

Abstract
Increased proliferation rates as well as resistance to apoptosis are considered major obstacles for the treatment of patients with chronic myelogenous leukemia (CML), thus highlighting the need for novel therapeutic approaches. Since senescence has been recognized as a physiological barrier against tumorigenesis, senescence-based therapy could represent a new strategy against CML. DNA demethylating agent 5-aza-2′-deoxycytidine (DAC) was reported to induce cellular senescence but underlying mechanisms remain to be elucidated. Here, we report that exposure to DAC triggers senescence in chronic leukemia cell lines as evidenced by increased senescence-associated β-galactosidase activity and lysosomal mass, accompanied by an up-regulation of cell cycle-related genes. We provide evidence that DAC is able to decrease telomere length, to reduce telomerase activity and to decrease human telomerase reverse transcriptase (hTERT) expression through decreased binding of c-myc to the hTERT promoter. Altogether, our results reveal the role of c-myc in telomere-dependent DAC-induced senescence and therefore provide new clues for improving chronic human leukemia treatments.
Abbreviations: C12FDG, fluorogenic substrate 5-dodecanoylaminofluorescein di-β-D-galactopyranoside; CDKN, cyclin-dependent kinase inhibitor; ChIP, chromatin immunoprecipitation; CML, chronic myeloid leukemia; CTCF, CCCTC-binding factor; DAC, 5-aza-2’-deoxycytidine; DSB, double strand breaks; FCS, fetal calf serum; FITC, fluorescein isothiocyanate; hTERT, human telomerase reverse transcriptase; M-PER®, Mammalian Protein Extraction Reagent; MSP, methylation-specific PCR; PBMC, peripheral blood mononuclear cell; SA-β-gal, senescence associated-β galactosidase; SIPS, stress-induced premature senescence; siRNA, small interfering RNA; TRAP, telomeric repeat amplification protocol; X-gal, 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder characterized by the Philadelphia chromosome translocation. This translocation results in the BCR-ABL oncogene that encodes a tyrosine kinase driving expansion of myeloid progenitors cells [1]. Increased proliferation as well as apoptosis resistance are among the mechanisms responsible for myeloid cell expansion [2]. Thus, there has been considerable interest in developing new strategies to override apoptosis resistance, eliminate tumor cells and consequently improve therapeutic outcome of resistant CML patients. Since senescence is now recognized as a crucial tumor suppressor mechanism that qualitatively equals programmed cell death [3], senescence-based therapy in CML could represent a new, largely unexplored, therapeutic approach.
In contrast to tumor stem cells, healthy somatic cells have a limited proliferative lifespan after which they stop dividing and enter a terminal state of irreversible growth arrest called replicative senescence. Senescent cells are both morphologically and functionally altered. Indeed, they are characterized by the acquisition of flattened and enlarged cell morphology, an increase in senescence associated-β-galactosidase (SA-β-gal) activity, and a modulation of cell cycle-regulated gene expression [4]. Stress-induced premature senescence (SIPS) closely resembles replicative senescence [5], and can be triggered by various stimuli such as oncogenic stress, DNA damage or cytotoxic drugs [6]. Interestingly, several observations indicate that telomere shortening to a critical length as well as uncapped telomere(s) may trigger SIPS [7].
Human telomerase is a ribonucleoprotein DNA polymerase that extends eukaryotic chromosome ends with telomeric (TTAGGG)n sequences to compensate for telomere erosion [8]. The enzyme is expressed in most human cancer and malignant cell lines, whereas in most normal somatic human cells its activity is weak or not detectable [9]. This enzyme consists of two essential components: the human telomerase reverse transcriptase (hTERT), which encodes the catalytic component, and the telomerase RNA, providing the template for telomere synthesis [10]. Telomerase activity is mainly regulated at hTERT transcriptional level. In particular, several groups have indicated that c-myc is a key transcriptional regulator of hTERT. Indeed, the hTERT proximal promoter harbors two E-boxes that are binding sites for c-myc [11]. In addition, c-myc overexpression has been reported to directly up-regulate hTERT mRNA levels [12].
Hypermethylation is considered one of the major epigenetic modifications involved in aberrant transcriptional silencing of tumor suppressor genes in human malignancies [13], [14]. The potential reversibility of epigenetic abnormalities encouraged the development of pharmacological inhibitors of DNA methylation such as decitabine (5-aza-2’-deoxycytidine, DAC) as an anti-cancer agent. DAC is currently undergoing preclinical and clinical trials for leukemia either use alone or in combination with other epigenetic or conventional anti-neoplastic drugs [15]. Encouraging results were achieved against hematologic malignancies and especially in CML patients [16], [17]. In vitro studies have shown the ability of this compound to induce DNA hypomethylation, re-expression of aberrantly silenced genes, cell cycle perturbation, apoptosis and differentiation, and increased sensitivity of cancer cells to conventional chemotherapeutic drugs [18], [19]. However, in vivo mechanisms involved in these processes have not been clearly elucidated.
Interestingly, we recently provided evidence that DAC has the potential to induce senescence in leukemia cells. In particular, we demonstrated that DAC-treated CML cells exhibit an increased SA-β-gal activity and specific changes in cell morphology [19]. In the current study, we confirmed the induction of cellular senescence after DAC exposure in CML cell lines and characterized the mechanisms involved in this process. We reported that DAC-treated CML cells display morphological features associated with a senescent phenotype, especially an enlarged morphology, a vacuole-rich cytoplasm and an up-regulation of cell cycle-dependent genes controlling the senescence program. We further pointed out that DAC-induced senescent cells exhibited telomere shortening, decreased telomerase activity as well as transcriptional down-regulation of hTERT due to reduced c-myc binding to the hTERT promoter.
Materials and Methods
Cell Culture and Drug Treatments
Cell lines were purchased from Deutsche Sammlung für Mikroorganismen und Zellkulturen (DSZM; Braunschweig, Germany). K-562, MEG-01 and U-937 cells were cultured in RPMI 1640 (Lonza, Verviers, Belgium), supplemented with 10% fetal calf serum (FCS; Lonza) and 1% antibiotic–antimycotic (Lonza). KBM-5 cells were cultured in IMDM (Lonza) supplemented with 15% FCS and 1% antibiotic–antimycotic. Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated and cultured as previously described [19]. DAC, 5-azacytidine, etoposide and doxorubicin were purchased from Sigma-Aldrich (Bornem, Belgium). Cells were treated daily with DAC and 5-azacytidine. Culture medium was changed every three days of treatment to maintain physiological growth rates.
SA-β-gal Assay
SA-β-gal activity was measured as previously reported [19]. K-562 cells treated with 80 nM doxorubicin for 72 hours were used as a positive control of senescence induction. SA-β-gal activity was also evaluated by flow cytometry, using 33 μM fluorogenic substrate 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG; Invitrogen, Tournai, Belgium) in cells pretreated with 100 nM bafilomycin A1 (Sigma-Aldrich).
Morphological Analysis
Cells were spun onto a glass slide for 4 min at 800 g using a Shandon Cytospin 4 (Thermofisher, Erembodegen, Belgium), fixed and stained with the Diff-Quick stain kit (Dade Behring S.A., Brussels, Belgium) according to the manufacturer’s procedure. Images were acquired using a Leica DM2000 equipped with a DFC420C camera and Leica FireCam software.
Immunofluorescence Microscopy
To assess lysosomal cellular distribution, 5.106 cells were stained for one hour with 50 nM Lysotracker Red (Invitrogen).
DNA damage accumulation was monitored by analysis of the γH2AX foci distribution. Immunostaining was performed as previously described [20]. Cells were incubated overnight at 4°C with anti-γH2AX antibody (Millipore, Brussels, Belgium), washed and subsequently stained with an Alexa Fluor® 488-conjugated anti-mouse antibody (Invitrogen) for 1 h at room temperature. Nuclei were stained by incubation with 10 μg/ml Hoechst 33342 (Invitrogen) for 15 min at 37°C. Fluorescence microscopy was realized with a cell^M imaging station from Olympus (Aartselaar, Belgium). Lysosomal area quantifications were performed using ImageJ software (rsbweb.nih.gov/ij/).
Cell Cycle Analysis
Samples were washed with 1X PBS and fixed with cold 70% ethanol. Then, cells were treated with RNase A (10 μg/ml) (Roche, Luxembourg, Luxembourg) and stained with 1 μg/ml of propidium iodide (BD Biosciences, Erembodegen, Belgium) for 30 min at room temperature in the dark. All samples were then processed using flow cytometry.
