

Abstract
The ATBF1/ZFHX3 gene at 16q22 is the second most frequently mutated gene in human prostate cancer and has reduced expression or mislocalization in several types of human tumors. Nonetheless, the hypothesis that ATBF1 has a tumor suppressor function in prostate cancer has not been tested. In this study, we examined the role of ATBF1 in prostatic carcinogenesis by specifically deleting Atbf1 in mouse prostatic epithelial cells. We also examined the effect of Atbf1 deletion on gene expression and signaling pathways in mouse prostates. Histopathologic analyses showed that Atbf1 deficiency caused hyperplasia and mouse prostatic intraepithelial neoplasia (mPIN) primarily in the dorsal prostate but also in other lobes. Hemizygous deletion of Atbf1 also increased the development of hyperplasia and mPIN, indicating a haploinsufficiency of Atbf1. The mPIN lesions expressed luminal cell markers and harbored molecular changes similar to those in human PIN and prostate cancer, including weaker expression of basal cell marker cytokeratin 5 (Ck5), cell adhesion protein E-cadherin, and the smooth muscle layer marker Sma; elevated expression of the oncoproteins phospho-Erk1/2, phospho-Akt and Muc1; and aberrant protein glycosylation. Gene expression profiling revealed a large number of genes that were dysregulated by Atbf1 deletion, particularly those that encode for secretory and cell membrane proteins. The four signaling networks that were most affected by Atbf1 deletion included those centered on Erk1/2 and IGF1, Akt and FSH, NF-κB and progesterone and β-estradiol. These findings provide in vivo evidence that ATBF1 is a tumor suppressor in the prostate, suggest that loss of Atbf1 contributes to tumorigenesis by dysregulating membrane and secretory proteins and multiple signaling pathways, and provide a new animal model for prostate cancer.
Introduction
The AT-motif binding factor 1/zinc finger homeobox 3 (ATBF1/ZFHX3) gene encodes a large protein structurally characterized by multiple zinc-finger motifs and four homeodomains [1]. ATBF1 appears to play a role in neuronal differentiation and cell death [2], [3], [4], atrial fibrillation [5], [6], and embryonic development [7]. ATBF1 could be a tumor suppressor in several organs including the prostate, breast, stomach, liver, and head and neck. Chromosomal deletion frequently occurs in cancer cells, which can inactivate tumor suppressor genes (TSGs) and thus contribute to tumorigenesis [8]. Our previous characterization of 16q22 deletion and mutational screening identified ATBF1 as a candidate TSG as it has frequent deletion and somatic mutations in prostate cancer [9]. Recent genome-wide sequencing of castration-resistant prostate cancer indicated that ATBF1 is the second most frequently mutated gene in human prostate cancer [10]. ATBF1 mutation is also frequent in endometrial cancers [11] and the ATBF1 locus is frequently deleted in breast cancers [12]. A germline variant of ATBF1 is linked to increased risk of sporadic prostate cancer [13] and ATBF1 expression is significantly reduced in breast cancer [12], [14], hepatocellular carcinoma [15], gastric cancer [16], [17], and two transgenic mouse models of prostate cancer, TRAMP-ARpe-T877A/Y and ARR2PB-c-myc[18], [19]. Reduced expression and/or mislocalization of ATBF1 protein has been detected in several types of cancer [14], [20], [21]. These alterations are associated with worse patient survival in breast cancer and histopathologic progression in head and neck cancer [14], [21].
While these studies suggest a tumor suppressor function for ATBF1 in human cancer, this hypothesis has not been tested in animal models. How ATBF1 might suppress carcinogenesis has not been determined, although as a transcription factor ATBF1 has been shown to regulate the expression of several differentiation or tumor-related genes including AFP, aminopeptidase N, neurod1, MUC5AC and p21WAF1[4], [22], [23], [24], [25]. Few mouse models are truly relevant to human prostate cancer by neoplastic phenotypes and/or genetic causes [26] but models still provide a powerful platform for understanding prostate cancer biology and developing novel therapies against prostate cancer.
In this study, we examined ATBF1 function in a knockout mouse model. Using the Cre-loxP system, we deleted Atbf1 specifically in mouse prostatic epithelium (PE) and found that inactivation of Atbf1 caused hyperplasia and mouse prostatic intraepithelial neoplasia (mPIN), primarily in the dorsal prostate (DP). A number of molecular alterations resembling those in human prostate cancer were detected in Atbf1 deletion-induced prostatic lesions. Furthermore, Atbf1 deletion dysregulated the expression of a large number of genes involved in multiple signaling pathways, particularly those that encode for secretory proteins and membrane proteins.
Methods and Materials
Ethics Statement
All animal work was performed in compliance with relevant regulatory standards and was approved by the Institutional Animal Care and Use Committee of Emory University.
Mouse Experiments
The PB-Cre4 transgenic mouse line was obtained from the NCI Mouse Models of Human Cancers Consortium (MMHCC, Frederick, MD, Cat#: 01XF5). Floxed Atbf1 mice (Atbf1flox/wt), in which the Atbf1 genomic DNA from exon 7 to exon 8 was flanked with loxP sequences, were generated in our previous study [7] and maintained on a mixed background of C57BL/6 J and 129Sv/J. Atbf1flox/+;PB-Cre+ (Atbf1PE +/-) mice were made by first crossing PB-Cre+/- male mice with Atbf1flox/+ female mice, then crossing Atbf1flox/+;PB-Cre+ male mice with Atbf1flox/+ female mice to obtain mice with all the desired genotypes. Atbf1+/+;PB-Cre+ (Atbf1PE +/+) male mice were examined as controls for different groups at different ages. Six Atbf1+/+;PB-Cre- male mice were also used as controls. All mice were genotyped by PCR using genomic DNA isolated from tail biopsies [7]. Alleles of Atbf1flox (1248 bp), Atbf1wt (1071 bp) and Atbf1∆7&8 (289 bp) were distinguished based on the size of their PCR products using primers 5’-GGCCCTTGACTGCATTTCTTTCCTGT-3’ and 5’-ATTCGTTAATGGGAAGGTGTCAGA-3’. Primers used for genotyping the Cre gene were 5’-CTGAAGAATGGGACAGGCATTG-3’ and 5’-CATCACTCGTTGCATCGACC-3’ (393 bp).
Histological Analysis, Immunohistochemical (IHC) Staining, Immunofluorescence (IF) Staining, and Special Staining
Prostatic tissues were surgically harvested and fixed in 4% formaldehyde overnight. Prostatic lobes were then dissected, embedded in paraffin, and sectioned at 5-μm thickness. Standard hematoxylin and eosin (H&E) staining was performed for histological analyses. Pathological diagnosis was performed by Dr. Henry F. Frierson and confirmed by Dr. Robert D. Cardiff via a paid service at the Center for Comparative Medicine, Department of Pathology, University of California at Davis. Previously established criteria for mouse prostatic lesions were followed [27], [28].
For IHC staining, tissue sections were deparaffinized, rehydrated, and washed in PBS. Antigen retrieval was done by heating slides in a microwave oven for 15 min or in a pressure cooker for 3 min in citrated buffer (pH 6.0, 10 mM trisodium citrate). After blocking with 5% normal goat serum in Tris-buffered saline with 0.1% Tween-20 (TBST), tissue sections were incubated with primary antibodies at 4°C overnight, followed by incubation with EnVision Polymer-HPR secondary antibodies (Dako, Glostrup, Denmark) at room temperature for 40 min. After the application of DAB-chromogen, tissue sections were mounted and visualized under microscopes. Representative slides were scanned with Nanozoomer 2.0HT (Hamamatsu, Bridgewater, NJ), and pictures were captured using NDP.view software (Hamamatsu). Primary antibodies used in this study included anti-Ki67 (Lab Vision, Fremont, CA), anti-cytokeratin 5 (Covance, Princeton, NJ), anti-cytokeratin18 (Abcam, Cambridge, MA), anti-Ar, anti-Sma (Sigma-Aldrich, St. Louis, MO), anti-synaptophysin (Invitrogen, Carlsbad, CA), anti-Cdh1, anti-phospho-Erk1/2, anti-phospho-Akt, and anti-Spink3 (Cell Signaling Technology, Billerica, MA), anti-Muc1 (Thermo Scientific, Waltham, MA), and anti-clusterin (Santa Cruz Biotechnology, Dallas, TX).
For immunofluorescent staining, tissue slides were prepared and incubated with primary antibodies as described above. Appropriate Alexa Fluor fluorochrome-conjugated secondary antibodies (Invitrogen) were used, and nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min. Slides were mounted and visualized under fluorescent microscopes (Zeiss, Thornwood, NY). Primary antibodies included anti-Atbf1, anti-Tn, and biotinylated-HPA [21], [29]. Special staining, including Periodic acid-Schiff (PAS) and Alcian blue, was performed by the Emory University Department of Pathology.
Proliferation Assay
Tissue sections of mouse prostates were immunostained with the anti-Ki67 antibody to detect cells that expressed the Ki67 proliferation marker. Slides were then scanned with the Nanozoomer 2.0HT scanner for cell counting. For each individual mouse, the number of Ki67-positive epithelial cells and the number of total epithelial cells in the dorsal prostate (DP) were determined using the ImageJ program [30]. The percentage of Ki67-positive epithelial cells in a DP was then calculated and plotted against the genotype. Student’s t-test was performed to evaluate the statistical significance for a difference between two genotypes.
Gene Expression Profiling and Signaling Pathway Analysis
Dorsal prostate (DP) tissue from 4 Atbf1flox/flox;PB-Cre+ (Atbf1PE-/-) and 4 Atbf1+/+;PB-Cre+ (Atbf1PE +/+) mice aged 13-15 months was used in the microarray analysis. After the whole prostate was surgically isolated, one piece of DP from each mouse was freshly isolated, weighed and snap frozen in liquid nitrogen, and the remaining prostatic lobes were collected for histological analyses.
The microarray experiment was performed by Beckman Counter Inc (Indianapolis, IN). Briefly, total RNA was extracted from DP tissues, and the quantity and quality were determined by spectrophotometry and an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA). Two hundred nanograms of total RNA was converted into labeled cRNA with nucleotides coupled to fluorescent dye Cy3 using the Low Input Quick Amp Kit (Agilent Technologies) following the manufacturer’s protocol. Cy3-labeled cRNA (600 ng) from each sample was hybridized to an Agilent Mouse Whole Genome 8x60k Microarray. The hybridized array was then washed and scanned, and data was extracted from the scanned image using Feature Extraction version 10.7 (Agilent Technologies).
