worm viability; (2) DAergic neurodegeneration; and (3) a functional behavioral response. For the viability assays, we generated transgenic worms overexpressing either SLC30A10–WT or L89P. These two SLC30A10 variants were driven by the sur-5 promoter, which directs strong gene expression to most C. elegans cells. For this assay, as transfection controls, we used worms transfected with a mixture that lacked SLC30A10 plasmids. Expression was confirmed by semiquantitative RT-PCR (Fig. 6). All worms were synchronized to the L4 stage before Mn treatment. Overexpression of SLC30A10–WT and L89P did not affect development or survival of C. elegans in the absence of Mn exposure (Fig. 7A). However, during Mn exposure (both at 100 and 200 mm; treatments in worms require high doses given the impermeability of the cuticle), SLC30A10–WT significantly increased nematode survival over transfection controls, whereas overexpression of the L89P mutant had no significant effect (Fig. 7B,C).
Figure 6.
Confirmation of SLC30A10 expression by semiquantitative RT-PCR. Primers used to detect SLC30A10 are described in Materials and Methods.
Figure 7.

SLC30A10 protects C. elegans against Mn toxicity. A–C, Control worms that were transfected with a mixture lacking SLC30A10 plasmids or worms transfected with SLC30A10–WT or L89P were synchronized in the L1 stage and grown to L4 stage. Worms were then exposed to 0, 100, or 200 mm Mn for 1 h. Viability was subsequently assessed as described in Materials and Methods (mean ± SE; n = 3 experiments with ∼30 worms per Mn treatment condition, per genotype, per experiment; *p < 0.05 and **p < 0.01 for the difference between control and other transfection conditions by one-way ANOVA, followed by Tukey–Kramer post hoc test).
Next, we tested the effect of SLC30A10 expression on Mn-induced neurodegeneration. Studies in human samples and mouse models indicate that the primary sites of neural injury in Mn-induced parkinsonism, including those attributable to SLC30A10 mutations, are the globus pallidus and striatum (Olanow, 2004; Liu et al., 2006; Perl and Olanow, 2007; Lechpammer et al., 2014), which contain GABAergic neurons (Gerfen and Surmeier, 2011). However, functional defects in the DAergic neurons of the substantia nigra pars compacta have also been reported in Mn-induced parkinsonism (Guilarte et al., 2006; Liu et al., 2006). Because C. elegans have only eight DAergic neurons, six in the head and two in the tail (Benedetto et al., 2010), it is a useful system to study DAergic neurodegeneration. Therefore, we used this model to investigate effects on DAergic neurons. Effects on GABAergic neurons were investigated later using a mammalian cell line and primary midbrain neurons as models (see Figs. 10–12). In the nematodes, we visualized the DAergic neurons by overexpression of dat-1::GFP (Fig. 8A). DAergic neurons in the L1 stage are more sensitive to Mn than in the L4 stage. Therefore, in this assay, all worms were synchronized to the L1 stage and then treated with Mn. Exposure of dat-1::GFP worms to Mn at 10–25 mm induced morphological changes characteristic of neurodegeneration in DAergic neurons, such as puncta, blebs, neuronal absence or shrinkage, presence of vacuoles, dorsal or ventral cord gaps, loss of cell bodies, or strand breaks in neuronal processes (Fig. 8B–E). At 10 mm, ∼80% of the worms had one or two degenerated neurons, and the remaining 20% had three or four damaged neurons. At 25 mm, ∼50% of affected worms had one or two degenerated neurons, and the other 50% had three or four damaged neurons. Note that, in neurons showing signs of neurodegeneration, GFP fluorescence was still clearly visible, indicating that the observed effect was not attributable to loss of GFP protein (Fig. 8B–E). To test the effect of SLC30A10, we coexpressed SLC30A10–WT or L89P in the DAergic neurons under the control of the dat-1 promoter along with dat-1::GFP. For this assay, worms transfected with dat-1::GFP only served as transfection controls. Expression of SLC30A10 variants was comparable by RT-PCR (Fig. 6). We focused our analyses on the six-head DAergic neurons. Overexpression of SLC30A10–WT and L89P did not affect the morphology of the neurons in the absence of Mn exposure (Fig. 8F). Importantly, compared with transection control dat-1::GFP worms, expression of SLC30A10–WT afforded protection from Mn at 10 and 25 mm, significantly attenuating DAergic neurodegeneration, whereas the L89P mutant failed to protect (Fig. 8G,H).