RNA Expression Analyses by Q-RT-PCR
Total RNA was extracted using NucleoSpin RNA II columns (Macherey-Nagel, Hoerdt France) according to the manufacturer’s instructions. cDNA was synthesized by reverse transcription of 1 μg total RNA with M-MLV Reverse Transcriptase, RNase H Minus, Point Mutant reverse transcriptase (Promega, Leiden, The Netherlands) using oligo(dT) primers (Promega). cDNAs were used as template for subsequent quantification by real-time PCR in a reaction mixture containing 1X Power SYBR® Green PCR Master Mix (Applied Biosystems, Halle, Belgium), and 0.1 μM of each primer (sequences available on request). Amplification was performed on an ABI 7300 real-time PCR system (Applied Biosystems). The amount of target gene was normalized to the endogenous level of β-actin using the 2-ΔCT method.
Protein Extraction and Western Blot
Histone isolation was performed in acidic conditions [21]. Whole cell extracts were prepared using M-PER® (Thermofisher) according to the manufacturer’s instructions. Protein concentration was determined using the Bradford assay (Bio-Rad, Nazareth Eke, Belgium). Proteins were resolved by SDS–PAGE and Western Blots were realized using the following primary antibodies: anti-β-actin (Sigma-Aldrich), anti-c-myc, anti-p16, anti-p19, anti-p21, anti-p27 (Santa Cruz, Boechout, Belgium), anti-γH2AX, anti-hTERT (Millipore) [22]. Proteins of interest were detected with ECL Prime Western Blotting Detection System reagent (GE Healthcare) using an ImageQuant LAS 4000 mini (GE Healthcare).
Relative Telomere Length Assays
Relative telomere length was assessed by flow-fluorescent in situ hybridization with a fluorescein-conjugated PNA probe using the Telomere PNA Kit/FITC (Dako Denmark A/S, Heverlee, Belgium) according to the manufacturer's protocol. Quantification of relative telomere length was carried out by flow cytometry.
Relative telomere length was also evaluated by a PCR-based method previously published with few modifications [23]. Genomic DNA was extracted using the DNeasy® Blood and Tissue Kit (Qiagen, Venlo, The Netherlands) according to manufacturer’s instructions. Twenty nanograms of DNA were used as template for subsequent quantification by real-time PCR in a reaction mixture containing 1X Power SYBR® Green PCR Master Mix (Applied Biosystems), using the primers (written from 5’ to 3’) for: telomere, forward: ACACTAAGGTTTGGGTTTGGGTTTGGGTTTGGGTTAGTGT and reverse: TGTTAGGTATCCCTATCCCTATCCCTATCCCTATCCCTAACA; RNA polymerase II promoter, forward: CTTCTTCCTCCCCCAAAAAC and reverse: GGGAAATAAGGAGCGAAAGG. PCR conditions used for the amplification with telomere primers were step 1: 95°C for 10 min; step 2: cycles of 95°C for 15 s, 49°C for 15 s; step 3: 40 cycles of 95°C for 15 s, 60°C for 15 s, 70°C for 1 min. PCR conditions used for the 40-cycle amplification with RNA polymerase II primers were as followed: 95°C for 15 s, 60°C for 1 min after an initial step of 95°C for 10 min. Amplification and detection were carried out using the Applied Biosystems 7300 Real-time PCR system. Relative telomere length was calculated as a telomere/RNA polymerase II ratio with the value for untreated cells set to 1.
Telomerase Activity Assay
Telomerase activity was measured using the Telomerase PCR ELISA kit (Roche) according to the manufacturer's protocol. The positive control (human kidney 293 cell extract) was supplied by the manufacturer.
Chromatin Immunoprecipitation (ChIP)
ChIP experiments were carried out using c-myc (Santa Cruz) and H3K4Me3 (Millipore) antibodies [20]. Primers were targeting the c-myc binding site within the basal region of hTERT promoter.
Flow Cytometry Acquisition and Analysis
Flow cytometry acquisitions were performed on a FACSCalibur (BD Biosciences) using Cellquest software (BD Biosciences). Data were analyzed using FlowJo software (Treestar, Ashland, OR, USA).
RNA Interference Assays
Small interfering RNAs (siRNAs) were obtained from Qiagen: CDKN2D-1 (SI00287707), CDKN2D-2 (SI00287714), c-myc-1 (SI00300902), c-myc-2 (SI02662611), control siRNA (AllStars Negative Control siRNA). Cells were transfected at 80% confluence with 10 nM siRNA and HiPerFect Transfection reagent (Qiagen) according to the manufacturer’s instructions.
Methylation-Specific PCR
Genomic DNA was isolated using the Dneasy® Blood and Tissue Kit (Qiagen, Venlo, The Netherlands) and then subjected to sodium bisulfite conversion using the EpiTect® Bisulfite Kit (Qiagen) according to manufacturer’s instructions. Methylation-specific PCR (MSP) were carried out as previously described [20]. DNA was amplified using the following primers specific for methylated (M) or unmethylated (U) sequences targeting: (i) the CCCTC-binding factor (CTCF) binding region in hTERT with hTERT-1 M (Forward 5’-GCGCGAGTTTTAGGTAGC-3’, reverse 5’-CTCGCGATAATAACTACGCA-3’) and hTERT-1UM (Forward 5’-GGTGTGAGTTTTAGGTAGT-3’, reverse 5’-CCTCACAATAATAACTACACA-3’); (ii) the basal promoter of hTERT with hTERT-2 M (Forward 5’-GGATTCGCGGGTATAGAC-3’, reverse 5’-CGCGAAAAAAACGAAACTA-3’) and hTERT-2UM (Forward 5’-AGTGGATTTGTGGGTATAGAT-3’, reverse 5’-CCACACAAAAAAAACAAAACTA-3’); (iii) the 1st exon of CDKN2D with CDKN2D-1 M (Forward 5’- ATTCGGTTTTTAAGGGGC-3’, reverse 5’-GACTACGCTACTCTCCCGTC-3’) and CDKN2D-1UM (Forward 5’-AGATTTGGTTTTTAAGGGGT-3’, reverse 5’-CCAACTACACTACTCTCCCATC-3’); and (iv) the basal promoter of CDKN2D with CDKN2D-2 M (Forward 5’-AAAATTTCGTTATTGTCGGC-3’, reverse 5’ATAACCAATAACTACGCGTCG-3’) and CDKN2D-2UM (Forward 5’-GAAAATTTTGTTATTGTTGGT-3’, reverse 5’-CATAACCAATAACTACACATCA-3’.
Statistical Analysis
Statistical analysis was performed using the GraphPad Prism 6.0 software. One-way and two-way ANOVA followed by the Holm-Sidak multiple comparison tests were used for statistical comparisons. p-values below 0.05 were considered as statistically significant.
Results
DAC Elicits Senescence in CML Cells
Previously, we highlighted a mixed population of suspension and adherent MEG-01 CML cells after DAC exposure [19]. Accordingly, to further examine the mechanisms of senescence induction by DAC in CML, we chose to analyze the two MEG-01 cell populations separately from the third day of treatment.
First, SA-β-gal activity was evaluated in K-562 CML cells with or without different doses of DAC from 0.125 to 2 μM up to 6 days. Senescent cells were identified as blue-stained cells by standard light microscopy. As shown in Figure 1A, microscopy analyses revealed that senescence induction was increased dose-dependently. The maximum of senescence was observed in K-562 cells exposed to 1 and 2 μM DAC for 6 days with 80 % of SA-β-gal positive cells. In this regard, we chose to treat CML cells with 2 μM of DAC for further experiments.
Figure 1.
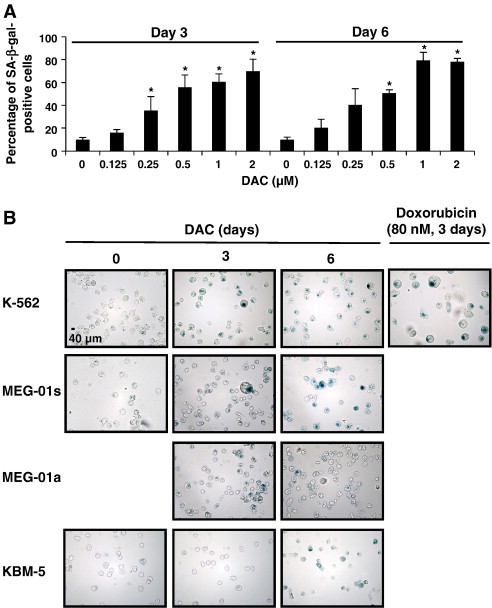
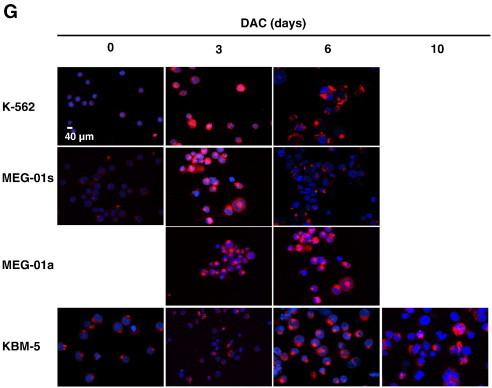
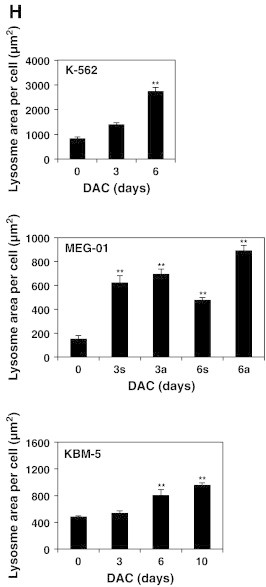
DAC elicits senescence in CML cells.