To remove array specific effects and ensure that signals from different arrays began at the same baseline, quantile normalization was conducted on each microarray. Two R packages, siggenes (based on Statistical Analysis of Microarrays t-test) and MAANOVA (MicroArray ANalysis Of VAriance) [31], were used to detect differentially expressed genes. Both software packages use a variance shrinkage approach to improve detection accuracy by combining information from all genes when estimating gene level variance. The final reported p-values were from R/MAANOVA. Genes with an expression fold change of 2 or greater and false discovery rate (FDR) less than 0.02 were considered differentially expressed. We then performed pathway analysis using the Ingenuity Pathway Analysis (IPA) program (http://www.ingenuity.com) (Ingenuity Systems, Redwood City, CA) to identify signaling pathways and networks that were attenuated by Atbf1 deletion. Subcellular localization of the corresponding proteins was determined based on the Genecards (http://www.genecards.org) and the Plasma Proteome (http://www.plasmaproteomedatabase.org) databases.
Validation of Differentially Expressed Genes by Quantitative RT-PCR
To validate differentially expressed genes, quantitative RT-PCR was performed in samples from both the 8 DPs used in the microarray analysis and additional DPs, including 7 of Atbf1PE +/+, 10 of Atbf1PE +/-, and 7 of Atbf1PE-/-. Briefly, DPs from mice aged at 12 to 15 months were harvested, and total RNAs were isolated using Trizol reagent (Invitrogen) following the manufacturer’s instructions. One μg of total RNA was used for cDNA synthesis using the iScript reverse transcription kit (Bio-Rad, Hercules, CA). Quantitative RT-PCR was performed with the SYBR Premix Ex Taq™ Kit (Clontech Laboratories, Mountain View, CA) and the ABI Prism 7500 Real-time PCR System (Applied Biosystems, Carlsbad, California). Relative fold changes were calculated using the 2-∆∆Ct method, with mouse β-actin mRNA as the internal control. For each gene, PCR primers were designed to generate PCR products spanning different exons to avoid possible interference from genomic DNA contamination. Primer sequences and PCR product sizes are shown in Table S1.
Results
Atbf1 Deletion Leads to Mouse Prostatic Intraepithelial Neoplasia (mPIN) with Increased Cell Proliferation
Mice with a tissue specific deletion of Atbf1 in prostatic epithelial cells (Atbf1PE-/-) were generated by breeding floxed Atbf1 mice (Atbf1flox/+) with PB-Cre4 transgenic mice (Figure 1A). Prostate-specific deletion of Atbf1 was confirmed by PCR-based genotyping. In 9-week-old Atbf1flox/+;PB-Cre+ (Atbf1PE +/-) mice, a smaller PCR product indicating the allele with deletion (Atbf1∆7&8) was observed in all prostatic lobes (anterior, dorsal, lateral and ventral prostates) but not in other organs examined (Figure 1B). Immunofluorescent staining demonstrated that Atbf1 was predominantly expressed in the nuclei of a majority of Atbf1PE +/+ prostatic epithelial cells, and Cre-mediated Atbf1 deletion dramatically decreased Atbf1 protein expression in the Atbf1PE-/- prostate (Figure 1C). Quantitative RT-PCR analysis with cDNA from the dorsal prostate (DP) at 12-15 weeks showed that hemizygous deletion of Atbf1 significantly reduced Atbf1 mRNA expression (P < 0.0001) (Figure 1D), indicating a haploinsufficiency of Atbf1.
Figure 1.
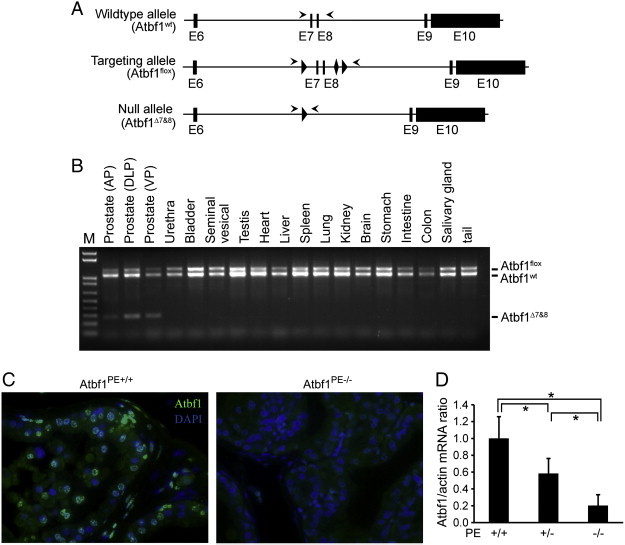
Design and confirmation of Atbf1 deletion in mouse prostates. (A) Schematic of PB-Cre-mediated Atbf1 deletion in mouse prostates. Engineered Atbf1 genomic DNA from exon 6 to exon 10 is shown at the top (wildtype allele). The LoxP and Frt sequences are marked by triangle- and diamond-shaped boxes respectively. Locations of genotyping primers are indicated by arrowheads. PB-Cre-mediated deletion of exons 7 and 8 generates the null allele (Atbf1Δ7&8). (B) Prostate-specific deletion of the Atbf1 gene was confirmed by genotyping PCR in a 9-week-old Atbf1flox/wt;PB-Cre+ (Atbf1PE+/-) mouse. While the Atbf1flox (1248 bp) and Atbf1wt (1071 bp) alleles were identified in all organs examined, the null allele Atbf1Δ7&8 (289 bp) was detectable only in the prostatic lobes. AP, anterior prostate. DLP, dorsal and lateral (dorsolateral) prostate. VP, ventral prostate. (C) Loss of Atbf1 expression in DP of Atbf1PE-/- mice as detected by immunofluorescent staining. Atbf1 (green) was expressed in the nuclei of prostatic epithelial cells in a 6-month-old Atbf1PE +/+ mouse (left panel), but was lost in the same type of cells in a 6-month-old Atbf1PE-/- mouse (right panel). The nuclei were counterstained with DAPI (blue). (D) Reduced Atbf1 mRNA levels in DPs of Atbf1PE +/- and Atbf1PE-/- mice as detected by quantitative RT-PCR. *, P < 0.0001.
To determine the consequence of Atbf1 deletion in the prostate, we examined a cohort of mice with wildtype Atbf1 (Atbf1PE +/+), hemizygously deleted Atbf1 (Atbf1PE +/-), and homozygously deleted Atbf1 (Atbf1PE-/-) aged 3 to 24 months. Most Atbf1PE +/+ mice had a histologically normal prostate phenotype, though a few developed hyperplasia after 18 months due to aging (Table 1). In Atbf1PE-/- mice, however, hyperplasia was seen in the dorsal prostate (DP) at as early as 4 months of age. In these DPs, tufted epithelial cells formed multiple layers and sometimes a solid bridge across the lumen. Atypical nuclei were observed at age 6 months (Figure 2A, panels f & j).
Table 1.
Differences in Atbf1 deletion-induced phenotype among prostatic lobes at different ages.
| Prostatic lobes | Age (Months) | Atbf1 alleles | Mice with different phenotypes (n) |
Total (n) | ||
|---|---|---|---|---|---|---|
| Normal | Hyperplasia | mPIN | ||||
| Dorsal | +/+ | 6 | 6 | |||
| 4-7 | +/- | 3 | 1 | 6 | ||
| -/- | 15 | 3 | 18 | |||
| +/+ | 21 | 21 | ||||
| 9-15 | +/- | 15 | 6 | 1 | 22 | |
| -/- | 8 | 13 | 21 | |||
| +/+ | 7 | 7 | 14 | |||
| 17-24 | +/- | 4 | 12 | 3 | 19 | |
| -/- | 1 | 16 | 17 | |||
| Lateral | +/+ | 6 | 6 | |||
| 4-7 | +/- | 6 | 6 | |||
| -/- | 9 | 4 | 13 | |||
| +/+ | 17 | 5 | 22 | |||
| 9-15 | +/- | 10 | 9 | 2 | 21 | |
| -/- | 4 | 14 | 2 | 20 | ||
| +/+ | 6 | 7 | 1 | 14 | ||
| 17-24 | +/- | 11 | 8 | 1 | 20 | |
| -/- | 1 | 4 | 10 | 15 | ||
| Anterior | +/+ | 6 | 6 | |||
| 4-7 | +/- | 5 | 1 | 6 | ||
| -/- | 8 | 10 | 18 | |||
| +/+ | 20 | 1 | 21 | |||
| 9-15 | +/- | 16 | 6 | 22 | ||
| -/- | 8 | 10 | 3 | 21 | ||
| +/+ | 9 | 6 | 15 | |||
| 17-24 | +/- | 6 | 10 | 3 | 19 | |
| -/- | 3 | 5 | 8 | 16 | ||
| Ventral | +/+ | 6 | 6 | |||
| 4-7 | +/- | 5 | 5 | |||
| -/- | 17 | 17 | ||||
| +/+ | 22 | 22 | ||||
| 9-15 | +/- | 21 | 21 | |||
| -/- | 12 | 8 | 20 | |||
| +/+ | 11 | 3 | 14 | |||
| 17-24 | +/- | 18 | 1 | 19 | ||
| -/- | 3 | 6 | 7 | 16 | ||
Notes: mPIN, mouse prostatic intraepithelial neoplasia; + and – indicate the presence and absence of an Atbf1 allele respectively.
Figure 2.
