

Abstract
The epithelial to mesenchymal transition (EMT) is implicated in many processes, ranging from tissue and organogenesis to cancer and metastatic spread. Understanding the key regulatory mechanisms and mediators within this process offers the opportunity to develop novel therapeutics with broad clinical applicability. To date, several components of EMT already are targeted using pharmacologic agents in fibrosis and cancer. As our knowledge of EMT continues to grow, the potential for novel therapeutics will also increase. This review focuses on the role of EMT both as a necessary part of development and a key player in disease progression, specifically the similarity in pathways used during both processes as targets for drug development. Also, the key role of the tumor microenvironment with EMT is outlined, focusing on both co-factors and cell types with the ability to modulate the progression of EMT in cancer and metastatic disease. Lastly, we discuss the current status of clinical therapies both in development and those progressed to clinical trial specifically targeting pathologic EMTs including small molecule inhibitors, non-coding RNAs, exogenous co-factors, and adjunctive therapies to current chemotherapeutics.
Keywords: Epithelial mesenchymal transition (EMT), Small molecule inhibitors, TGFβ, Cancer associated fibroblasts
1 Introduction
The epithelial to mesenchymal transition (EMT) was first described in the context of being a fundamental component in the differentiation of tissues and organs within the developing embryo [1]. Beyond tissue and organogenesis, EMT has been described as a crucial mediator of wound healing, a feature of EMT necessary for tissue remodeling post-injury [2]. The characteristics of EMT that are essential to wound healing also connect EMT to organ fibrosis, including pulmonary fibrosis, renal fibrosis, and hepatic fibrosis. In these settings, EMT is intimately linked to diseased states. The integral role of EMT in disease was taken one step further with evidence supporting a key role in tumor initiation and, subsequently, the progression of metastatic disease [3].
EMT is contemporarily classified into 3 classes: type 1 referring to embryogenesis; type 2 referring to wound healing; type 3 referring to its role in cancer and metastases. A hallmark of each is the process through which epithelial cells lose their cell-cell junctions, apical-basal polarity, and re-organize their cytoskeleton by e-configuring their gene expression. These processes are highly regulated in the setting of type 1 and 2 EMT. In type 3 EMT, dysregulation leads to diseased states [4].
On a molecular level, EMT is defined by several intermediates responsible for influencing gene expression. These include transcription factors, multiple interacting signal pathways, promoter sequence modifications, and autocrine signaling. Of these, the principal regulators of EMT are transcription factors classified into 3 families: SNAIL, TWIST, and zinc-finger E-box-binding (ZEB). The fundamental event in EMT is the repression of epithelial cadherin (E-cadherin), which is responsible for maintaining adherens junctions and cell-cell adhesion. SNAIL, TWIST, and ZEB factors all can repress E-cadherin while also activating key mesenchymal genes including N-cadherin (cell-cell adhesion), vimentin (an intermediate filament protein that regulates cell motility), and fibronectin 1 (cell growth and migration). Together, these transcription factors orchestrate the activation of mesenchymal gene expression while repressing epithelial genes through diverse downstream targets.
The same mediators used for modifying gene expression are used as biomarkers to identify a cell as epithelial or mesenchymal. For example, expression of E-cadherin is used to describe a cell as being in an epithelial state whereas expression of N-cadherin, vimentin, and/or fibronectin 1 are used to describe the mesenchymal phenotype (see Figure 1). The importance and clinical relevance of these pathways and mediators will be discussed throughout this review [5].
Figure 1.
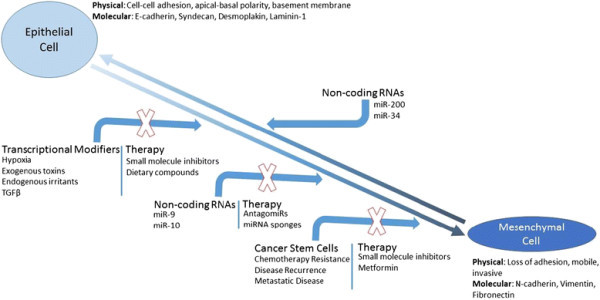
Schematic of EMT, EMT inducers, and potential or existing areas within the framework for therapeutic intervention.
The focus of this review will be on 3 major areas. First, the role of EMT as a necessary pathway to development and healing. Next, the pathophysiologic impact of EMT on diseased states (primarily fibrotic phenotypes and cancer). Finally, discussing the areas within the spectrum of EMT which allow for novel therapeutics and the current progress towards clinical application.
2 Review
2.1 SECTION 1: The role of EMT in development and diseased states
2.1.1 EMT and development
EMTs are pathways followed during normal development; most organs and adult tissues are generated via these transitions embryologically. Each of these transitions rely on signal cascades with multiple regulatory and feedback loops allowing both plasticity and cooperation during development. EMT plays a prominent role during gastrulation and many of the key molecular players during these processes have been noted to be important in disease progression during adulthood. For example, members of the TGFβ superfamily are powerful inductors of gastrulation [6]. SNAIL1 and SNAIL2, known mediators of EMT in adults, are dependent on TGFβ activation and required for gastrulation to proceed [7],[8].
The importance of key EMT mediators in early gastrulation is further emphasized in SNAIL-mutant mice where cells are unable to migrate from the primitive streak, effectively halting development [9]. TGFβ signaling and the SNAIL family of transcription factors are two important influences during dysregulated EMT in adult disease, as well. Understanding the signaling targets during embryologic EMT and comparing them to those found in the progression of disease is an area of research that may offer new therapeutic targets.
2.1.2 EMT and diseased states
EMT has a role in several disease states including pathologic organ degradation (ie fibrosis) and cancer. How EMT fits into these processes will be reviewed, including the initiating signal pathways, downstream targets, and mechanisms for regulating these pathologic EMTs.
In all pathologic EMTs, a series of steps are conserved and are crucial for defining an epithelial cell’s conversion to a mesenchymal phenotype. Epithelial cells are broadly characterized as those positioned on the basement membrane (either as single layered or multilayered tissues), show polarity, adhere, and communicate using cell-cell junctions [10]. Loss of these characteristics places epithelial cells on a spectrum. On one end is the epithelial phenotype and on the other end is the mesenchymal phenotype. At the furthest extreme, the mesenchymal phenotype, the result is cells which are invasive and resistant to apoptosis [11]. These features of EMT are consistent between cell types and tissue environments.
One such example can be seen in the pathways regulating cell-cell junctions and their dissolution during EMT. Cell-cell junctions are critical for maintaining basement membranes and the integrity of epithelial layers. In vertebral cells, tight junctions, gap junctions, and desmosomes are the primary regulators of these connections [11],[12]. Several EMT activators act either jointly or independently on each of these cell contact mediators and their impact on cell-cell junctions is nearly the same regardless of cell type [5]. The importance of factors on the cell surface to EMT is further illustrated by recent findings illustrating that alterations in the glycosylation of cell surface glycoproteins impacts cellular adhesive properties. The characteristics of altered O- and N-glycans through mRNA expression during tumor progression is extremely relevant and well described in a recent review [13].
EMT and organ fibrosis
Fibrotic tissues are characterized by an up-regulation of extracellular matrix (ECM) protein deposition eventually leading to compromise of organ function, and in the most severe cases, organ failure [14]. The cells responsible for ECM protein deposition are myofibroblasts typically in response to epithelial or endothelial injury [15]. As tissue repair progresses, the role of myofibroblasts shifts from creating a scaffold for healing into facilitating wound contraction and closure [16]. Myofibroblasts originate from several cell types most notably existing epithelial cells, endothelial cells, interstitial fibroblasts, and myeloid precursor cells in bone marrow [17]-[20].
Inflammatory cytokines, specifically TGFβ and IL-1, are critical in the differentiation of cells from any site into myofibroblasts [21]. In wound healing, myofibroblasts are key to creating wound tension and depositing important co-factors required for wound contraction. This process is tightly regulated, and at its completion, the population of myofibroblasts recruited to the site of injury to promote healing undergoes population-wide apoptosis [22]. In situations where myofibroblasts persist in the extracellular matrix, a fibrotic phenotype develops. In wounds, this is manifested as hypertrophic scar or keloid [23]. In organs, the result is fibrosis [24],[25].
