

Abstract
Minimally differentiated acute myeloid leukemia (AML-M0) is a rare subtype of AML with poor prognosis. Although genetic alterations are increasingly reported in AML, the gene mutations have not been comprehensively studied in AML-M0. We aimed to examine a wide spectrum of gene mutations in patients with AML-M0 to determine their clinical relevance. Twenty gene mutations including class I, class II, class III of epigenetic regulators (IDH1, IDH2, TET2, DNMT3A, MLL-PTD, ASXL1, and EZH2), and class IV (tumor suppressor genes) were analyzed in 67 patients with AML-M0. Mutational analysis was performed with polymerase chain reaction–based assays followed by direct sequencing. The most frequent gene mutations from our data were FLT3-ITD/FLT3-TKD (28.4%), followed by mutations in IDH1/IDH2 (28.8%), RUNX1 (23.9%), N-RAS/K-RAS (12.3%), TET2 (8.2%), DNMT3A (8.1%), MLL-PTD (7.8%), and ASXL1 (6.3%). Seventy-nine percent (53/67) of patients had at least one gene mutation. Class I genes (49.3%) were the most common mutated genes, which were mutually exclusive. Class III genes of epigenetic regulators were also frequent (43.9%). In multivariate analysis, old age [hazard ratio (HR) 1.029, 95% confidence interval (CI) 1.013-1.044, P = .001) was the independent adverse factor for overall survival, and RUNX1 mutation (HR 2.326, 95% CI 0.978-5.533, P = .056) had a trend toward inferior survival. In conclusion, our study showed a high frequency of FLT3, RUNX1, and IDH mutations in AML-M0, suggesting that these mutations played a role in the pathogenesis and served as potential therapeutic targets in this rare and unfavorable subtype of AML.
Introduction
Minimally differentiated acute myeloid leukemia (AML-M0) accounts for approximately 2% to 5% of all AMLs according to the French-American-British classification [1]. It frequently occurs in elderly patients and confers a poor prognosis [2]. Morphologically, the leukemic cells are large and agranular blasts mimicking lymphoblasts and negative for cytochemical reactions of myeloperoxidase (MPO), Sudan Black B, or nonspecific esterase [1]. The immunophenotypic characteristics of AML-M0 blasts are low expression of MPO, positive for at least one myeloid antigen (CD13, CD33, CD15, or CD11b), frequent expression of stem cell–associated antigens (CD34, HLA-DR, CD117), TdT, and occasional coexpression of lymphoid-associated antigens (CD7 or CD19) [1], [3]. As for cytogenetic abnormalities, despite that the incidence of abnormal, complex, or unbalanced chromosomal changes has been reported to be more frequent, there are no recurrent or specific cytogenetic abnormalities in AML-M0 [3]. In AML, gene mutations not only have an implication in molecular pathogenesis but also provide a prognostic relevance in addition to the cytogenetic subtypes [4].
Previous studies have focused on class I and class II mutations in AML-M0 [5], [6], [7], [8]. The development of AML was oftentimes caused by at least two-hit process mostly by class I and class II mutations. The class I mutation is defined by activating mutations of receptor tyrosine kinases and RAS signaling pathways, and the class II mutation is loss-of-function mutations of hematopoietic transcription factors [9]. RUNX1 mutation was the most common gene mutation described in AML-M0 [5]. FLT3 mutation was also reported as a recurrent gene mutation, whereas RAS and PTPN11 mutations were less frequent in AML-M0 [6], [7], [8]. Other gene mutations with prognostic relevance have not been studied comprehensively in AML-M0, including mutated genes of epigenetic regulators, such as IDH1, IDH2, TET2, DNMT3A, ASXL1, and EZH2 genes [10], [11], [12], [13].
We thus examined a wide spectrum of gene mutations, including class I genes of activated signaling pathways (FLT3-ITD, FLT3-TKD, C-FMS, KIT, N-RAS, K-RAS, PTPN11, and JAK2V617F), class II genes affecting hematopoietic transcription and differentiation (RUNX1, NPM1, and CEBPα), class III genes of epigenetic regulators (IDH1, IDH2, TET2, DNMT3A, MLL-PTD, ASXL1, and EZH2), and class IV genes of tumor suppressors (WT1 and TP53) from the bone marrow cells of patients with AML-M0 at the initial diagnosis. The status of gene mutations was also correlated with the clinicohematological features to determine their clinical relevance in patients with AML-M0.