Figure 8.
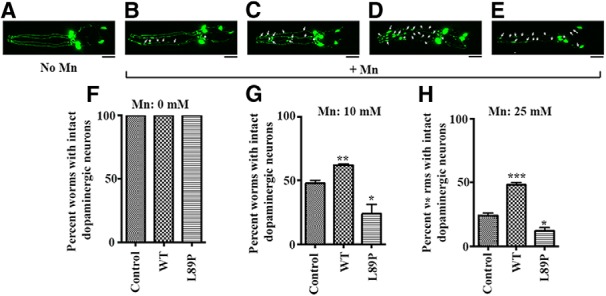
SLC30A10 protects DAergic neurons from neurodegeneration induced by Mn exposure. A–E, Worms expressing dat-1::GFP were synchronized in the L1 stage and treated with 0 mm Mn (A) or 10 mm Mn for 1 h (B–E). DAergic neurons were then imaged as described in Materials and Methods. In B–E, arrowheads indicate degeneration in neuronal processes, and asterisks indicate degeneration in cell bodies. Scale bars, 10 μm. F–H, Worms expressing dat-1::GFP only (control) or dat-1::GFP along with dat-1::SLC30A10–WT or L89P were grown, treated with 0, 10, or 25 mm Mn, and imaged as described above. For each genotype, the number of worms in which DAergic neurons were “intact” (i.e., did not show any evidence of neurodegeneration) was calculated as described in Materials and Methods (mean ± SE; n = 3 experiments with at least 20 worms per Mn treatment condition, per genotype, per experiment; *p < 0.05, **p < 0.01, and ***p < 0.001 for the difference between control and other transfection conditions by one-way ANOVA, followed by Tukey–Kramer post hoc test).
To further confirm that the morphological results correlated with functional changes, we validated the integrity of DAergic neurotransmission with a functional behavioral assay, the basal slowing response. Food sensing in C. elegans is regulated by a DAergic neural circuit, which slows down nematodes in the presence of bacteria. This locomotion can be quantified as the number of bends in the anterior body region during a 20 s interval. The same three groups of worms used for the morphologic assessment of DAergic neurodegeneration were used for this functional assay (i.e., worms expressing dat-1::GFP were transfection controls, and worms in the experimental groups expressed SLC30A10–WT or L89P along with dat-1::GFP). Well fed transfection control animals moved quickly (∼26 bends/20 s) in search for food on plates lacking bacteria; in contrast, they moved slowly (∼17 bends/20 s) upon reentering the bacterial lawn when transferred to bacteria-containing plates (Fig. 9A). This change in locomotion reflects the integrity of DAergic neurons. When DAergic neuron function is compromised, the change in the number of body bends becomes smaller. In nematodes carrying a tyrosine hydroxylase (TH) mutant [cat-2 (e1112)], the difference of body bends per 20 s between the bacteria-containing plate and the empty plate is almost eliminated, indicating dramatic loss of DAergic neuronal activity (Sawin et al., 2000). Expression of the SLC30A10 genes did not affect the basal slowing response in the absence of exposure to Mn (Fig. 9B; change of ∼9 bends/20 s in each genotype). In transfection control worms, treatment with Mn (10 or 25 mm) significantly reduced the change in the number of body bends to ∼3–4/20 s (Fig. 9C,D), indicating that Mn affected the integrity of the DAergic neuronal circuit. Importantly, in SLC30A10–WT-expressing worms, the change in the number of body bends after Mn treatment was significantly greater than Mn-treated transfection control worms (Fig. 9C,D), indicating improved basal slowing response and protection of the DAergic neuronal circuit. In contrast, no significant effect was observed in SLC30A10–L89P worms (Fig. 9C,D). Overall, the studies in C. elegans complement the cell culture results and show that SLC30A10 protects against Mn toxicity and induced neurodegeneration at the organismal level.
Figure 9.

SLC30A10 improves basal slowing response in C. elegans during Mn exposure. A, The basal slowing response was assayed in control worms that expressed dat1::GFP as described in Materials and Methods (mean ± SE; n = 3 experiments with 5–10 worms per Mn treatment condition, per experiment. ****p < 0.0001 by Student's t test). B–D, Worms transfected with dat1::GFP (control) or those transfected with dat1::GFP and dat-1::SLC30A10–WT or L89P were synchronized at the L1 stage and then exposed to 0, 10, or 25 mm Mn for 1 h. Worms were kept on NGM plates for recovery. The next day, the basal slowing response was measured (mean ± SE; n = 3 experiments with 5–10 worms per Mn treatment condition, per experiment, per genotype; *p < 0.05 and ****p < 0.0001 by one-way ANOVA, followed by Tukey–Kramer post hoc test).
