

Abstract
Background
Nephrogenic diabetes insipidus (NDI) is a rare disease whose complications include polyuria, renal dysfunction, growth disorder and mental retardation. The details of NDI’s clinical course have been unclear. To address this uncertainty, we performed a large investigation of the clinical course of NDI in Japan.
Methods
Between December 2009 and March 2011, we provided a primary questionnaire to 26,282 members of the Japan Endocrine Society, the Japanese Urological Association, the Japanese Society for Pediatric Endocrinology, the Japanese Society for Pediatric Nephrology, the Japanese Society of Nephrology, the Japanese Society of Neurology and the Japanese Society of Pediatric Urology. In addition, we provided a secondary questionnaire to 121 members who reported experience with cases of NDI. We asked about patient’s age at onset, diagnosis, complications, effect of treatment and patient’s genotype.
Results
We enrolled 173 patients with NDI in our study. Of these NDI patients, 143 were congenital and 30 were acquired. Of the 173, 73 patients (42%) experienced urologic complications. Among the 143 with congenital NDI, 20 patients (14%) had mental retardation. Patients with NDI mainly received thiazide diuretics, and some patients responded to treatment with desmopressin acetate (DDAVP). Gene analyses were performed in 87 patients (61%) with congenital NDI, revealing that 65 patients had an arginine vasopressin receptor type 2 (AVPR2) gene mutation and that 8 patients (9.2%) had an aquaporin 2 (AQP2) gene mutation. Patients with the AVPR2 mutation (D85N) generally showed a mild phenotype, and we found that DDAVP was generally an effective treatment for NDI among these patients.
Conclusion
We suggest that adequate diagnosis and treatment are the most important factors for improving prognoses. We further suggest that gene analysis should be performed for optimal treatment selection and the early detection of NDI among siblings.
Keywords: aquaporin 2, deamino arginine vasopressin, nephrogenic diabetes insipidus, thiazide diuretics, vasopressin v2 receptor
Nephrogenic diabetes insipidus (NDI) is a rare disease that is characterized by an inability to concentrate urine due to insensitivity of the collecting tubule to arginine vasopressin.1 NDI can be either congenital or acquired. Common cases of congenital NDI (CNDI) result from mutations in the arginine vasopressin receptor 2 (AVPR2) gene, which has X-linked recessive inheritance. 1, 2 Some other congenital cases result from mutations in the aquaporin 2 (AQP2) gene, which can have autosomal recessive or dominant inheritance.1, 2 Acquired NDI is commonly caused by drugs, such as lithium and demeclocycline.3 However, the clinical status of NDI has remained unknown.
CNDI is commonly diagnosed as a result of characteristic symptoms, such as polyuria, polydipsia, fever of unknown etiology, convulsion, vomiting and constipation in early infancy. 4 However, it has been reported that some cases of CNDI exhibit far milder symptoms such as night enuresis, and that the diagnosis is only made later through gene analyses.5 Further, the long-term effects of treatment for NDI, the prognosis of NDI and the correlation between the phenotype and genotype in cases of NDI all remain unknown.
In Japan, Imura et al.6 carried out a nationwide survey for 5 “hormone receptor diseases” including NDI from 1977 to 1982. It was reported that there were 78 cases of NDI, which seemed to be comprised of at least 2 subtypes; early onset type and late onset type.6 Because the responsible genes of CNDI such as AVPR2 and AQP2 genes had not been detected at that time, they were unable to provide a molecular biological diagnosis of CNDI with a mild phenotype and in female carriers.
Hence, to ascertain the characteristics of NDI in the general population, we conducted the nationwide survey of NDI in Japan. Here, we report the results of this survey, including clinical findings at onset, complications, the effects of treatment and genotypes in Japan.