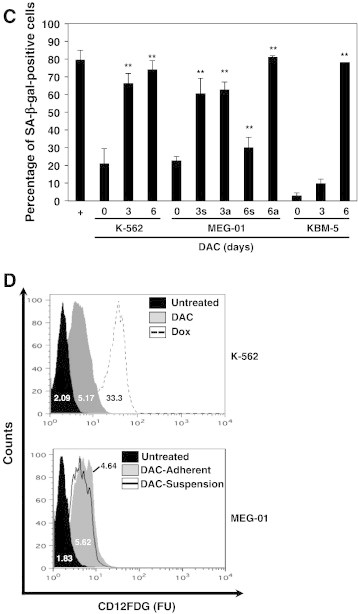
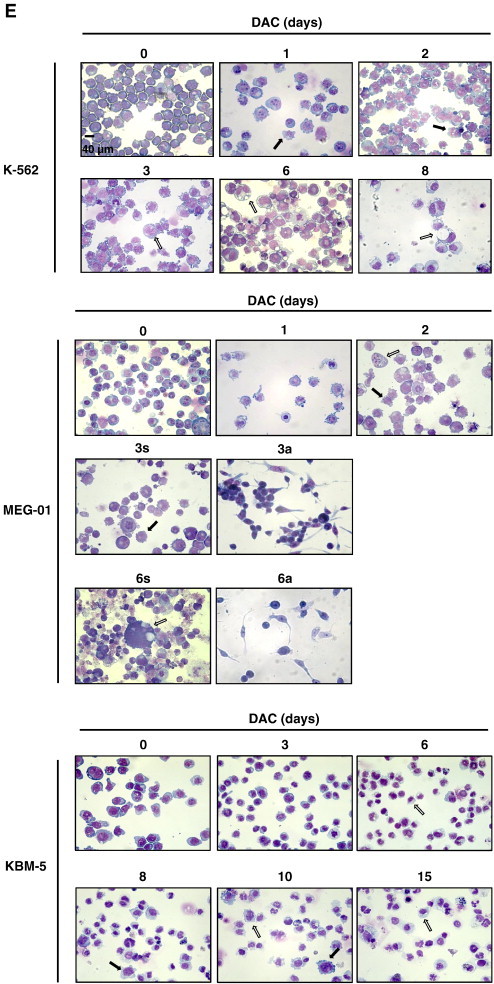
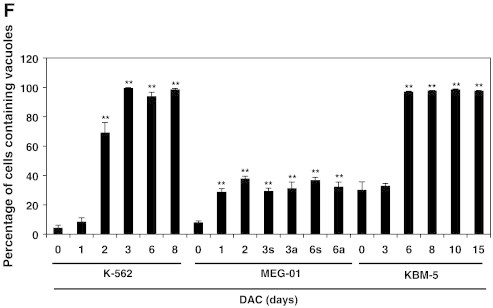
(A) DAC increases SA-ß-gal activity in K-562 cells. Data are the mean (±SD) of 3 independent experiments. * P < 0.05, ** P < 0.01 versus control (two-way ANOVA). (B to H) K-562, MEG-01 and KBM-5 cells were cultured in presence or absence of 2 μM DAC for the indicated period of time. “a” and “s” refer to adherent and suspension MEG-01 populations, respectively. (B) Cytochemical detection of SA-ß-gal activity. (C) Percentage of SA-β-gal-positive cells. The positive control (+) corresponds to K-562 cells treated with 80 nM doxorubicin for 72 hours. (D) Flow cytometry determination of SA-ß-gal. Numbers represent mean fluorescence intensity. (E) Morphological analysis. White arrows indicate vacuoles and black arrows indicate bulbous protrusions. (F) Number of cells exhibiting vacuoles as a percentage of the total cell population. (G) Analysis of lysomal mass with pictures representing the merged signal of LysoTracker Red (lysosomes) and Hoechst (nuclei). (H) Lysosomal mass area quantification. Results represent the mean ± SD of three independent experiments. All pictures are representative of three independent experiments. * P < 0.05, ** P < 0.01 versus control (one-way ANOVA).
SA-β-gal activity was compared in K-562, MEG-01 and KBM-5 CML cells with or without DAC treatment. K-562 cells treated with doxorubicin, a known inducer of cellular senescence [24], were used as a positive control. As expected, doxorubicin induced an increase in SA-β-gal activity in K-562 cells compared to untreated cells (Figure 1, B and C). Similarly, DAC treatment resulted in a time-dependent increase of SA-β-gal activity in K-562, MEG-01 and KBM-5 cells. Indeed, up to 74, 30, 81 and 85% of K-562, suspension MEG-01, adherent MEG-01 and KBM-5 cells, respectively, were significantly positive for SA-β-gal staining after 6 days of culture with DAC. Remarkably, the kinetic of induction of SA-β-gal activity in KBM-5 cells is slower than in K-562 and MEG-01 cells. Indeed, while only 7% of KBM-5 cells were positive for SA-β-gal staining after 3 days of DAC treatment, 60% of K-562 and MEG-01 cells were positive. Nevertheless, in the three CML cell lines, approximately 80% of cells were SA-β-gal positive after 6 days of treatment.
SA-β-gal activity was further quantified in K-562 and MEG-01 cells by flow cytometry using C12FDG (Figure 1D). Results showed that, after 4 days of DAC treatment, K-562, adherent and suspension MEG-01 cells exhibited a 2.5, 3.1 and 2.5-fold increase in SA-β-gal compared to untreated cells, respectively. Therefore, these data support our previous results (Figure 1, B and C).
Senescence induction was confirmed by assessment of cell morphology after May-Grünwald Giemsa staining in DAC-treated K-562, MEG-01 and KBM-5 cells. As shown in Figure 1D, the size of CML cells increased and bulbous protrusions became visible after one day (Figure 1E, black arrows). Two-day-treated K-562 cells exhibit numerous small cytoplasmic vacuoles (Figure 1E). After longer treatment, more than 94% of K-562 cells exhibit vacuoles (Figure 1F). In addition, vacuole number decreased while their volume increased. Analysis of May-Grunwald Giemsa-stained KBM-5 cells after 3-day exposure to DAC indicate that the relatively scarce cytoplasm of the cells contained also many little vacuoles (Figure 1E). After longer treatments, cells presented either bulbous protrusions or little cytoplasmic protrusions (white arrows). In addition, the percentage of cells containing vacuoles as well as the vacuole number was remarkably increased in a time-dependent manner (Figure 1, E and F). Similarly, approximately 30% of DAC-treated MEG-01 cells in suspension exhibited cytoplasmic vacuoles from day 1 (Figure 1F). These cells were larger than untreated cells and also characterized by the presence of bulbous protrusions (Figure 1E, black arrows).
Since senescent cells typically exhibit an increase in lysosome size and number, we further assessed the cellular distribution of lysosomes in CML cells. K-562, MEG-01 and KBM-5 cells with or without DAC treatment were stained with Lysotracker Red and microscopy observations indicate that the lysosomal compartment is more prominent in K-562 and MEG-01 cells after 3 days of treatment (Figure 1G). Indeed, 6 day-DAC-treated K-562 and MEG-01 cells exhibited a 3.3 and 5.8-fold increase in the lysosomal area compared to untreated cells, respectively (Figure 1H). DAC-treated KBM-5 cells also exhibited an increased lysosomal mass from day 6 compared to control cells (Figure 1, G and H). After 10 days of treatment, KBM-5 cells showed a 2-fold increase in the lysosomal mass.
DAC Induces Increased Expression of Key Cell Cycle Regulators of the Senescence Program
Senescence is commonly associated with changes in the expression levels of various cyclin-dependent kinase inhibitors resulting in an irreversible cell cycle arrest [25]. To assess the effect of DAC on cell cycle, DNA content of DAC-treated K-562 and MEG-01 cells exposed to DAC up to 10 and 8 days, respectively, was analyzed by flow cytometry. As shown in Figure 2, A and B, DAC treatment induced a transitional accumulation in G2/M cell cycle phase in K-562 cells at day 1 and day 2 with an equal percentage of cells in G2/M at day 1 and day 4. Results also revealed that the sub-G1 population was increased in a time-dependent manner after 8 days of exposure with DAC. Indeed, the number of K-562 cells in sub-G1 phase was increased from 6.3 to 62.8% in control and 10-day treated K-562 cells, respectively. No cell cycle phase-specific accumulation was observed in both suspension and adherent MEG-01 populations after DAC exposure (Figure 2, A and B). Nevertheless, flow cytometry analyses indicate a time-dependent accumulation of suspension cells in sub-G1 phase from day 6, reaching 60.8% after 10 days of treatment.