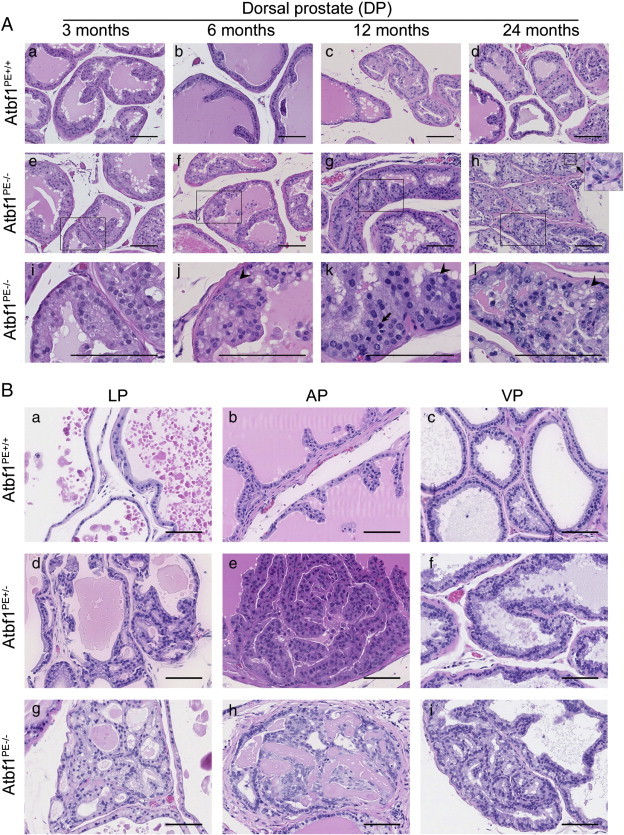
Development of mouse prostatic intraepithelial neoplasia (mPIN) in Atbf1PE-/- prostates. (A) Representative hematoxylin and eosin (HE)-stained mouse tissue sections from dorsal prostates (DPs) with wildtype (Atbf1PE +/+) (a-d) and null Atbf1 (Atbf1PE-/-) (e-h) at 3, 6, 12 and 24 months. Images in i-l are magnified areas of the marked rectangles in images e-h respectively. Note that DPs with Atbf1 deletion (Atbf1PE-/-) show normal prostate (e & i), hyperplasia (f & j) and mPIN (g, h, k, & l) at 3, 6, 12 and 24 months respectively. A mitotic figure is indicated by an arrow in k. The inset at the upper right corner of h is the magnified area of the nearby square, showing intraepithelial red blood cells. Arrowheads in j-l indicate cytoplasmic hyaline associated with atypia. (B) Representative HE-stained tissue sections from mouse lateral prostates (LPs), anterior prostates (APs) and ventral prostates (VPs). Atbf1PE +/+ mice had normal histological phenotypes in these lobes (a-c). Hyperplasia was sometimes detected in Atbf1PE +/- mice (d and e), whereas mPIN was observed in all three lobes with homozygous deletion of Atbf1 (Atbf1PE-/-) (g-i). All scale bars are 100 μm.
By 15 months of age, 13 of 21 Atbf1PE-/- mice further developed mouse prostatic intraepithelial neoplasia (mPIN) (Figure 2A, panels g & h). The DP showed tufted atypical luminal cells, prominent nucleoli, and abundant pale cytoplasm in the mPIN lesions. Hyperchromasia was commonly detected and mitotic figures were seen in some cases (Figure 2A, panel k). Nearly all Atbf1PE-/- mice at age 17-24 months developed mPIN lesions, and a few showed more severe mPIN lesions as indicated by poor orientation of unorganized atypical cells, severe pleomorphism, association with small intraepithelial blood vessels, and host inflammatory responses (Figure 2A, panels h & l). Simple atrophy with cystic dilation was also observed in Atbf1PE-/- mice. In addition, cytoplasmic hyaline was frequently associated with atypia (Figure 2A). The hyaline structure was not associated with accumulation of glycogen, since they were negative to Periodic acid-Schiff (PAS) and Alcian blue (AB) staining (Figure S1). These results indicate that Atbf1 deletion causes progressive development of precancerous lesions in mouse DPs.
In addition to DP, Atbf1 deletion also caused precancerous lesions in other lobes of the prostate (Figure 2B). At 9-15 months of age, most Atbf1PE-/- mice developed hyperplasia or mPIN in the lateral prostate (LP) and anterior prostate (AP), while the ventral prostate (VP) was least affected (Table 1). In addition, mice without the Cre transgene (2 each of Atbf1+/+, Atbf1flox/+ or Atbf1flox/flox), which maintained the wildtype Atbf1, had no visible hyperplasia or dysplasia by age 12 months, indicating that Atbf1 deletion caused the histopathologic phenotypes in Atbf1PE-/- mice. The majority of human prostate cancers (70%) arise in the peripheral zone and the most analogous part in mice is the DP and LP [32]. Therefore we focused on the DP for further characterization.
We examined cell proliferation, which is a hallmark for both cancerous and precancerous lesions, in DP by IHC staining of the Ki67 proliferation marker. Whereas Ki67 positive cells were rare in DPs with wildtype Atbf1 (Atbf1PE +/+) or hemizygous deletion of Atbf1 (Atbf1PE +/-) (0.48% and 0.35% of cells, respectively), they were significantly more frequent (1.73%) in Atbf1-null (Atbf1PE-/-) DPs (Figure 3A). The increase in Ki67-positive cells in Atbf1PE-/- mice indicates accelerated cell proliferation in hyperplasia and mPIN lesions caused by the Atbf1 deletion (P < or = 0.0001) (Figure 3A).
Figure 3.
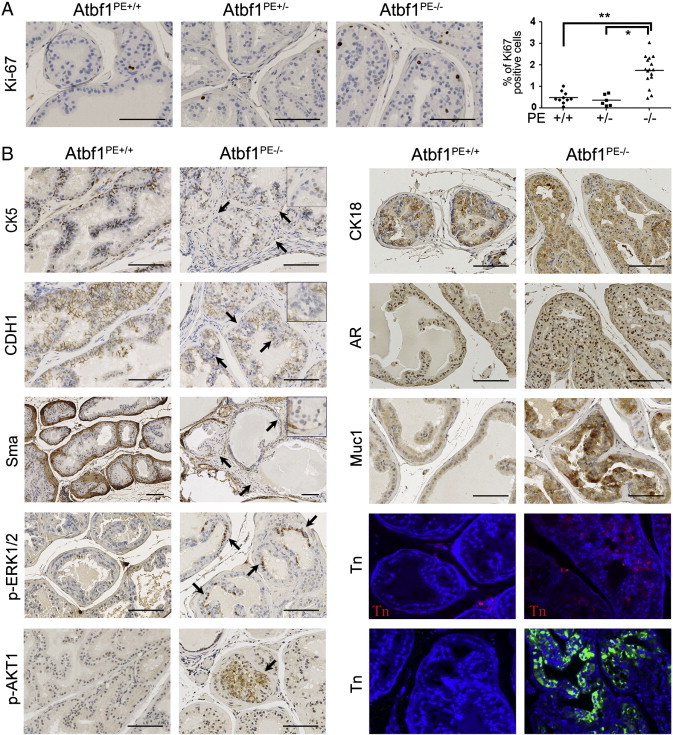
Cell proliferation analysis and molecular characterization of mPIN lesions induced by homozygous deletion of Atbf1. (A) Evaluation of cell proliferation by detecting Ki67-positive cells with immunohistochemicial staining. The percentage of Ki67-positive cells in the epithelium of a DP was plotted against Atbf1 genotype (circular-, squared- and triangle-shaped boxes for Atbf1PE +/+, Atbf1PE +/- and Atbf1PE-/- respectively), and mean values are indicated by horizontal lines. An increase in Ki67-positive cells is seen in hyperplasia and mPIN lesions. *, P = 0.0001. **, P < 0.0001. (B) Homozygous deletion of Atbf1 (Atbf1PE-/-) changed the expression of molecules involved in different characteristics of prostate and prostate cancer, including luminal-basal-stroma architectural markers cytokeratin 5 (Ck5), Ck18, E-cadherin (Cdh1) and smooth muscle actin (Sma); epithelial differentiation markers androgen receptor (Ar); oncogenic signaling molecules phospho-Erk1/2 (p-Erk1/2) and phospho-Akt (p-Akt); and glycoprotein mucin 1 (Muc1) and glycosylation marker Tn antigen (the upper and lower Tn panels were detected by the antibody and HPA methods respectively). DPs with mPIN from 24-month-old mice were used for Ck5, Ck18, E-cadherin, Sma and Ar, while those of 12-month-old mice were used for p-Erk1/2, p-Akt, Muc1 and Tn antigen. Immunofluorescent staining with nuclei counterstained with DAPI (blue) was used for Tn detection, while immunohistochemcial staining was performed for the rest of the molecules. Note the attenuation or loss of Ck5, Cdh1 and Sma, as indicated by arrows and magnified regions at the upper right corner of respective images. Also note the appearance of both p-Erk1/2 and p-Akt (pointed by arrows) and significant increase in Muc1 and Tn antigen expression in mPIN lesions with Atbf1 deletion. All scale bars are 100 μm.
Atbf1 Deletion Attenuates Basal Cells and the Smooth Muscle Layer while Maintaining the Luminal Characteristics
Normal prostate contains luminal and basal epithelial cells that are supported by the fibromuscular layer. To determine whether Atbf1 deletion-induced neoplastic lesions interrupt the structure of the prostate, we determined the expression of several molecular markers for different types of cells in the prostate by IHC staining (Figure 3B). Compared to normal prostates, Atbf1 deletion-induced mPIN lesions had reduced or absent expression of the basal cell marker cytokeratin 5 (Ck5), suggesting the attenuation of basal cells by Atbf1 deletion. The epithelial adhesion protein E-cadherin (Cdh1), which was uniformly detected in prostatic epithelia of wildtype mice, was also decreased or absent once Atbf1 was deleted (Figure 3B). The expression pattern of smooth muscle marker Sma (smooth muscle actin) was also decreased or absent in the smooth muscle layer, indicating its attenuation by Atbf1 deletion (Figure 3B).
In the tufted cells of mPIN induced by Atbf1 deletion, luminal cell markers cytokeratin 18 (Ck18) and androgen receptor (Ar) were expressed as in normal prostatic epithelial cells (Figure 3B), indicating a luminal characteristic of mPIN lesions induced by Atbf1 deletion. Neuroendocrine cells, which were detected by synaptophysin (Syn) staining, were rarely seen in both genotypes.