MATERIALS AND METHODS
The questionnaire and data analysis
Between December 2009 and March 2011, we provided a primary questionnaire to 26,282 members of the Japan Endocrine Society, the Japanese Urological Association, the Japanese Society for Pediatric Endocrinology, the Japanese Society for Pediatric Nephrology, the Japanese Society of Nephrology, the Japanese Society of Neurology and the Japanese Society of Pediatric Urology. On the primary questionnaire, we asked about experiences in clinical practice and the number of patients who had NDI. In addition, we provided a secondary questionnaire to 121 members (96 hospitals) who reported experience with cases of NDI. We asked about patient’s age at onset, diagnosis, complications, effect of treatment, patient’s genotype and administration of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) during the fetal period. Gene analyses were performed in individual facilities. We used the results of gene analyses. Attending physicians determined the type of NDI and the presence of secondary urologic complications, based on disease’s clinical manifestation and patient’s laboratory data. When the same case was reported by multiple members, we excluded each of the redundant reports based on the age and sex of the patient or the name of the healthcare facility. In the survey, we asked for information regarding patients of all ages, complications, genders and races. We carefully analyzed the results of the secondary questionnaire. At least 2 researchers checked each report, and several team meetings were conducted to ensure the accuracy of our survey results. To assess the coverage rate of our survey, we compared the number of NDI patients aged less than 20 years in our survey with the number of patients who were registered as having CNDI in the Medical Aid Program for Chronic Pediatric Diseases of Specified Categories in Japan, from fiscal 2009 to fiscal 2011. The study was approved by the Ethics Committee of Tottori University (approval number 1384).
Statistical analysis
For our statistical analyses, we relied on the Mann-Whitney U-test and Fisher’s exact test. Values of P less than 0.05 were defined as significant.
RESULTS
We received 5,139 responses to our primary questionnaire from 26,282 members. Together, the primary questionnaires reported 210 NDI patients. We provided a secondary questionnaire to 121 members (96 hospitals) who had reported experience with cases of NDI. Respondents to the secondary questionnaire reported 183 patients with NDI. Ten patients were excluded because of insufficient information; we analyzed 173 patients. Of these, 143 patients had CNDI (124 males and 19 females) and 30 patients had acquired NDI (14 males and 16 females). There was no duplicated case. Our study included 96 CNDI patients who were under 20 years as of 2009. In the database of the Medical Aid Program for Chronic Pediatric Diseases of Specified Categories in Japan, a yearly mean of 131 CNDI patients were reported from fiscal 2009 to fiscal 2011. Accordingly, the coverage achievement rate of our survey was estimated to be 73%, which is high.
Congenital NDI
In this study, 143 patients had CNDI (124 males and 19 females), out of a total of 173 patients with any form of NDI. Details of age at diagnosis are presented in Fig. 1. The median age of CNDI diagnosis was 2.0 months (n = 125), and the mean [s] age was 1.2 [4.6] years (n = 125). Ninety-eight patients (69%) with CNDI were diagnosed within the 1st year of life, and 19 patients (13%) were diagnosed between 1 year and 4 years of age. Eight patients with CNDI were diagnosed between 5 and 29 years of age, half of whom had siblings or children who were diagnosed with NDI beforehand. We were unable to obtain the information on the age of CNDI diagnosis in 18 patients. Three patients were diagnosed as with CNDI in their 20s, 1 of whom had the mutation p.D85N in the AVPR2 gene, which resulted in a mild phenotype, as was described subsequently. 7
Fig. 1.
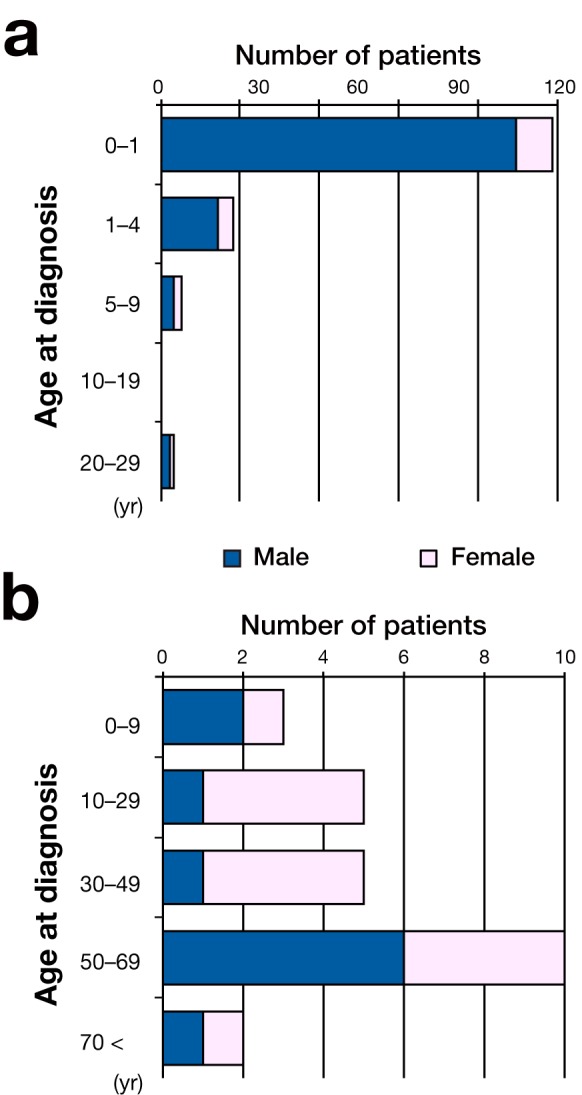
Histogram of the age at diagnosis with CNDI (a) and acquired NDI (b). The number of male patients is depicted with a closed bar. The number of female patients is depicted with an open bar. CNDI, congenital NDI; NDI, nephrogenic diabetes insipidus.