Figure 2.
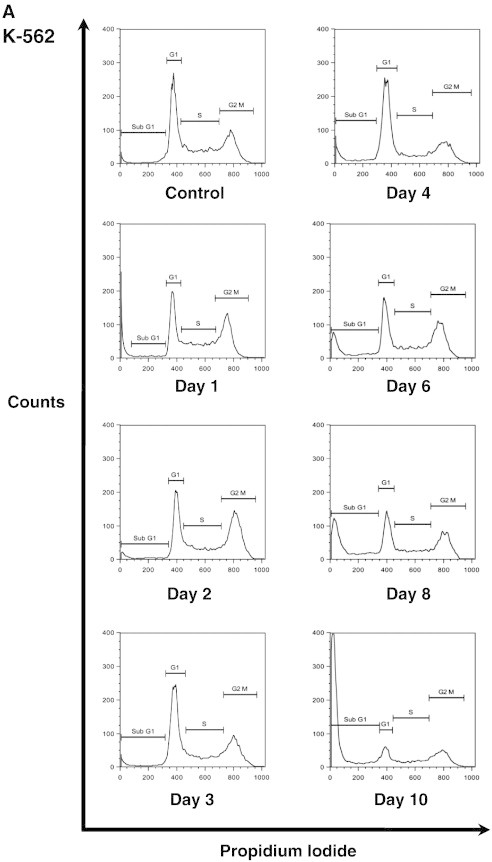
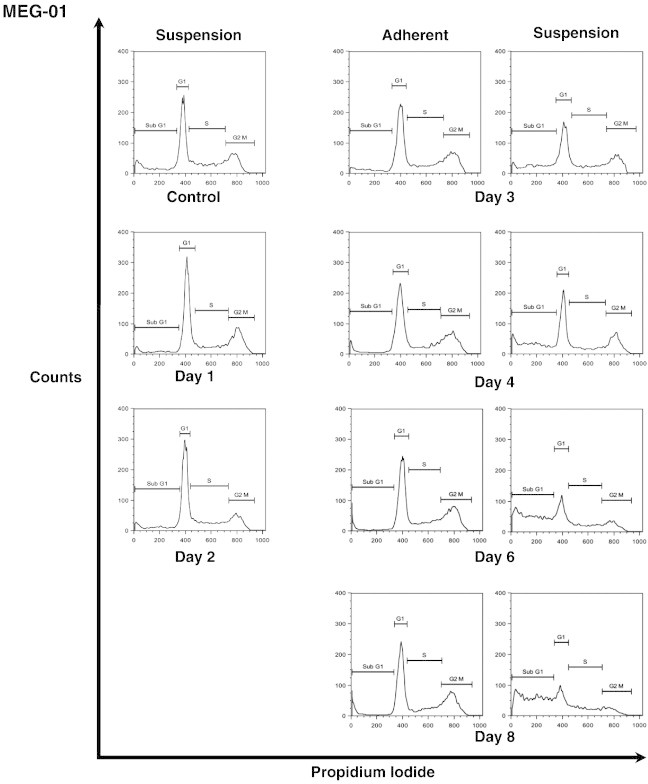
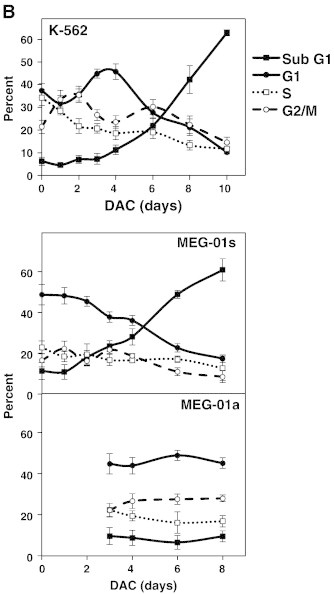
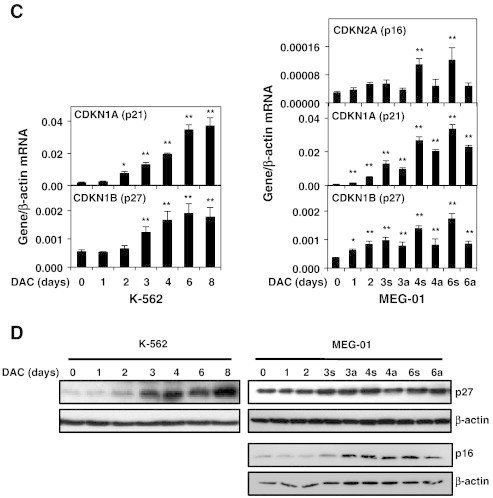
DAC induces an increase in the expression of key cell cycle regulators of the senescence program.
K-562 and MEG-01 cells were treated with 2 μM DAC for the indicated period of time. “a” and “s” refer to adherent and suspension MEG-01 populations, respectively. (A, B) Cell cycle analysis. (C, D) Analyses of mRNA and protein expression levels of cell cycle regulators. Cell cycle graphs and blots are representative of three independent experiments. Data are the mean ± SD of three independent experiments. * P < 0.05, ** P < 0.01 versus control (one-way ANOVA).
Then, the expression of key cell cycle-related genes involved in the senescence process was evaluated in K-562 and MEG-01 cells by real-time PCR. As shown in Figure 2C, DAC-treated K-562 cells displayed a significant increased expression of CDKN1A (p21WAF1/CIP1) and CDKN1B (p27Kip1) genes, two G1-S and G2-M phase-checkpoint regulators. Nevertheless, K-562 cells do not express CDKN2A (p16INK4A) gene due to a locus deletion (data not shown). Similarly, DAC treatment induced a time-dependent increase of CDKN1A and CDKN1B in MEG-01 cells. Expression of the G1 cell-cycle inhibitor CDKN2A was increased in DAC-treated MEG-01 cells. In addition, p27 protein levels were increased in a time-dependent manner from day 3, in K-562 and MEG-01 cells exposed to DAC (Figure 2D). Similarly, p16 protein expression was increased in DAC-treated K-562 cells in a time-dependent manner. Nevertheless, Western Blot analyses did not show any induction of p21 in the two CML cell lines (data not shown). Therefore, senescence induction was associated with an increased expression of multiple cell cycle regulators in K-562 and MEG-01 cells.
DAC-Induced Senescence is not Associated with DNA Damage Accumulation
We further addressed whether DAC-induced senescence in CML is associated with the activation of DNA damage signals. Thus, we examined the γH2AX level, the earliest known cellular response to DNA damage localized at double strand breaks (DSB) sites [26], in K-562 cells exposed to DAC. Microscopy (Figure 3A) and Western Blot (Figure 3B) analyses revealed that the proportion of γH2AX-positive cells in DAC-treated K-562 cells was not changed up to 6 days of DAC treatment compared to untreated K-562 cells. In contrast, γH2AX expression could be clearly detected after senescence induction since it appeared after 8 days of treatment.
Figure 3.
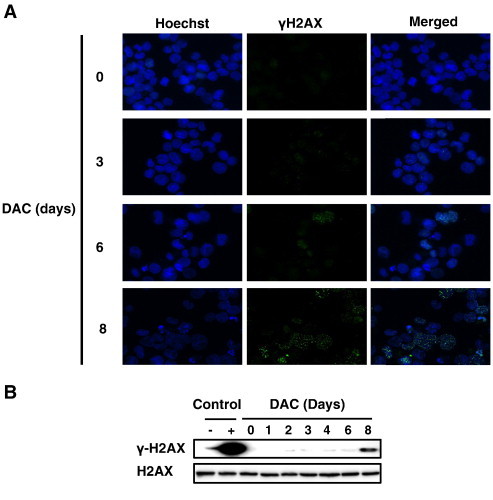
DAC-induced senescence is independent of DNA damage accumulation.
K-562 were cultured in presence of 2 μM DAC for the indicated period of time. (A) Cells were assessed for γH2AX phosphorylation (green) and counterstained with Hoechst 33342 (blue). Pictures are representative of three different experiments. (B) Western Blot analysis of γH2AX expression. U-937 cells left untreated (−) or treated with 100 μM etoposide for 4 h (+). Blots are representative results of three independent experiments.