Validation of results in GABAergic AF5 cells
Physiologic differences likely exist between neurons in C. elegans and mammalian systems. Furthermore, C. elegans do not express SLC30A10 homologs, and we observed that HeLa cells express SLC30A10 mRNA at very low levels. The lack of expression of SLC30A10 in the above systems was ideal for overexpression studies because it ensured that results obtained were not confounded by endogenous protein. However, it was also important to validate our findings in mammalian model systems that express endogenous SLC30A10. Although our goal was to test the relevance of our results in primary cultures of mammalian neurons, before working with primary neurons, as an intermediate step, we decided to verify our results in a rat-derived neural cell line, differentiated AF5 cells. AF5 cells are useful for studies on Mn toxicity because, after differentiation, they assume a GABAergic lineage (Sanchez et al., 2006; Crooks et al., 2007). As described above, during Mn toxicity, GABAergic neurons are primarily affected (Olanow, 2004; Liu et al., 2006; Crooks et al., 2007; Perl and Olanow, 2007; Stanwood et al., 2009). In AF5 cells, SLC30A10–WT trafficked to the cell surface, whereas the Δ105-107 mutant was trapped in the endoplasmic reticulum (Fig. 10A,B). After transfection, transcript levels of SLC30A10–WT and Δ105-107 were comparable and modestly (twofold to fourfold) elevated over endogenous. Having verified this, we proceeded to perform cell viability assays. We observed that expression of SLC30A10–WT or Δ105-107 did not affect viability in the absence of Mn treatment (Fig. 10C). Importantly, after a 16 h treatment with Mn, viability of cells expressing SLC30A10–WT was ∼50% higher than that of cells expressing the control construct (Fig. 10D). There was no difference between the viability of Mn-treated transfection control and Δ105-107-expressing cells (Fig. 10D). Thus, assays in AF5 cells replicated findings observed in HeLa cells and C. elegans.
Next, we decided to take advantage of the endogenous expression of SLC30A10 in AF5 cells to determine the requirement of SLC30A10 in mediating Mn detoxification. Our idea was to deplete SLC30A10 using siRNA and then perform viability assays. To achieve this, we designed four siRNAs against rat SLC30A10 and tested them individually. Of the four siRNAs tested, we identified one siRNA that reproducibly reduced SLC30A10 expression by ∼50% compared with cells transfected with a control siRNA that did not target any rat gene (Fig. 10E). Knockdown of SLC30A10 did not affect cell viability in the absence of Mn (Fig. 10F). Importantly, after a 16 h exposure to 500 μm Mn, viability of SLC30A10-depleted cells was significantly less than that of cells transfected with the control siRNA (Fig. 10G). As an additional confirmatory step, we used ICP-MS to measure intracellular Mn in AF5 cells transfected with anti-SLC30A10 or control siRNAs and exposed for 16 h to 125 μm Mn. Consistent with the results of the viability assay, in cells depleted of SLC30A10, intracellular Mn was greater than that in transfection control cells (5.87 ± 0.03 ppb/μg lysate after knockdown compared with 1.02 ± 0.007 ppb/μg lysate in controls). The knockdown results complement our overexpression studies and indicate that SLC30A10 plays an essential role in mediating Mn detoxification and protecting against Mn toxicity.
Validation of results in primary midbrain neurons
Encouraged by the results in AF5 cells, we proceeded with assays in primary neurons. We generated cultures of primary midbrain neurons from newborn mice and first substantiated our findings about SLC30A10 localization. For this, we transfected the cultures with SLC30A10–WT or Δ105-107 and assessed localization using immunofluorescence. Importantly, consistent with results in HeLa and AF5 cells and in C. elegans, SLC30A10–WT trafficked to the cell surface, whereas the Δ105-107 mutant was trapped in the endoplasmic reticulum (Fig. 11A).