The symptoms and signs at the onset of CNDI are presented in Table 1. Polyuria was the most common symptom at onset (79 of the 143 patients; 55%). Polydipsia was the second common symptom (69 of the 143; 48%). It is well known that many patients with CNDI have fever at onset;5 53 patients (37%) had fever at onset and 30 patients with CNDI (21%) presented no symptoms at onset except for fever. Although it has been reported that vomiting and anorexia occur at the onset of 70% of CNDI patients,5 vomiting and anorexia were found in only 18 patients (13%) in our study. On the other hand, infant patients (diagnosed before 6 months of age, n = 90) were more likely to present with fever at onset (49 of the 90; 54%) than the total survey sample. Approximately 40% of the patients who were less than 6 months had either poor weight gain, or polydipsia and polyuria. Eight patients (5.6%) with CNDI were diagnosed among those who already had mental retardation (MR).
Table 1.
Symptoms and signs at the onset of congenital NDI (n = 143)
| Symptoms and signs | n |
| Polyuria | 79 |
| Polydipsia | 69 |
| Thirst | 19 |
| Nocturnal enuresis | 15 |
| Fever | 53 |
| Poor weight gain | 41 |
| Failure to thrive | 14 |
| Mental retardation | 8 |
| Vomiting | 12 |
| Anorexia | 6 |
| Convulsion | 3 |
| Disturbance of consciousness | 1 |
| Constipation | 1 |
| Others* | 9 |
NDI, nephrogenic diabetes insipidus.
*Urinary tract infection, dysphoria, weight loss, bradycardia.
The incidence of MR is critically related to prognosis such as quality of life. Twenty patients with CNDI (14%) were complicated with MR. In 3 of the 20, MR was severe. Because of the limited sample size, we could not find any significant differences in age (P = 0.908, Mann-Whitney U-test) or the serum sodium levels in CNDI patients with and without MR at the time of diagnosis. However, patients with MR were diagnosed at an age range from 0.5 to 2.5 years, and generally appeared to be older than patients with other complications at the time of diagnosis (the mean age of CNDI diagnosis was 2.0 months overall).
Genetic analysis
Analyses of the AVPR2 and/or AQP2 genes were performed for 87 (61%) of all CNDI patients. For 10 of the 87, we were unable to obtain the name of the gene in which a mutation was detected. Most of them (65 of 87; 75%) had AVPR2 gene mutations, surprisingly including 10 female patients. On the other hand, AQP2 gene abnormalities were identified in 8 patients (4 males and 4 females; 9.2%). In 4 patients (2 males and 2 females; 4.6%), AVPR2 or AQP22 mutation was not detected by mutation analysis. Our survey results included 35 distinct AVPR2 mutations and 4 distinct AQP2 mutations that caused CNDI (Tables 2 and 3). Of the 39 mutations, 14 (12 in AVPR2 and 2 in AQP2) have not been reported previously. These novel AVPR2 and AQP2 mutations are marked in Tables 2 and 3. Missense mutations in the AVPR2 gene were the most common mutations. The p.D85N mutation was detected in 7 patients (11%) of the 65, and p.R106C was detected in 5 (7.7%). In previous reports, p.D85N, p.V88M, p.R104C, p.R106C, p.Y128S, p.N317K, p.P322S, p.S333del AVPR2 gene mutations were reported as being associated with a mild phenotype;7, 8 these were detected in 22 patients (34%) of the 65 in our study. Of the 22, 19 patients (81%) were diagnosed at the age younger than 3 years, and 11 patients (50%), at the age younger than 6 months. Ten patients were diagnosed after 3 years of age; p.D85N, p.R104C, and p.Y128S were among the mutations detected in these patients, as was W99X (1 case), a previously unreported mutation. Four of the 10 patients did not receive gene analyses.
Table 2.