DAC-Induced Telomere Shortening in CML Cell Lines is Associated With Transcriptional Down-Regulation of hTERT
In order to assess involvement of telomere length in DAC-induced senescence, we evaluated the relative telomere length by flow cytometry (Figure 4A), and by real-time PCR (Figure 4B). Flow cytometry analyses indicate that DAC causes telomere shortening in K-562 cells after 3 days of treatment. Indeed, relative telomere length was reduced by 40% in 3-day-DAC treated K-562 cells compared to control cells. Remarkably, relative telomere length in DAC-treated K-562 cells after 6 days and PBMCs were similar. Real-time PCR analyses confirmed that telomere shortening was significantly increased after DAC exposure. Flow cytometry analyses demonstrated that a 6-day DAC treatment period induced a decrease by 50 and 32% of relative telomere length in MEG-01 and KBM-5 cells, respectively. Real-time PCR analyses indicate that telomere shortening was time-dependent in both cell lines.
Figure 4.
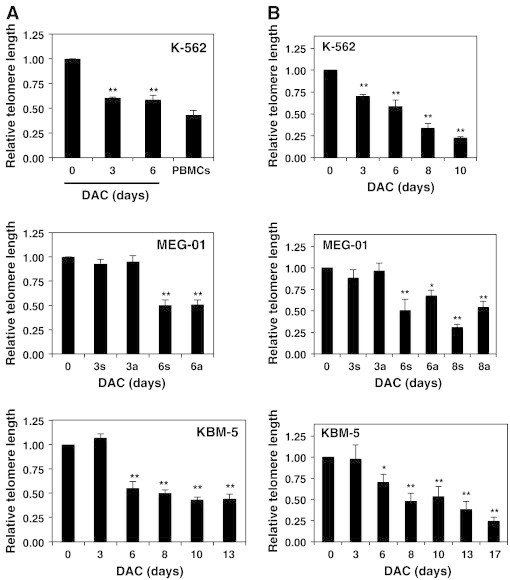
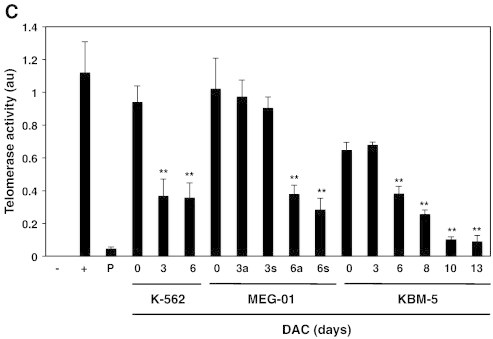
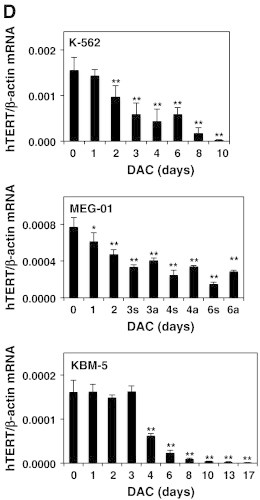
DAC-induced telomere shortening in CML cell lines is associated with transcriptional down-regulation of hTERT.
K-562, MEG-01 and KBM-5 cells treated in the presence or absence of 2 μM DAC for the indicated periods. “a” and “s” refer to adherent and suspension MEG-01 populations, respectively. CML cells and PBMCs were assayed for relative telomere length by flow cytometry (A) and by Q-RT-PCR (B). (C) Telomerase activity measurement in CML cells, PBMCs (P) and positive control (+). (D) Q-RT-PCR analyses. Data are the mean ± SD of three independent experiments. * P < 0.05, ** P < 0.01 versus control (one-way ANOVA).
Telomerase activity was then assessed in K-562, MEG-01 and KBM-5 cells exposed to DAC up to 6 days using a modified TRAP assay to evaluate if the reduction of telomere length in DAC-treated cells was due to telomerase modulation. K-562 cells presented a 2.6-fold decrease in telomerase activity after 3 days of treatment (Figure 4C). A decrease in telomerase activity was also observed in DAC-treated KBM-5 and MEG-01 cells but it occurred only after 6 days of treatment. Indeed, KBM-5 as well as adherent and suspension MEG-01 cells exhibited a 2.2-, 2.6- and 3.5-fold reduced telomerase activity, respectively. According to reduced telomere length in PBMCs (Figure 4A), telomerase activity in PBMCs was lower than in K-562, MEG-01 and KBM-5 cells.
The expression pattern of hTERT is considered to be a rate-limiting determinant of the enzymatic activity of human telomerase. Thus, hTERT expression was assessed by real-time PCR. DAC induced a significant time-dependent down-regulation of hTERT expression from 2 days of DAC treatment in K-562 and MEG-01 cells (Figure 4D). In addition, the level of hTERT mRNA is drastically reduced after 4 days in DAC-treated KBM-5 cells. Consequently, these results suggest that DAC reduced hTERT expression in CML cells and that telomerase activity closely correlated with hTERT expression.
DAC Reduces hTERT Expression Through Decreased Binding of c-myc to the hTERT Promoter in CML Cells
Since c-myc acts as a key transcriptional activator of hTERT [27], we further elucidated the mechanism leading to hTERT down-regulation after DAC treatment by analyzing the binding of c-myc to the hTERT promoter (Figure 5A). ChIP assays revealed that the histone methylation mark H3K4me3, a hallmark of active promoters and actively transcribed regions, was enriched at the hTERT promoter in untreated K-562 cells. However, H3K4Me3 decreased at the hTERT promoter after 4 days of DAC treatment, and correlated with decreased hTERT expression. We also demonstrated a decreased binding of c-myc to the hTERT promoter after 3 days of DAC treatment. This binding was dramatically impaired after longer exposure to DAC. These results suggest that DAC reduces hTERT expression at least in part through decreased binding of c-myc to hTERT promoter in CML cells.
Figure 5.
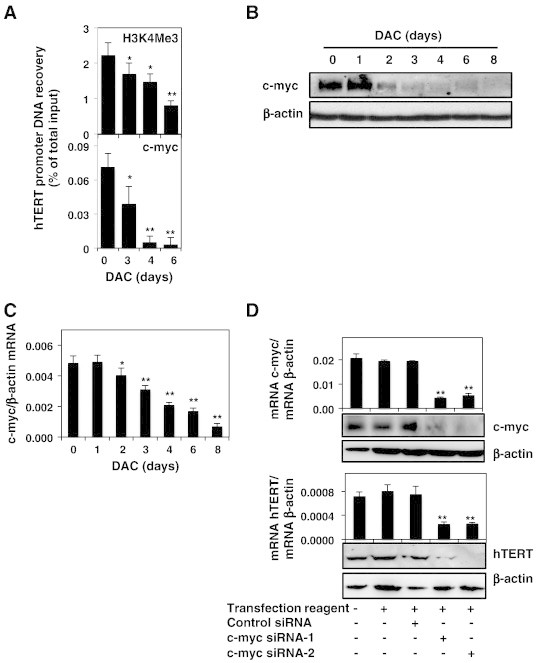
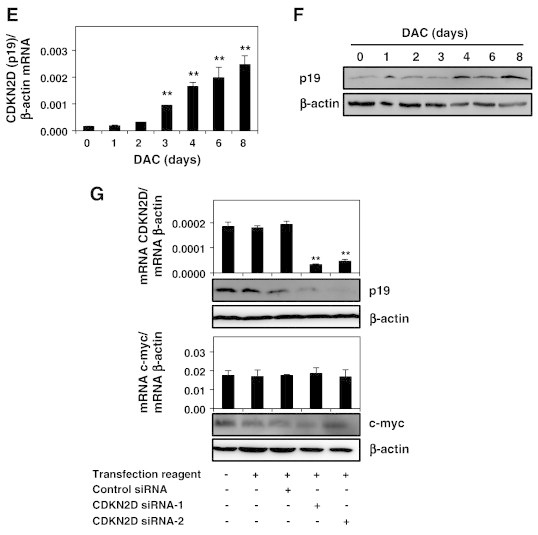
DAC reduces hTERT expression through decreased binding of c-myc to the hTERT promoter in CML cells.
K-562 cells were cultured in presence or absence of 2 μM DAC. (A) ChIP analysis of H3K4Me3 and c-myc recruitment to hTERT promoter. (B and C) Analysis of c-myc expression at protein and mRNA levels. (D) c-myc and hTERT expression at mRNA and protein levels in K-562 cells transfected with or without control siRNA or c-myc siRNAs. (E and F) Analysis of CDKN2D (p19) expression at protein and mRNA levels. (G) CDKN2D and c-myc expression were evaluated at mRNA and protein levels in K-562 cells transfected with or without control siRNA or CDKN2D siRNAs. Data are the mean (±SD) of three independent experiments. Blots are representative of three independent experiments. * P < 0.05, ** P < 0.01 versus control (one-way ANOVA).
We further attempted to determine whether decreased binding of c-myc to the hTERT promoter could be due to an alteration of c-myc expression in DAC-treated K-562 cells. As shown in Figure 5B, c-myc protein expression remained unaffected after one day of DAC treatment but was drastically decreased after 2 days in K-562 cells. The decrease of c-myc protein was due to a transcriptional down-regulation as shown by real-time PCR from day 2 in K-562 cells (Figure 5C).