Alveolar epithelial cells, hepatocytes, lens epithelium, endothelial cells, mesothelial cells, and cardiomyocytes all have the potential to undergo EMT and resemble myofibroblasts, subsequently contributing to organ fibrosis [26]. In pulmonary fibrosis models, TGFβ stimulates EMT in alveolar epithelial cells. Similarly, CCL4-induced liver fibrosis is linked to EMT in hepatocytes. Recent evidence shows a role for EMT in peritoneal fibrosis in patients that receive peritoneal dialysis in a MAPK and SNAIL1 dependent fashion [27]. EMT in lens epithelial cells causes progressive capsular opacification and can be prevented by inhibiting TGFβ with Smad7 [26].
In renal fibrosis, mice that do not express Smad3 (a target of TGFβ) are protected from development of fibrosis. The crucial role of TGFβ in renal fibrosis was taken a step further by Zeisberg et al. who demonstrated that inhibiting TGFβ with an antagonist (BMP7) actually could revert renal fibrosis in mice [28].
EMT and cancer
The pathologic potential of EMT is seen in organ fibrosis and similar mechanisms are seen in the development of cancer, evolution of disease, and metastatic progression. In cancer, EMTs rely on: 1) exogenous induction agents acting through transcriptional control 2) Non-coding RNAs and 3) creating and maintaining cancer stem cells [29].
1. Transcriptional control mechanisms
The discovery of the transcription factor SNAIL1 interacting with the CDH1 promoter to repress the production of E-cadherin began our understanding of how EMT is regulated [30],[31]. Numerous other transcription factors have been found to play a role in the progression of epithelial cells to a mesenchymal phenotype including SNAIL2, ZEB1, ZEB2, E47, KLF8, TWIST1, and FOXC2, all which have the ability to promote EMT in various cell cancer lines [32],[33]. Of these, the nuclear factors which seem to be most important are SNAIL, ZEB, and TWIST proteins and these can be modulated to initiate EMT by exogenous stimuli including hypoxia, bile acids, and nicotine [34]-[36].
The association between disease progression and solid tumor oxygenation is seen in various malignancies. It is apparent that tumor hypoxia contributes to several features of aggressive cancers by enhancing invasive growth, local spread, and distant deposits [37]. The mechanism for hypoxic influence of these features is likely multifactorial, however there is clear interplay with EMT. In pancreatic tumor cells, hypoxia (induced by culturing cells in a hypoxic chamber) resulted in expression of EMT makers when compared to normal conditions [34]. This observation is seen in other solid tumors including lung and breast [38].
Cholestatic disorders seem to promote hepatic fibrosis and to the progression of carcinogenesis in cholangiocytes and hepatocytes. Studies have shown that chendeocycholic acid (CDCA), a bile acid, can activate EGFR to mediate gene expression. In a cDNA microarray, CDCA increased the expression of SNAIL to repress E-cadherin both in vitro and in vivo in liver cancer models, initiating an EMT program [36]. The ability for bile acids to play an activating role is just one of many examples where exogenous substrates can induce EMT.
Another substrate which activates EMT through a similar mechanism is nicotine. Specifically, in breast and lung cancer cell lines (A549, MDA-MB-468, MCF-7), treatment with nicotine increased the proliferation, invasion, and migration of both lung and breast cancer cells in a dose-dependent fashion. Notably, expression of E-cadherin was repressed while the expression of fibronectin and vimentin, both mesenchymal markers, increased. The mechanism for these changes occurred through signaling by nicotinic acetylcholine receptors (nAChRs) [35].
Recent data demonstrates a pathway for cigarette smoke to stimulate EMT in small bronchi of patients demonstrating a mechanism contributing to the progression of disease. In primary human bronchial epithelial cells (HBEC), exposure to cigarette smoke led to expression of EMT markers by increased production of intracellular ROS, release of TGFβ, and TGFβ activation of the transcription factors Smad3 and ERK1/2 [39]. This kind of interaction between an exogenous substrate and the cellular machinery activating EMT has significant clinical implication as treatments can be designed to potentially halt or reverse the progression of disease.
2. Non-coding RNAs
Non-coding RNAs and, in particular, microRNAs (miRNAs) have gained significant attention for their ability to modify gene expression allowing for quick cellular adaptation to new environments and conditions [29],[40]. The regulatory association of miRNAs is seen in many settings including development, maintaining cell homeostasis, and cell-cell interaction [41]. Li et al., in 2009, showed a role for miRNAs in reversing EMT in pancreatic tumor cells that were resistant to common chemotherapeutics [42].
Since that time, numerous miRNAs have been found to interact either directly or indirectly with EMT machinery in several cell models [43]. Interestingly, these miRNAs can act as either tumor suppressors or tumor activators based on the cell type and environment they interact with. Additionally, their adaptive properties play a role in chemotherapy resistance.
Two major classes of miRNAs have been studied extensively in the setting of EMT: the miR-200 family and the miR-34 family. Other families include the miR-205, miR-9, and miR-10 families, respectively. The miR-200 family has five named members located on human chromosome 1 (mir-200a, mir-200b, mir-429) and 12 (mir-200c, mir-141). Both the miR-200 and miR-34 families are typically considered to be “tumor suppressive” as their members all stabilize the epithelial phenotype [44].
The interplay between miR-200 and the ZEB family of transcription factors to maintain E-cadherin expression has been validated in several systems. In a TGFβ-induced model of EMT in a murine mammary cell line, miR-200 family member expression was repressed. This same cell line was transfected with synthetic pre-miRNAs to stimulate overexpression of corresponding miR-200 members in vitro. The result was inhibiting EMT in the transfected cell lines, when compared to control, by increasing the expression of E-cadherin. Furthermore, using an elegantly designed luciferase assay, miR-200 family members were noted to interact directly with the transcription factors ZEB1 and ZEB2 [45]. ZEB1 and ZEB2 typically act as EMT inducers by suppressing E-cadherin expression, however in the presence of miR-200, ZEB1 and ZEB2 could not interact with E-cadherin and the epithelial phenotype was maintained [45],[46].
The clinical relevance of miR-200 family members can be seen in malignant breast, prostate, and colon cancer cells. Silencing of the miR-200 family induces early transformation of epithelial cells leading to EMT, reinforcing the role of these miRNAs in tumor suppression [47].
An interesting link between the p53 tumor suppressor gene, SNAIL, and the miR-34 was described by Siemans et al. in 2011. In colon cancer cells, activation of p53 notably induced the expression of miR-34 family genes, resulting in the inhibition of SNAIL activation. As would be predicted, a correlate decrease in EMT markers was seen in the absence of SNAIL activation in this cell line. Treatment of cells with miR-34 halted TGFβ dependent EMT. It was noted, however, that the action of miR-34 family members was not unidirectional. The miR-34 family promoter sequences contain E-boxes which can be bound by SNAIL, resulting in decreased expression of miR-34. This double negative feedback loop between miR-34 and SNAIL offers an interesting regulatory mechanism for EMT creating unique challenges in drug development targeting either of these factors [48].
In contrast to the tumor suppressive functions of miR-200 and miR-34, the miR-9 family appears to be tumor activating through a direct interaction with the CDH1 promoter which subsequently initiates EMT [49]. miR-9 expression is increased in primary breast, gastric, and brain cancers [29]. Additionally, increased expression of miR-9 is seen in metastatic disease further supporting a tumorigenic role for this family of miRNAs.
It must be emphasized that work with non-coding RNAs is far from complete and their relevance to cancer (and cancer therapy) remains imprecise. While the miR-200 family appears to have compelling data for a tumor suppressive function, as discussed above, there is are also data indicating a pro-metastatic niche for miR-200 family members.
In samples from colon cancer patients, there is lowered expression of miR-200 within the primary tumor which fits the impression that miR-200 is tumor suppressive. However, in metastatic lesions, miR-200 expression is high [47],[50]. This allows for argument that miR-200 plays a pro-metastatic role in cancer by stabilizing the mesenchymal phenotype after deposition to distant sites (i.e. an important regulatory element in the mesenchymal to epithelial transition). Other evidence for a pro-metastatic role for miR-200 is seen in pancreatic, breast, and gastric cancer.
