

Abstract
Telomeric proteins have an essential role in the regulation of the length of the telomeric DNA tract and in protection against end-to-end chromosome fusion. Telomere organization and how individual proteins are involved in different telomere functions in living cells is largely unknown. By using green fluorescent protein tagging and photobleaching, we investigated in vivo interactions of human telomeric DNA-binding proteins with telomeric DNA. Our results show that telomeric proteins interact with telomeres in a complex dynamic fashion: TRF2, which has a dual role in chromosome end protection and telomere length homeostasis, resides at telomeres in two distinct pools. One fraction (∼73%) has binding dynamics similar to TRF1 (residence time of ∼44 s). Interestingly, the other fraction of TRF2 binds with similar dynamics as the putative end-protecting factor hPOT1 (residence time of ∼11 min). Our data support a dynamic model of telomeres in which chromosome end-protection and telomere length homeostasis are governed by differential binding of telomeric proteins to telomeric DNA.
Telomeres, nucleoprotein structures at chromosome ends, have a key role in the processes of cell senescence and immortalization (for a review, see references 3 and 4). In each cell division, telomere length is reduced as a result of incomplete end replication and degradation by a putative nuclease (29). In somatic cells, this telomere shortening ultimately induces cell senescence (5). In cancer and germ line cells, telomere shortening is compensated by elongation mechanisms involving the enzyme telomerase and alternative mechanisms (10, 24). These compensation mechanisms allow for the immortality of cancer cells (16). In addition to the regulatory role in maintenance of chromosome length, telomeres are essential for genetic stability since they protect against chromosome end-to-end fusion and subsequent chromosome loss (15, 35).
Telomeric DNA-binding proteins have an essential role both in regulation of the length of the telomeric DNA tract and in protection against chromosome end-to-end fusion (for a review, see references 8, 9, and 13). In mammals, TTAGGG repeat-binding factors TRF1 and TRF2 bind directly to the double-stranded telomeric DNA (6). Although TRF1 and TRF2 are similar in protein structure, including DNA-binding domains, they are significantly different in function. After expression of dominant-negative (DN) TRF mutants, chromosome end-to-end fusions are immediately induced in cells expressing DN-TRF2 (33, 35) but not in cells expressing DN-TRF1. These results suggest that TRF2, in contrast to TRF1, has a direct role in protection against end-to-end fusion. In mice, targeted deletion of TRF1 induces early embryonic lethality (22). Telomere fusions were not observed in early embryos. However, targeted deletion in embryonic stem cells induces temporary growth inhibition that seems to be associated with telomere fusions (21). Therefore, results are conflicting with regard to a potential function of TRF1 in chromosome end protection. Chromosomes terminate with a single-stranded overhang of telomeric DNA (29, 36). In Oxytricha nova, the telomeric end-binding protein associates with this single-stranded overhang to protect the ultimate chromosome end (18). Recently, POT1 (named for protection of telomeres) was identified as a telomeric end-binding protein homolog in Schizosaccharomyces pombe and in humans (2). In S. pombe, POT1 binds single-stranded telomeric DNA and has a direct role in end protection. In humans POT1 also binds single-stranded telomeric DNA, and a role in end protection has been suggested. Unexpectedly, deletion of the putative single-stranded binding domain of hPOT1 does not result in end-to-end chromosome fusions but in rapid, telomerase-mediated telomere elongation (28). These studies suggest that hPOT1 may have a role in protecting the single-stranded overhang from accessibility to telomerase. In contrast, hPOT1 may be critical in the recruitment of telomerase to the single-stranded ultimate chromosome end since overexpression of hPOT1 induces telomere elongation in telomerase-positive cells and not in telomerase negative cells (7).
Telomere length regulation involves a “protein counting” mechanism since this regulatory process is set on the number of binding sites for double-stranded telomeric DNA-binding proteins at individual telomeres in yeast (30). In mammals, a similar “protein counting” is suggested, since overexpression of TRF1 and TRF2 induces gradual shortening of telomeres, likely related to increased accumulation of TRF1 and TRF2 at telomeres (33, 34). Moreover, introduction of artificial TRF1 or TRF2 binding sites in a subtelomeric region results in shortening of telomeric DNA at that specific chromosome end (1). This indicates a cis-acting mechanism of telomere length homeostasis. In mice, the set point is different for individual chromosome ends (39). It is unknown whether this regulation process requires formation of (relatively) stable nucleoprotein structures in which the ultimate end of long telomeres may be shielded from telomerase action. Alternatively, regulation may require dynamic protein-DNA interactions to allow accumulation and release of regulatory complexes at long telomeres to inhibit telomerase activity. Here, we investigate in living cells telomere residence times of the currently known telomeric proteins that have direct telomeric DNA-binding capacity (TRF1, TRF2, and hPOT1) by using fluorescence recovery after photobleaching (FRAP) and fluorescence loss in photobleaching (FLIP) (20, 37). We provide evidence that telomeric proteins interact with telomeres in a complex, dynamic fashion: two distinct residence times of the various proteins were observed that may relate to distinct telomere functions.