In order to assess whether c-myc is involved in the transcriptional regulation of hTERT in our cell model, we down-regulated c-myc expression using siRNAs and evaluated its effect on hTERT expression. Results demonstrated a tight correlation between c-myc and hTERT expression levels at both RNA and protein levels (Figure 5D).
CDKN2D (p19INK4D) has been described as a negative regulator of c-myc [28]. In this context, we investigated whether DAC may affect CDKN2D expression in K-562 cells. Results indicate that CDKN2D expression is significantly increased in DAC treated K-562 cells in a time-dependent manner (Figure 5, E and F).
We further attempted to determine whether c-myc repression in DAC-treated K-562 cells could be related to CDKN2D up-regulation. For this purpose, the effect of CDKN2D silencing using siRNAs on c-myc expression was investigated. Surprisingly, we found that CDKN2D knockdown did not alter c-myc at both RNA and protein levels in our cell model (Figure 5G).
The Expression of hTERT, but not CDKN2D, is Modulated Through Epigenetic Mechanisms in DAC-Treated K-562 Cells
Since DAC is a demethylating agent, we hypothesized that DAC could trigger senescence in CML cells via epigenetic mechanisms. To verify this hypothesis, we further analyzed whether 5-azacytidine, another demethylating agent, could induce the senescence program in K-562 cells. As shown in Figure 6A, 5-azacytidine treatment triggers a dose-dependent increase in SA-β-gal activity. Nevertheless, the kinetic of senescence induction was slower with 5-azacytidine than with DAC treatment. Indeed, K-562 cells display a significant increased SA-β-gal activity from 0.125 μM 5-azacytidine after 6 days of treatment.
Figure 6.
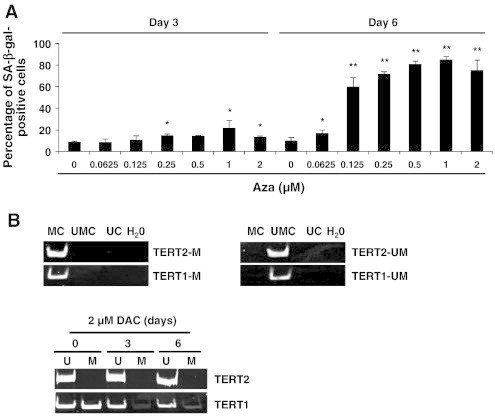
DAC-mediated hTERT down-regulation is partially due to an epigenetic regulation.
(A) SA-ß-gal activity was assessed in K-562 cells treated with various concentrations of 5-azacytidine. Data are the mean (±SD) of 3 independent experiments. ** P < 0.01 versus control (two-way ANOVA). (B) Analysis of hTERT promoter methylation in K-562 cells treated with DAC. “U” and “M” refer to primers specific for methylated (M) or unmethylated (U) sequences. “UMC”, “MC” and “UC” correspond to unmethylated converted, methylated converted and un converted unmethylated DNA, respectively. Pictures are representative of three independent experiments.
In order to determine whether hTERT down-regulation could also be due to an epigenetic regulation, we assessed the methylation status of hTERT promoter as well as the CTCF binding region of hTERT by MSP. Methylation specificity was demonstrated with fully unmethylated, methylated converted as well as with unmethylated unconverted DNA (Figure 6B). MSP revealed that the basal promoter of hTERT was not methylated in K-562 cells and that DAC did not change this state. In contrast, the CTCF binding region in hTERT was partially methylated and, interestingly, DAC treatment reduced dramatically its methylation from day 3.
Since we provided evidence that CDKN2D expression was significantly increased in DAC treated K-562 cells, we then attempted to elucidate the mechanisms leading to CDKN2D modulation. For this purpose, CDKN2D expression was monitored in K-562 cells treated with various concentrations of DAC from 0.0625 to 2 μM. As shown in Figure 7A, K-562 cells exposed to increasing doses of DAC exhibit increased CDKN2D gene expression.
Figure 7.
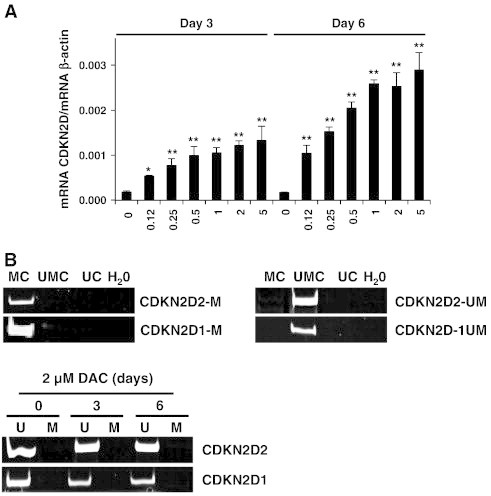
CDKN2D up-regulation is not due to a direct epigenetic action of DAC.
(A) CDKN2D mRNA expression levels in K-562 cells cultured in presence of various concentrations of DAC. Data are the mean (±SD) of three independent experiments. * P < 0.05, ** P < 0.01 versus control (one-way ANOVA). (B) Analysis of CDKN2D promoter methylation in K-562 cells treated with DAC. “U” and “M” refer to primers specific for methylated (M) or unmethylated (U) sequences. “UMC”, “MC” and “UC” correspond to unmethylated converted, methylated converted and unconverted unmethylated DNA, respectively. Pictures are representative of three independent experiments.
Since DAC triggered an increase in CDKN2D expression in a dose-dependent manner, we hypothesized that methylation of CDKN2D promoter region could correlate with the lack of CDKN2D expression in K-562 cells. To test this hypothesis, CDKN2D methylation status was assessed by MSP. Methylation specificity was demonstrated with fully unmethylated, methylated converted as well as with unmethylated unconverted DNA (Figure 7B). Results revealed that DAC treatment did not have any effect on CDKN2D promoter methylation, which is already unmethylated in untreated cells.
Discussion
Altogether, we provided evidence that DAC-induced senescence in CML cells was associated with decreased telomerase activity as well as reduced hTERT transcriptional expression through decreased binding of c-myc to the hTERT promoter.
Senescence induction was revealed by the significant increase in SA-β-gal activity, the enlarged and flattened morphology as well as the markedly increase of the lysosomal compartment, which consist of characteristic markers for senescent cells [29]. To our knowledge, no study has focused on senescence induction after DAC treatment in leukemia cells. Although other studies have suggested that DAC may induce senescence in different solid cancer cells, the authors did not investigate in details the mechanisms involved in the senescence program [30], [31].
Several lines of evidence indicate that the senescence program may be accompanied by DNA damage response, triggered either by dysfunctional telomeres or DSBs [32]. In this study, γH2AX foci formation was distinguished only after senescence induction in the senescent DAC-treated CML cells suggesting that the DNA damage response was not involved in the senescence program. We have previously reported that the DAC-induced senescence was followed by apoptosis induction after sustained exposure to DAC [19]. Accordingly, here, we pointed out that the formation of γH2AX foci occurred only after eight days of treatment in CML cells concomitantly with apoptosis induction.
Cyclin-dependent kinase inhibitors such as CDKN2A (p16INK4A), CDKN1A (p21WAF1/CIP1) and CDKN1B (p27Kip1) are key effectors of the cellular senescence program [25]. Consistently, we further showed that DAC-induced senescence was accompanied by significant increase in the expression of p16 and p27 at RNA and protein levels. Accordingly, up-regulation of CDKN1A, CDKN1B mRNA as well as p16 protein levels were also reported in the previous studies highlighting the ability of DAC to induce senescence in cancer cells in the same range of concentrations [30], [31]. Although DAC-treated K-562 and MEG-01 cells exhibit increased CDKN1A mRNA levels, p21 was not detectable at the protein level and was not induced by DAC exposure (data not shown). There is increasing evidence that p21 expression may be post-transcriptionally regulated by microRNAs. In addition, post-translational regulations and specifically ubiquitin-dependent proteasomal degradation play a critical role in p21 expression and activity [33]. Therefore, the lack of p21 protein expression in DAC-treated CML cells could be due to these mechanisms. Nevertheless, these results demonstrated that p21 overexpression is probably not implicated in DAC-induced senescence.
Although senescence is mainly associated with G1/G0 or G2/M arrest [34], we provided evidence that DAC elicits a transitional accumulation in G2/M in K-562 senescent cells only at day 1 and day 2. Abrogation of cell cycle checkpoint after longer exposure to DAC may be due to p53 gene inactivation in K-562 cells [35]. No specific cell cycle arrest was observed in DAC-induced MEG-01 senescent cells, likely due to the up-regulation of genes involved in the various stages of cell-cycle progression after DAC exposure. Indeed, in particular, p21WAF1/CIP1 and p27Kip1 may regulate the G1/S as well as the G2/M checkpoints [36], [37].