Summary of AVPR2 mutations reported in this study
| Gene | Type | Mutation | Frequency |
| AVPR2 | Complete deletion | – | 2 |
| Splicing | Intron 2† | 1 | |
| Missense | A37P§ | 1 | |
| L44F | 1 | ||
| D85N | 7 | ||
| V88M | 1 | ||
| R104C | 2 | ||
| R106C | 5 | ||
| G107E | 1 | ||
| G107R§ | 1 | ||
| R113W | 1 | ||
| Y128S | 2 | ||
| W164R | 1 | ||
| P178L§ | 1 | ||
| R181C | 2 | ||
| R202C | 3 | ||
| P286S | 1 | ||
| N317K | 1 | ||
| P322S | 2 | ||
| S329G | 1 | ||
| L336P§ | 1 | ||
| Nonsense | W99X§ | 2 | |
| W156X§ | 1 | ||
| L161X | 1 | ||
| W200X | 1 | ||
| R337X | 1 | ||
| Deletion | c.528delG§ | 1 | |
| V278del§ | 1 | ||
| S333del | 1 | ||
| c.727-728delAG | 1 | ||
| 5’UTR-AVPR2 DEL 32,787 | 2 | ||
| Others | 3 | ||
| Insertion | c.299 300insA§ | 1 | |
| c.498 499insTC (L168RfsX45)§ | 1 | ||
| c.855 856insCGCA§ | 1 | ||
| Frameshift | L35fs.§ | 1 | |
| Unknown‡ | – | 8 |
AVPR2, arginine vasopressin receptor type 2.
†Splice acceptor AG ARROW CG.
‡There was a mutation but detail was unknown.
§Novel mutation.
Table 3.
Summary of AQP2 mutations reported in this study
| Gene | Type | Mutation | Frequency |
| AQP2 | Missense | A130V§ | 1 |
| N184H§ | 1 | ||
| R254Q | 2 | ||
| Deletion | c.763 722del | 2 | |
| Unknown‡ | – | 2 |
AQP2, aquaporin 2.
‡There was mutation but detail was unknown.
§Novel mutation.
Acquired NDI
The age of onset of acquired NDI ranged widely among patients. The peak age of onset was at approximately 50 to 69 years. In 15 patients (50%), acquired NDI was caused by administration of lithium carbonate. Between 10 and 49 years of age, the ratio of female patients with acquired NDI was higher. This sex difference may be due to the fact that both depression and bipolar disorder, which are treated with lithium carbonate, are more common in females. Two patients acquired NDI by ACEI or ARB treatments for their mothers during the fetal period. In 13 patients, the underlying cause was unknown. Seven patients with acquired NDI (23%) had MR as a complication. MR was severe in 3 of them.
Urologic complications of NDI
In 73 patients (42%) of the 173, NDI was accompanied by urologic complications. The frequencies of various urologic complications are presented in Table 4. Eleven patients (6.4%) had renal failure. We found no significant difference in the rate of whole urologic complication between CNDI and acquired NDI (P = 0.536, Fisher’s exact test). However, CNDI was more frequently accompanied by hydronephrosis and/or hydroureter (P = 0.0454, Fisher’s exact test). Two cases lacked information on urologic complications. No patients required clean intermittent catheterizations.
Table 4.
Urologic complications in patients with NDI (n = 173)
| Symptoms and signs | n |
| Hydronephrosis | 47 |
| Hydroureter | 24 |
| Renal failure | 11 |
| Vesicoureteral reflux | 11 |
| Slightly dilatation of renal pelvic | 7 |
| Dilatation of bladder | 3 |
| Renal atrophy/dwarf kidney | 3 |
| Neurogenic bladder | 2 |
| Acidosis | 1 |
NDI, nephrogenic diabetes insipidus.
Treatment
Details of treatment efficacy are presented in Fig. 2. Most patients with NDI were treated with thiazide diuretics (hydrochlorothiazide or trichlormethiazide) with or without potassium supplementation, low-sodium diet, non-steroidal anti-inflammatory agents (NSAIDs) and aldosterone antagonists (amiloride or spironolactone).
Fig. 2.