Materials and Methods
Patients and Materials
From 1991 to 2010, a total of 67 patients fulfilling the diagnostic criteria of de novo AML-M0 at Chang Gung Memorial Hospital and Mackay Memorial Hospital was enrolled. The diagnosis of AML-M0 was made according to the French-American-British criteria: > 30% blasts in bone marrow, < 3% of blasts positive for MPO or Sudan Black B, and expression of at least one myeloid antigen [1]. Patients with leukemia blasts expressing specific lymphoid markers (cytoCD3, cytoCD79a, or cytoCD22) were excluded in this study. G-banding method was used for karyotypic analysis, and results were interpreted according to the International System for Human Cytogenetic Nomenclature. Cytogenetic categorization of favorable-, intermediate-, and adverse-risk groups was accorded to the criteria recommended by European LeukemiaNet (ELN) Guidelines [4]. A panel of monoclonal antibodies including myeloid-associated antigens (CD13, CD33, CD11b, CD14, CD15, and/or CD41a), lymphoid-associated antigens (CD7, CD19, cytoCD3, and cytoCD22 or cytoCD79a if necessary), as well as lineage-nonspecific antigens (CD34, CD117, HLA-DR, TdT, or CD56) was used to determine the immunophenotypes of leukemia cells. The study was approved by the Institutional Review Boards of Chang Gung Memorial Hospital and Mackay Memorial Hospital.
Cell Fractionation
The mononuclear cells were obtained from bone marrow samples by Ficoll-Hypaque density gradient centrifugation (1.077 g/ml; Amersham Pharmacia, Buckinghamshire, United Kingdom). The mononuclear cells were then cryopreserved in medium containing 10% DMSO and 20% FBS at − 70°C or in liquid nitrogen until test.
DNA, RNA Extraction, and cDNA Preparation
Genomic DNA was extracted from frozen mononuclear cells of bone marrow samples by using the QIAamp DNA mini kit (Qiagen GmbH, Hilden, Germany) following the manufacturer’s instructions. The TRIzol Reagent (Life Technology, Carlsbad, CA) was used to extract RNA that was reversely transcribed to cDNA with the Superscript II RNase H2 reverse transcriptase kit (Invitrogen Corporation, Carlsbad, CA).
Detection of Gene Mutations
FLT3-ITD, FLT3-TKD, KIT, C-FMS, K-RAS, N-RAS, JAKV617F, PTPN11, RUNX1, CEBPα, NPM1, TP53, WT1, IDH1, IDH2, TET2, DNMT3A, MLL-PTD, ASXL1, and EZH2 mutations were analyzed. The genomic DNA–polymerase chain reaction or reverse transcription–polymerase chain reaction assays followed by direct sequencing were used to detect FLT3-ITD [14], point mutations at tyrosine kinase domain of FLT3 (FLT3-TKD) [15], KIT and C-FMS mutations [16], point mutations at codons 12, 13, and 61 in exons 1 and 2 of N-RAS and K-RAS genes [17], exons 3 to 8 of RUNX1 mutations [18], CEBPα mutations [19], MLL-PTD [20], PTPN11, TET2, IDH1, IDH2, DNMT3A, and ASXL1 [21] as previously described. The detection of mutated genes in TP53, exons 1 to 3 and exons 7 to 9 of WT-1, JAK2V617F, and NPM1 was performed according to the previously reported methods of other investigators with some modification [22], [23], [24], [25]. The detection of EZH2 mutation was carried out using a self-designed and/or previously reported method, which was described in detail in the Supplementary Materials (Tables W1–W3 and Figure W1).
Statistical Analysis
Fisher exact test, χ2 analysis, and Wilcoxon rank-sum test were used whenever appropriate to make comparisons between groups. Estimates of survival were calculated according to the Kaplan-Meier method. Comparisons of estimated survival curves were analyzed by the log-rank test. For multivariate analysis, a Cox regression model was used to identify prognostic variables. Variables with P values of .2 or less in univariate analysis were included in the model. In all analysis, the P values were two-sided and considered statistically significant when values were lower than .05. Statistical analysis was carried out by SPSS version 17.0 (SPSS Inc, Chicago, IL).