Another important miRNA family is the miR-10 group including miR10b. In the study of metastatic breast cancer cells, miR-10b was noted to be highly expressed and positively regulate cell invasion. It was also able to initiate tumor formation and played a role in distant metastases. Taken together, these properties are highly suggestive that miR-10b is implicated in EMT [51].
3. Cancer stem cells
The concept of the cancer stem cell is based on the hypothesis that a single cell type or subset of cells can act as tumor-initiators in various cancers ranging from hematopoietic to solid organ. By definition, these cancer stem cells (CSC) have features which give them “stem-like” properties allowing for self-renewal and differentiation leading to the histologic heterogeneity displayed by tumors [52].
Seminal work done in the mid-1990s provided compelling evidence supporting the role of CSCs in tumor initiation and progression in acute myeloid leukemia (AML). The AML-initiating cells, defined by their cell surface marker expression (CD34+,CD38-), were able to proliferate, disseminate, and maintain an AML phenotype similar to the patients they were derived from after transplantation into severe combined immune-deficient (SCID) mice [53].
Furthermore, the AML-initiating cells appeared to be derived from normal hematopoietic stem cells directly linking stem cells to tumorigenic cells. In other words, normal primitive cells underwent leukemic transformation resulting in the first experimentally described CSC population [54]. This pattern of tumor-initiation has subsequently been described in various solid tumors including brain, breast, pancreatic, prostate, and others [55]-[59].
Ultimately, CSCs exert their pathophysiologic impact in several ways. Their ability to self-renew and differentiate is at least partly responsible for the heterogeneity seen within tumors. One potential consequence of this is chemotherapy and radiotherapy resistance within populations of tumors. Additionally, CSCs can regenerate a tumor when transplanted into an appropriate host in experimental settings. This offers a proposed mechanism for metastatic disease and cancer recurrence. These properties have made CSCs, irrespective of their tumor of origin, extremely important for cancer therapy and an important new cancer target.
The machinery of EMT appears to connect directly to the formation and maintenance of CSCs. Mani et al. induced EMT in human, non-tumorigenic mammary epithelial cells using either TWIST or SNAIL transcription factors. As seen in prior studies, exposed cells acquired a mesenchymal phenotype [30]. Studying these cells using flow cytometry revealed a pattern of cell surface expression that was seen in breast cancer stem cells (CD44High/CD24Low). EMT made non-tumorigenic epithelial cells, at least in breast tissue, look like cancer stem cells.
While it was interesting that EMT made epithelial cells look like stem cells, more important was the discovery that these cells acted like stem cells. CD44High/CD24Low cells were able to self-renew and give rise to multiple cell lineages. Furthermore, it was noted that CD44High/CD24Low cells expressed a mesenchymal phenotype and up-regulated the expression of EMT transcription factors [60]. Together, these findings connected two important cancer-related processes – CSCs and EMT.
Other important EMT signaling pathways are also implicated in the transformation and maintenance of CSCs. For example, Wnt signaling is a well-studied pathway necessary for a variety of EMTs, both developmental and pathologic [61]. Wnt signaling is classically described as being canonical or non-canonical based on the involvement of β-catenin [62]. Hyperactivity of canonical and non-canonical Wnt pathways can trigger EMT programs and, more recently, play key roles in CSC biology [61].
In fact, Wnt activity may be used as a marker for defining CSCs in some malignancies. Using colon CSCs derived from patient samples, Wnt signaling was assessed using a GFP reporter system. Cells with high GFP expression notably had increased clonogenicity compared to those cells with low GFP expression. Stated another way, colon CSCs with high Wnt expression were highly potent at inducing tumors following injection into mice. The extent of Wnt activation was positively correlated with expression of well-studied cell surface makers for CSCs [63].
Notch signaling is another important pathway required for the conversion and maintenance of CSCs using EMT. Multiples studies show the importance of Notch signaling in multiple breast cancer subtypes and its role in invasive disease. Notch signaling, specifically Notch4 receptor activation, is increased in breast CSCs. In addition, Notch signal pathway inhibition decreases the number of breast CSCs both in vitro and in vivo [64].
The importance of Notch signaling is also seen in pancreatic CSCs. The γ-secretase inhibitor IX can be used to inactivate Notch signaling. After treating pancreatic CSCs with this secretase inhibitor, cell proliferation, migration, and invasion are all decreased. The mechanism for this appears to be through inhibiting the mesenchymal transition of pancreatic cells lines demonstrating the importance of Notch signaling to EMT [65]. Treatments directed at Wnt and Notch signal pathways may allow for regulation of EMT in tumors and modulation of CSCs.
A property of CSCs with important clinical relevance is the observation that they demonstrate resistance to both chemotherapy and radiotherapy [59]. Imatinib mesylate is a tyrosine kinase inhibitor used for the treatment of chronic myeloid leukemia (CML) with excellent clinical result. Early studies, following its approval for use by the Food and Drug Administration in 2001, showed a complete response to therapy in 76% of patients at a median follow-up of 19 months [66].
However, these results have since been somewhat tempered by the finding that patients with an early response to imatinib show evidence of disease recurrence [67]. The mechanism for this appears to be supported by a significant role for CSCs in resisting initial treatment and leading the progression to cancer recurrence as CSCs isolated from patients with CML show insensitivity to imatinib [68]. Some authors describe this as the “weed” or “dandelion” hypothesis as it may appear the problem is solved, but until the root is removed the weed can and will come back [69].
The “dandelion” hypothesis was tested by Li et al. in a breast cancer model. Cells were taken from patients with HER2+ breast cancer before and after treatment with lapatinib. Following chemotherapy, the percentage of CD44High/CD24Low cells increased. These cells, with a molecular signature consistent with breast CSCs, were able to self-renew and had a propensity for tumor formation [70]. CD44High/CD24Low breast CSCs appear to also show resistance to conventional radiation therapies [71]. The implication of these data are two-fold. First, chemotherapy and radiotherapy appear to select for the survival CSCs. Second, chemotherapy and radiotherapy do not impair the ability of CSCs to grow and replicate.
The focus of this review to this point has been on EMT, however an important process that must be discussed is its “opposite” pathway, mesenchymal to epithelial transition (MET). Just as EMT plays a critical role in development, MET also is a process first recognized in development. One specific example is the formation of epithelial nephrons which require the transcription factor Pax-2 to initiate MET. This illustrates that while MET functions to transition cells to an epithelial phenotype, the regulatory factors required are not necessarily the same for both processes [72]. MET is an expansive topic and the migrating cancer stem cell (MCSC) model will be discussed to briefly emphasize the role MET plays in metastatic disease and distant seeding of tumor cells.
The (MCSC) model attempts to integrate the concepts of CSC, EMT, MET, and metastatic disease. CSCs are generated via an EMT allowing them the ability to migrate and disseminate. Upon reaching a distant site, the CSC is exposed to a new environment which can either promote dormancy or undergo a MET to recapture epithelial characteristics. The next step would be the formation of a metastatic lesion with conserved features from the primary tumor [73].
The relevance to this model on a clinical level cannot be understated as therapies targeting EMT could inadvertently promote MET and progression of disease. Overcoming these challenges will likely define the next steps in cancer drug development directed at EMTs.
2.2 SECTION 2: Targeting EMT and developing novel therapies
Several mechanisms have been proposed to target EMT to develop novel cancer therapeutics. These targets include transcriptional regulators (ie SNAIL, TGFβ), non-coding RNAs, and CSCs. Additionally, a new focus of research centers on the interactions between the tumor microenvironment and its role in initiation, propagation, and termination of EMT. Novel therapeutic targets have emerged within the tumor microenvironment which have promising implications for cancer therapy.
As the key players within the epithelial-mesenchymal transition (EMT) continue to be elucidated, novel cancer targets are being identified within this framework to direct future clinical applications. Broadly, these therapeutic targets can be classified into 2 large categories: (1) preventing EMT (2) altering the tumor microenvironment.
2.2.1 Preventing EMT
Targeting mediators of transcriptional control
The first target for preventing EMT is developing interventions against very specific intermediates of the transition. Key players, as discussed, include TGFβ and transcription factors, and most current clinically-applicable therapies are based on these targets. TGFβ activity is modulated with several strategies including small molecule inhibitors and exogenous compounds.