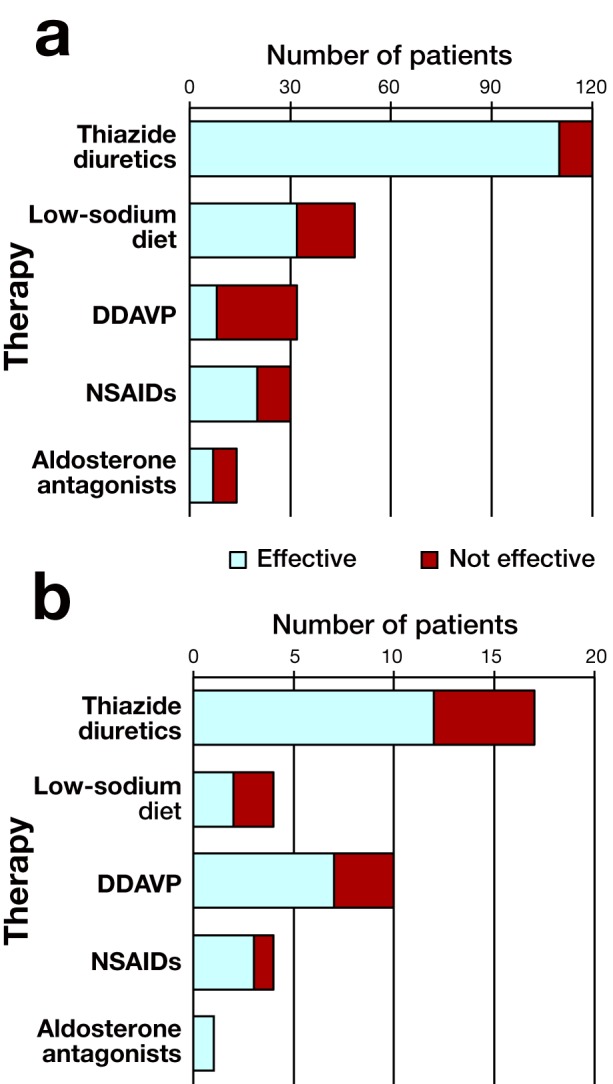
The efficacy of therapy for CNDI (a) and acquired NDI (b). The patients in whom the treatment reduced urine volume are depicted with an open bar, and patients in whom treatment did not reduce urine volume are depicted with a closed bar. CNDI, congenital NDI; DDAVP, desmopressin acetate; NDI, nephrogenic diabetes insipidus; NSAID, non-steroidal anti-inflammatory agent.
Thiazide diuretics were effective in 110 (92%) of 120 patients with CNDI. All 15 patients treated with aldosterone antagonists were also treated with thiazide diuretics. A low-sodium diet was useful for 32 (65%) of 49 patients with CNDI. Desmopressin acetate (DDAVP) was provided in 32 patients (28 males and 4 females), which was effective in 8 patients [6 males (21%) and 2 females (50%)] of them. The dose of DDAVP was not investigated in the questionnaire. All 8 patients who were effectively treated with DDAVP had AVPR2 gene mutations (p.A37P, p.D85N, p.R104C, and p.Y128S) or AQP2 gene mutations (p.R254Q and c.763_722del).
DISCUSSION
To our knowledge, we have conducted the nationwide survey of NDI in Japan, which has allowed us to evaluate the characteristics of NDI—especially CNDI—in Japan. Our survey is believed to include almost 3/4 of CNDI patients in the Medical Aid Program for Chronic Pediatric Diseases of Specified Categories in Japan during fiscal 2009.
NDI is characterized by resistance of the kidney to AVP due to genetic or acquired causes.9 CNDI is rare, and is known to be caused by mutations in AVPR2 (Xq28) or AQP2 (12q13.12). 1, 2, 3 In contrast, acquired NDI is generally an adverse effect to drugs including lithium, demeclocycline, and amphotericin. As can be expected from their different etiologies, most patients with CNDI are diagnosed in infancy,5, 10 and most of acquired NDI are diagnosed in adulthood. In our extensive study, we also found that 7.0% of patients with CNDI were diagnosed after the age of 3 years, including 3 patients who were diagnosed in their 20s. In general, patients with CNDI show polyuria and polydipsia from birth, as well as fever, poor weight gain and vomiting within the 1st week of life.11, 12 However, polyuria and polydipsia are not always the chief complaints associated with CNDI,5, 10 and other various symptoms are usually found during infancy, such as fever, poor weight gain, failure to thrive, vomiting and anorexia. Furthermore, the results of our survey have revealed that 21% of CNDI patients present no symptoms except for fever at the time of onset. Our findings suggest that many clinicians already consider CNDI when facing a fever of unknown etiology in an infant, but that all clinicians should do so.