Results
Patient Characteristics
The baseline characteristics of the 67 patients with AML-M0 are listed in the Supplemental Materials (Table W4). The median age was 49.0 years (range 0.3-97.9 years). Twenty-two of the 67 (32.8%) patients were female. The estimated median overall survival (OS) was 5.1 months (Figure W2A). Thirty-nine patients received standard AML induction protocol (60 mg/m2 daunomycin for 3 days and 150 mg/m2 cytarabine for 7 days for adults and TPOG-AML97A for children with an age younger than 18 years) [26]. Six patients (age 64.3-81.2 years) received low dose therapy including low dose cytarabine, hydroxyurea, or melphalan. Seventeen patients with a median age of 73.1 years old received supportive care only. Five (7.5%) patients were treated as acute lymphoblastic leukemia. Of the 39 patients who received standard AML therapy, 7 had early death during the induction therapy and 29 achieved complete remission yet with a relapse rate of 72.4% (21/29). Three patients from those who relapsed underwent hematopoietic stem cell transplantation (HSCT) after second complete remission. The median OS of the 39 patients who received chemotherapy was 11.1 months (Figure W2B). The median follow-up time for this cohort was 4.8 months (range 0-176.6 months). Fifty-nine patients died and eight were still alive.
Frequency and Types of Mutations in AML-M0
Of the 67 patients with de novo AML-M0, 79.1% (53/67) patients were found to have at least one gene mutation among the genes examined. The diagram of the gene mutation status in patients with AML-M0 is illustrated in Figure 1. The pairwise cooperativeness between gene mutations is depicted in Figure 2.
Figure 1.
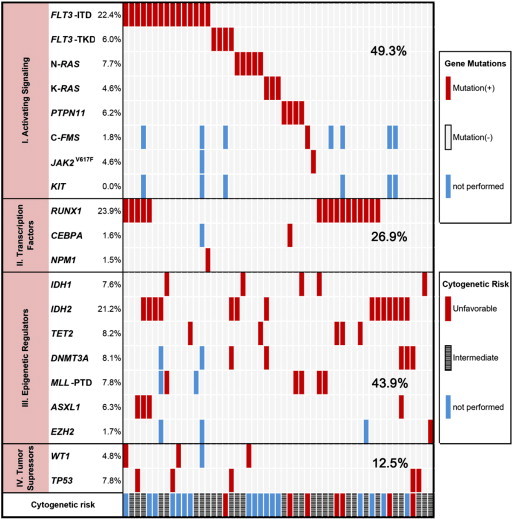
Diagram of AML-M0 patients with gene mutations. The gene mutation status, cooperating mutations, and cytogenetic risk groups in AML-M0 patients at the initial diagnosis are illustrated.
Figure 2.
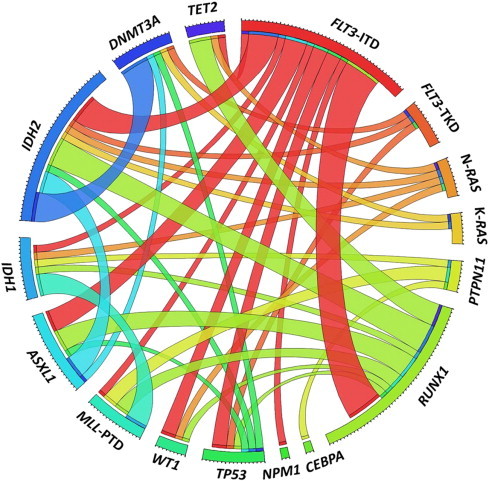
Pairwise cooperativeness between gene mutations in AML-M0 patients. A circus diagram depicts the pairwise cooperativeness of gene mutation in AML-M0 patients. The length of the arc represents the number of mutations in the first gene, and the width of the ribbon represents the number of patients with a mutation in the second gene.
Class I mutations
FLT3-ITD mutations were detected in 22.4% (15/67) and FLT3-TKD in 6.0% (4/67) patients. RAS mutations were present in 12.3% (8/65) patients, with three K-RAS and five N-RAS mutations. The frequency of PTPN11, C-FMS, and JAK2V617F mutations were 6.2% (4/65), 1.8% (1/56), and 1.6% (1/64), respectively. None had KIT mutation.