MATERIALS AND METHODS
Constructs and cell lines.
Green fluorescent protein (GFP) fusions to the N terminus of TRF1, TRF2, and hPOT1 were made. To generate the GFP-TRF1 fusion, PCR with the forward primer TTTGAGCTCAAATTGCGGAGGATGTT and the reverse primer TTTCTGCAGTCAGTCTTCGCTGTC was used to amplify the hTRF1 gene from an hTRF1-containing vector (kindly provided by T. de Lange). The amplified TRF1 was digested with SacI and PstI and cloned into pEGFP-C1 (BD Biosciences, Clontech) digested with identical enzymes. To generate the GFP-TRF2 fusion, a hTRF2-containing vector (kindly provided by T. de Lange) was digested with BssHII, made blunt and digested EcoRI. The hTRF2-containing fragment was isolated and cloned into pEGFP-C1 that was digested with BglII, made blunt and digested with EcoRI. To generate the GFP-POT1 fusion, hPOT1-containing vector (kindly provided by T. Cech) was digested with BamHI and XbaI. The hPOT1-containing fragment was isolated and cloned into pEGFP-C1 that was digested with the same enzymes.
Cell lines.
HeLa cells were transfected with these constructs by using calcium phosphate and stable clones were selected with G418 (1 mg/ml) and enriched by fluorescence-activated cell sorting (FACSCalibur; BD Biosciences). Cells were cultured in HEPES-containing Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (Life Technologies) at 37°C. GFP fusion protein expression level and subcellular localization was analyzed by immunoblotting and immunofluorescence, respectively, with rabbit polyclonal antibodies against TRF1 (antibody 371) and TRF2 (antibody 508) (34, 35) and against hPOT1 (antibody 978) (28). Secondary antibodies used were horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) and horseradish peroxidase-conjugated swine anti-rabbit IgG (DakoCytomation) for immunoblotting and TRITC (tetramethyl rhodamine isothiocyanate)-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories) for immunofluorescence. Immunoblots were visualized by using enhanced chemiluminescence (Amersham Biosciences). Immunofluorescence images were obtained by using a Leica DMRXA microscope equipped with a Sensys charge-coupled device camera (Photometrics) and QFluoro software (Leica). To demonstrate colocalization of GFP-tagged proteins with telomeric DNA fluorescence in situ hybridization with a Cy3-coupled PNA probe for telomeric DNA was performed (39). For each GFP fusion protein, three independent clones with relatively low GFP fusion protein expression level were selected for bleaching experiments. Since expression levels of individual cells within a clone were variable, we selected only cells with intermediate expression levels for FRAP and FLIP-FRAP analysis.
Live cell microscopy.
Live-cell microscopy was performed with a Zeiss LSM 410 confocal laser scanning microscope equipped with a heated (37°C) scan stage and a Plan-Apochromat oil immersion objective (40×, numerical aperture [NA] 1.3, for diffusion measurements in the nucleoplasm or 63×, NA 1.4, for FRAP and FLIP at telomeres). GFP fluorescence was detected by using the 488-nm line of a fiber-coupled 60-mW argon laser, a dichroic beamsplitter (488/543) and a 510- to 545-bandpass emission filter.
Photobleaching assays.
To determine effective diffusion coefficients (Deff) of GFP-TRF1 and GFP-TRF2 in the nucleoplasm, a 2-μm wide strip through the nucleus was bleached for 200 ms at a 500-μW laser intensity, measured at focal plane. Redistribution of fluorescence to the bleached strip was monitored with 100-ms intervals at 10-μW laser intensity. Deff was calculated by fitting the experimental data to a one-dimensional diffusion model (11, 12, 19).
Association kinetics of the GFP-tagged telomeric proteins to telomeres were assessed in two complementary photobleaching methods.
(i) FRAP on individual telomeres.