As of October 2012, more than 220 mutations in the AVPR2 gene and more than 50 mutations in the AQP2 gene have been identified as causing NDI. 2, 7, 9, 13 Spanakis et al.7 found 13 hotspot mutations in the AVPR2 gene. These mutations were reported in more than 6 families with NDI. 7 To evaluate whether there are clustered mutations among patients with CNDI in Japan, we conducted a survey concerning the detection of mutations in the AVPR2 and AQP2 genes. Although some mutations were especially common (AVPR2 gene: p.D85N, 11% of patients with the gene; p.R106C, 7.7% of patients with the gene), we could not find clearly clustered mutations. However, analyses of mutations in AVPR2 and AQP2 were not performed in 1/3 of the CNDI patients in our study. If all CNDI patients had received gene analyses, it is possible that p.D85N and p.R106C would have appeared as more common mutations in the AVPR2 gene. Moreover, it has been reported that 14 mutations in the AVPR2 gene (p.D85N, p.V88M, p.R104C, p.R106C, p.Y128S, p.G201D, p.F287L, p.M311V, p.N317K, p.N317S, p.N321Y, p.P322S, p.S329R, p.S333del) are associated with partial CNDI, in which mild dysfunction of AVPR2 is exhibited in vitro.8 In our survey, these were detected in 22 (34%) of the 65 patients. Eleven (50%) of the 22 were early diagnosed at age less than 6 months. Because of the small sample size, we could not analyze the relationship between age of diagnosis and genotype. However, AVPR2 gene mutations (p.A37P, p.D85N, p.R104C, and p.Y128S) or AQP2 gene mutations (p.R254Q and c.763_722del) were present in each of the 8 patients with CNDI who were effectively treated with DDAVP. These AVPR2 gene mutations have previously been reported as being associated with a mild phonotype of CNDI. 7, 8 It has been reported that the majority of the subjects with AQP2 gene mutations did not respond to DDAVP. Surprisingly, 4 (50%) of 8 patients with AQP2 gene mutation had response to DDAVP. 14 Accordingly, we suggest that gene analysis can play important roles in treatment selection and early detection of CNDI in siblings.
Commonly, female patients who have CNDI with the AVPR2 mutation are asymptomatic because the mutation is heterozygous. However, our survey surprisingly included 10 female patients with CNDI and the AVPR2 mutations (10 of 86 cases; 12%). Some studies have found that female carriers show a clinical phenotype similar to that of male carriers because of skewed X-inactivation or Turner syndrome.15– 17 Because our form questionnaires did not include details of genotype or X-inactivation, we could not compare clinical form with genotype. However, our survey might reveal that there are more female cases with the AVPR2 mutation than has previously been expected. Further investigation is needed to clarify this issue.
The optimal goals of treatments for NDI are to reduce urine volume and adequately replace lost water through increased water intake. These goals are achieved to some extent by using low-sodium diets, thiazide diuretics, potassium-sparing diuretics (such as amiloride and aldosterone antagonists) and NSAIDs (such as indomethacin and selective cyclooxygenase-2 inhibitors).11, 18– 20 In some reports,18– 21 urine volume decreased by 30% to 70% among patients who received diuretic therapy. If sufficient control of urine volume is not achieved through a single treatment, different treatments are often combined. Although treatment of NDI with DDAVP is generally considered to be ineffective, it has been reported that high doses of DDAVP (10–40 μg) are effective for p.D85N, p.V88M, or p.R104C AVPR2 gene mutations,5, 11, 21 which have mild phenotypes7, 8 as a consequence of the retained function of the AVPR2 receptor. In our study, DDAVP was effective in 4 patients with AVPR2 mutations that caused mild phenotypes (p.A37P, p.D85N, p.R104C, and p.Y128S), and in 4 patients with AQP2 mutations (p. R254Q, c.763_722del). Accordingly, if one of the above mutations is detected, treatment with DDAVP should be considered.
MR is considered as a major complication of NDI, and is critically related to prognosis. In the original studies of NDI, it was reported that MR was prevalent in 70% to 90% of the patients. 3 On the other hand, a report demonstrated that the majority of patients with NDI have normal intelligence.5, 22 In the present study, we found that 14% of CNDI patients are accompanied by MR. The results suggest that the actual prevalence of this complication is located between the findings of these 2 studies,3, 5 and MR may be avoided through treatment. Indeed, it has been reported that NDI leads to recurrent severe hyperosmotic dehydration1, 11 and brain edema brought about by attempts to rehydrate too quickly.11 These events damage both endothelial and other brain cells, and deposit calcium phosphate and other substances, particularly within or around the wall of small vessels,23 resulting in MR. In our study, 8 patients (5%) already had MR at the time of CNDI diagnosis. Moreover, patients with CNDI and MR at onset tend to be diagnosed at an age that is older than the mean age of CNDI diagnosis. These findings indicate that the early diagnosis and careful treatment play an important role in preventing MR.