Class II mutation
A total of 17 RUNX1 mutations was detected in 23.9% (16/67) patients, including two nonsense mutations, seven frameshift, seven missense mutations, and one insertion mutation. One patient had CEBPα and NPM1 mutations each.
Class III mutations
IDH mutations were present in 28.8% (19/66) patients. All IDH mutations were missense mutations, with IDH1-R132 in five, IDH2-R140 in five, and IDH2-R172 in nine patients. TET2, DNMT3A, MLL-PTD, and ASXL1 mutations were detected less frequently, occurring in 8.2% (5/61), 8.1% (5/62), 7.8% (5/64), and 6.3% (4/64) patients, respectively. EZH2 mutation was detected in only 1 of 60 patients examined.
Class IV mutations
TP53 and WT1 occurred in 7.8% (5/64) and 4.8% (3/63) patients, respectively.
On the basis of the functional class of gene groups, 49.3% (33/67) patients had class I mutations that were mutually exclusive, 26.9% (18/67) had class II, 43.9% (29/66) had class III, and 12.5% (8/64) had class IV mutations. Twenty-four patients had gene mutations within a single class, with 12 in class I, 4 in class II, 7 in class III, and 1 in class IV.
Karyotypes and the Association with Gene Mutations
Cytogenetic data were available for 49 patients. Twenty-nine (59.2%) patients had normal karyotypes, 7 (14.3%) patients had a single abnormality, 3 (6.1%) patients had two abnormalities, and 10 (20.4%) patients had complex cytogenetic aberrations. Monosomal karyotypes were found in eight (16.3%) patients. Trisomy 21, trisomy 8, monosomy 8, and del(5q) were found in four, three, three, and two patients, respectively. Only one patient with isolated trisomy 13 had concomitant RUNX1 mutation. Thirty-eight patients (77.6%) were in the intermediate-risk group, 11 (22.4%) patients in the unfavorable-risk group, and none had favorable cytogenetics by the ELN criteria. All gene mutations examined were not associated with specific ELN cytogenetic risk groups or normal karyotypes.
Correlations of Gene Mutations with Clinicohematological Features of Patients with AML-M0
We correlated gene mutations with clinical parameters, including age, sex, hemoglobin level, platelet counts, white blood cell counts, percentages of circulating blasts and marrow blasts, and cytogenetic risk groups (Table 1). RUNX1 and IDH mutations were significantly associated with older age (median 68.0 vs 46.7 years for RUNX1, P = .011; 66.0 vs 44.1 for IDH, P = .016). Patients with MLL-PTD were younger than those without MLL-PTD (37.0 vs 54.0, P = .044). ASXL1 and DNMT3A mutations were associated with lower WBC counts (2.1 × 109/l vs 15.6 × 109/l for ASXL1-mutated and wild-type, P = .021; 2.3 × 109/l vs 15.9 × 109/l for DNMT3A-mutated and wild-type, P = .032). RAS mutations were associated with higher percentage of circuiting blasts (88.0% vs 49.3%, P = .012). Other gene mutations, including FLT3 mutations, had no correlation with any clinical features.
Table 1.
Clinical Characteristics of AML-M0 Patients with RUNX1, FLT3, IDH, and RAS Mutations.