Telomere shortening has been proposed as an important initiator of stress-induced premature senescence [38]. Interestingly, our results indicate that DAC elicits telomere shortening in CML cells. We have also established that DAC-induced senescence was associated with hTERT down-regulation resulting in a significant reduction of telomerase activity in CML cells. So far, only a few studies have focused on hTERT expression in premature senescent cells. Specifically, recently, indole-3-carbinol, which is derived by hydrolysis from glycobrassicin produced in cruciferous vegetables, was shown to trigger senescence by down-regulating hTERT expression and telomerase activity in human breast cancer cells [39]. In addition, our results were in agreement with previous data indicating that DAC exposure may induce hTERT demethylation, leading to reduced telomerase activity and hTERT expression [40]. DAC was also described to inhibit telomerase activity through transcriptional repression of hTERT in human prostate cancer cells [41]. Therefore, we provided evidence that DAC decreased telomere length by reducing telomerase activity as well as hTERT expression in senescent CML cells.
Telomerase counterbalances telomere loss by synthetizing telomeric DNA repeats to the 3' end of DNA strands [42]. Malignant cells are recognized to exhibit telomerase activity in contrast to normal somatic cells where the activity of this enzyme is mostly inactive thus leading to a progressive shortening of telomeres [43]. Cancer cells are described to have short telomeres and to maintain their telomeres through their increased telomerase activity thus conferring immortalization [44]. Accordingly, we provided evidence that telomerase activity was very low in human normal cells compared to control CML cells. Our data also indicate that the average telomere length in K-562 cells was higher than that in normal blood cells. To the best of our knowledge, only a few studies have compared the telomere length in CML and normal cells. In contrast to our data, telomere length of leukemia cells from CML patients was reported to be shorter compared to the lymphocytes of human normal controls [45]. This study focused on CML patient cells whereas we evaluated telomere length in CML cancer cell lines. This consideration could explain the discrepancy between studies. However, another study in line with our results has reported that cells from breast cancer patients display significantly longer telomeres compared to normal cells [46].
Several investigators published that the oncogene c-myc acts as a key transcriptional activator of hTERT during carcinogenesis [27], [47]. In the present study, we demonstrated using siRNA experiments that c-myc is involved in hTERT transcription in K-562 cells. In addition, we provided evidence that DAC-mediated c-myc down-regulation triggers telomere-dependent senescence by regulating hTERT in CML. Accordingly, c-myc down-regulation was reported to trigger cellular senescence in solid cancers and lymphoma [48]. Furthermore, genistein, a potent DNA methyltransferase inhibitor, was described to abrogate hTERT transcriptional activity through c-myc down-regulation in prostate cancer cells [49].
Here, we provided evidence that DAC reduces hTERT expression through decreased binding of c-myc to the hTERT promoter. Nevertheless, we do not exclude the contribution of other epigenetic changes that would result in hTERT down-regulation. Indeed, hTERT is regulated by several epigenetic alterations at promoter sites including histone acetylation and promoter methylation [27]. Specifically, several studies have pointed out that hTERT is hypermethylated in various adenocarcinoma cell line models and that demethylation of this gene by 5-azacytidine, another classical demethylating agent, inhibits its transcription through reactivation of the binding of the transcriptional repressor CTCF [50]. Here, we showed that DAC induces demethylation of hTERT promoter within CTCF binding site region. Therefore, besides abrogating hTERT expression through decreased binding of c-myc to the hTERT promoter in CML cells, DAC could further reduce hTERT expression by DNA demethylation-mediated CTCF recruitment.
Recent reports demonstrated that CDKN2D may negatively regulate c-myc transcriptional activity through their physical interaction [28]. Here, our results indicate that CDKN2D is up-regulated in DAC-treated CML cells. In this regard, we suggest that c-myc repression could be due to CDKN2D up-regulation. Nevertheless, surprisingly, the siRNA-mediated knockdown of CDKN2D did not alter c-myc at both RNA and protein levels in K-562 cells. Since CDKN2D basal level is very low in K-562 cells, it is possible that CDKN2D knockdown is not enough to alter c-myc expression. Further studies are required to clarify whether CDKN2D physically interacts with c-myc in DAC-induced senescent CML cells. Nonetheless, besides p19, we cannot rule out that other regulators of c-myc expression are modulated by DAC treatment. Indeed, Mxi1, a transcriptional repressor of c-myc, has been reported to be silenced by DNA methylation in cancer [51]. Similarly, several miRNAs including miR-34a, 34c and Let-7a targeting c-myc regulation are reported to be silenced by DNA methylation [13], [14], [52]. Therefore, further investigations are required to elucidate the mechanisms of DAC-mediated c-myc down-regulation.
K-562 cells exposed to increasing doses of DAC exhibit CDKN2D gene overexpression suggesting that effects induced by DAC on this gene could be related to its epigenetic action. We hypothesized that the increased expression of CDKN2D could result of direct promoter demethylation. However, MSP analysis did not reveal any evidence of CDKN2D promoter methylation in K-562 cells. Consequently, the mechanism of enhanced CDKN2D expression in DAC-treated K-562 cells remains to be elucidated. In this context, DNA demethylation may reactivate genes or transcription factors involved in the regulation of CDKN2D thus leading to up-regulation of this gene after DAC treatment. Nevertheless, we showed that overexpression of p19 per se is not sufficient to induce cellular senescence in K-562 cells (supplementary information, Figure SI1).
There is increasing evidence that epigenetic mechanisms such as DNA methylation are involved in the establishment and/or the maintenance of senescence program [53]. Here, we provided evidence that DAC treatment triggers cellular senescence in CML cells. We additionally showed that K-562 cells exposed to increasing doses of DAC or 5-azacytidine become senescent in a dose-dependent manner suggesting that senescence induced by these compounds could be related to demethylating effects. Consistently with our findings, a recent study reported that DNMT inhibition by 5-azacytidine induces senescence in stem cells [54].
Standard chemotherapeutic regimens are now recognized to exert their therapeutic potential not only via forcing cancer cells to die but also by promoting a terminal arrest program that contributes to the outcome of cancer therapy as well [55]. It became clear that senescence functions as a physiological in vivo mechanism to counteract tumor progression from the early steps of carcinogenesis. Importantly, senescent tumor cells may be cleared, in vivo, by the innate immune system, thus resulting in tumor regression [56]. In this context, the concept of pro-senescence therapy has emerged over the past few years as a novel therapeutic approach to treat cancer with or without traditional chemotherapies [6].
Acknowledgments
TK is a recipient of a doctoral grant from the «Ministère de la Culture, de l’Enseignement supérieur et de la Recherche du Luxembourg». CG, MS and AG were supported by a Télévie Luxembourg fellowship. MS is supported by a “Waxweiler grant for cancer prevention research” from the Action Lions “Vaincre le Cancer”. This work was supported by Télévie Luxembourg, the «Recherche Cancer et Sang» fundation and «Recherches Scientifiques Luxembourg» association. The authors thank «Een Häerz fir Kriibskrank Kanner» association and the Action Lions “Vaincre le Cancer” for generous support. MD is supported by the National Research Foundation of Korea (NRF) grant for the Global Core Research Center (GCRC) funded by the Korea government, Ministry of Science, ICT & Future Planning (MSIP) (No. 2011–0030001).
Footnotes
Conflict of interest The authors declare no conflict of interest.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.05.009.
Appendix A. Supplementary data
Supplementary material.