We then went on to test whether expression of SLC30A10–WT protected neurons against Mn toxicity. Primary midbrain neurons develop extensive neuritic trees (Fig. 11B). Treatment with 100–800 μm Mn causes a dramatic loss of neurites, which can be used to quantify the extent of neurotoxicity [Stanwood et al., 2009; treatment with higher amounts of Mn induces nonspecific cell death (Stanwood et al., 2009)]. For our assay, we transfected neurons with SLC30A10–WT or Δ105-107 and then treated these neurons with 0 (control) or 200 μm Mn for 16 h. After the Mn treatment, we stained the neurons with an antibody against tubulin, which marks the cytoskeleton and allows visualization of the neuritic tree (Sepulveda et al., 2009) along with an anti-FLAG antibody to detect transfection (SLC30A10 constructs were FLAG tagged). Under control (0 μm Mn) conditions, both SLC30A10–WT- and Δ105-107-expressing neurons had well developed neuritic trees (Fig. 11B; for presence of the transfection marker in imaged neurons, see Fig. 12). Quantification on a per neuron basis revealed that, in SLC30A10–WT-expressing neurons, the longest primary neurite had an average size of 57.4 ± 2.4 μm, and the average length of the total neuritic tree was 290.6 ± 28.5 μm (Fig. 11C,D). There was no statistical difference in either of the above parameters between neurons expressing SLC30A10–WT or Δ105-107 (Fig. 11C,D). Moreover, between SLC30A10–WT- and Δ105-107-expressing neurons, there was no difference in the total number of neurites and total number of primary neurites per neuron (Fig. 11E,F). Thus, expression of SLC30A10–Δ105-107 by itself did not affect neural morphology. Treatment with Mn had a dramatic effect on Δ105-107-expressing neurons. The length of the longest primary neurite decreased by ∼76% to 13.6 ± 2 μm, and the total length of the neuritic tree decreased by ∼89% to 26.9 ± 5.8 μm (Fig. 11C,D). Significant decreases were also observed in the total number of neurites and total number of primary neurites per neuron (Fig. 11E,F). Remarkably, SLC30A10–WT-expressing neurons were completely protected against this Mn-induced neurotoxicity, and, in WT-expressing cells, there was no effect of Mn treatment on the length of the longest primary neurite, total length of the neuritic tree, total number of neurites, and total number of primary neurites (Fig. 11B–F). Thus, expression of SLC30A10–WT, which reduces intracellular Mn, offers a profound level of protection against Mn-induced neurotoxicity in primary mammalian neurons, whereas a disease-causing SLC30A10 mutant that cannot mediate Mn efflux does not. Note that many of the neurons in our primary cultures were positively stained with an antibody against GAD 65/67 (Fig. 11G), which are markers of GABAergic neurons (Stephenson et al., 2005). We failed to detect TH-positive DAergic neurons in the cultures. This was not surprising given that DAergic neurons are fragile in culture and, without the addition of neuroprotective factors such as brain or glial-derived neurotrophic factors, constitute <10% of total neurons in a primary culture (Orme et al., 2013). Because the major morphological changes in Mn-induced parkinsonism occur in the GABAergic neurons of the globus pallidus, we did not attempt further to enrich our primary cultures for DAergic neurons in this study.
In summary, the data obtained in cell culture, C. elegans, and primary neurons identify SLC30A10 to be a surface-localized Mn efflux transporter that reduces cellular Mn levels and safeguards cells and neurons from Mn toxicity. Furthermore, the results indicate that familial parkinsonism caused by SLC30A10 mutations occurs as a result of a specific block in the Mn efflux activity of SLC30A10. Finally, these findings underline the therapeutic potential of enhanced efflux in the management of Mn-induced parkinsonism.
Discussion
Our results shed novel light on the mechanisms by which mutations in SLC30A10 cause familial parkinsonism. SLC30A10 is a cell surface-localized Mn efflux transporter. Mutations in SLC30A10 that cause parkinsonism block the Mn efflux function of SLC30A10. Expression of these mutants by themselves does not induce deleterious effects. However, SLC30A10 is widely expressed in the nervous system, including in the basal ganglia (Quadri et al., 2012), and in individuals homozygous for SLC30A10 mutations, neurons of the basal ganglia will be highly sensitive to Mn toxicity.