|
RUNX1 (n = 67) |
FLT3-ITD or FLT3-TKD (n = 67) |
IDH1/IDH2 (n = 66) |
N-RAS/K-RAS (n = 65) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | Wild type | P | Mutation | Wild type | P | Mutation | Wild type | P | Mutation | Wild type | P | |
| Number of patients | 16 | 51 | 19 | 48 | 19 | 47 | 8 | 57 | ||||
| Age (years)⁎ | 68.0 (5.5-97.9) | 46.7 (0.3-84.2) | .011 | 46.7 (11.0-75.8) | 54.3 (0.3-97.9) | .461 | 66.0 (5.5-84.2) | 44.1 (0.3-97.9) | .016 | 49.1 (16.0-73.5) | 49.0 (0.3-97.9) | .905 |
| Sex (male/female) | 4/12 | 18/33 | .550 | 7/12 | 15/33 | .660 | 7/12 | 15/32 | .701 | 3/5 | 19/38 | 1.000 |
| Hemoglobin (g/l) ⁎ | 77 (40-110) | 74 (38-124) | .600 | 68 (40-117) | 77 (38-124) | .519 | 75 (40-121) | 75 (38-124) | .756 | 89 (38-124) | 75 (40-127) | .340 |
| Platelet (× 109/l) ⁎ | 4.5 (0.8-13.9) | 3.2 (0.1-59.8) | .394 | 4.4 (0.8-13.5) | 3.0 (0.2-59.8) | .681 | 3.3 (0.1-19.9) | 3.0 (0.2-59.8) | .851 | 2.8 (.2-59.8) | 3.3 (0.1-23.6) | .761 |
| WBC (× 109/l) ⁎ | 22.9 (1.1-187.2) | 10.7 (0.6-397.8) | .595 | 15.9 (0.6-204.6) | 13.0 (0.7-379.2) | .761 | 5.4 (0.6-63.6) | 15.2 (0.7-379.2) | .184 | 31.5 (5.4-156.6) | 10.7 (0.6-379.2) | .123 |
| Circulating blasts (%)⁎ | 61.8 (0-98.2) | 53.0 (0-99.3) | .899 | 63 (1-98.0) | 53.5 (0-99.3) | .882 | 46.0(0-98.0) | 55.0 (0-99.3) | .817 | 88.0 (30.0-98.5) | 49.3 (0-99.3) | .012 |
| Marrow blasts (%) ⁎ | 88.4 (76.0-99.0) | 90.1 (29.4-98.8) | .857 | 91.8 (72.2-99.0) | 88.5 (29.4-98.4) | .557 | 88.2 (43.5-95.3) | 89.4 (29.4-99.0) | .954 | 94.6 (69.2-96.0) | 88.9 (29.4-99.0) | .104 |
| Karyotype | .708 | .252 | .246 | 1.000 | ||||||||
| Favorable | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| Intermediate | 10 | 28 | 11 | 27 | 12 | 26 | 3 | 35 | ||||
| Poor | 2 | 9 | 1 | 10 | 1 | 10 | 1 | 9 | ||||
| Trisomy 13 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | ||||
| Monosomy | 1 | 7 | 2 | 6 | 1 | 7 | 0 | 8 | ||||
| EFS† | 1.9 (0-5.2) | 2.2 (0-5.5) | .133 | 5.3 (4.1-6.5) | 1.9 (0.8-3.0) | .207 | 1.0 (0.5-1.5) | 3.0 (0-6.1) | .863 | 1.0 (0.7-1.3) | 3.0 (0-6.0) | .139 |
| OS† | 2.6 (0-6.2) | 5.3 (0-12.8) | .168 | 15.2 (6.8-23.6) | 3.0 (0.2-5.8) | .171 | 5.1 (0.4-9.8) | 5.3 (0-13.2) | .897 | 1.0 (0-3.1) | 6.3 (0.8-11.8) | .096 |
Values are expressed as medians (range).
Values are expressed as medians (95% CI).
Outcome Analysis
We assessed the impact of cytogenetic risk group and FLT3, IDH, RUNX1, RAS, ASXL1, DNMT3A, TET2, and MLL-PTD mutations on OS and event-free survival (EFS; Table 2). Less prevalent gene mutations were excluded from statistical analysis. Cytogenetics had prognostic significance, with a median OS of 13.8 months [95% confidence interval (CI) 3.2-24.4 months] and 4.4 months (95% CI 1.6-7.2 months) for intermediate- and unfavorable-risk groups, respectively (P = .043). The mutational status of FLT3 (P = .171), IDH (P = .897), RUNX1 (P = .168), RAS (P = .096), ASXL1 (P = .760), TET2 (P = .076), DNMT3A (P = .996), or MLL-PTD (P = 0.247) did not have significant influence on OS or EFS by the log rank test (Figure W3). When the gene mutations were categorized according to their functional class groups, no impact on OS or EFS was observed with regard to the functional class groups.
Table 2.