1. Small molecule inhibitors
TGFβ signaling has classically been targeted using antisense oligonucleotides, monoclonal antibodies, and chemical inhibitors for treatment of various inflammatory and neoplastic conditions [74]. Small molecule inhibitors offer a novel approach to modulating the effects of TGFβ at multiple levels [75].
CX-4945 is a small molecule inhibitor of CK2 (protein kinase CK2, a serine/threonine kinase) and is the first orally bioavailable inhibitor in its class. Previously, a link between CK2 and TGFβ signaling was proposed, specifically by inducing EMT [76]. Treatment of a human lung adenocarcinoma cell line (A549) with CX-4945 demonstrated a potent inhibition on the signaling of TGFβ and down-regulation of EMT markers. CX-4945 inhibited the TGFβ1 cadherin switch, preventing progression towards a mesenchymal phenotype by A549 cells. Additionally, CX-4945 halted TGFβ mediated signaling in both canonical Smad pathways and non-Smad pathways (Akt and ERK1/2), Wnt pathways, and focal adhesion pathways (FAK, Src, MMP, NF-kB) [77].
CX-4945 (Silmitasertib) is currently in clinical trial for the treatment of advanced solid tumors and multiple myeloma. It has reached phase 2 study for treatment of non-resectable cholangiocarcinoma in combination with gemcitabine and cisplatin compared to standard care of gemcitabine plus cisplatin. The study is presently active and recruiting patients. CX-4945 is also in phase 1 study for the treatment of relapsed or refractory multiple myeloma.
Another group of small molecules targeting TGFβ signaling are ALK5 kinase inhibitors. Several have been described including EW-7195, EW-7203, EW-7197, IN-1130, SB-431542, SD-208, SD-093, Ki26894, LY580276, LY-573636, LY-2109761, and LY2152799 (see Table 1) [78],[79]. Each are novel small molecule inhibitors of ALK5 kinase (or TGFβ type 1 receptor kinase), required for TGFβ 1 signaling.
Table 1.
Small molecule inhibitors, mechanism, clinical targets, delivery, and status in clinical application
| Inhibitors | Mechanism | Target | Delivery | Clinical status |
|---|---|---|---|---|
| CX-4945 |
CK2 inhibitor |
Lung, myeloma |
Oral, IV |
Phase 1/2 |
| EW-7195 |
TGFβ Inhibitor |
Breast, Lung metastases |
Orally available |
Phase 1 Clinical Trial |
| EW-7203 |
TGFβ 1 and 2 receptor kinase inhibitor |
Breast, Lung metastases |
IV |
Pre-Clinical |
| EW-7197 |
TGFβ 1 and 2 receptor kinase inhibitor |
Breast, Lung metastases |
IV |
Pre-Clinical |
| IN-1130 |
TGFβ 1 and 2 receptor kinase inhibitor |
Breast, Lung metastases |
IV |
Pre-Clinical |
| SB-431542 |
Inhibition of ATP binding to ALK5 kinase |
Glioma |
IV |
Pre-Clinical |
| SD-208 |
TGFβ 1 receptor kinase inhibitor |
Metastatic breast cancer |
IV |
Pre-Clinical |
| SD-093 |
TGFβ 1 receptor kinase inhibitor |
Metastatic breast cancer |
IV |
Pre-Clinical |
| LY-2152799 |
TGFβ 1 receptor kinase antagonist |
Breast, Myelodysplastic syndromes, Hepatocellular Carcinoma, Pancreatic Cancer, Malignant Glioma |
Orally available |
Clinical trial: Phase 2/3; Phase 2; Phase 1b/2; Phase 1b/2a |
| LGK974 |
Porcupine inhibitor (Wnt signaling) |
Breast |
Orally available |
Phase 1 Clinical Trial |
| CWP232291 |
Wnt signaling inhibitor |
Hematologic malignancies (AML) |
Oral, IV |
Phase 1 Clinical Trial |
| MK-0752 | ϒ-secretase inhibitor | Cranial tumors, solid tumors | Oral, IV | Phase 1/2 Clinical Trial |
TGFβ ligand binds to type 1 and type II receptor kinases on the cell surface bringing them together allowing for phosphorylation of receptor 1 kinase. Following phosphorylation, signaling proceeds in the cell through Smad proteins (a total of at least 8 unique Smad proteins) ultimately translocating to the nucleus and regulating the expression of target genes [80]. Blocking the receptor kinase at the cell surface is an attractive target for drug development and has shown promise in blocking EMT for therapeutic benefit [81].
The potential clinical applicability of EW-7195 was seen in preventing TGF1-β-dependent breast tumor metastasis to the lung. EW-7195 inhibited both TGFβ induced EMT and cell motility in vitro while inhibiting lung metastasis in vivo [82]. EW-7203, IN-1130, and EW-7197 were developed by the same group and demonstrate a similar function in halting the TGF1β-dependent EMT and also inhibiting metastatic spread of primary tumor (breast to lung) [82].
SB-431542 is another small molecule inhibitor exerting its affect through a highly specific interaction with ALK5 kinase. SB-431542 competitively inhibits binding to the ATP site of ALK5 kinase preventing downstream activation of TGFβ targets. It does not act on other kinase-dependent pathways including ERK, JNK, or p38 [83]. In a human glioma cell model, SB-431542 was able to reduce cell proliferation and motility by repressing the expression of EMT proteins [84]. Despite this promising data, SB-431542, developed by GlaxoSmithKline (GSK), has not made the transition to the clinical setting. Building on the work with SB-431542, an orally available small molecular inhibitor with similar mechanism of action, GW-788388 is currently being studied. GW-788388 has shown promise for application in halting the progression of renal fibrosis by inhibiting EMT [85]. Interestingly, GW-788388 also shows potential as a treatment for reversing cardiac fibrosis in patients with Chagas disease [86].
SD-208 and SD-093 are two additional small-molecule inhibitors which target TGFβ-dependent EMT. SD-208, an orally bioavailable compound, was noted to inhibit the development of pulmonary fibrosis in a rat model. Subsequently, SD-208 was used in vivo in mouse metastatic breast cancer models. SD-208 conferred a strong anti-tumor effect in treated mice with no obvious signs of toxicity. SD-208 remains in the pre-clinical phase at this time, however these data support a move to clinical trial in the near future [87].
More selective small molecule inhibitors targeting ALK5 kinase with specific activity towards either Smad or non-Smad pathways have also been studied for their ability to reverse or halt EMT in cancer progression. The ALK5 inhibitor A-83-01 appears to inhibit Smad signaling with little effect on non-canonical pathways. Similar to the other ALK5 small molecule inhibitors, A-83-01 demonstrates the ability to decrease expression of EMT markers in a TGFβ-dependent fashion. A-83-01 notably showed significantly more potency than other similar small molecule inhibitors. [88].
Another TGFβ small molecule inhibitor, LY2157299, has gained attention as a possible adjunctive treatment for chemotherapy resistant cancers. In the case of triple negative breast cancer, many are initially responsive to traditional chemotherapeutic agents; however, the likelihood of recurrence is high, typically with a more aggressive, less responsive tumor. One hypothesis for this pattern of disease is the resistance of a small population of cells within the initial tumor to traditional chemotherapy agents. This specialized group of cells, often described as being cancer stem cells, relies on mechanisms involving EMT to continue to survive [52].
Based on this background, LY2157299 (TGFβ kinase antagonist) was used to treat a population of triple negative breast cancer cells previously treated with the chemotherapeutic agent paclitaxel. LY2157299 treatment reduced the size of paclitaxel-resistant cells, slowed the growth rate of tumors, and reduced the tumor forming potential of residual cancer cells [89].
LY2157299, currently in the Lily Oncology pipeline, is being studied in clinical trial for application in myelodysplastic syndromes (phase 2/3), hepatocellular carcinoma (phase 2), unresectable pancreatic cancer (phase 1b/2), and malignant glioma (phase 1b/2a). The progression of LY2157299 into a potential clinical therapeutic highlights the potential of using small molecule inhibitors that target EMT to treat advances malignancies.