Although Mn is required for life, elevated cellular levels are toxic and induce apoptosis (Roth et al., 2002; Aschner et al., 2009; Milatovic et al., 2009). When systemic levels of Mn increase, Mn is preferentially deposited in neurons of the basal ganglia, and damage to these neurons leads to the observed parkinsonism (Olanow, 2004; Perl and Olanow, 2007; Aschner et al., 2009). Interestingly, the major morphological changes seen in Mn-induced parkinsonism occurs in the globus pallidus and striatum (Olanow, 2004; Liu et al., 2006; Perl and Olanow, 2007), which contain GABAergic neurons (Gerfen and Surmeier, 2011). This pathologic picture is distinct from idiopathic Parkinson's disease in which DAergic neurons of the substantia nigra pars compacta are degraded (Olanow, 2004; Perl and Olanow, 2007). However, decreases in total striatal dopamine levels and agonist-induced dopamine release have been reported in animal models of Mn-induced parkinsonism, suggesting that the DAergic neurons are functionally compromised (Guilarte et al., 2006; Liu et al., 2006). Whether the Mn-induced damage initiates as a functional defect in DAergic neurons which then progresses to GABAergic neurons or vice versa is not clear. Because the goal of our current study was to understand the functional differences between SLC30A10–WT and mutants, we wanted to ensure that our results were not influenced by neuron-specific effects. Therefore, we assayed SLC30A10 function using DAergic and GABAergic model systems.
Interestingly, development of parkinsonism in patients with SLC30A10 mutations does not require exposure to elevated Mn (Tuschl et al., 2008, 2012; Quadri et al., 2012). How then are the neurons of the basal ganglia in these individuals exposed to high Mn? Mn is ubiquitously available through diet, and the excretion of Mn primarily occurs by secretion into bile (Aschner et al., 2009; Butterworth, 2013). SLC30A10 is also expressed in the liver (Quadri et al., 2012) and likely plays a major role in transporting Mn into bile because patients with SLC30A10 mutations exhibit increased serum Mn levels, suggesting that they have significantly lowered Mn excretion (Quadri et al., 2012; Tuschl et al., 2012). This retained Mn is expected to cross the blood–brain barrier and eventually induce neuronal damage.
The five disease-causing SLC30A10 mutants that we tested exhibited defective trafficking (Fig. 1). Several other human diseases occur as a result of altered trafficking of ion transporters. As examples, defective trafficking of the chloride transporter cystic fibrosis transmembrane conductance regulator (CFTR), the water transporter aquaporin, and the Cu transporter ATP7A cause cystic fibrosis, nephrogenic diabetes insipidus, and Wilson's disease, respectively (Sato et al., 1996; Rubenstein et al., 1997; Rubenstein and Zeitlin, 1998; Tamarappoo and Verkman, 1998; Burrows et al., 2000; Roth et al., 2012). Interestingly, CFTRΔF508, the most common CFTR mutation that causes cystic fibrosis, and several disease-causing aquaporin mutations are trapped in the endoplasmic reticulum (Sato et al., 1996; Rubenstein et al., 1997; Rubenstein and Zeitlin, 1998; Tamarappoo and Verkman, 1998). Chemical chaperones, which are small-molecular-weight compounds that aid the folding of proteins and help them escape the quality-control systems of the endoplasmic reticulum (Papp and Csermely, 2006; Roth et al., 2012), have been used to rescue the trafficking and function of CFTR and aquaporin mutants (Sato et al., 1996; Rubenstein et al., 1997; Rubenstein and Zeitlin, 1998; Tamarappoo and Verkman, 1998). Importantly, rescuing the trafficking of CFTRΔF508 with a chemical chaperone reversed the disease phenotype in a human trial (Rubenstein and Zeitlin, 1998). Although it is not yet clear whether the defect in Mn efflux seen with SLC30A10 mutants is solely attributable to a block in trafficking, it is likely that some, if not most, of these mutants will retain the capability to mediate Mn transport. Therefore, a translationally important area of research emerging from our work is to identify chemical chaperones or small molecules that can restore trafficking of SLC30A10 mutants and subsequently determine whether restoring trafficking also rescues function. Identification of a chaperone or molecule that rescues SLC30A10 function will be a therapeutic breakthrough.
An interesting observation in the C. elegans experiments was that expression of the L89P mutant exerted a dominant-negative effect in the DAergic neurodegeneration assay. That is, after Mn treatment, the extent of DAergic neurodegeneration in L89P-expressing worms was more than that seen in transfection controls (Fig. 8G,H). This dominant-negative effect was not observed in the viability and basal slowing response assays. The likely explanation for the difference in outcome between assays is that the L89P-expressing DAergic neurons undergo some form of recovery with time. Indeed, for the DAergic neurodegeneration assay, worms were allowed to recover for 4 h after a 1 h exposure to Mn, whereas for the viability and basal slowing responses, the recovery period was 24 h. Nonetheless, because the disease-causing SLC30A10 mutants exhibit altered cellular localization, the possibility that their expression somehow alters the trafficking and/or activity of other proteins required for Mn detoxification thereby increasing neurodegeneration needs to be considered in discussions of the pathobiology of this disease.