Atbf1 Deletion in Mouse Prostates Dysregulates a Number of Genes, Particularly those that Encode for Secretory and Cell Membrane Proteins
As a transcription factor, Atbf1 regulates the expression of many genes. To understand what genes are dysregulalted by Atbf1 deletion, we conducted a microarray analysis using DPs from 8 mice at 13-15 months of age, 4 with wildtype Atbf1 (Atbf1PE +/+) and 4 with homozygous deletion of Atbf1 (Atbf1PE-/-). Histological analysis confirmed normal prostatic phenotypes in the Atbf1PE +/+ mice and hyperplasia and mPIN in the Atbf1PE-/- mice. Probes were synthesized from total RNA and hybridized to an Agilent mouse whole genome 8x60k chip, which covers 39,430 Entrez genes and 16,251 lincRNAs. After quantile normalization, gene expression was analyzed by two R computer programs, siggenes and MAANOVA, and the two programs generated similar results with a correlation of 0.97. A supervised gene list was generated using R/MAANOVA. With a false discovery rate at 0.02 and a fold change of 2 or greater, a total of 445 probes were differentially expressed in Atbf1PE-/- DPs, representing 176 upregulated and 215 downregulated) genes (some genes were represented by two or more probes on the microarray) (Table S2).
To validate differential expression of the genes between normal prostates and mPIN, we performed quantitative RT-PCR on a total of 32 samples, including 11 for Atbf1PE +/+, 10 for Atbf1PE +/-, and 11 for Atbf1PE-/- mice. Twenty-six of 30 up-regulated and 5 of 6 down-regulated genes were confirmed with statistical significance (P < 0.05) (Figure 4A, B and Table S3). A number of these molecules have been associated with cancer development. We selected two up-regulated genes, Spink3 and Clu, and confirmed their elevated protein expression in Atbf1PE-/- DP by IHC (Figure 4C). Among the 30 validated genes, 7 (Mcm10, Gabra4, Gabrb1, Rgs2, Serpinb11, St3gal3 and Col4a6) were also differentially expressed in Atbf1PE +/- mice compared to wildtype mice, further indicating the haploinsufficiency of Atbf1 in the regulation of a subset of its target genes (Figure 4A).
Figure 4.
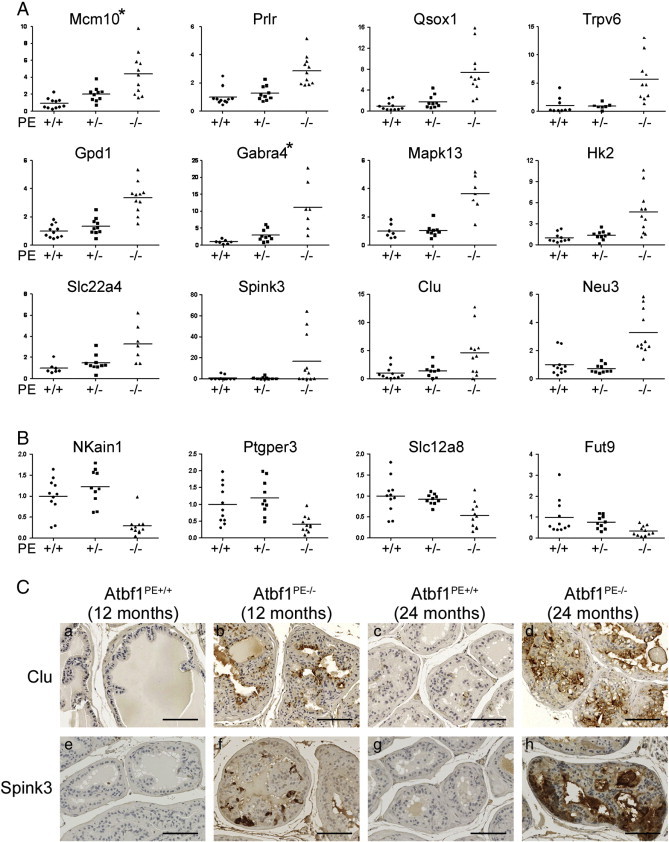
Validation of dysregulated genes by homozygous deletion of Atbf1 in mouse dorsal prostates (DPs). Expression of upregulated (A) and downregulated genes (B) was analyzed by real-time RT-PCR (A, B), with 2 genes’ protein expression also evaluated by immunohistochemical staining (C). In the real-time RT-PCR assay, the relative gene expression levels were plotted as circular (Atbf1PE +/+), squared (Atbf1PE +/-) and triangle (Atbf1PE-/-) shaped boxes respectively, and mean values are indicated by lines. For each gene, the average expression level in Atbf1PE +/+was set to 1, and levels in each individual were adjusted accordingly. Asterisks indicate two genes (Gabra4 and Mcm10) expressed at intermediate levels in Atbf1PE +/- mice, which indicates haploinsufficiency for these genes. Scale bars in panel C are 100 μm.
Notably, a number of the dysregulated genes encode secretory proteins or cell membrane proteins. Among the 391 differentially expressed proteins, 64 (16.4%) were annotated as secretory proteins, including Clu, Timp4, Wnt4, C1ra and Col4a6, and 101 (25.8%) were reportedly localized to the cytoplasmic membrane, including Trpv6, Prlr, Tacstd2, Slc12a8 and Ptger3 (Table S2). In addition, 117 of the 391 differentially expressed proteins (29.9%) have been detected in the plasma or serum, according to the Plasma Proteome database (Table S2).
Membrane proteins play essential roles in different cellular processes, and their abnormal expression and modification often occur during cancer development. One family of such proteins are mucins, which have an aberrant and unique expression pattern in cancer cells and thus have been used as diagnostic markers as well as therapeutic targets [33], [34]. Mucin 1 (MUC1) is an O-glycosylated transmembrane glycoprotein which primarily hydrates, lubricates and protects the epithelial luminal surfaces of ducts [35]. Its core peptide is attached with a number of O-glycans in normal cells but the level of glycosylation is reduced in tumors, leading to the exposure of epitopes for immunodetection and cytoplasmic localization of MUC1 [35], [36]. Therefore, we examined Muc1 expression, and found that Muc1 was significantly upregulated in Atbf1 deletion-induced neoplastic lesions (Figure 3B). The increase in Muc1 expression in the cytoplasm has been associated with alterations in its glycosylation [35], and the Tn antigen, a truncated mucin-type O-glycan, is often overexpressed in human malignancies including prostate cancer [37]. We examined the expression of Tn antigen by immunostaining of prostates with both Helix pomatia agglutinin (HPA), a snail lectin which recognizes Tn antigen of glycoproteins, and an anti-Tn monoclonal antibody. Strong staining for Tn antigen was detected in Atbf1PE-/- DP but not in Atbf1PE +/+ DP, indicating that Atbf1 deletion increased Tn antigen expression (Figure 3B).
Atbf1 Deletion Attenuates Multiple Signaling Pathways Including the Activation of Erk1/2 AKT Oncogenic Signaling
We then input genes differentially expressed between prostates with and without Atbf1 deletion, along with their expression levels, into the Ingenuity Pathway Analysis (IPA) program to identify functional networks and signaling pathways that are affected by Atbf1 deletion. Several were identified to have statistically significant P values, and multiple biological processes were affected, including cell differentiation, tissue development, cell death, cell movement, secretion of bodily fluid, cation transport, atrial fibrillation and blood pressure regulation (Table S4). All these functions are associated with signals mediated by extracellular and cell membrane molecules. The four networks with the smallest P values are shown in Figure 5, which included those centered ERK1/2 and IGF1 (Figure 5A), Akt and FSH (Figure 5B), NF-κB (Figure 5C) and progesterone and β-estradiol (Figure 5D). Although no canonical pathways were predicted with statistical significance, all four of these networks play a role in cancer development.
Figure 5.
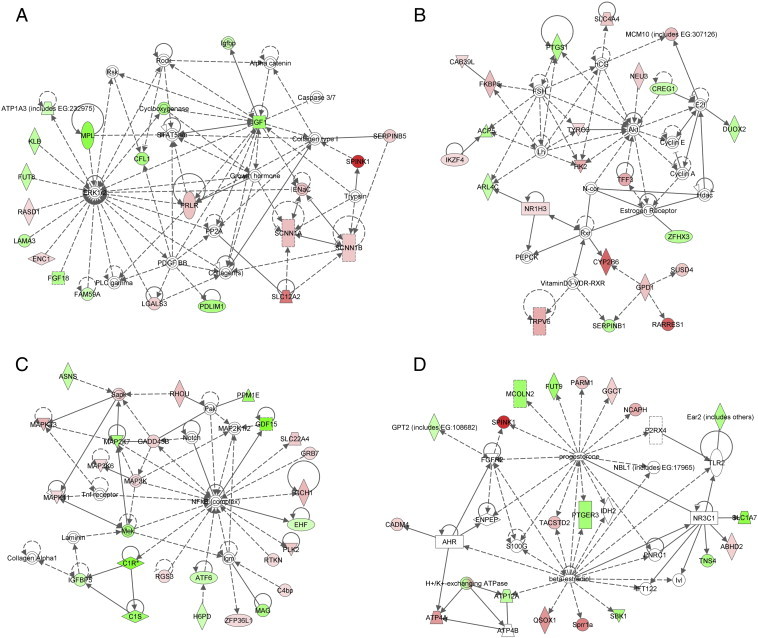
Identification of molecular pathways that are altered by Atbf1 inactivation in mouse prostates. Genes differentially expressed between Atbf1-positive and Atbf1–negative prostates, along with their expression levels, were imported to the Ingenuity Pathway Analysis (IPA) program to construct interacting pathways. The four networks with the smallest P values are shown. The intensity of the node color indicates the degree of upregulation (red) or downregulation (green) in Atbf1-null prostates. Solid and dashed lines indicate direct and indirect interactions respectively. Different shapes of the nodes, including square, circle, diamond, rectangles etc., represent functional classification of the genes.
ERK and AKT are frequently activated during carcinogenesis, so we examined whether they are activated by Atbf1 deletion. Using IHC staining, we found that phosphorylated Erk1/2 and Akt, which represent the activated Erk1/2 and Akt, were clearly detected in Atbf1-null DPs but were undetectable in DPs with wildtype Atbf1 (Figure 3B).