Univariate and Multivariate Analyses with Respect to EFS and OS.
| EFS |
OS |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate |
Multivariate |
Univariate |
Multivariate |
|||||||||
| HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | |
| Age | 1.015 | 1.005-1.024 | .003 | 1.016 | 1.000-1.033 | .055 | 1.019 | 1.008-1.029 | < .001 | 1.029 | 1.013-1.044 | .001 |
| Karyotype risk | 1.613 | 0.799-3.253 | .182 | 1.250 | 0.515-3.036 | .622 | 2.059 | 1.004-4.224 | .049 | 1.735 | 0.712-4.227 | .225 |
| Platelet counts | 0.976 | 0.941-1.012 | .191 | 0.967 | 0.894-1.047 | .413 | 0.969 | 0.930-1.009 | .124 | 0.923 | 0.840-1.014 | .093 |
| Marrow blasts (%) | 1.007 | 0.991-1.024 | .392 | 1.015 | 0.997-1.033 | .108 | 1.001 | 0.980-1.022 | .956 | |||
| FLT3 mutation | 0.693 | 0.389-1.234 | .213 | 0.665 | 0.369-1.201 | .176 | 0.414 | 0.163-1.055 | .065 | |||
| RUNX1 mutation | 1.549 | 0.867-2.768 | .140 | 2.365 | 0.946-5.915 | .066 | 1.508 | 0.835-2.724 | .173 | 2.326 | 0.978-5.533 | .056 |
| IDH mutation | 0.950 | 0.531-1.702 | .864 | 0.963 | 0.540-1.716 | .897 | ||||||
| RAS mutation | 1.767 | 0.816-3.824 | .149 | 2.405 | 0.679-8.518 | .174 | 1.893 | 0.877-4.087 | .104 | 1.728 | 0.500-5.975 | .388 |
| MLL-PTD | 0.492 | 0.177-1.366 | .173 | 0.525 | 0.143-1.921 | .300 | 0.553 | 0.199-1.536 | .256 | |||
| ASXL1 mutation | 0.898 | 0.279-2.888 | .857 | 0.835 | 0.260-2.681 | .762 | ||||||
| DNMT3A mutation | 1.375 | 0.425-4.442 | .595 | 1.002 | 0.399-2.518 | .996 | ||||||
| P53 mutation | 2.106 | 0.747-5.936 | .159 | 0.942 | 0.263-3.373 | .927 | 1.762 | 0.693-4.481 | .234 | |||
| PTPN11 mutation | 0.61 | 0.190-1.960 | .407 | 0.707 | 0.221-2.265 | .559 | ||||||
| TET2 mutation | 0.445 | 0.174-1.142 | .092 | 1.183 | 0.297-4.716 | .812 | 0.437 | 0.169-1.128 | .087 | 1.308 | 0.314-5.441 | .712 |
| Class I mutation | 0.797 | 0.481-1.320 | .378 | 0.714 | 0.426-1.196 | .200 | ||||||
| Class II mutation | 1.492 | 0.850-2.617 | .163 | 1.447 | 0.819-2.557 | .204 | ||||||
| Class III mutation | 1.063 | 0.639-1.769 | .813 | 1.150 | 0.686-1.927 | .595 | ||||||
| Class IV mutation | 1.985 | 0.884-4.456 | .097 | 1.712 | 0.804-3.645 | .163 | ||||||
To assess whether the gene mutations had independent prognostic values in patients with AML-M0 in context of other clinical and molecular parameters, we performed multivariate analysis for FLT3, RUNX1, RAS, TP53, and TET2 mutations adjusting for age, platelet counts, percentage of marrow blasts, and/or ELN cytogenetic risk groups (Table 2). The result showed that age [hazard ratio (HR) 1.029, 95% CI 1.013-1.044, P = .001] was the most significant adverse factor for OS, and patients with RUNX1 mutation (HR 2.326, 95% CI 0.978-5.533, P = .056) had a trend toward an inferior survival. For EFS, age (HR 1.016, 95% CI 1.000-1.033, P = .055) was the independent unfavorable risk factor after adjusting for cytogenetic risk group, platelet counts, RAS, MLL-PTD, TET2, and TP53 gene mutation status.