References
- 1.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- 2.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 3.Kahlem P, Dorken B, Schmitt CA. Cellular senescence in cancer treatment: friend or foe? J Clin Investig. 2004;113:169–174. doi: 10.1172/JCI20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marcotte R, Wang E. Replicative senescence revisited. J Gerontol. 2002;57:B257–B269. doi: 10.1093/gerona/57.7.b257. [DOI] [PubMed] [Google Scholar]
- 5.Frippiat C, Chen QM, Remacle J, Toussaint O. Cell cycle regulation in H(2)O(2)-induced premature senescence of human diploid fibroblasts and regulatory control exerted by the papilloma virus E6 and E7 proteins. Exp Gerontol. 2000;35:733–745. doi: 10.1016/s0531-5565(00)00167-4. [DOI] [PubMed] [Google Scholar]
- 6.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23:2919–2933. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 7.Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim Sh SH, Kaminker P, Campisi J. Telomeres, aging and cancer: in search of a happy ending. Oncogene. 2002;21:503–511. doi: 10.1038/sj.onc.1205077. [DOI] [PubMed] [Google Scholar]
- 9.Shay JW, Zou Y, Hiyama E, Wright WE. Telomerase and cancer. Hum Mol Genet. 2001;10:677–685. doi: 10.1093/hmg/10.7.677. [DOI] [PubMed] [Google Scholar]
- 10.Nugent CI, Lundblad V. The telomerase reverse transcriptase: components and regulation. Genes Dev. 1998;12:1073–1085. doi: 10.1101/gad.12.8.1073. [DOI] [PubMed] [Google Scholar]
- 11.Kyo S, Takakura M, Taira T, Kanaya T, Itoh H, Yutsudo M, Ariga H, Inoue M. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT) Nucleic Acids Res. 2000;28:669–677. doi: 10.1093/nar/28.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, Dalla-Favera R. Direct activation of TERT transcription by c-MYC. Nat Genet. 1999;21:220–224. doi: 10.1038/6010. [DOI] [PubMed] [Google Scholar]
- 13.Florean C, Schnekenburger M, Grandjenette C, Dicato M, Diederich M. Epigenomics of leukemia: from mechanisms to therapeutic applications. Epigenomics. 2011;3:581–609. doi: 10.2217/epi.11.73. [DOI] [PubMed] [Google Scholar]
- 14.Schnekenburger M, Diederich M. Epigenetics Offer New Horizons for Colorectal Cancer Prevention. Curr Colorectal Cancer Rep. 2012;8:66–81. doi: 10.1007/s11888-011-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seidel C, Florean C, Schnekenburger M, Dicato M, Diederich M. Chromatin-modifying agents in anti-cancer therapy. Biochimie. 2012;94:2264–2279. doi: 10.1016/j.biochi.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 16.Kantarjian HM, O'Brien S, Cortes J, Giles FJ, Faderl S, Issa JP, Garcia-Manero G, Rios MB, Shan J, Andreeff M. Results of decitabine (5-aza-2'deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer. 2003;98:522–528. doi: 10.1002/cncr.11543. [DOI] [PubMed] [Google Scholar]
- 17.Issa JP, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, Talpaz M, Garcia-Manero G, Kantarjian HM. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23:3948–3956. doi: 10.1200/JCO.2005.11.981. [DOI] [PubMed] [Google Scholar]
- 18.Charlet J, Schnekenburger M, Brown KW, Diederich M. DNA demethylation increases sensitivity of neuroblastoma cells to chemotherapeutic drugs. Biochem Pharmacol. 2012;83:858–865. doi: 10.1016/j.bcp.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Schnekenburger M, Grandjenette C, Ghelfi J, Karius T, Foliguet B, Dicato M, Diederich M. Sustained exposure to the DNA demethylating agent, 2'-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem Pharmacol. 2011;81:364–378. doi: 10.1016/j.bcp.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Karius T, Schnekenburger M, Ghelfi J, Walter J, Dicato M, Diederich M. Reversible epigenetic fingerprint-mediated glutathione-S-transferase P1 gene silencing in human leukemia cell lines. Biochem Pharmacol. 2011;81:1329–1342. doi: 10.1016/j.bcp.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 21.Nunes MJ, Milagre I, Schnekenburger M, Gama MJ, Diederich M, Rodrigues E. Sp proteins play a critical role in histone deacetylase inhibitor-mediated derepression of CYP46A1 gene transcription. J Neurochem. 2010;113:418–431. doi: 10.1111/j.1471-4159.2010.06612.x. [DOI] [PubMed] [Google Scholar]
- 22.Schnekenburger M, Morceau F, Duvoix A, Delhalle S, Trentesaux C, Dicato M, Diederich M. Expression of glutathione S-transferase P1-1 in differentiating K562: role of GATA-1. Biochem Biophys Res Commun. 2003;311:815–821. doi: 10.1016/j.bbrc.2003.10.072. [DOI] [PubMed] [Google Scholar]
- 23.Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009;37:e21. doi: 10.1093/nar/gkn1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, Roninson IB. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–4818. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- 25.Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000;35:317–329. doi: 10.1016/s0531-5565(00)00083-8. [DOI] [PubMed] [Google Scholar]
- 26.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kyo S, Takakura M, Fujiwara T, Inoue M. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008;99:1528–1538. doi: 10.1111/j.1349-7006.2008.00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature. 2004;431:712–717. doi: 10.1038/nature02958. [DOI] [PubMed] [Google Scholar]
- 29.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timmermann S, Hinds PW, Munger K. Re-expression of endogenous p16ink4a in oral squamous cell carcinoma lines by 5-aza-2'-deoxycytidine treatment induces a senescence-like state. Oncogene. 1998;17:3445–3453. doi: 10.1038/sj.onc.1202244. [DOI] [PubMed] [Google Scholar]
- 31.Amatori S, Bagaloni I, Viti D, Fanelli M. Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer. 2011;71:113–115. doi: 10.1016/j.lungcan.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 32.d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 33.Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010;22:1003–1012. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102:1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Law JC, Ritke MK, Yalowich JC, Leder GH, Ferrell RE. Mutational inactivation of the p53 gene in the human erythroid leukemic K562 cell line. Leuk Res. 1993;17:1045–1050. doi: 10.1016/0145-2126(93)90161-d. [DOI] [PubMed] [Google Scholar]
- 36.Niculescu AB, III, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bashir T, Pagano M. Cdk1: the dominant sibling of Cdk2. Nat Cell Biol. 2005;7:779–781. doi: 10.1038/ncb0805-779. [DOI] [PubMed] [Google Scholar]
- 38.Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Investig. 2004;113:8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marconett CN, Sundar SN, Tseng M, Tin AS, Tran KQ, Mahuron KM, Bjeldanes LF, Firestone GL. Indole-3-carbinol downregulation of telomerase gene expression requires the inhibition of estrogen receptor-alpha and Sp1 transcription factor interactions within the hTERT promoter and mediates the G1 cell cycle arrest of human breast cancer cells. Carcinogenesis. 2011;32:1315–1323. doi: 10.1093/carcin/bgr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guilleret I, Benhattar J. Demethylation of the human telomerase catalytic subunit (hTERT) gene promoter reduced hTERT expression and telomerase activity and shortened telomeres. Exp Cell Res. 2003;289:326–334. doi: 10.1016/s0014-4827(03)00281-7. [DOI] [PubMed] [Google Scholar]
- 41.Kitagawa Y, Kyo S, Takakura M, Kanaya T, Koshida K, Namiki M, Inoue M. Demethylating reagent 5-azacytidine inhibits telomerase activity in human prostate cancer cells through transcriptional repression of hTERT. Clin Cancer Res. 2000;6:2868–2875. [PubMed] [Google Scholar]
- 42.Blackburn EH, Collins K. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Granger MP, Wright WE, Shay JW. Telomerase in cancer and aging. Crit Rev Oncol Hematol. 2002;41:29–40. doi: 10.1016/s1040-8428(01)00188-3. [DOI] [PubMed] [Google Scholar]
- 44.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 45.Samassekou O, Ntwari A, Hebert J, Yan J. Individual telomere lengths in chronic myeloid leukemia. Neoplasia. 2009;11:1146–1154. doi: 10.1593/neo.09836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svenson U, Nordfjall K, Stegmayr B, Manjer J, Nilsson P, Tavelin B, Henriksson R, Lenner P, Roos G. Breast cancer survival is associated with telomere length in peripheral blood cells. Cancer Res. 2008;68:3618–3623. doi: 10.1158/0008-5472.CAN-07-6497. [DOI] [PubMed] [Google Scholar]
- 47.Ducrest AL, Szutorisz H, Lingner J, Nabholz M. Regulation of the human telomerase reverse transcriptase gene. Oncogene. 2002;21:541–552. doi: 10.1038/sj.onc.1205081. [DOI] [PubMed] [Google Scholar]
- 48.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104:13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jagadeesh S, Kyo S, Banerjee PP. Genistein represses telomerase activity via both transcriptional and posttranslational mechanisms in human prostate cancer cells. Cancer Res. 2006;66:2107–2115. doi: 10.1158/0008-5472.CAN-05-2494. [DOI] [PubMed] [Google Scholar]
- 50.Renaud S, Loukinov D, Abdullaev Z, Guilleret I, Bosman FT, Lobanenkov V, Benhattar J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007;35:1245–1256. doi: 10.1093/nar/gkl1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lazcoz P, Munoz J, Nistal M, Pestana A, Encio IJ, Castresana JS. Loss of heterozygosity and microsatellite instability on chromosome arm 10q in neuroblastoma. Cancer Genet Cytogenet. 2007;174:1–8. doi: 10.1016/j.cancergencyto.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 52.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Korner H, Knyazev P, Diebold J, Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–2600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 53.Bandyopadhyay D, Medrano EE. The emerging role of epigenetics in cellular and organismal aging. Exp Gerontol. 2003;38:1299–1307. doi: 10.1016/j.exger.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 54.So AY, Jung JW, Lee S, Kim HS, Kang KS. DNA methyltransferase controls stem cell aging by regulating BMI1 and EZH2 through microRNAs. PLoS ONE. 2011;6:e19503. doi: 10.1371/journal.pone.0019503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmitt CA. Cellular senescence and cancer treatment. Biochim Biophys Acta. 2007;1775:5–20. doi: 10.1016/j.bbcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11:503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.