The observation that overexpression of SLC30A10–WT protects cells and worms from Mn toxicity is also of clinical significance, because it provides the first evidence that increasing efflux can protect against Mn toxicity at the organismal level. Implicit to this observation is the possibility that increasing efflux may be a viable strategy for the management of Mn-induced parkinsonism attributable to occupational or environmental exposure to elevated Mn levels in individuals who do not carry mutations in SLC30A10. Although gene therapy with SLC30A10 is challenging, small molecules or drugs that increase efflux either by altering SLC30A10 levels or activity may be therapeutically promising. Expression of SLC30A10 transcript was enhanced by Mn (Quadri et al., 2012), suggesting that molecular mechanisms to increase SLC30A10 expression exist in cells and may be amenable to therapeutic modulation. Furthermore, efflux enhancing small molecules may also act by mechanisms independent of SLC30A10. We demonstrated previously that two other transporters, SPCA1 and ferroportin, play a role in mediating Mn efflux (Yin et al., 2010; Mukhopadhyay and Linstedt, 2011). Mutations in SPCA1 and ferroportin are known to occur in humans but do not cause a Mn toxicity phenotype (Van Baelen et al., 2004; Brissot et al., 2011), suggesting that SLC30A10 is the primary transporter responsible for mediating Mn efflux. However, it is feasible that a particular drug may increase the Mn transport activity of SPCA1 or ferroportin by changing their expression, localization, or activity. Identification of a drug that increases efflux independent of SLC30A10 will be beneficial for patients who suffer from parkinsonism attributable to mutations in SLC30A10, as well as those who develop parkinsonism solely attributable to Mn toxicity.
In conclusion, our results elucidate the function of SLC30A10 and further characterize the mechanism by which mutations in this protein alter its cellular activity and contribute to the development of a familial parkinsonian syndrome. These findings highlight the importance of interactions between genetic and environmental factors in the development of parkinsonian syndromes and identify enhanced Mn efflux to be a potentially viable strategy for the management of Mn-induced parkinsonism.
Footnotes
This work was funded by National Institutes of Health Grants R01-ES010563 (M.A. and A.B.B.) and R00-ES020488 (S.M.). We thank Dr. Andrey Selyunin for technical assistance in microscopy assays, Dr. Donald Smith for providing us with the AF5 cells, Dr. Ruth Valentine for the FLAG-tagged SLC30A10–WT construct, Dr. Nathan Miller (director of the ICP-MS core facility at University of Texas at Austin) for performing the Mn measurement assays, Dr. M. Diana Neely for providing cDNAs from human induced stem cell-derived progenitors, Dr. Karin Tuschl for providing L89P SLC30A10 plasmid, Drs. Guy Caldwell and David Miller for providing gateway cloning vectors and microinjection markers, and Drs. Adam D. Linstedt and Manoj Puthenveedu for critical reading of this manuscript.
References
- Aschner M, Erikson KM, Herrero Hernández E, Tjalkens R. Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med. 2009;11:252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachert C, Lee TH, Linstedt AD. Lumenal endosomal and Golgi-retrieval determinants involved in pH-sensitive targeting of an early Golgi protein. Mol Biol Cell. 2001;12:3152–3160. doi: 10.1091/mbc.12.10.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. Extracellular dopamine potentiates Mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet. 2010;6:e1001084. doi: 10.1371/journal.pgen.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosomworth HJ, Thornton JK, Coneyworth LJ, Ford D, Valentine RA. Efflux function, tissue-specific expression and intracellular trafficking of the Zn transporter ZnT10 indicate roles in adult Zn homeostasis. Metallomics. 2012;4:771–779. doi: 10.1039/c2mt20088k. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Brissot P, Bardou-Jacquet E, Jouanolle AM, Loréal O. Iron disorders of genetic origin: a changing world. Trends Mol Med. 2011;17:707–713. doi: 10.1016/j.molmed.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Burkhard PR, Delavelle J, Du Pasquier R, Spahr L. Chronic parkinsonism associated with cirrhosis: a distinct subset of acquired hepatocerebral degeneration. Arch Neurol. 2003;60:521–528. doi: 10.1001/archneur.60.4.521. [DOI] [PubMed] [Google Scholar]
- Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97:1796–1801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth RF. Parkinsonism in cirrhosis: pathogenesis and current therapeutic options. Metab Brain Dis. 2013;28:261–267. doi: 10.1007/s11011-012-9341-7. [DOI] [PubMed] [Google Scholar]
- Cao S, Gelwix CC, Caldwell KA, Caldwell GA. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci. 2005;25:3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Burdette AJ, Porter JC, Ricketts JC, Fox SA, Nery FC, Hewett JW, Berkowitz LA, Breakefield XO, Caldwell KA, Caldwell GA. The early-onset torsion dystonia-associated protein, torsinA, is a homeostatic regulator of endoplasmic reticulum stress response. Hum Mol Genet. 2010;19:3502–3515. doi: 10.1093/hmg/ddq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks DR, Welch N, Smith DR. Low-level manganese exposure alters glutamate metabolism in GABAergic AF5 cells. Neurotoxicology. 2007;28:548–554. doi: 10.1016/j.neuro.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LM, Maiya R, Sullivan MA, Han Y, Walton HA, Boehm SL, 2nd, Bergeson SE, Mayfield RD, Morrisett RA. Sindbis viral-mediated expression of eGFP-dopamine D1 receptors in situ with real-time two-photon microscopic detection. J Neurosci Methods. 2004;139:25–31. doi: 10.1016/j.jneumeth.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Dokmanić I, Sikić M, Tomić S. Metals in proteins: correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr D Biol Crystallogr. 2008;64:257–263. doi: 10.1107/S090744490706595X. [DOI] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- Guilarte TR, Chen MK, McGlothan JL, Verina T, Wong DF, Zhou Y, Alexander M, Rohde CA, Syversen T, Decamp E, Koser AJ, Fritz S, Gonczi H, Anderson DW, Schneider JS. Nigrostriatal dopamine system dysfunction and subtle motor deficits in manganese-exposed non-human primates. Exp Neurol. 2006;202:381–390. doi: 10.1016/j.expneurol.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Huang L, Tepaamorndech S. The SLC30 family of zinc transporters - a review of current understanding of their biological and pathophysiological roles. Mol Aspects Med. 2013;34:548–560. doi: 10.1016/j.mam.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM., Jr Pathology of inherited manganese transporter deficiency. Ann Neurol. 2014;75:608–612. doi: 10.1002/ana.24131. [DOI] [PubMed] [Google Scholar]
- Liu X, Sullivan KA, Madl JE, Legare M, Tjalkens RB. Manganese-induced neurotoxicity: the role of astroglial-derived nitric oxide in striatal interneuron degeneration. Toxicol Sci. 2006;91:521–531. doi: 10.1093/toxsci/kfj150. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lu M, Fu D. Structure of the zinc transporter YiiP. Science. 2007;317:1746–1748. doi: 10.1126/science.1143748. [DOI] [PubMed] [Google Scholar]
- Lucchini RG, Guazzetti S, Zoni S, Donna F, Peter S, Zacco A, Salmistraro M, Bontempi E, Zimmerman NJ, Smith DR. Tremor, olfactory and motor changes in Italian adolescents exposed to historical ferro-manganese emission. Neurotoxicology. 2012;33:687–696. doi: 10.1016/j.neuro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros VR. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatovic D, Zaja-Milatovic S, Gupta RC, Yu Y, Aschner M. Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol Appl Pharmacol. 2009;240:219–225. doi: 10.1016/j.taap.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Linstedt AD. Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity. Proc Natl Acad Sci U S A. 2011;108:858–863. doi: 10.1073/pnas.1013642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science. 2012;335:332–335. doi: 10.1126/science.1215930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Bachert C, Smith DR, Linstedt AD. Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol Biol Cell. 2010;21:1282–1292. doi: 10.1091/mbc.E09-11-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Redler B, Linstedt AD. Shiga toxin-binding site for host cell receptor GPP130 reveals unexpected divergence in toxin-trafficking mechanisms. Mol Biol Cell. 2013;24:2311–2318. doi: 10.1091/mbc.E13-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatomo S, Umehara F, Hanada K, Nobuhara Y, Takenaga S, Arimura K, Osame M. Manganese intoxication during total parenteral nutrition: report of two cases and review of the literature. J Neurol Sci. 1999;162:102–105. doi: 10.1016/S0022-510X(98)00289-5. [DOI] [PubMed] [Google Scholar]