2. Exogenous compounds
Several exogenous compounds, including dietary and herbal chemopreventative agents may act in a TGFβ-dependent fashion to impair EMT. Resveratrol is a natural polyphenol found most commonly in grapes and red wine. It has been discussed in the oncology literature based on reports of its ability to suppress cancer metastasis and invasion [90],[91]. Many mechanisms have been suggested for its mode of action, however compelling data suggest resveratrol may inhibit EMT. In a lung cancer model (A549 cells), treatment with TGFβ alone compared with TGFβ and resveratrol showed no initiation of EMT in the latter group. Cells treated with resveratrol increased expression of epithelial markers while repressing fibronectin and vimentin. The mechanism appeared to rely on modulating EMT transcription factors SNAIL1 and SLUG [92].
Another dietary agent EGCG, the major polyphenolic compound in green tea, demonstrates anti-proliferative effects in several cancers [93]. EGCG inhibits the induction of p300/CBP, a histone acetyltransferase with known activity on Smad2 and Smad3. In lung cancer cells treated with EGCG, TGFβ induction of EMT was potently halted in a Smad2/Smad3-dependent fashion. EGCG, acting through p300/CBP was able to prevent EMT and blocked invasion of lung cancer cells [94].
The translation of bench findings to clinical trial for both resveratrol and EGCG has been challenging. At this stage, resveratrol has been studied in phase 1 clinical trial and EGCG remains in pre-clinical study. Other dietary and herbal chemopreventative agents also have been noted to inhibit EMT through non-TGFβ-dependent mechanisms. β-elemene, an active component of the herbal medicine Curcuma wenyujin, acts to stop EMT through Smad3 [95].
Curcumin, the active component of the spice turmeric, is an inhibitor of EMT by altering the SNAIL1 transcription factor [12]. 2-Hydroxycinnamaldehyde also inhibits EMT via a SNAIL-dependent mechanism in breast cancer cells and also prevents lung metastasis in a mouse orthotopic breast cancer model [96]. The next steps for each is devising the best mode of drug delivery, further drug development, and devising treatments to synergize with existing cancer therapeutics.
Non-coding RNAs
Studying miRNAs and their potential role in cancer is an extremely fast-growing area of research. In parallel, several research groups are working on ways to modulate the effect of miRNAs in tumor biology with the intent of developing new and novel therapeutic targets. Antagomirs and miRNA sponges each show promise for future clinical application and will be discussed.
1. Antagomirs
As described earlier, miRNAs can play a role in cancer progression and metastases leading to them often being referred to as “onco-miRs” [97]. Antagomirs were first described and designed by Krϋtzfeldt et al. in 2005. They were developed as pharmacologic agents to silence miRNAs in vivo and defined as being single-stranded RNAs complementary to miRNAs. Krϋtzfeldt’s group demonstrated antagomir-122, selective against miR-122, completely silenced its target in multiple tissues types [98]. Similar findings were seen with other antagomirs showing a broad bioavailability and potent effect across tissue types.
The same group responsible for determining the important role played by miR-10b in breast cancer invasion and metastatic progression also utilized an antagomir (antagomir-10b) to silence miR-10b in vitro and in vivo. Interestingly, antagomir-10b did not reduce the bulk or size of the primary mammary tumor. However, it significantly suppressed the formation of metastatic lesions in a mouse. Overall it was well tolerated by treated animals laying the groundwork for further study of antagomirs as anti-cancer drugs [51],[99].
2. miRNA sponges
miRNA sponges are another tool increasingly utilized to modify the activity of miRNAs [100]. These sponges are constructed by utilizing 3’UTR mRNA sites complementary to targeted miRNA. By doing so, they act as a binding site for specific miRNAs and functionally inhibit miRNA binding to native targets [101]. Early work demonstrates that miRNA sponges can down-regulate tumorigenic miRNAs in vivo [102]. The important question that remains to be answered is how well this strategy would work as a cancer therapy and if it would be tolerated by patients.
As illustrated, this remains a relatively young area of work with few advancements to the clinical setting to date. However, strategies including the development of antagomirs and miRNA sponges have the potential to make an impact on a patient level.
Cancer stem cells
As discussed, EMT signaling pathways have important roles in the transformation of epithelial cells into CSCs. Several groups have started the process towards drug development to inhibit this process using large-scale screening techniques [103],[104]. The results of these screens allows for the identification of potential pharmacologic agents for eliminating CSCs. Understanding the properties of these agents and assessing their biologic activity is the first step towards drug development as stand-alone agents or as adjunctive therapies [103].
Another strategy for eliminating CSCs is specifically targeting EMT pathways important for their generation and maintenance. Physiologic factors already exist to prevent mesenchymal transition, likely secreted by epithelial cells in order to remain in a fixed state. These autocrine factors include Dickkopf-1 (DKK1) and secreted frizzle-related protein 1 (SFRP1), both known inhibitors of Wnt pathways. These proteins could be pharmacologically mimicked to produce a similar effect in diseased states [105].
1. Small molecule inhibitors
Wnt pathway inhibitors currently in development for the purpose of inhibiting CSC formation include LGK974 and CWP232291 (see Table 1). LGK974 is a small molecule inhibitor of Porcupine, a Wnt-specific acyltransferase, and strongly inhibits Wnt signaling both in vitro and in vivo. In rodent breast cancer models, LGK974 demonstrated dose dependent tumor regression with acceptable tolerance and oral bioavailability [106]. LGK974 is presently in phase 1 clinical trial. CWP232291 is another small molecule Wnt pathway inhibitor which notably produced marked cytotoxicity against CSCs in hematologic malignancies while showing minimal effect on normal bone marrow cells. These findings led to its application for treating AML. CWP232291 is currently in recruitment for a phase 1 clinical trial for treatment of relapsed or recurrent AML [107].
Notch signaling has also been targeted with small molecule inhibitors, both as independent and adjunctive therapies. MK-0752 is a potent γ-secretase inhibitor with strong efficacy for regulating Notch signaling, particularly in cells with stem-like properties. MK-0752 is also orally available making it an attractive potential chemotherapeutic. Phase 1 studies show acceptable tolerability, however low efficacy as a single treatment for extra-cranial tumors [108]. MK-0752 is presently in clinical trial assessing its potential for use in multidrug regimens for advanced solid tumors.
2. Metformin
Small molecule inhibitors offer the potential for future therapeutic options against difficult to treat malignancies, particularly those which demonstrate treatment resistance. The transition from the bench to wide-spread use, however, is often long and difficult. Utilizing pharmacologic agents already used in clinical practice to target CSCs would have the potential to more quickly impact patients. Recent work with the oral hypoglycemic metformin offers a possible model to follow for applying this strategy. Metformin, a mainstay in contemporary management of type 2 diabetes mellitus, is extremely well tolerated and extensively used. The link between cancer and metformin was first reported in 2005 after studying a large group of patients using a national database [109].
Based on this observation, several groups tested the biologic impact of metformin on tumor progression in mice models. Interestingly, metformin can inhibit the growth of solid tumors, including breast tumors, in both diabetic and non-diabetic mice [110],[111]. Several mechanisms have been proposed to explain this result, however it appears that metformin can selectively inhibit the growth of breast CSCs by modifying the EMT machinery [112]. When combined with conventional chemotherapeutics, the effect is potentiated [113]. This is an important example of leveraging existing therapies to modify EMTs. Additional EMT pathway targets and novel pharmacologic agents targeting CSCs are well-summarized in another recent review [114].
2.2.2 Altering the tumor microenvironment
To this point, EMT has largely been discussed as a process at the tumor level, with the transition to a mesenchymal phenotype occurring in tightly regulated fashion. Inducers of EMT come from many sources including the tissue environment in which the cell originates. With travel to distant sites, as is the case in metastatic disease, a new microenvironment in encountered. Clearly, the interplay between the tumor microenvironment (TMEN) and the tumor – including the microenvironment as a stimulator of EMTs – is important in cancer progression [115]. Targeting and altering the TMEN offers another approach to direct therapies to prevent EMT.
Components of the TMEN
The TMEN is a specialized extracellular matrix (ECM) wherein a cancer cell proliferates [116]. The components, therefore, are similar to those seen in any inflammatory ECM and include soluble factors, cytokines, chemokines, neovascular elements, immune cells, and mesenchymal stem cells (and their derivatives) [117],[118] Within the microenvironment, several components are clearly cancer-promoting elements [119].