Discussion
Atbf1 Deletion Induces mPIN in Mouse Prostates
While a tumor suppressor function of ATBF1 has been suggested by its frequent chromosomal deletion at 16q22 and somatic mutations in human cancers [8], [9], [10], the effect of Atbf1 inactivation on tumorigenesis has not been tested in a mouse model because conventional deletion of Atbf1 in mice is embryonic lethal and loss of even one allele of Atbf1 results in preweaning mortality and partial embryonic lethality [7]. In this study, we used the Cre-loxP system to specifically delete Atbf1 in mouse prostatic epithelium, and found that Atbf1 deletion induced mPIN in mice, providing for the first time direct evidence for a tumor suppressor function of Atbf1. Hyperplasia was detected as early as 4 months of age, and mPIN occurred when Atbf1PE-/- mice aged. Atbf1 deletion-induced mPIN had established mPIN features such as prominent nucleoli, abundant pale cytoplasm, severe pleomorphism, association with host inflammatory responses, and discontinuity of the fibromuscular sheath [27]. Another feature in the Atbf1-null mPIN was severe atypic cytoplasmic hyaline, which is seen sometimes in human prostate cancer [38] but rarely in mPIN induced by the knockout of other tumor suppressor genes [27]. In human prostates, PIN has been widely considered as a precursor of prostate adenocarcinoma [39], and many known oncogenic genetic events induce mPIN rather than invasive carcinoma in mouse prostates [40], [41]. Therefore, induction of mPIN by Atbf1 deletion provides functional evidence for Atbf1 tumor suppressor activity.
No invasive prostate cancer was detected in any of the Atbf1PE-/- mice even at 25 months old, which has been common for many known tumor suppressor genes including p53, Rb, Nkx3.1, Maspin, Brca2 and p27Kip, whose deletion alone also only causes mPIN lesions [42], [43], [44], [45], [46], [47]. It is well recognized that multiple genetic and epigenetic alterations are usually required for an invasive cancer to develop. While Atbf1 deletion alone appears to be insufficient to induce invasive prostate cancer, it could cooperate with other oncogenic events to induce invasive carcinoma. We are in the process of testing whether Atbf1 deletion cooperates with Pten deletion, a well characterized oncogenic event in prostate cancer [48], in the development and progression of prostate cancer.
Atbf1 Deletion-Induced mPIN Lesions Share Multiple Morphological and Molecular Characteristics with Human Prostate Cancer
Mouse DP is anatomically and biochemically closest to the peripheral zone of human prostate [49], and most human prostate cancers arise from the peripheral zone of prostate [50]. Thus finding of the most severe histopathological lesions in the DP of Atbf1PE-/- mice (Figures 1, 2) indicates a reasonable relevance of Atbf1 deletion-induced mPIN to human prostate cancer. Another histological change that also occurs in human prostate cancer is the interruption of the fibromuscular layer in Atbf1 deletion-induced mPIN lesions, as indicated by attenuated expression of the Sma smooth muscle marker (Figure 3B).
Shared cellular and molecular characteristics further indicate the relevance of Atbf1 deletion-induced mPIN lesion to human prostate cancer. One shared characteristic is increased cell proliferation, as indicated by the increase in Ki67-positive cells in Atbf1-null prostates (Figure 3A). In human prostate cancer, an increased Ki67 proliferation index correlates with higher tumor grade and worse patient survival [51]. Atbf1 deletion-induced mPIN lesions also maintained luminal characteristics as seen in most human prostate cancers, indicated by the positive staining of luminal markers Ck18 and Ar and decreased staining of the Ck5 basal cell marker (Figure 3B).
Multiple molecular alterations reported in human prostate cancer also occurred in Atbf1 deletion-induced mPIN lesions. For example, the cell-cell adhesion molecule E-cadherin, which is often downregulated in human prostate cancer [52], was also downregulated in mPIN lesions induced by Atbf1 deletion (Figure 3B). Increased expression of activated Erk1/2 and Akt (i.e., phosphorylated Erk1/2 and Akt), which mediate two common oncogenic signaling pathways and often occur in human prostate cancer and other mouse models of prostate cancer [53], [54], [55], [56], was also detected in Atbf1-null mPIN (Figure 3B). Another example is the upregulation of Muc1 by Atbf1 deletion (Figure 3B), which also occurs in human primary and metastatic prostate cancers and suggests Muc1 as a potential therapeutic target for cancer treatment [33], [35], [36]. MUC1 is a major cell surface protein that functions as a physical and biological barrier to protect mucous epithelia, and interacts with a number of proteins to trigger diverse signaling pathways including the MAPK/ERK pathway [33], [57]. Overexpression of Muc1 alone in mouse mammary glands can also lead to increased ERK1/2 activity [58]. Thirdly, Atbf1 deletion led to the expression of the Tn antigen (Figure 3B), an O-glycan that is usually found on cancer cells but not on normal cells [29], [59]. Altered glycosylation in cancer cells could affect MUC1 distribution and signaling [35]. These molecular similarities between Atbf1 deletion-induced mPIN lesions and human prostate cancer indicate the model’s intrinsic relevance.
Atbf1 Deficiency Attenuates the Secretory Profile of Mouse Prostates
ATBF1 is a transcription factor, and we would thus expect that Atbf1 deletion in mouse prostates dysregulates a number of genes. This is indeed the case, as hundreds of genes, 391 of which are protein-coding, were differentially expressed between normal prostates and Atbf1-null prostates (Table S2). Some differentially expressed genes, including Clu, Qsox1, Hk2 and Tacstd2, were also identified in mouse prostates with the knockout of Nkx3.1 or Pten, both of which have a tumor suppressor function in prostate cancer [48], [60], [61]. Interestingly, 64 of the 391 (16%) differentially expressed proteins were annotated as secretory proteins and 117 of them (29.9%) have been detected in the plasma or serum (Table S2). We verified the elevation of two secretory proteins, Clu and Spink3, by IHC staining in mouse DPs. Clu was also up-regulated in Nkx3.1 and Pten-deficient mouse prostates and has been implicated in increased prostate cancer resistance to chemotherapy [48], [61], [62]. SPINK1, the human homolog of Spink3, was overexpressed in a subset of ETS rearrangement-negative prostate cancers, and its reduced expression in 22Rv1 prostate cancer cells attenuates cell invasion [63], [64]. In a preliminary experiment, we used liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) to identify secretory proteins in mouse prostates, and found several secretory proteins that were differentially expressed between the wildtype and Atbf1PE-/- mice (data not shown), supporting the conclusion from the microarray study. Dysregulation of a large number of secretory proteins by Atbf1 deletion thus suggests an important role of Atbf1 in the structure and function of the adult prostate. For example, Atbf1 could be necessary for the induction and/or maintenance of differentiated secretory luminal cells in the prostate. Supporting this idea, a role for ATBF1 has been detected in neuronal differentiation [65], [66].
Atbf1 Deletion Leads to Abnormalities in Multiple Signaling Pathways Involved in Carcinogenesis
Some of the secretory proteins dysregulated by Atbf1 deletion, including Igf1 (Tables S2), are known signaling molecules. In addition, 101 of the 391 (25.8%) differentially expressed proteins between normal and Atbf1-null prostates have been reported as membrane proteins (Table S2), including receptors (e.g., Prlr and Ptger3), cell surface antigens (e.g., Ly6e), and ion channels and transporters (e.g., Slc22a4, Trpv6, and Atp12a) (Table S2). Ion channel and transporter proteins affect the concentration of extracellular and intracellular cations to regulate a broad range of biological events related to cancer, including cell proliferation, apoptosis and migration [67]. Whereas some of the ion channel proteins are dysregulated in human prostate cancer, e.g., TRPV6 is upregulated in advanced prostate tumors to increase Ca2 + entry [68], many of the differentially expressed proteins such as PrlR and RGS3 are known to influence cellular signaling. It is thus possible that Atbf1 deletion attenuates multiple signaling pathways and networks, and the pathway analyses supports this predication (Figure 5, Table S4).
When differentially expressed genes between normal and Atbf1-null prostates and their expression levels were examined with the Ingenuity Pathway Analysis (IPA) program, a number of signaling pathways/networks were identified (Table S4), and these pathways regulate multiple biological processes including cell differentiation, tissue development, cell death, cell movement, secretion of bodily fluid, cation transport, atrial fibrillation and blood pressure regulation (Table S4). The four with the highest statistical significance centered on ERK1/2-IGF1, AKT-FSH, NF-κB and progesterone-β-estradiol (Figure 5), and each of these four networks has been implicated in cancer development. For example, activated ERK1/2 and AKT, two well established oncogenic molecules that can be activated by a number of genetic events during carcinogenesis, were also detectable in Atbf1 deletion-induced mPIN lesions (Figure 3B).
For the network centered on ERK1/2 and IGF1, there were multiple molecules linking them (Figure 5A), but whether and how these two nodes truly interact are unknown, although higher serum concentrations of IGF1 are associated with an increased risk of several types of cancers including prostate cancer [69], and IGF1 signaling involves the activation of ERK1/2 [70]. For the network centered on follicle-stimulating hormone (FSH) and AKT (Figure 5B), the FSH hormone is known to act on granulosa cells within the immature follicle to promote proliferation, inhibit apoptosis, and stimulate hormone production; and multiple signaling pathways including the PI3K/AKT pathway are involved in FSH function [71], [72]. FSH also stimulates the PI3K-dependent pathway in the proliferation and differentiation of Sertoli cells [73], [74] and meiotic maturation of oocytes [75]. Interestingly, in addition to androgens, FSH is another hormone that influences the pathogenesis and progression of prostate cancer, as FSH and its receptors are upregulated in prostate cancer and the serum level of FSH is associated with extraprostatic extension of prostate cancer [76], [77]. Therefore, increased FSH hormonal signaling activity and subsequent PI3K/AKT activation could be one of the major pathways that mediate the effect of Atbf1 deletion on prostatic tumorigenesis.
Another signaling network attenuated by Atbf1 deletion focused on NF-κB (Figure 5C), which often has increased activity in human cancers and thus has been considered a therapeutic target in cancer treatment [78]. In human prostate cancer, NF-κB is constitutively active in a subset of castration-resistant prostate cancers, and NF-κB overexpression significantly associates with shorter patient survival [79], [80]. Human prostate cancer cell lines also have constitutively active NF-κB [81], and inhibition of NF-κB activity inhibited their growth in xenograft models, decreased bone resorption of prostate cancer cells co-cultured with bone marrow, and reduced their invasive capability [82], [83]. Interestingly, ATBF1 is the second most frequently mutated gene in castration-resistant human prostate cancer [9], [10]. Taken together, it is possible that inactivation of ATBF1 in human prostate cancer activates NF-κB signaling to induce and/or promote the progression of prostate cancer to an androgen-independent and/or metastatic state.