Photobleaching was applied to a small area covering a single telomere for 1 s at a 50-μW laser intensity. Redistribution of fluorescence was monitored with 10-s time intervals at 7.5 μW starting at 2 s after the bleach pulse. Images were analyzed by using KS400 software (Zeiss). The relative fluorescence intensity of individual telomeres, normalized to total nuclear fluorescence, was calculated at each time interval as follows: Irel(t) = (Tt/T0)/(Nt/N0), where Tt is the intensity of the telomere at time point t after bleaching, T0 is the intensity of the telomere before bleaching, Nt is the total nuclear intensity at time point t after bleaching, and N0 is the total nuclear intensity before bleaching. The telomere intensities are corrected for nucleoplasmic signal, and the nuclear intensity is corrected for background. The experimental data were fitted (least-squares best fit) to the following equation: Irel(t) = f1(1 − 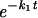 ) + f2(1 −
) + f2(1 − 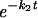 ), where f1 and f2 are the fractions and k1 and k2 are the corresponding rate constants of those fractions (25). Half lives were calculated as t1/2 = ln2/k, and residence times were taken at 97% of recovery.
), where f1 and f2 are the fractions and k1 and k2 are the corresponding rate constants of those fractions (25). Half lives were calculated as t1/2 = ln2/k, and residence times were taken at 97% of recovery.
(ii) Simultaneous FRAP and FLIP in a single nucleus.
Complementary to the FRAP experiments, simultaneous FRAP and FLIP in a single nucleus was used, mainly to assess fractions with more stable binding more accurately. Photobleaching was applied to about half the nucleus for 1 s at a 100-μW laser intensity. Redistribution of fluorescence was monitored with 20-s time intervals at 7.5 μW starting at 2 s after the bleach pulse. The difference between relative fluorescence intensities of bleached (FRAP) and unbleached (FLIP) telomeres was calculated as ΔIrel(t) = [(Tt/T0)unbleached − (Tt/T0)bleached] and normalized to the first datum point after bleaching. Half-times and recovery times were calculated by fitting experimental data (least-squares best fit) to the equation ΔIrel(t) = f1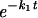 + f2
+ f2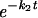 .
.
RESULTS
Characterization of cell lines expressing GFP-TRF1, GFP-TRF2, and GFP-POT1.
HeLa cells were transfected with GFP fusion constructs. We selected clones that were characterized by GFP-TRF1, GFP-TRF2, or GFP-POT1 expression levels similar to the endogenous protein level as judged by immunoblotting (Fig. 1A, C, and E, respectively). Expression levels of cells in an individual clone were sometimes variable. In analyzing these clones, the use of confocal microscopy allows for selection of cells with intermediate expression levels in the various assays (see below). GFP-TRF1 clearly colocalized with endogenous TRF2 at telomeres (Fig. 1B). Similarly, GFP-TRF2 colocalized with endogenous TRF1 (Fig. 1D), and GFP-POT1 colocalized with endogenous TRF2 (Fig. 1F). These results suggest that tagging TRF1, TRF2, and POT1 with GFP does not inhibit binding to telomeres. In addition, the immunofluorescence studies suggest a similar ratio of telomere-bound versus nucleoplasmic unbound proteins for GFP-tagged and untagged proteins (comparison of telomere versus nucleoplasmic fluorescence intensity in Fig. 1B for endogenous TRF2 [red] and in Fig. 1D for GFP-tagged TRF2 [green] and similarly for TRF1). This indicates that fusion proteins are not displaced from telomeres by endogenous proteins in cells that express both in similar amounts. These results also suggest that telomere-binding characteristics of tagged and nontagged proteins are not very different.
FIG. 1.

Expression level and localization of GFP-TRF1, GFP-TRF2, and GFP-POT. (A) Immunoblot of whole-cell extracts of untransfected HeLa cells (lane 1) and HeLa cells with stable expression of GFP-TRF1 (lane 2), probed with anti-TRF1 antibody, indicating similar expression levels of GFP-tagged TRF1 and endogenous TRF1. (B) In formaldehyde-fixed HeLa cells expressing GFP-TRF1 (green), endogenous TRF2 was detected by indirect immunofluorescence with anti-TRF2 antibodies (red). GFP-tagged TRF1 colocalizes with endogenous TRF2 at telomeres (arrowheads). (C) Immunoblot of whole-cell extracts of untransfected HeLa cells (lane 1) and HeLa cells with stable expression of GFP-TRF2 (lane 2), probed with anti-TRF2 antibody, indicating similar expression levels of GFP-tagged TRF2 and endogenous TRF2. (D) In formaldehyde-fixed HeLa cells expressing GFP-TRF2 (green), endogenous TRF1 was detected by indirect immunofluorescence with anti-TRF1 antibodies (red). GFP-tagged TRF2 colocalizes with endogenous TRF1 at telomeres (arrowheads). (E) Immunoblot of whole-cell extracts of untransfected HeLa cells (lane 1) and HeLa cells with stable expression of GFP-POT (lane 2), probed with anti-POT antibody, indicating similar expression levels of GFP-tagged POT and endogenous POT. (F) In formaldehyde-fixed HeLa cells expressing GFP-POT1 (green), endogenous TRF2 was detected with anti-TRF2 antibodies (red). GFP-tagged POT1 colocalizes with endogenous TRF2 at the telomeres (arrowheads). Scale bar, 10 μm.