The cytokines IL-1, IL-8, IL-10, and TGFβ all demonstrate effects on either tumor growth or metastasis through their interactions with cancer cells. The major cell types responsible for these mediators in the TMEN include the primary tumor cell and stromal cells including macrophages, adipocytes, and cancer associated fibroblasts (CAFs) [118]. A key cell type in the TMEN are mesenchymal stem cells (MSCs) which exhibit the ability to differentiate into nearly all stromal cell lineages. The plasticity of MSCs and their importance in maintaining the TMEN make them a critical target in cancer therapy [120].
Microenvironmental regulators of EMT
Mesenchymal stem cells (MSCs) have the ability to induce EMTs in several models and play a role in both pre-metastatic and metastatic sites. Breast cancer cell lines (MDA-MB-231, T47D, SK-Br3) cultured with MSCs increase expression of EMT markers (N-cadherin, Vimentin, Twist, SNAIL) with concomitant decrease in E-cadherin expression. This increase in EMT markers corresponded with an increase in protein expression as well, with increased Vimentin and SNAIL compared to untreated cells. Gene expression assays also revealed upregulation of TGFβ-receptor implicating TGFβ mechanistically as a regulator of this EMT. This occurred with only co-culturing these breast cancer cell lines with MSCs and no further co-stimulatory factors [121].
A similar role for MSCs initiating EMT is seen in hepatocellular carcinoma (HCC). HCC cell lines (SMMC-7721 and Hep-3B) expressed EMT markers after exposure to IFNy and TNF-alpha stimulated MSCs. The mechanism of action appeared to be via TGFβ expressed by MSCs, similar to results seen in breast cancer cell lines. These findings were correlated to clinical samples revealing over-expression of SSEA-4 (a marker for MSCs in clinical HCC tissues) was found to confer a shorter cancer-free interval and worse overall survival [122].
While MSCs have the ability to initiate EMTs, they also differentiate into other cell types within the TMEN to potentiate their action as tumor promoters. One particular cell type, the cancer-associated fibroblast (CAF) has garnered significant attention as a target for cancer therapy [123]. CAFs are defined by their similarities with myofibroblasts based on their origin (bone marrow or by EMT of resident fibroblasts) and pattern of surface expression (most notably alpha-SMA, MMP1, MMP3) [124]. Unlike myofibroblasts, CAFs are not removed by apoptosis and cannot be “deactivated”. Given that CAFs are the most abundant cell type in the TMEN in several cancers (breast, prostate, pancreatic), that they can stimulate EMT, and they are important mediators of chemotherapy resistance, eliminating CAFs offers an exciting drug target.
Therapeutic targets of TMEN-stimulated EMTs
The most significant challenge of designing therapies to eliminate CAFs are their similarities with myofibroblasts – which are essential for wound healing. Predictably, the pathways used in cancer progression are also essential for wound healing [125]. Osteopontin, a secreted phosphoprotein implicated in tumor metastasis both mechanistically and as a marker, is able to induce MSCs to adopt a CAF phenotype through autocrine signaling of TGFβ-1 [126]. This pathway, first observed in breast cancer, was also noted to be conserved in bone fracture healing (unpublished data). Unlike in cancer, where this signaling pathway is pathologic, fracture healing is dependent on the differentiation of MSCs into myofibroblasts to ensure appropriate bone remodeling. Cancer drugs must be specific to CAFs without negatively affecting myofibroblasts or other cell lines important for healing.
Despite these inherent challenges, strategies to inhibit CAFs have been tried at numerous potential points of intervention. Imatinib mesylate, a tyrosine kinase inhibitor currently used in the clinical setting for treatment of numerous malignancies, decreased the viability of CAFs in vitro by inhibiting expression of alpha-SMA [127]. Another approach targeting TGFβ uses the antisense oligonucleotide AP-12009 for the treatment of glioma [128]. At present, AP-12009 (Trabedersen) is being prepared to be tested in a phase II clinical trial for systemic IV administration in malignant melanoma and pancreatic cancer.
Another potential site for intervention within the TMEN include blocking the interaction between MSCs and cancer cells by preventing MSC recruitment to tumor sites. Zoledronic acid (ZA), a bisphosphonate, appears to show promise in this role. Specifically, in a breast cancer model, treatment with ZA prevented MSC migration by decreasing the expression of RANTES and IL-6, both markers for MSC migration. These data suggest a role for ZA as an adjunctive therapy to conventional chemotherapeutics [129].
3 Conclusions
EMT plays a considerable role both in normal development and diseased states. Understanding the interplay between EMT and development, both embryologically and in wound healing, has offered unique perspectives in determining the key regulatory steps for progression to disease. Shared molecular pathways amongst all EMTs allows for the development of novel therapies to potentially halt or reverse EMT prior to metastatic spread. Small molecule inhibitors and exogenous compounds appear to offer the most promise at this stage.
Additionally, the interplay between the TMEN and induction of EMT creates yet another opportunity to design targeted therapies in cancer. Modulating the key effectors within the tumor stroma is an area ripe for drug development. Undoubtedly, EMTs and their regulators have significant clinical implication across the oncologic spectrum.
Abbreviations
EMT: Epithelial-mesenchymal transition
TGF: Transforming growth factor
EGFR: Epidermal growth factor receptor
ECM: Extracellular matrix
IL: Interleukin
MAPK: Mitogen-activated protein kinase
nAChRs: Nicotinic acetylcholine receptor
ROS: Reactive oxygen species
miR: micro-RNA
ALK5: TGFβ type 1 receptor
CK2: Protein kinase 2
INF: Interferon
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
AK and PK conceived the format for the review and participated in its design and drafting. Literature and data acquisition were conducted by AK. Final manuscript editing and review were conducting by PK. All authors read and approved the final manuscript.
Contributor Information
Anai N Kothari, Email: ankothari@lumc.edu.
Zhiyong Mi, Email: zhmi@luc.edu.
Matthew Zapf, Email: mzapf@luc.edu.
Paul C Kuo, Email: pkuo@lumc.edu.