Surprisingly, the fourth most affected signaling network by Atbf1 deletion was the estrogen-progesterone signaling network (Figure 5D). The presence of estrogen and progesterone receptors in prostate cancer has been documented [84], and so has the role of estrogen in the development and progression of prostate cancer [85], [86]. For example, androgen-responsive LNCaP prostate cancer cells are stimulated by estradiol for growth via estrogen receptors while the androgen-insensitive PC-3 prostate cancer cell proliferation is inhibited by estrogen [85]. Human prostate expresses both ERα and ERβ, which are dysregulated during the development and progression of prostate cancer. ERα is often upregulated to mediate the oncogenic effects of estradiol, which also involves the estrogen-regulated progesterone receptor (PR), during prostate cancer progression [87]. On the other hand, ERβ is downregulated in castration-resistant prostate cancer and thus serves as a tumor suppressor [87]. ERβ mediates the inhibitory effect of antiestrogens on the development of castration-resistant prostate cancer by interacting with other transcription factors to upregulate FOXO1, inducing anoikis and thus suppressing the growth of prostate cancer [88].
The role of estrogen and progesterone in prostate cancer is better documented in mouse models [86]. Further supporting the effect of Atbf1 deletion on estrogen signaling, our previous studies demonstrated that in breast cancer cells estrogen signaling upregulates ATBF1 transcription but causes ATBF1 protein degradation [89], [90], while ATBF1 inhibits ERα function by selectively competing with one of its coactivators for the binding to ERα [91]. Atbf1 deletion increased cell proliferation only in ERα-positive but not in ERα-negative cells [92] and the Pg-progesterone signaling upregulates ATBF1 in mammary epithelial cells [93]. All these findings suggest the possibility that Atbf1 suppresses ERα signaling in normal prostates and Atbf1 deletion leads to more active estrogen signaling that could promote the development and progression of prostate cancer.
The fifth most significant network based on the P value was the network of 9 genes involved in atrial fibrillation (Table S4), a common heart rhythm disorder. The role of ATBF1 in atrial fibrillation has been suggested by a genetic association of ATBF1 sequence variants with this disorder [5], [6], and the 9 genes regulated by Atbf1 provide a clue for how ATBF1 mutation may modulate atrial fibrillation.
In summary, we found that deletion of Atbf1 in mouse prostates caused mPIN lesions, and the mPIN lesions shared a number of histopathologic and molecular features with human PIN and prostate cancer, providing functional evidence for a tumor suppressor activity of ATBF1 in human prostate cancer and establishing a mouse model of prostatic carcinogenesis that is relevant to human prostate cancer. In addition, a number of genes, particularly those encoding for cell membrane and secretory proteins, were dysregulated by Atbf1 deletion, and the most affected signaling networks centered on Erk1/2 and IGF1, Akt and FSH, NF-κB and progesterone and β-estradiol, all of which have been implicated in human cancer development. These findings provide in vivo evidence that ATBF1 is a tumor suppressor in the prostate, suggest that loss of Atbf1 contributes to tumorigenesis by dysregulating membrane and secretory proteins and multiple signaling pathways, and provide a new and clinically relevant animal model for prostate cancer.
Acknowledgments
The authors thank Dr. Robert Cardiff of University of California at Davis for pathological diagnosis; Drs. Leon Bernal-Mizrachi and Jeanne Kowalski for their help in the design and analysis of microarray experiments; Drs. Wei Zhou, Michael M. Shen, and Song-Qing Fan for valuable discussions; and Drs. Lori King and Anthea Hammond for manuscript editing. This work was supported by National Institutes of Health grant CA121459 (to JT Dong).
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.05.001.
Appendix A. Supplementary Data
Periodic acid-Schiff (PAS) and Alcian blue (AB) staining in mouse dorsal prostates. Genotypes are indicated at the top. All scale bars are 100 μm.
Primer sequences and PCR product sizes for gene expression validation by real-time RT-PCR.
Differentially expressed genes between dorsal prostates with and without homozygous deletion of Atbf1, with fold changes, protein localization to the cellular membrane (CM), and whether their proteins are secretory (S) or have been detected in the plasma or serum (Yes), as identified by microarray analysis.
Validation of differentially expressed genes between dorsal prostates with normal Atbf1 (+/+) and those with hemizygous (+/-) or homozygous deletion (-/-) of Atbf1 by real-time RT-PCR.
Functional annotation of differentially expressed genes between prostates with and without homozygous deletion of Atbf1 by Ingenuity Pathways Analysis (IPA).
References
- 1.Miura Y, Tam T, Ido A, Morinaga T, Miki T, Hashimoto T, Tamaoki T. Cloning and characterization of an ATBF1 isoform that expresses in a neuronal differentiation-dependent manner. J Biol Chem. 1995;270:26840–26848. doi: 10.1074/jbc.270.45.26840. [DOI] [PubMed] [Google Scholar]
- 2.Ishii Y, Kawaguchi M, Takagawa K, Oya T, Nogami S, Tamura A, Miura Y, Ido A, Sakata N, Hashimoto-Tamaoki T. ATBF1-A protein, but not ATBF1-B, is preferentially expressed in developing rat brain. J Comp Neurol. 2003;465:57–71. doi: 10.1002/cne.10807. [DOI] [PubMed] [Google Scholar]
- 3.Jung CG, Uhm KO, Miura Y, Hosono T, Horike H, Khanna KK, Kim MJ, Michikawa M. Beta-amyloid increases the expression level of ATBF1 responsible for death in cultured cortical neurons. Mol Neurodegener. 2011;6:47. doi: 10.1186/1750-1326-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jung CG, Kim HJ, Kawaguchi M, Khanna KK, Hida H, Asai K, Nishino H, Miura Y. Homeotic factor ATBF1 induces the cell cycle arrest associated with neuronal differentiation. Development. 2005;132:5137–5145. doi: 10.1242/dev.02098. [DOI] [PubMed] [Google Scholar]
- 5.Benjamin EJ, Rice KM, Arking DE, Pfeufer A, van Noord C, Smith AV, Schnabel RB, Bis JC, Boerwinkle E, Sinner MF. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–881. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gudbjartsson DF, Holm H, Gretarsdottir S, Thorleifsson G, Walters GB, Thorgeirsson G, Gulcher J, Mathiesen EB, Njolstad I, Nyrnes A. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009;41:876–878. doi: 10.1038/ng.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun X, Fu X, Li J, Xing C, Martin DW, Zhang HH, Chen Z, Dong JT. Heterozygous deletion of Atbf1 by the Cre-loxP system in mice causes preweaning mortality. Genesis. 2012;50:819–827. doi: 10.1002/dvg.22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong JT. Chromosomal deletions and tumor suppressor genes in prostate cancer. Cancer Metastasis Rev. 2001;20:173–193. doi: 10.1023/a:1015575125780. [DOI] [PubMed] [Google Scholar]
- 9.Sun X, Frierson HF, Chen C, Li C, Ran Q, Otto KB, Cantarel BL, Vessella RL, Gao AC, Petros J. Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat Genet. 2005;37:407–412. doi: 10.1038/ng1528. [DOI] [PubMed] [Google Scholar]
- 10.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun X, Zhou Y, Otto KB, Wang M, Chen C, Zhou W, Subramanian K, Vertino PM, Dong JT. Infrequent mutation of ATBF1 in human breast cancer. J Cancer Res Clin Oncol. 2007;133:103–105. doi: 10.1007/s00432-006-0148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J, Sauvageot J, Ewing CM, Sun J, Liu W, Isaacs SD, Wiley KE, Diaz L, Zheng SL, Walsh PC. Germline ATBF1 mutations and prostate cancer risk. Prostate. 2006;66:1082–1085. doi: 10.1002/pros.20430. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, Hamaguchi M, Kawaguchi M, Miura Y, Iwase H. ATBF1-A messenger RNA expression is correlated with better prognosis in breast cancer. Clin Cancer Res. 2005;11:193–198. [PubMed] [Google Scholar]
- 15.Kim CJ, Song JH, Cho YG, Cao Z, Lee YS, Nam SW, Lee JY, Park WS. Down-regulation of ATBF1 is a major inactivating mechanism in hepatocellular carcinoma. Histopathology. 2008;52:552–559. doi: 10.1111/j.1365-2559.2008.02980.x. [DOI] [PubMed] [Google Scholar]
- 16.Kataoka H, Miura Y, Joh T, Seno K, Tada T, Tamaoki T, Nakabayashi H, Kawaguchi M, Asai K, Kato T. Alpha-fetoprotein producing gastric cancer lacks transcription factor ATBF1. Oncogene. 2001;20:869–873. doi: 10.1038/sj.onc.1204160. [DOI] [PubMed] [Google Scholar]
- 17.Cho YG, Song JH, Kim CJ, Lee YS, Kim SY, Nam SW, Lee JY, Park WS. Genetic alterations of the ATBF1 gene in gastric cancer. Clin Cancer Res. 2007;13:4355–4359. doi: 10.1158/1078-0432.CCR-07-0619. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi S, Watanabe T, Okada M, Inoue K, Ueda T, Takada I, Watabe T, Yamamoto Y, Fukuda T, Nakamura T. Noncanonical Wnt signaling mediates androgen-dependent tumor growth in a mouse model of prostate cancer. Proc Natl Acad Sci U S A. 2011;108:4938–4943. doi: 10.1073/pnas.1014850108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, Sawyers CL. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 20.Mabuchi M, Kataoka H, Miura Y, Kim TS, Kawaguchi M, Ebi M, Tanaka M, Mori Y, Kubota E, Mizushima T. Tumor suppressor, AT motif binding factor 1 (ATBF1), translocates to the nucleus with runt domain transcription factor 3 (RUNX3) in response to TGF-beta signal transduction. Biochem Biophys Res Commun. 2010;398:321–325. doi: 10.1016/j.bbrc.2010.06.090. [DOI] [PubMed] [Google Scholar]
- 21.Sun X, Li J, Sica G, Fan SQ, Wang Y, Chen Z, Muller S, Chen ZG, Fu X, Dong XY. Interruption of nuclear localization of ATBF1 during the histopathologic progression of head and neck squamous cell carcinoma. Head Neck. 2013;35:1007–1014. doi: 10.1002/hed.23077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yasuda H, Mizuno A, Tamaoki T, Morinaga T. ATBF1, a multiple-homeodomain zinc finger protein, selectively down-regulates AT-rich elements of the human alpha-fetoprotein gene. Mol Cell Biol. 1994;14:1395–1401. doi: 10.1128/mcb.14.2.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kataoka H, Joh T, Miura Y, Tamaoki T, Senoo K, Ohara H, Nomura T, Tada T, Asai K, Kato T. AT motif binding factor 1-A (ATBF1-A) negatively regulates transcription of the aminopeptidase N gene in the crypt-villus axis of small intestine. Biochem Biophys Res Commun. 2000;267:91–95. doi: 10.1006/bbrc.1999.1911. [DOI] [PubMed] [Google Scholar]
- 24.Mori Y, Kataoka H, Miura Y, Kawaguchi M, Kubota E, Ogasawara N, Oshima T, Tanida S, Sasaki M, Ohara H. Subcellular localization of ATBF1 regulates MUC5AC transcription in gastric cancer. Int J Cancer. 2007;121:241–247. doi: 10.1002/ijc.22654. [DOI] [PubMed] [Google Scholar]
- 25.Miura Y, Kataoka H, Joh T, Tada T, Asai K, Nakanishi M, Okada N, Okada H. Susceptibility to killer T cells of gastric cancer cells enhanced by Mitomycin-C involves induction of ATBF1 and activation of p21 (Waf1/Cip1) promoter. Microbiol Immunol. 2004;48:137–145. doi: 10.1111/j.1348-0421.2004.tb03491.x. [DOI] [PubMed] [Google Scholar]
- 26.Valkenburg KC, Williams BO. Mouse models of prostate cancer. Prostate Cancer. 2011;2011:895238. doi: 10.1155/2011/895238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, Shen MM, Cardiff RD. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161:727–735. doi: 10.1016/S0002-9440(10)64228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, Humphrey PA, Sundberg JP, Rozengurt N, Barrios R. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 29.Ju T, Otto VI, Cummings RD. The Tn antigen-structural simplicity and biological complexity. Angew Chem Int Ed Engl. 2011;50:1770–1791. doi: 10.1002/anie.201002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H, Kerr KM, Cui X, and Churchill GA (2003). MAANOVA: a software package for the analysis of spotted cDNA microarray experiments In Book ed.(Eds). Springer, New York, pp. 313-341.