To demonstrate colocalization of GFP-tagged proteins with telomeric DNA, we analyzed HeLa cells transiently expressing CFP-POT1 and YFP-TRF2 with subsequent fluorescence in situ hybridization by using a Cy3-coupled PNA probe for telomeric DNA (39). These studies clearly demonstrate colocalization of tagged hPOT1 and TRF2 proteins with telomeric DNA (Fig. 2). Similar results were obtained by expression of CFP- and YFP-tagged combinations of TRF1 and TRF2, TRF1 and hPOT1, and TRF2 and hRap1 (data not shown). Since these cells transiently overexpress the tagged proteins over the endogenous proteins, these results also suggest that tagging does not inhibit interactions with known binding partners, i.e., TRF1 with hPOT1 (28) and TRF2 with hRap1 (27).
FIG. 2.

Colocalization of CFP-tagged POT1 and YFP-tagged TRF2 with telomeric DNA. (A and B) Fluorescence image of HeLa cell transiently transfected with CFP-POT1 and YFP-TRF2, by using CFP-specific filter set (A) or by using YFP-specific filter set (B). (C) HeLa cell shown in panels A and B subsequently hybridized with a Cy3-tagged probe for telomeric DNA. CFP-POT1 and YFP-TRF2 colocalize with telomeric DNA (arrowheads). Scale bar, 10 μm.
GFP-TRF1 and GFP-TRF2 have highly dynamic interactions with telomeres in living cells.
Binding sites of telomeric proteins, i.e., telomeres, were readily identified in living cells that express GFP-TRF1 or GFP-TRF2 (Fig. 1B and D). This allowed distinct investigation of diffusion and binding processes of the proteins in the nucleus. Intranuclear diffusion was assessed in regions in between telomeres. Telomere binding was assessed by specific analysis of telomeric spots. Association of telomeric proteins with telomeres was assessed by photobleaching of a small region covering a single telomere and subsequent quantitative analysis of fluorescence redistribution specifically at the bleached telomere. The efficiency and irreversibility of bleaching was ascertained in fixed cells that express GFP-TRF1 (Fig. 3A). In living cells, fluorescent proteins already show significant redistribution to the bleached nuclear region and telomere at 2 s after bleaching (Fig. 3B). FRAP analysis indicated that the binding kinetics of TRF1 (t1/2 = 9.4 ± 1.3 s) and most of the TRF2 (t1/2 = 8.1 ± 1.5 s) at bleached telomeres were similar (Fig. 3C). These results indicate that the association of TRF1 and TRF2 with telomeres is highly dynamic. The association of TRF1 and TRF2 contrasts strongly to the association of histone H2B to DNA. By using similar FRAP analysis of HeLa cells expressing GFP-tagged H2B, we observed no exchange with unbleached H2B over an interval of 3 min (data not shown). Similar residence times (∼44 s) of TRF1 and TRF2 may relate to the similarities in DNA-binding domains (6).
FIG. 3.

FRAP analysis of telomere-bound GFP-TRF1 and GFP-TRF2. (A and B) FRAP on fixed and living HeLa cells expressing GFP-TRF1. A small region covering a single telomere was photobleached (indicated by a white box). Images were acquired before bleaching and at 10-s intervals after bleaching, starting at 2 s. Scale bar, 2.5 μm. Redistribution of GFP fusion protein was not observed in fixed cells (A) but was observed in living cells (B). (C) Quantitative analysis of redistribution of GFP-TRF1 and GFP-TRF2 at individual bleached telomeres in living cells. The circle in panel B indicates the area used to calculate fluorescence redistribution. The relative telomere intensity was calculated with corrections for fluorescence loss in the process of bleaching and imaging. Values represent means ± the standard error of the mean (SEM) from at least 40 cells. The analysis indicated similar redistribution rates for TRF1 and TRF2.
Intranuclear diffusion of TRF1 and TRF2 is in distinct complexes.