Megacystis, trabeculated bladder wall, hydroureter, hydronephrosis and other urinary tract abnormalities are major complications of NDI that result from persistent polyuria. 12 Although Uribarri et al. reported that urinary tract abnormalities were found in 67% of patients with NDI in the United States,24 such abnormalities were found in only 73 patients (42%) in our study. Eleven patients (6.4%) experience renal failure in our study. Because ultrasonic examination is widely recognized as important tool for the early detection of urologic complications, we suggest that ultrasonographic follow-up should be performed for all NDI patients. Further, adequate treatment and control of NDI should be performed to prevent the complication of urinary tract abnormalities. Additionally, adequate treatment of urinary tract abnormalities is also needed to prevent acquired NDI.
It is widely recognized that several drugs can cause acquired NDI, including lithium, demeclocycline and amphotericin. Of the drugs that are capable of inducing NDI, lithium is prescribed most frequently.18 It is reported that over 40% of patients treated with lithium develop NDI as an adverse effect.25 In our study, 15 patients (50%) with lithium-induced NDI were detected. Our sample size was too limited to investigate the actual conditions of acquired NDI because we were unable to provide this survey to members of the Japanese Society of Psychiatry and Neurology. However, our findings suggest that all patients receiving lithium treatment should be checked for acquired NDI.
In conclusion, we have presented the results of the nationwide survey of NDI in Japan. Our findings suggest the existence of a form of CNDI with mild symptoms, which is often diagnosed at older ages. We suggest that adequate diagnosis and treatment are the most important factors for improving prognoses. We further suggest that gene analysis should be performed for optimal treatment selection and the early detection of NDI among siblings.
Acknowledgments
Acknowledgments: We thank the members of the Japan Endocrine Society, the Japanese Urological Association, the Japanese Society for Pediatric Endocrinology, the Japanese Society for Pediatric Nephrology, the Japanese Society of Nephrology, the Japanese Society of Neurology and the Japanese Society of Pediatric Urology.
This work was supported in part by a Grant-in-Aid for Health Labour Sciences Research Grants (Number: 21211001) from the Ministry of Health, Labour and Welfare of Japan. This work was supported in part by Grans-in-Aid for Scientific Research, Japan Society for the Promotion of Science (Grant Number: 24591512 and 23591568).
The authors declare no conflict of interest.
REFERENCES
- 1.Bichet DG . Nephrogenic diabetes insipidus. Am J Med. 1998; 105: -42 [DOI] [PubMed] [Google Scholar]
- 2.Fujiwara TM , Bichet DG . Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol. 2005; 16: 2836-46 [DOI] [PubMed] [Google Scholar]
- 3.Sands JM , Bichet DG ; American College of Physicians; American Physiological Society. Nephrogenic diabetes insipidus. Ann Intern Med. 2006; 144: 186-94 [DOI] [PubMed] [Google Scholar]
- 4. Crawford JD, Bode HH Disorders of the posterior pituitary in children In: Gardner LI , editor. Endocrine and genetic diseases of childhood and adolescence. 2nd ed. Philadelphia: W.B. Saunders; 1975. p. 126-58 [Google Scholar]
- 5.van Lieburg AF , Knoers NV , Monnens LA . Clinical presentation and follow-up of 30 patients with congenital nephrogenic diabetes insipidus. J Am Soc Nephrol. 1999; 10: 1958-64 [DOI] [PubMed] [Google Scholar]
- 6.Imura H , Matsumoto K , Ogata E , Yoshida S , Igarashi Y , Kono T . [“Hormone receptor diseases” in Japan: a nation-wide survey for testicular feminization syndrome, pseudohypoparathyroidism, nephrogenic diabetes insipidus, Bartter’s syndrome and congenital adrenocortical unresponsiveness to ACTH]. Nihon Naibunpi Gakkai Zasshi. 1980; 56: 1031-49 [DOI] [PubMed] [Google Scholar]