- 32.Lee CH, Akin-Olugbade O, Kirschenbaum A. Overview of prostate anatomy, histology, and pathology. Endocrinol Metab Clin North Am. 2011;40:565–575. doi: 10.1016/j.ecl.2011.05.012. [viii-ix] [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Cozzi PJ. MUC1 is a promising therapeutic target for prostate cancer therapy. Curr Cancer Drug Targets. 2007;7:259–271. doi: 10.2174/156800907780618338. [DOI] [PubMed] [Google Scholar]
- 34.Scholfield DP, Simms MS, Bishop MC. MUC1 mucin in urological malignancy. BJU Int. 2003;91:560–566. doi: 10.1046/j.1464-410x.2003.04132.x. [DOI] [PubMed] [Google Scholar]
- 35.Singh R, Bandyopadhyay D. MUC1: a target molecule for cancer therapy. Cancer Biol Ther. 2007;6:481–486. doi: 10.4161/cbt.6.4.4201. [DOI] [PubMed] [Google Scholar]
- 36.Kufe DW. Functional targeting of the MUC1 oncogene in human cancers. Cancer Biol Ther. 2009;8:1197–1203. doi: 10.4161/cbt.8.13.8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hakomori SI, Cummings RD. Glycosylation effects on cancer development. Glycoconj J. 2012;29:565–566. doi: 10.1007/s10719-012-9448-4. [DOI] [PubMed] [Google Scholar]
- 38.Rioux-Leclercq NC, Epstein JI. Unusual morphologic patterns of basal cell hyperplasia of the prostate. Am J Surg Pathol. 2002;26:237–243. doi: 10.1097/00000478-200202000-00011. [DOI] [PubMed] [Google Scholar]
- 39.Clouston D, Bolton D. In situ and intraductal epithelial proliferations of prostate: definitions and treatment implications. Part 1: Prostatic intraepithelial neoplasia. BJU Int. 2012;109(Suppl 3):22–26. doi: 10.1111/j.1464-410X.2012.11040.x. [DOI] [PubMed] [Google Scholar]
- 40.Kasper S. Survey of genetically engineered mouse models for prostate cancer: analyzing the molecular basis of prostate cancer development, progression, and metastasis. J Cell Biochem. 2005;94:279–297. doi: 10.1002/jcb.20339. [DOI] [PubMed] [Google Scholar]
- 41.Jeet V, Russell PJ, Khatri A. Modeling prostate cancer: a perspective on transgenic mouse models. Cancer Metastasis Rev. 2010;29:123–142. doi: 10.1007/s10555-010-9212-9. [DOI] [PubMed] [Google Scholar]
- 42.Maddison LA, Sutherland BW, Barrios RJ, Greenberg NM. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004;64:6018–6025. doi: 10.1158/0008-5472.CAN-03-2509. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, Nikitin AY. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66:7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 44.Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, Desai N, Wang Y, Hayward SW, Cunha GR, Cardiff RD, Shen MM, Abate-Shen C. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 2002;62:2999–3004. [PubMed] [Google Scholar]
- 45.Shao LJ, Shi HY, Ayala G, Rowley D, Zhang M. Haploinsufficiency of the maspin tumor suppressor gene leads to hyperplastic lesions in prostate. Cancer Res. 2008;68:5143–5151. doi: 10.1158/0008-5472.CAN-08-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Francis JC, McCarthy A, Thomsen MK, Ashworth A, Swain A. Brca2 and Trp53 deficiency cooperate in the progression of mouse prostate tumourigenesis. PLoS Genet. 2010;6:e1000995. doi: 10.1371/journal.pgen.1000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 48.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 49.Berquin IM, Min Y, Wu R, Wu H, Chen YQ. Expression signature of the mouse prostate. J Biol Chem. 2005;280:36442–36451. doi: 10.1074/jbc.M504945200. [DOI] [PubMed] [Google Scholar]
- 50.McNeal JE. Cancer volume and site of origin of adenocarcinoma in the prostate: relationship to local and distant spread. Hum Pathol. 1992;23:258–266. doi: 10.1016/0046-8177(92)90106-d. [DOI] [PubMed] [Google Scholar]
- 51.Bubendorf L, Tapia C, Gasser TC, Casella R, Grunder B, Moch H, Mihatsch MJ, Sauter G. Ki67 labeling index in core needle biopsies independently predicts tumor-specific survival in prostate cancer. Hum Pathol. 1998;29:949–954. doi: 10.1016/s0046-8177(98)90199-x. [DOI] [PubMed] [Google Scholar]
- 52.De Marzo AM, Knudsen B, Chan-Tack K, Epstein JI. E-cadherin expression as a marker of tumor aggressiveness in routinely processed radical prostatectomy specimens. Urology. 1999;53:707–713. doi: 10.1016/s0090-4295(98)00577-9. [DOI] [PubMed] [Google Scholar]
- 53.Gao H, Ouyang X, Banach-Petrosky WA, Gerald WL, Shen MM, Abate-Shen C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci U S A. 2006;103:14477–14482. doi: 10.1073/pnas.0606836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jeong JH, Wang Z, Guimaraes AS, Ouyang X, Figueiredo JL, Ding Z, Jiang S, Guney I, Kang GH, Shin E. BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PLoS One. 2008;3:e3949. doi: 10.1371/journal.pone.0003949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oka H, Chatani Y, Kohno M, Kawakita M, Ogawa O. Constitutive activation of the 41- and 43-kDa mitogen-activated protein (MAP) kinases in the progression of prostate cancer to an androgen-independent state. Int J Urol. 2005;12:899–905. doi: 10.1111/j.1442-2042.2005.01164.x. [DOI] [PubMed] [Google Scholar]
- 56.Shen MM, Abate-Shen C. Pten inactivation and the emergence of androgen-independent prostate cancer. Cancer Res. 2007;67:6535–6538. doi: 10.1158/0008-5472.CAN-07-1271. [DOI] [PubMed] [Google Scholar]
- 57.Bafna S, Kaur S, Batra SK. Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene. 2010;29:2893–2904. doi: 10.1038/onc.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schroeder JA, Thompson MC, Gardner MM, Gendler SJ. Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation in the mouse mammary gland. J Biol Chem. 2001;276:13057–13064. doi: 10.1074/jbc.M011248200. [DOI] [PubMed] [Google Scholar]
- 59.Slovin SF, Ragupathi G, Musselli C, Olkiewicz K, Verbel D, Kuduk SD, Schwarz JB, Sames D, Danishefsky S, Livingston PO. Fully synthetic carbohydrate-based vaccines in biochemically relapsed prostate cancer: clinical trial results with alpha-N-acetylgalactosamine-O-serine/threonine conjugate vaccine. J Clin Oncol. 2003;21:4292–4298. doi: 10.1200/JCO.2003.04.112. [DOI] [PubMed] [Google Scholar]
- 60.Magee JA, Abdulkadir SA, Milbrandt J. Haploinsufficiency at the Nkx3.1 locus. A paradigm for stochastic, dosage-sensitive gene regulation during tumor initiation. Cancer Cell. 2003;3:273–283. doi: 10.1016/s1535-6108(03)00047-3. [DOI] [PubMed] [Google Scholar]
- 61.Song H, Zhang B, Watson MA, Humphrey PA, Lim H, Milbrandt J. Loss of Nkx3.1 leads to the activation of discrete downstream target genes during prostate tumorigenesis. Oncogene. 2009;28:3307–3319. doi: 10.1038/onc.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rizzi F, Bettuzzi S. The clusterin paradigm in prostate and breast carcinogenesis. Endocr Relat Cancer. 2010;17:R1–R17. doi: 10.1677/ERC-09-0140. [DOI] [PubMed] [Google Scholar]
- 63.Tomlins SA, Rhodes DR, Yu J, Varambally S, Mehra R, Perner S, Demichelis F, Helgeson BE, Laxman B, Morris DS. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell. 2008;13:519–528. doi: 10.1016/j.ccr.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jhavar S, Brewer D, Edwards S, Kote-Jarai Z, Attard G, Clark J, Flohr P, Christmas T, Thompson A, Parker M. Integration of ERG gene mapping and gene-expression profiling identifies distinct categories of human prostate cancer. BJU Int. 2009;103:1256–1269. doi: 10.1111/j.1464-410X.2008.08200.x. [DOI] [PubMed] [Google Scholar]
- 65.Ido A, Miura Y, Tamaoki T. Activation of ATBF1, a multiple-homeodomain zinc-finger gene, during neuronal differentiation of murine embryonal carcinoma cells. Dev Biol. 1994;163:184–187. doi: 10.1006/dbio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 66.Hemmi K, Ma D, Miura Y, Kawaguchi M, Sasahara M, Hashimoto-Tamaoki T, Tamaoki T, Sakata N, Tsuchiya K. A homeodomain-zinc finger protein, ZFHX4, is expressed in neuronal differentiation manner and suppressed in muscle differentiation manner. Biol Pharm Bull. 2006;29:1830–1835. doi: 10.1248/bpb.29.1830. [DOI] [PubMed] [Google Scholar]