To investigate complex formation of TRF1 and TRF2 in the nucleoplasm, we studied diffusion of GFP-TRF1 and GFP-TRF2 by using FRAP in a strip bleaching technique (11, 12, 19). Since telomeres are easily identified in these cells (Fig. 1B and D), strip regions without telomeres were selected for bleaching. The effective diffusion coefficients (Deff) of GFP-TRF1 and GFP-TRF2 were 3.3 ± 0.6 and 2.0 ± 0.5 μm2/s, respectively (for free GFP in human fibroblasts, the Deff was 18 μm2/s). Although it is possible that complexes containing both TRF1 and TRF2 were present, the observed difference in Deff suggests that a major fraction of both molecules move in the nucleoplasm in complexes of different compositions. In addition, TRF1 and TRF2 diffuse slower than the freely mobile DNA repair proteins measured in the same experimental setup in one of our labs (12, 19), suggesting that TRF1 and TRF2 move in larger complexes in the nucleoplasm.
Association of TRF2 with telomeres is in two distinct kinetic pools.
To investigate whether fractions of TRF1 and TRF2 associate with telomeres more stably over time, we applied FRAP and FLIP simultaneously within a single nucleus. The laser was used to photobleach about one-half of the nucleus (Fig. 4A). Next, redistribution of fluorescence was analyzed at several telomeric spots in the bleached (Fig. 4A, pink regions), as well as in the unbleached half of the nucleus (Fig. 4A, blue regions). This type of analysis has the advantage of allowing a direct comparison of bleached and unbleached telomeres within a single cell, avoiding potential errors due to, e.g., loss of fluorescence by the bleach pulse and by monitor bleaching. Simultaneous FLIP-FRAP analysis indicated that GFP-TRF1 is completely redistributed over bleached and unbleached telomeres within 2 min after bleaching (Fig. 4B and D). In contrast, GFP-TRF2 redistribution is not complete within this time interval (Fig. 4C and D). Least-squares best-fit analysis of the experimental data indicated rapid exchange of TRF1 at telomeres in a single fraction (Fig. 4E). In contrast, the binding kinetics of TRF2 were characterized by a secondary slow component, in addition to a rapidly exchanging fraction with an association constant remarkably similar to that of TRF1 (Table 1). The smallest fraction (∼27%) of TRF2 forms more stable complexes with telomeres (with t1/2 ≈ 138 s and a residence time of ∼11 min). These results suggest that TRF2 associates with telomeres in two distinct complexes. Dual binding kinetics may relate to the dual role of TRF2 both in telomere length regulation and in chromosome end protection (33-35).
FIG. 4.

Simultaneous FLIP-FRAP of telomere-bound GFP-TRF1 and GFP-TRF2. (A) FLIP-FRAP on living HeLa cells expressing GFP-TRF1. Cells are photobleached over a region covering about one-half of the nucleus (indicated by a white box). The images were acquired before bleaching and at 20-s intervals after bleaching, starting at 2 s. The pink circles in the bleached area and blue circles in the unbleached area indicate the regions that are used to calculate fluorescence redistribution. Scale bar, 5 μm. (B and C) Quantitative analysis of redistribution of GFP-TRF1 (B) and GFP-TRF2 (C) at telomeres separately in bleached (pink) and unbleached (blue) half of the nucleus. Values are means ± the SEM from at least 40 cells. (D) Difference (Δ) in telomere intensity in bleached and unbleached part of cell, calculated from the data shown in panels B (TRF1) and C (TRF2). (E) A fitting analysis of the experimental data in panel D to the equation ΔIrel(t) = f1e−k1t + f2e−k2t indicated a good fit with the single binding kinetics of GFP-TRF1 (green line). In contrast, GFP-TRF2 redistribution does not fit with single binding kinetics (blue dashed line) but does fit with dual binding kinetics (red lines). Note the similarity between the fitted curves of the fast fraction of TRF2 and of TRF1. For percentages of fractions and association constants, see Table 1.
TABLE 1.
Results of random fitting analysis of telomeric protein exchange at telomeres (FLIP-FRAP analysis)
| Protein | Fraction (%) | Rate constant (s−1) | Half-time (s)a |
|---|---|---|---|
| TRF1 | 100 | 0.028 | 24.7* |
| TRF2 (fast fraction) | 73 | 0.028 | 24.7* |
| TRF2 (slow fraction) | 27 | 0.0050 | 138 |
| POT1 | 100 | 0.0054 | 128 |
*, the half-times of the fast fractions calculated by FLIP-FRAP methodology are higher than the half-times calculated by the FRAP method. Since half of the nucleus was bleached, a significant amount of time is required for redistribution of telomeric proteins compared to bleached and unbleached nuclear halves.
hPOT1 has telomere-binding kinetics similar to the slow fraction of TRF2.