- 7.Spanakis E , Milord E , Gragnoli C . AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J Cell Physiol. 2008; 217: 605-17 [DOI] [PubMed] [Google Scholar]
- 8.Neocleous V , Skordis N , Shammas C , Efstathiou E , Mastroyiannopoulos NP , Phylactou LA . Identification and characterization of a novel X-linked AVPR2 mutation causing partial nephrogenic diabetes insipidus: a case report and review of the literature. Metabolism. 2012; 61: 922-30 [DOI] [PubMed] [Google Scholar]
- 9.Oksche A , Rosenthal W . The molecular basis of nephrogenic diabetes insipidus. J Mol Med (Berl). 1998; 76: 326-37 [DOI] [PubMed] [Google Scholar]
- 10.Mizuno H , Sugiyama Y , Ohro Y , Imamine H , Kobayashi M , Sasaki S .Clinical characteristics of eight patients with congenital nephrogenic diabetes insipidus. Endocrine. 2004; 24: 55-9 [DOI] [PubMed] [Google Scholar]
- 11.Moeller HB , Rittig S , Fenton RA . Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev. 2013; 34: 278-301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wesche D , Deen PM , Knoers NV . Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012; 27: 2183-204 [DOI] [PubMed] [Google Scholar]
- 13.Sasaki S , Chiga M , Kikuchi E , Rai T , Uchida S . Hereditary nephrogenic diabetes insipidus in Japanese patients: analysis of 78 families and report of 22 new mutations in AVPR2 and AQP2. Clin Exp Nephrol. 2013; 17: 338-44 [DOI] [PubMed] [Google Scholar]
- 14.Shida Y , Matsuoka H , Chiga M , Uchida S , Sasaki S , Sugihara S . Characterization of AQP-2 gene mutation (R254Q) in a family with dominant nephrogenic DI. Pediatr Int. 2013; 55: 105-7 [DOI] [PubMed] [Google Scholar]
- 15.Nomura Y , Onigata K , Nagashima T , Yutani S , Mochizuki H , Nagashima K . Detection of skewed X-inactivation in two female carriers of vasopressin type 2 receptor gene mutation. J Clin Endocrinol Metab. 1997; 82: 3434-7 [DOI] [PubMed] [Google Scholar]
- 16.Kinoshita K , Miura Y , Nagasaki H , Murase T , Bando Y , Oiso Y . A novel deletion mutation in the arginine vasopressin receptor 2 gene and skewed X chromosome inactivation in a female patient with congenital nephrogenic diabetes insipidus. J Endocrinol Invest. 2004; 27: 167-70 [DOI] [PubMed] [Google Scholar]
- 17.Balkin MS , Hoffman R , Willner J . Idiopathic diabetes insipidus occurring in a patient with Turner’s syndrome. Mt Sinai J Med. 1978; 45: 742-7 [PubMed] [Google Scholar]
- 18.Soylu A , Kasap B , Ogun N , Ozturk Y , Turkmen M , Hoefsloot L . Efficacy of COX-2 inhibitors in a case of congenital nephrogenic diabetes insipidus. Pediatr Nephrol. 2005; 20: 1814-7 [DOI] [PubMed] [Google Scholar]
- 19.Sasaki S . Nephrogenic diabetes insipidus: update of genetic and clinical aspects. Nephrol Dial Transplant. 2004; 19: 1351-3 [DOI] [PubMed] [Google Scholar]
- 20.Jakobsson B , Berg U . Effect of hydrochlorothiazide and indomethacin treatment on renal function in nephrogenic diabetes insipidus. Acta Paediatr. 1994; 83: -5 [DOI] [PubMed] [Google Scholar]
- 21.Mizuno H , Fujimoto S , Sugiyama Y , Kobayashi M , Ohro Y , Uchida S . Successful treatment of partial nephrogenic diabetes insipidus with thiazide and desmopressin. Horm Res. 2003; 59: 297-300 [DOI] [PubMed] [Google Scholar]
- 22.Hoekstra JA , van Lieburg AF , Monnens LA , Hulstijn-Dirkmaat GM , Knoers VV . Cognitive and psychosocial functioning of patients with congenital nephrogenic diabetes insipidus. Am J Med Genet. 1996; 61: 81-8 [DOI] [PubMed] [Google Scholar]
- 23.Schofer O , Beetz R , Kruse K , Rascher C , Schutz C , Bohl J . Nephrogenic diabetes insipidus and intracerebral calcification. Arch Dis Child. 1990; 65: 885-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uribarri J , Kaskas M . Hereditary nephrogenic diabetes insipidus and bilateral nonobstructive hydronephrosis. Nephron. 1993; 65: 346-9 [DOI] [PubMed] [Google Scholar]
- 25.Grunfeld JP , Rossier BC . Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009; 5: 270-6 [DOI] [PubMed] [Google Scholar]