- 67.Arcangeli A, Yuan JX. American Journal of Physiology-Cell Physiology theme: ion channels and transporters in cancer. Am J Physiol Cell Physiol. 2011;301:C253–C254. doi: 10.1152/ajpcell.00159.2011. [DOI] [PubMed] [Google Scholar]
- 68.Van Haute C, De Ridder D, Nilius B. TRP channels in human prostate. Sci World J. 2010;10:1597–1611. doi: 10.1100/tsw.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Furstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol. 2002;3:298–302. doi: 10.1016/s1470-2045(02)00731-3. [DOI] [PubMed] [Google Scholar]
- 70.Zhu C, Qi X, Chen Y, Sun B, Dai Y, Gu Y. PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in IGF-1-induced VEGF-C upregulation in breast cancer. J Cancer Res Clin Oncol. 2011;137:1587–1594. doi: 10.1007/s00432-011-1049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hunzicker-Dunn M, Maizels ET. FSH signaling pathways in immature granulosa cells that regulate target gene expression: branching out from protein kinase A. Cell Signal. 2006;18:1351–1359. doi: 10.1016/j.cellsig.2006.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hunzicker-Dunn ME, Lopez-Biladeau B, Law NC, Fiedler SE, Carr DW, Maizels ET. PKA and GAB2 play central roles in the FSH signaling pathway to PI3K and AKT in ovarian granulosa cells. Proc Natl Acad Sci U S A. 2012;109:E2979–E2988. doi: 10.1073/pnas.1205661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dupont J, Musnier A, Decourtye J, Boulo T, Lecureuil C, Guillou H, Valet S, Fouchecourt S, Pitetti JL, Nef S. FSH-stimulated PTEN activity accounts for the lack of FSH mitogenic effect in prepubertal rat Sertoli cells. Mol Cell Endocrinol. 2010;315:271–276. doi: 10.1016/j.mce.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 74.Meroni SB, Riera MF, Pellizzari EH, Galardo MN, Cigorraga SB. FSH activates phosphatidylinositol 3-kinase/protein kinase B signaling pathway in 20-day-old Sertoli cells independently of IGF-I. J Endocrinol. 2004;180:257–265. doi: 10.1677/joe.0.1800257. [DOI] [PubMed] [Google Scholar]
- 75.Hoshino Y, Yokoo M, Yoshida N, Sasada H, Matsumoto H, Sato E. Phosphatidylinositol 3-kinase and Akt participate in the FSH-induced meiotic maturation of mouse oocytes. Mol Reprod Dev. 2004;69:77–86. doi: 10.1002/mrd.20150. [DOI] [PubMed] [Google Scholar]
- 76.Porter AT, F ACRO, Ben-Josef E. Humoral mechanisms in prostate cancer: A role for FSH. Urol Oncol. 2001;6:131–138. doi: 10.1016/s1078-1439(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 77.Ide H, Terado Y, Sakamaki K, Inoue M, Nakajima A, Lu Y, Hisasue S, Yamaguchi R, Muto S, Horie S. Serum level of follicle-stimulating hormone is associated with extraprostatic extension of prostate cancer. Prostate Int. 2013;1:109–112. doi: 10.12954/PI.13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen HM, Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14:348–363. doi: 10.1007/s10495-009-0315-0. [DOI] [PubMed] [Google Scholar]
- 79.McCall P, Bennett L, Ahmad I, Mackenzie LM, Forbes IW, Leung HY, Sansom OJ, Orange C, Seywright M, Underwood MA. NFkappaB signalling is upregulated in a subset of castrate-resistant prostate cancer patients and correlates with disease progression. Br J Cancer. 2012;107:1554–1563. doi: 10.1038/bjc.2012.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suh J, Payvandi F, Edelstein LC, Amenta PS, Zong WX, Gelinas C, Rabson AB. Mechanisms of constitutive NF-kappaB activation in human prostate cancer cells. Prostate. 2002;52:183–200. doi: 10.1002/pros.10082. [DOI] [PubMed] [Google Scholar]
- 81.Sweeney C, Li L, Shanmugam R, Bhat-Nakshatri P, Jayaprakasan V, Baldridge LA, Gardner T, Smith M, Nakshatri H, Cheng L. Nuclear factor-kappaB is constitutively activated in prostate cancer in vitro and is overexpressed in prostatic intraepithelial neoplasia and adenocarcinoma of the prostate. Clin Cancer Res. 2004;10:5501–5507. doi: 10.1158/1078-0432.CCR-0571-03. [DOI] [PubMed] [Google Scholar]
- 82.Kikuchi E, Horiguchi Y, Nakashima J, Kuroda K, Oya M, Ohigashi T, Takahashi N, Shima Y, Umezawa K, Murai M. Suppression of hormone-refractory prostate cancer by a novel nuclear factor kappaB inhibitor in nude mice. Cancer Res. 2003;63:107–110. [PubMed] [Google Scholar]
- 83.Andela VB, Gordon AH, Zotalis G, Rosier RN, Goater JJ, Lewis GD, Schwarz EM, Puzas JE, O'Keefe RJ. NFkappaB: a pivotal transcription factor in prostate cancer metastasis to bone. Clin Orthop Relat Res. 2003;S75–85 doi: 10.1097/01.blo.0000093048.96273.aa. [DOI] [PubMed] [Google Scholar]
- 84.Srinivasan G, Campbell E, Bashirelahi N. Androgen, estrogen, and progesterone receptors in normal and aging prostates. Microsc Res Tech. 1995;30:293–304. doi: 10.1002/jemt.1070300405. [DOI] [PubMed] [Google Scholar]
- 85.Castagnetta LA, Carruba G. Human prostate cancer: a direct role for oestrogens. Ciba Found Symp. 1995;191:269–286. doi: 10.1002/9780470514757.ch16. [discussion 286–269] [DOI] [PubMed] [Google Scholar]
- 86.McNamara KM, Handelsman DJ, Simanainen U. The mouse as a model to investigate sex steroid metabolism in the normal and pathological prostate. J Steroid Biochem Mol Biol. 2012;131:107–121. doi: 10.1016/j.jsbmb.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 87.Bonkhoff H, Berges R. The evolving role of oestrogens and their receptors in the development and progression of prostate cancer. Eur Urol. 2009;55:533–542. doi: 10.1016/j.eururo.2008.10.035. [DOI] [PubMed] [Google Scholar]
- 88.Nakajima Y, Akaogi K, Suzuki T, Osakabe A, Yamaguchi C, Sunahara N, Ishida J, Kako K, Ogawa S, Fujimura T. Estrogen regulates tumor growth through a nonclassical pathway that includes the transcription factors ERbeta and KLF5. Sci Signal. 2011;4:ra22. doi: 10.1126/scisignal.2001551. [DOI] [PubMed] [Google Scholar]
- 89.Dong XY, Guo P, Sun X, Li Q, Dong JT. Estrogen up-regulates ATBF1 transcription but causes its protein degradation in estrogen receptor-alpha-positive breast cancer cells. J Biol Chem. 2011;286:13879–13890. doi: 10.1074/jbc.M110.187849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dong XY, Fu X, Fan S, Guo P, Su D, Dong JT. Oestrogen causes ATBF1 protein degradation through the oestrogen-responsive E3 ubiquitin ligase EFP. Biochem J. 2012;444:581–590. doi: 10.1042/BJ20111890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong XY, Sun X, Guo P, Li Q, Sasahara M, Ishii Y, Dong JT. ATBF1 inhibits ER function by selectively competing with AIB1 for binding to ER in ER-positive breast cancer cells. J Biol Chem. 2010;285:32801–32809. doi: 10.1074/jbc.M110.128330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li M, Fu X, Ma G, Sun X, Dong X, Nagy T, Xing C, Li J, Dong JT. Atbf1 regulates pubertal mammary gland development likely by inhibiting the pro-proliferative function of estrogen-ER signaling. PLoS One. 2012;7:e51283. doi: 10.1371/journal.pone.0051283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li M, Zhao D, Ma G, Zhang B, Fu X, Zhu Z, Fu L, Sun X, Dong JT. Upregulation of ATBF1 by progesterone-PR signaling and its functional implication in mammary epithelial cells. Biochem Biophys Res Commun. 2013;430:358–363. doi: 10.1016/j.bbrc.2012.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Periodic acid-Schiff (PAS) and Alcian blue (AB) staining in mouse dorsal prostates. Genotypes are indicated at the top. All scale bars are 100 μm.
Primer sequences and PCR product sizes for gene expression validation by real-time RT-PCR.
Differentially expressed genes between dorsal prostates with and without homozygous deletion of Atbf1, with fold changes, protein localization to the cellular membrane (CM), and whether their proteins are secretory (S) or have been detected in the plasma or serum (Yes), as identified by microarray analysis.
Validation of differentially expressed genes between dorsal prostates with normal Atbf1 (+/+) and those with hemizygous (+/-) or homozygous deletion (-/-) of Atbf1 by real-time RT-PCR.
Functional annotation of differentially expressed genes between prostates with and without homozygous deletion of Atbf1 by Ingenuity Pathways Analysis (IPA).