To investigate interactions at the ultimate chromosome end, we studied telomere association of hPOT1. hPOT1 has a single-stranded telomeric DNA-binding domain (2). Therefore, hPOT1 is binding to the single-strand overhang at the ultimate chromosome end. GFP-POT1 clearly colocalized with endogenous TRF2 at telomeres, indicating GFP-POT1 is still functional in binding to telomeres (Fig. 1F). FRAP analysis of individual telomeres demonstrates that telomere binding of GFP-POT1 is not very dynamic compared to TRF1 and TRF2 (Fig. 5A). Least-squares best-fit analysis of FLIP-FRAP data indicated that POT1 binds telomeres as a single fraction with binding dynamics strikingly similar to the slow fraction of TRF2 (Fig. 5B and C and Table 1). Similar association kinetics suggests that TRF2 and hPOT1 may interact in the formation of a chromosome end protection complex at human telomeres.
FIG. 5.

Analysis of FRAP and FLIP-FRAP experiments of telomere-bound GFP-POT1. (A) Quantitative analysis of the redistribution of GFP-POT1 at individual bleached telomeres in living HeLa cells by using FRAP; (B) quantitative analysis of redistribution of GFP-POT1 at telomeres separately in the bleached and unbleached region of the nucleus of living HeLa cells by using FLIP-FRAP. Values are means ± the SEM from at least 40 cells. (C) Fitting analysis of experimental data from panel B to the equation ΔIrel(t) = f1e−k1t + f2e−k2t indicates single binding kinetics of GFP-POT1. Note the similarity between the fitted curves of POT1 (green line) and the slow fraction of TRF2 (dotted red line). The percentages of fractions and association constants are summarized in Table 1.
DISCUSSION
The process of telomere length regulation involves coordinated activity of enzymes that effectuate lengthening and shortening of the telomeric DNA tract, i.e., telomerase and a putative nuclease, respectively (5, 23, 32). This regulation process is set on the number of binding sites for telomeric double-stranded DNA-binding proteins in yeast (30) and in mammals (1). In this cis-acting regulation mechanism, more binding sites for TRF1 inhibit telomerase-mediated elongation, whereas more TRF2-binding sites stimulate nuclease-mediated shortening (1, 23). Our in vivo binding studies suggest that telomere length homeostasis is effectuated in a process that involves highly dynamic binding of TRF1 and TRF2. The association times of TRF1 and most TRF2 proteins (half times of ∼8 s) are much shorter than the association times of core histones in the nucleosomal complex (25), linker histones (26, 31), or those of RAD51, RAD52, and TFIIH in the foci of DNA repair (12, 17). Highly dynamic binding of TRF1 and TRF2 may be essential for fast adaptation in the process of telomerase-mediated elongation or nuclease-mediated shortening of telomeric DNA. Alternatively, dynamic binding may be essential for accumulation of telomerase-inhibiting or nuclease-stimulating complexes at longer telomeres.
TRF1 interacts with hPOT1 and the single-strand DNA-binding domain of hPOT1 is critical to inhibition of telomerase-mediated telomere elongation (28). These observations suggest that hPOT1 is the terminal transducer of TRF1 length control. However, our studies suggest that the majority of hPOT1 proteins have much longer residence times at telomeres in comparison to the majority of TRF1 proteins. Therefore, our observations argue against a straightforward model of more TRF1 binding sites at telomeric DNA, resulting in more TRF1 protein complexes at telomeres, resulting in more hPOT1 proteins, resulting in more telomerase inhibition. Our observations suggest instead that hPOT1 residence time at telomeres is regulated by its single-strand binding domain or by interaction with other telomeric proteins (e.g., TRF2). Thus, rather than recruiting more hPOT1 proteins at telomeres, the role of TRF1 may be to modulate hPOT1 function, i.e., increase telomerase inhibiting activity. This process may involve the telomeric protein PinX1, a TRF1-interacting protein that has telomerase inhibiting activity (38).
In addition to its role in telomere length homeostasis, TRF2 is critical in chromosome end protection (33, 35). Our studies of telomere association times demonstrate an additional slow fraction of TRF2. Remarkably, this slow fraction of TRF2 has an association time similar to hPOT1. Since hPOT1 binds single-stranded telomeric DNA and since chromosomes end in a single-strand overhang, chromosome end protection may involve a complex of TRF2 and hPOT1 at the ultimate end of chromosomes. However, interaction between TRF2 and hPOT1 is not supported by positive coimmunoprecipitation experiments (28). This may relate to involvement of a minor fraction of TRF2. In agreement, our results suggest that the slow fraction is only 27% of telomere bound TRF2. Alternatively, interaction between TRF2 and hPOT1 may be indirect or only in association with telomeric DNA. In a proposed telomere loop model, the single-stranded overhang at the ultimate end invades the double-stranded telomeric DNA at the base of the telomere (14). A single protein complex, including the single-stranded DNA-binding protein hPOT1 and the double-stranded DNA-binding protein, TRF2 may have the capacity to stabilize such a proposed structure.