- Ohana E, Hoch E, Keasar C, Kambe T, Yifrach O, Hershfinkel M, Sekler I. Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter. J Biol Chem. 2009;284:17677–17686. doi: 10.1074/jbc.M109.007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW. Manganese-induced parkinsonism and Parkinson's disease. Ann N Y Acad Sci. 2004;1012:209–223. doi: 10.1196/annals.1306.018. [DOI] [PubMed] [Google Scholar]
- Orme RP, Bhangal MS, Fricker RA. Calcitriol imparts neuroprotection in vitro to midbrain dopaminergic neurons by upregulating GDNF expression. PLoS One. 2013;8:e62040. doi: 10.1371/journal.pone.0062040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp E, Csermely P. Chemical chaperones: mechanisms of action and potential use. Handb Exp Pharmacol. 2006:405–416. doi: 10.1007/3-540-29717-0_16. [DOI] [PubMed] [Google Scholar]
- Perl DP, Olanow CW. The neuropathology of manganese-induced Parkinsonism. J Neuropathol Exp Neurol. 2007;66:675–682. doi: 10.1097/nen.0b013e31812503cf. [DOI] [PubMed] [Google Scholar]
- Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90:467–477. doi: 10.1016/j.ajhg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JA, Horbinski C, Higgins D, Lein P, Garrick MD. Mechanisms of manganese-induced rat pheochromocytoma (PC12) cell death and cell differentiation. Neurotoxicology. 2002;23:147–157. doi: 10.1016/S0161-813X(01)00077-8. [DOI] [PubMed] [Google Scholar]
- Roth SD, Schüttrumpf J, Milanov P, Abriss D, Ungerer C, Quade-Lyssy P, Simpson JC, Pepperkok R, Seifried E, Tonn T. Chemical chaperones improve protein secretion and rescue mutant factor VIII in mice with hemophilia A. PLoS One. 2012;7:e44505. doi: 10.1371/journal.pone.0044505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med. 1998;157:484–490. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100:2457–2465. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JF, Crooks DR, Lee CT, Schoen CJ, Amable R, Zeng X, Florival-Victor T, Morales N, Truckenmiller ME, Smith DR, Freed WJ. GABAergic lineage differentiation of AF5 neural progenitor cells in vitro. Cell Tissue Res. 2006;324:1–8. doi: 10.1007/s00441-005-0094-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/S0896-6273(00)81199-X. [DOI] [PubMed] [Google Scholar]
- Sepúlveda MR, Vanoevelen J, Raeymaekers L, Mata AM, Wuytack F. Silencing the SPCA1 (secretory pathway Ca2+-ATPase isoform 1) impairs Ca2+ homeostasis in the Golgi and disturbs neural polarity. J Neurosci. 2009;29:12174–12182. doi: 10.1523/JNEUROSCI.2014-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanwood GD, Leitch DB, Savchenko V, Wu J, Fitsanakis VA, Anderson DJ, Stankowski JN, Aschner M, McLaughlin B. Manganese exposure is cytotoxic and alters dopaminergic and GABAergic neurons within the basal ganglia. J Neurochem. 2009;110:378–389. doi: 10.1111/j.1471-4159.2009.06145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson DT, Li Q, Simmons C, Connell MA, Meglasson MD, Merchant K, Emborg ME. Expression of GAD65 and GAD67 immunoreactivity in MPTP-treated monkeys with or without L-DOPA administration. Neurobiol Dis. 2005;20:347–359. doi: 10.1016/j.nbd.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998;101:2257–2267. doi: 10.1172/JCI2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M, Clayton PT. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia—a new metabolic disorder. J Inherit Metab Dis. 2008;31:151–163. doi: 10.1007/s10545-008-0813-1. [DOI] [PubMed] [Google Scholar]
- Tuschl K, Clayton PT, Gospe SM, Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Chong WK, Mills PB. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90:457–466. doi: 10.1016/j.ajhg.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Baelen K, Dode L, Vanoevelen J, Callewaert G, De Smedt H, Missiaen L, Parys JB, Raeymaekers L, Wuytack F. The Ca2+/Mn2+ pumps in the Golgi apparatus. Biochim Biophys Acta. 2004;1742:103–112. doi: 10.1016/j.bbamcr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Yin Z, Jiang H, Lee ES, Ni M, Erikson KM, Milatovic D, Bowman AB, Aschner M. Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J Neurochem. 2010;112:1190–1198. doi: 10.1111/j.1471-4159.2009.06534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]