References
- Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- Weber CE, Li NY, Wai PY, Kuo PC. Epithelial-mesenchymal transition, TGF-beta, and osteopontin in wound healing and tissue remodeling after injury. J Burn Care Res. 2012;33:311–318. doi: 10.1097/BCR.0b013e318240541e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymkowsky MW, Savagner P. Epithelial-mesenchymal transition: a cancer researcher’s conceptual friend and foe. The Am J Pathol. 2009;174:1588–1593. doi: 10.2353/ajpath.2009.080545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg C, Solnica-Krezel L. Back and forth between cell fate specification and movement during vertebrate gastrulation. Curr Opin Genet Dev. 2008;18:311–316. doi: 10.1016/j.gde.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- Ciruna B, Rossant J. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell. 2001;1:37–49. doi: 10.1016/s1534-5807(01)00017-x. [DOI] [PubMed] [Google Scholar]
- Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol. 2001;21:8184–8188. doi: 10.1128/MCB.21.23.8184-8188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci. 2012;125:4417–4422. doi: 10.1242/jcs.099697. [DOI] [PubMed] [Google Scholar]
- Huang T, Chen Z, Fang L. Curcumin inhibits LPS-induced EMT through downregulation of NF-κB-Snail signaling in breast cancer cells. Oncol Rep. 2013;29:117–124. doi: 10.3892/or.2012.2080. [DOI] [PubMed] [Google Scholar]
- Lange T, Samatov TR, Tonevitsky AG, Schumacher U. Importance of altered glycoprotein-bound N- and O-glycans for epithelial-to-mesenchymal transition and adhesion of cancer cells. Carbohydr Res. 2014;389:39–45. doi: 10.1016/j.carres.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101:830–839. doi: 10.1002/jcb.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, Toews GB, White ES, Lynch Iii JP, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- Redd MJ, Cooper L, Wood W, Stramer B, Martin P. Wound healing and inflammation: embryos reveal the way to perfect repair. Philos Trans R Soc Lond B Biol Sci. 2004;359:777–784. doi: 10.1098/rstb.2004.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittan M, Hunt T, Jeffery R, Poulsom R, Forbes SJ, Hodivala-Dilke K, Goldman J, Alison MR, Wright NA. Bone marrow derivation of pericryptal myofibroblasts in the mouse and human small intestine and colon. Gut. 2002;50:752–757. doi: 10.1136/gut.50.6.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SJ, Russo FP, Rey V, Burra P, Rugge M, Wright NA, Alison MR. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology. 2004;126:955–963. doi: 10.1053/j.gastro.2004.02.025. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Kubo T, Murakami T, Takahashi M, Hakamata Y, Kobayashi E, Yoshida S, Hosokawa K, Yoshikawa K, Itami S. Bone marrow cells differentiate into wound myofibroblasts and accelerate the healing of wounds with exposed bones when combined with an occlusive dressing. Br J Dermatol. 2005;152:616–622. doi: 10.1111/j.1365-2133.2005.06402.x. [DOI] [PubMed] [Google Scholar]
- Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti-fibrotic therapies. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2013;1832:1088–1103. doi: 10.1016/j.bbadis.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DL, Kao WW, Greenhalgh DG. Apoptosis down-regulates inflammation under the advancing epithelial wound edge: delayed patterns in diabetes and improvement with topical growth factors. Surgery. 1997;121:372–380. doi: 10.1016/s0039-6060(97)90306-8. [DOI] [PubMed] [Google Scholar]
- Sayah DN, Soo C, Shaw WW, Watson J, Messadi D, Longaker MT, Zhang X, Ting K. Downregulation of apoptosis-related genes in keloid tissues. J Surg Res. 1999;87:209–216. doi: 10.1006/jsre.1999.5761. [DOI] [PubMed] [Google Scholar]
- Rastaldi MP. Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J Nephrol. 2006;19:407. [PubMed] [Google Scholar]
- Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Strippoli R, Benedicto I, Perez Lozano ML, Cerezo A, Lopez-Cabrera M, del Pozo MA. Epithelial-to-mesenchymal transition of peritoneal mesothelial cells is regulated by an ERK/NF-kappaB/Snail1 pathway. Dis Model Mech. 2008;1:264–274. doi: 10.1242/dmm.001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, de Herreros AG. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Becker KF, Rosivatz E, Blechschmidt K, Kremmer E, Sarbia M, Hofler H. Analysis of the E-cadherin repressor Snail in primary human cancers. Cells Tissues Organs. 2007;185:204–212. doi: 10.1159/000101321. [DOI] [PubMed] [Google Scholar]
- Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–3456. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikov AV, Liu L, Platen M, Gladkich J, Salnikova O, Ryschich E, Mattern J, Moldenhauer G, Werner J, Schemmer P. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PLoS One. 2012;7:e46391. doi: 10.1371/journal.pone.0046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D. Nicotine induces cell proliferation, invasion and epithelial‐mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukase K, Ohtsuka H, Onogawa T, Oshio H, Ii T, Mutoh M, Katayose Y, Rikiyama T, Oikawa M, Motoi F. Bile acids repress E‐cadherin through the induction of Snail and increase cancer invasiveness in human hepatobiliary carcinoma. Cancer Sci. 2008;99:1785–1792. doi: 10.1111/j.1349-7006.2008.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- Chia SK, Wykoff CC, Watson PH, Han C, Leek RD, Pastorek J, Gatter KC, Ratcliffe P, Harris AL. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol. 2001;19:3660–3668. doi: 10.1200/JCO.2001.19.16.3660. [DOI] [PubMed] [Google Scholar]
- Milara J, Peiro T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68:410–420. doi: 10.1136/thoraxjnl-2012-201761. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S, Sarkar FH. Targeting miRNAs involved in cancer stem cell and EMT regulation: an emerging concept in overcoming drug resistance. Drug Resist Updat. 2010;13:109–118. doi: 10.1016/j.drup.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- Li Y, Vanden Boom TG, 2nd, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ma L. MicroRNA control of epithelial–mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012;31:653–662. doi: 10.1007/s10555-012-9368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos V, Moutinho C, Villanueva A, Boque R, Silva P, Carneiro F, Esteller M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene. 2011;31:2062–2074. doi: 10.1038/onc.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10:4256–4271. doi: 10.4161/cc.10.24.18552. [DOI] [PubMed] [Google Scholar]
- Gravgaard KH, Lyng MB, Laenkholm A, Søkilde R, Nielsen BS, Litman T, Ditzel HJ. The miRNA-200 family and miRNA-9 exhibit differential expression in primary versus corresponding metastatic tissue in breast cancer. Breast Cancer Res Treat. 2012;134:207–217. doi: 10.1007/s10549-012-1969-9. [DOI] [PubMed] [Google Scholar]
- Hur K, Toiyama Y, Takahashi M, Balaguer F, Nagasaka T, Koike J, Hemmi H, Koi M, Boland CR, Goel A. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut. 2013;62:1315–1326. doi: 10.1136/gutjnl-2011-301846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly J, Chandra D, Zhou J, Claypool K. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–1708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- Biddle A, Mackenzie IC. Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev. 2012;31:285–293. doi: 10.1007/s10555-012-9345-0. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao M, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincan E, Barker N. The upstream components of the Wnt signalling pathway in the dynamic EMT and MET associated with colorectal cancer progression. Clin Exp Metastasis. 2008;25:657–663. doi: 10.1007/s10585-008-9156-4. [DOI] [PubMed] [Google Scholar]
- Stemmer V, De Craene B, Berx G, Behrens J. Snail promotes Wnt target gene expression and interacts with β-catenin. Oncogene. 2008;27:5075–5080. doi: 10.1038/onc.2008.140. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, Felipe De S, Melo E, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–718. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palagani V, El Khatib M, Kossatz U, Bozko P, Müller MR, Manns MP, Krech T, Malek NP, Plentz RR. Epithelial mesenchymal transition and pancreatic tumor initiating CD44 /EpCAM cells are inhibited by γ-secretase inhibitor IX. PLoS One. 2012;7:e46514. doi: 10.1371/journal.pone.0046514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target? J Natl Cancer Inst. 2004;96:583–585. doi: 10.1093/jnci/djh095. [DOI] [PubMed] [Google Scholar]