In conclusion, telomeric proteins have a critical and complex role in regulating telomere function. Our studies in living human cells indicate complex dynamic interactions of human telomeric DNA-binding proteins with telomeric DNA. Considering known functions and interactions of these telomeric proteins, our results clearly provide further insight into relations of telomere structure to telomere function.
Acknowledgments
We thank J. H. J. Hoeijmakers, W. Vermeulen, and R. Kanaar for helpful discussions.
This study was supported by Dutch Cancer Society (KWF) grant NKB98-1856.
REFERENCES
- 1.Ancelin, K., M. Brunori, S. Bauwens, C. E. Koering, C. Brun, M. Ricoul, J. P. Pommier, L. Sabatier, and E. Gilson. 2002. Targeting assay to study the cis functions of human telomeric proteins: evidence for inhibition of telomerase by TRF1 and for activation of telomere degradation by TRF2. Mol. Cell. Biol. 22:3474-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumann, P., and T. R. Cech. 2001. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292:1171-1175. [DOI] [PubMed] [Google Scholar]
- 3.Blackburn, E. H. 2000. Telomere states and cell fates. Nature 408:53-56. [DOI] [PubMed] [Google Scholar]
- 4.Blasco, M. A. 2003. Mammalian telomeres and telomerase: why they matter for cancer and aging. Eur. J. Cell Biol. 82:441-446. [DOI] [PubMed] [Google Scholar]
- 5.Bodnar, A. G., M. Ouellette, M. Frolkis, S. E. Holt, C. P. Chiu, G. B. Morin, C. B. Harley, J. W. Shay, S. Lichtsteiner, and W. E. Wright. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279:349-352. [DOI] [PubMed] [Google Scholar]
- 6.Broccoli, D., A. Smogorzewska, L. Chong, and T. de Lange. 1997. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 17:231-235. [DOI] [PubMed] [Google Scholar]
- 7.Colgin, L. M., K. Baran, P. Baumann, T. R. Cech, and R. R. Reddel. 2003. Human POT1 facilitates telomere elongation by telomerase. Curr. Biol. 13:942-946. [DOI] [PubMed] [Google Scholar]
- 8.de Lange, T. 2002. Protection of mammalian telomeres. Oncogene 21:532-540. [DOI] [PubMed] [Google Scholar]
- 9.Dubrana, K., S. Perrod, and S. M. Gasser. 2001. Turning telomeres off and on. Curr. Opin. Cell Biol. 13:281-289. [DOI] [PubMed] [Google Scholar]
- 10.Dunham, M. A., A. A. Neumann, C. L. Fasching, and R. R. Reddel. 2000. Telomere maintenance by recombination in human cells. Nat. Genet. 26:447-450. [DOI] [PubMed] [Google Scholar]
- 11.Ellenberg, J., E. D. Siggia, J. E. Moreira, C. L. Smith, J. F. Presley, H. J. Worman, and J. Lippincott-Schwartz. 1997. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 138:1193-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Essers, J., A. B. Houtsmuller, L. van Veelen, C. Paulusma, A. L. Nigg, A. Pastink, W. Vermeulen, J. H. Hoeijmakers, and R. Kanaar. 2002. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 21:2030-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans, S. K., and V. Lundblad. 2000. Positive and negative regulation of telomerase access to the telomere. J. Cell Sci. 113:3357-3364. [DOI] [PubMed] [Google Scholar]
- 14.Griffith, J. D., L. Comeau, S. Rosenfield, R. M. Stansel, A. Bianchi, H. Moss, and T. de Lange. 1999. Mammalian telomeres end in a large duplex loop. Cell 97:503-514. [DOI] [PubMed] [Google Scholar]
- 15.Hackett, J. A., D. M. Feldser, and C. W. Greider. 2001. Telomere dysfunction increases mutation rate and genomic instability. Cell 106:275-286. [DOI] [PubMed] [Google Scholar]
- 16.Hahn, W. C., S. A. Stewart, M. W. Brooks, S. G. York, E. Eaton, A. Kurachi, R. L. Beijersbergen, J. H. Knoll, M. Meyerson, and R. A. Weinberg. 1999. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 5:1164-1170. [DOI] [PubMed] [Google Scholar]
- 17.Hoogstraten, D., A. L. Nigg, H. Heath, L. H. F. Mullenders, R. van Driel, J. H. Hoeijmakers, W. Vermeulen, and A. B. Houtsmuller. 2002. Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol. Cell 10:1163-1174. [DOI] [PubMed] [Google Scholar]
- 18.Horvath, M. P., V. L. Schweiker, J. M. Bevilacqua, J. A. Ruggles, and S. C. Schultz. 1998. Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell 95:963-974. [DOI] [PubMed] [Google Scholar]
- 19.Houtsmuller, A. B., S. Rademakers, A. L. Nigg, D. Hoogstraten, J. H. Hoeijmakers, and W. Vermeulen. 1999. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 284:958-961. [DOI] [PubMed] [Google Scholar]
- 20.Houtsmuller, A. B., and W. Vermeulen. 2001. Macromolecular dynamics in living cell nuclei revealed by fluorescence redistribution after photobleaching. Histochem. Cell Biol. 115:13-21. [DOI] [PubMed] [Google Scholar]
- 21.Iwano, T., M. Tachibana, M. Reth, and Y. Shinkai. 2004. Importance of TRF1 for functional telomere structure. J. Biol. Chem. 279:1442-1448. [DOI] [PubMed] [Google Scholar]
- 22.Karlseder, J., L. Kachatrian, H. Takai, K. Mercer, S. Hingorani, T. Jacks, and T. de Lange. 2003. Targeted deletion reveals an essential function for the telomere length regulator Trf1. Mol. Cell. Biol. 23:6533-6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlseder, J., A. Smogorzewska, and T. de Lange. 2002. Senescence induced by altered telomere state, not telomere loss. Science 295:2446-2449. [DOI] [PubMed] [Google Scholar]
- 24.Kim, N. W., M. A. Piatyszek, K. R. Prowse, C. B. Harley, M. D. West, P. L. Ho, G. M. Coviello, W. E. Wright, S. L. Weinrich, and J. W. Shay. 1994. Specific association of human telomerase activity with immortal cells and cancer. Science 266:2011-2015. [DOI] [PubMed] [Google Scholar]
- 25.Kimura, H., and P. R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lever, M. A., J. P. Th'ng, X. Sun, and M. J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature 408:873-876. [DOI] [PubMed] [Google Scholar]
- 27.Li, B., S. Oestreich, and T. de Lange. 2000. Identification of human Rap1: implications for telomere evolution. Cell 101:471-483. [DOI] [PubMed] [Google Scholar]
- 28.Loayza, D., and T. De Lange. 2003. POT1 as a terminal transducer of TRF1 telomere length control. Nature 424:1013-1018. [DOI] [PubMed] [Google Scholar]
- 29.Makarov, V. L., Y. Hirose, and J. P. Langmore. 1997. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 88:657-666. [DOI] [PubMed] [Google Scholar]
- 30.Marcand, S., E. Gilson, and D. Shore. 1997. A protein-counting mechanism for telomere length regulation in yeast. Science 275:986-990. [DOI] [PubMed] [Google Scholar]
- 31.Misteli, T., A. Gunjan, R. Hock, M. Bustin, and D. T. Brown. 2000. Dynamic binding of histone H1 to chromatin in living cells. Nature 408:877-881. [DOI] [PubMed] [Google Scholar]
- 32.Pennock, E., K. Buckley, and V. Lundblad. 2001. Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell 104:387-396. [DOI] [PubMed] [Google Scholar]
- 33.Smogorzewska, A., B. van Steensel, A. Bianchi, S. Oelmann, M. R. Schaefer, G. Schnapp, and T. de Lange. 2000. Control of human telomere length by TRF1 and TRF2. Mol. Cell. Biol. 20:1659-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Steensel, B., and T. de Lange. 1997. Control of telomere length by the human telomeric protein TRF1. Nature 385:740-743. [DOI] [PubMed] [Google Scholar]
- 35.van Steensel, B., A. Smogorzewska, and T. de Lange. 1998. TRF2 protects human telomeres from end-to-end fusions. Cell 92:401-413. [DOI] [PubMed] [Google Scholar]
- 36.Wellinger, R. J., A. J. Wolf, and V. A. Zakian. 1993. Saccharomyces telomeres acquire single-strand TG1-3 tails late in S phase. Cell 72:51-60. [DOI] [PubMed] [Google Scholar]
- 37.White, J., and E. Stelzer. 1999. Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol. 9:61-65. [DOI] [PubMed] [Google Scholar]
- 38.Zhou, X. Z., and K. P. Lu. 2001. The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell 107:347-359. [DOI] [PubMed] [Google Scholar]
- 39.Zijlmans, J. M., U. M. Martens, S. S. Poon, A. K. Raap, H. J. Tanke, R. K. Ward, and P. M. Lansdorp. 1997. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc. Natl. Acad. Sci. USA 94:7423-7428. [DOI] [PMC free article] [PubMed] [Google Scholar]