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- Oravecz-Wilson KI, Philips ST, Yilmaz ÖH, Ames HM, Li L, Crawford BD, Gauvin AM, Lucas PC, Sitwala K, Downing JR. Persistence of leukemia-initiating cells in a conditional knockin model of an imatinib-responsive myeloproliferative disorder. Cancer Cell. 2009;16:137–148. doi: 10.1016/j.ccr.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. Mesenchyme to epithelium transition during development of the mammalian kidney tubule. Cells Tissues Organs (Print) 1996;156:187–201. doi: 10.1159/000147846. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ, Koteliansky V, Arteaga CL. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551–1559. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-β signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011–1022. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- Zou J, Luo H, Zeng Q, Dong Z, Wu D, Liu L. Protein kinase CK2alpha is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J Transl Med. 2011;9:97. doi: 10.1186/1479-5876-9-97. 5876-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim SH. CK2 inhibitor CX-4945 blocks TGF-β1-induced epithelial-to-mesenchymal transition in A549 human lung adenocarcinoma cells. PLoS One. 2013;8:e74342. doi: 10.1371/journal.pone.0074342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinto O, Yashiro M, Kawajiri H, Shimizu K, Shimizu T, Miwa A, Hirakawa K. Inhibitory effect of a TGFβ receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous gastric cancer cells. Br J Cancer. 2010;102:844–851. doi: 10.1038/sj.bjc.6605561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga CL. Inhibition of TGFβ signaling in cancer therapy. Curr Opin Genet Dev. 2006;16:30–37. doi: 10.1016/j.gde.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Fabregat I, Fernando J, Mainez J, Sancho P. TGF-beta signaling in cancer treatment. Curr Pharm Des. 2014;20:2934–2947. doi: 10.2174/13816128113199990591. [DOI] [PubMed] [Google Scholar]
- Park C, Son J, Jin CH, Nam J, Kim D, Sheen YY. EW-7195, a novel inhibitor of ALK5 kinase inhibits EMT and breast cancer metastasis to lung. Eur J Cancer. 2011;47:2642–2653. doi: 10.1016/j.ejca.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Hjelmeland MD, Hjelmeland AB, Sathornsumetee S, Reese ED, Herbstreith MH, Laping NJ, Friedman HS, Bigner DD, Wang XF, Rich JN. SB-431542, a small molecule transforming growth factor-beta-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther. 2004;3:737–745. [PubMed] [Google Scholar]
- Petersen M, Thorikay M, Deckers M, Van Dinther M, Grygielko E, Gellibert F, De Gouville A, Huet S, Ten Dijke P, Laping N. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2007;73:705–715. doi: 10.1038/sj.ki.5002717. [DOI] [PubMed] [Google Scholar]
- de Oliveira FL, Araújo-Jorge TC, de Souza EM, de Oliveira GM, Degrave WM, Feige J, Bailly S, Waghabi MC. Oral administration of GW788388, an inhibitor of transforming growth factor beta signaling, prevents heart fibrosis in Chagas disease. PLoS Negl Trop Dis. 2012;6:e1696. doi: 10.1371/journal.pntd.0001696. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ge R, Rajeev V, Ray P, Lattime E, Rittling S, Medicherla S, Protter A, Murphy A, Chakravarty J, Dugar S, Schreiner G, Barnard N, Reiss M. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-beta type I receptor kinase in vivo. Clin Cancer Res. 2006;12:4315–4330. doi: 10.1158/1078-0432.CCR-06-0162. [DOI] [PubMed] [Google Scholar]
- Tojo M, Hamashima Y, Hanyu A, Kajimoto T, Saitoh M, Miyazono K, Node M, Imamura T. The ALK‐5 inhibitor A‐83‐01 inhibits Smad signaling and epithelial‐to‐mesenchymal transition by transforming growth factor‐β. Cancer Sci. 2005;96:791–800. doi: 10.1111/j.1349-7006.2005.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, Stanford J, Cook RS, Arteaga CL. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:2783–2840. [PubMed] [Google Scholar]
- Wang H, Zhang H, Tang L, Chen H, Wu C, Zhao M, Yang Y, Chen X, Liu G. Resveratrol inhibits TGF-β1-induced epithelial-to-mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology. 2013;303:139–146. doi: 10.1016/j.tox.2012.09.017. [DOI] [PubMed] [Google Scholar]
- Jankun J, Selman SH, Swiercz R, Skrzypczak-Jankun E. Why drinking green tea could prevent cancer. Nature. 1997;387:561. doi: 10.1038/42381. [DOI] [PubMed] [Google Scholar]
- Ko H, So Y, Jeon H, Jeong M, Choi H, Ryu S, Lee S, Yoon H, Choi K. TGF-β1-induced epithelial–mesenchymal transition and acetylation of Smad2 and Smad3 are negatively regulated by EGCG in human A549 lung cancer cells. Cancer Lett. 2013;335:205–213. doi: 10.1016/j.canlet.2013.02.018. [DOI] [PubMed] [Google Scholar]
- Zhang X, Li Y, Zhang Y, Song J, Wang Q, Zheng L, Liu D. Beta-elemene blocks epithelial-mesenchymal transition in human breast cancer cell line MCF-7 through Smad3-mediated down-regulation of nuclear transcription factors. PLoS One. 2013;8:e58719. doi: 10.1371/journal.pone.0058719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail IA, Kang HS, Lee H, Chang H, Yun J, Lee CW, Kim NH, Kim HS, Yook JI, Hong S. 2-Hydroxycinnamaldehyde inhibits the epithelial-mesenchymal transition in breast cancer cells. Breast Cancer Res Treat. 2013;137:697–708. doi: 10.1007/s10549-012-2388-7. [DOI] [PubMed] [Google Scholar]
- Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ’antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–347. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nana-Sinkam SP, Croce CM. MicroRNAs as therapeutic targets in cancer. Transl Res. 2011;157:216–225. doi: 10.1016/j.trsl.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szász AM, Wang ZC, Brock JE, Richardson AL, Weinberg RA. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–1046. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody LC, Germain AR, VerPlank L, Nag PP, Munoz B, Perez JR, Palmer MA. Phenotypic high-throughput screening elucidates target pathway in breast cancer stem cell-like cells. J Biomol Screen. 2012;17:1204–1210. doi: 10.1177/1087057112458317. [DOI] [PubMed] [Google Scholar]
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah K, Bell G, Guo W, Rubin J, Richardson AL. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–940. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, Li J, Tompkins C, Pferdekamper A, Steffy A, Cheng J, Kowal C, Phung V, Guo G, Wang Y, Graham MP, Flynn S, Brenner JC, Li C, Villarroel MC, Schultz PG, Wu X, McNamara P, Sellers WR, Petruzzelli L, Boral AL. et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110:20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JC, Lorenzi MV. Drug Discovery Approaches to Target Wnt Signaling in Cancer Stem Cells. Oncotarget. 2010;1(7):552–566. doi: 10.18632/oncotarget.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, Giannotti M, Hing J, Beckman R, Lorusso P. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012;30:2307–2313. doi: 10.1200/JCO.2011.39.1540. [DOI] [PubMed] [Google Scholar]
- Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–915. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–2040. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cello ntogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;9:3831–3838. [PubMed] [Google Scholar]
- Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse JC, Kalluri R. Mechanisms of metastasis: epithelial‐to‐mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007;101:816–829. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- Weber CE, Kuo PC. The tumor microenvironment. Surg Oncol. 2012;21:172–177. doi: 10.1016/j.suronc.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem. 2007;101:805–815. doi: 10.1002/jcb.21159. [DOI] [PubMed] [Google Scholar]
- Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121:3804–3809. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, Driver J, Franzen C, Mascarenhas J, Mi Z, Gupta G, Wai P, Kuo P. The constituents and potential targets of the extracellular matrix: implications for carcinogenesis and cancer treatment. J Carcinogene Mutagene S. 2013;13:006. doi:10.4172/2157-2518.S13-006. [Google Scholar]
- Hu M, Polyak K. Molecular characterisation of the tumour microenvironment in breast cancer. Eur J Cancer. 2008;44:2760–2765. doi: 10.1016/j.ejca.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Dwyer R, Kelly J, Khan S, Murphy J, Curran C, Miller N, Hennessy E, Dockery P, Barry F. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT) Breast Cancer Res Treat. 2010;124:317–326. doi: 10.1007/s10549-010-0734-1. [DOI] [PubMed] [Google Scholar]
- Jing Y, Han Z, Liu Y, Sun K, Zhang S, Jiang G, Li R, Gao L, Zhao X, Wu D. Mesenchymal stem cells in inflammation microenvironment accelerates hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition. PLoS One. 2012;7:e43272. doi: 10.1371/journal.pone.0043272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micke P. Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer. 2004;45:S163–S175. doi: 10.1016/j.lungcan.2004.07.977. [DOI] [PubMed] [Google Scholar]
- Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1:482–497. [PMC free article] [PubMed] [Google Scholar]
- Wai PY, Kuo PC. Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev. 2008;27:103–118. doi: 10.1007/s10555-007-9104-9. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, McArthur GA, Demetri GD, Joensuu H, Bono P, Herrmann R, Hirte H, Cresta S, Koslin DB, Corless CL, Dirnhofer S, van Oosterom AT, Nikolova Z, Dimitrijevic S, Fletcher JA. Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor) J Clin Oncol. 2006;24:1195–1203. doi: 10.1200/JCO.2005.04.0717. [DOI] [PubMed] [Google Scholar]
- Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, Balasubramaniam A, Nair S, Oliushine V, Parfenov V, Poverennova I, Zaaroor M, Jachimczak P, Ludwig S, Schmaus S, Heinrichs H, Schlingensiepen KH. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. 2011;13:132–142. doi: 10.1093/neuonc/noq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo M, De Luca A, Lamura L, Normanno N. Zoledronic acid blocks the interaction between mesenchymal stem cells and breast cancer cells: implications for adjuvant therapy of breast cancer. Ann Oncol. 2012;23:597–604. doi: 10.1093/annonc/mdr159. [DOI] [PubMed] [Google Scholar]