Discussion
Previous studies reporting the clinical features or gene mutation patterns in patients with AML-M0 were mainly from the western population and a few from the Japanese population [6]. In this study, we examined 20 gene mutations in 67 patients with AML-M0 from the Taiwanese ethnicity, and it showed a wider range of gene mutations in AML-M0 compared to the western and other Asian population. It has been described that RUNX1 mutations were common in AML-M0 with reported frequencies ranging from 12.7% to 46% [5], [6], [27], [28] and up to 65.4% in those with normal karyotypes [29]. Another common mutation in patients with AML-M0 was FLT3-ITD, with a frequency of 22% to 29% [6], [7]. We found that FLT3-ITD, RUNX1, IDH1, IDH2, and RAS were the recurrent mutations with frequencies between 10% and 30% in our series, and our results of FLT3-ITD and RUNX1 mutations were mostly in line with previous studies. However, the frequency of RUNX1 mutation seemed to be slightly higher in our cohort compared to the Japanese population (23.9% vs 15.7%) [6]. The frequency of RAS (12.3%) and PTPN11 (6.2 %) mutations was slightly higher in our series compared with the study of Roumier et al. [7], which showed that RAS and PTPN11 mutations occurred less frequently in patients with AML-M0. Of note, we found a high occurrence of IDH2 (21.2%), which has not been previously described. Other gene mutations of epigenetic regulators including TET2, DNMT3A, MLL-PTD, and ASXL1 mutations occurred less frequently and EZH2 mutation was rare. If we took the gene functional groups into consideration, approximately half of the patients with AML-M0 had class I gene mutations (49.3%) and class II gene mutations involving hematopoietic differentiation occurred in one fourth of patients, suggesting that the receptor tyrosine kinase/RAS signaling was the most important pathway involved in AML-M0, having FLT3 mutations being the main causal factor. Mutations of epigenetic regulator genes (class III) detected in about 40% of patients also played an important role in the leukemogenesis of AML-M0.
In univariate analysis, age was the most significant adverse factor for OS and EFS. Unfavorable cytogenetic subgroup also conferred a poor risk factor for OS. Most of the genes with high occurrence in the present series have been reported to be poor prognostic molecular markers in patients with AML, especially in the cytogenetically normal group, including FLT3-ITD [30], [31], IDH2 [12], [32], and RUNX1 mutations [29], [33]. However, we did not observe such correlation in our patients. NPM1 mutation occurred in about 50% of AML patients with normal karyotypes and was associated with a favorable survival [34], [35]. About 60% of the patients had normal karyotypes, but only one patient had NPM1 mutation in our study. The poor outcome of our AML-M0 patients might be attributed partly to the near absence of NPM1 mutation along with the high occurrence of FLT3-ITD, which is a poor prognostic factor in normal karyotype AML. The lack of prognostic impact of FLT3 mutations in the present study might be attributed to the limited number of AML-M0 patients with general short survival, which avert a confirmed conclusion. IDH2 had a higher occurrence than IDH1 in AML, but the location of IDH2 mutation was more frequent on R140 than on R172 in the previous studies, in which M0 accounted for less than 5% of the studied population [12], [32], [36], [37], [38]. Interestingly, we observed that IDH2-R172 occurred more frequently than IDH2-R140 or IDH1-R132 in the present AML-M0 series. IDH2 mutation on R172 has been reported to confer a poorer outcome compared to IDH2-R140 or IDH1-R132 in AML [36], [38], [39]. The number of each subtype of IDH mutants was very small to make a meaningful statistic analysis. Findings from the multivariate analysis showed consistency with the previous study on that RUNX1 mutation was the poor prognostic factor for OS and EFS [29].
Approximately half of the patients had multiple cooperating gene mutations. More than 80% of patients harboring mutations of epigenetic regulator genes had other coexisted gene mutations. Although ASXL1 mutations occurred in only four patients with AML-M0, remarkably, three of them were associated with both RUNX1 and FLT3-ITD mutations. Likewise, all of the five patients with MLL-PTD carried other mutated genes. Our findings supported a multiple-hit model of leukemogenesis in AML [40]. Previous studies have shown that RUNX1 mutations frequently coexisted with FLT3-ITD or FLT3-TKD [6], trisomy 13 (the locus of the FLT3 gene), or FLT3 overexpression [27], [28], [29]. We found only one patient harboring both RUNX1 mutation and trisomy 13 and five patients with coexisting RUNX1 and FLT3-ITD mutations. The underlying mechanism of their cooperating roles in leukemogenesis merits further investigation.
Most AML-M0 patients succumb to the disease despite current standard treatment. For an aggressive AML subtype as AML-M0, having HSCT early in the course of the disease had improved the outcome in some small series [41], [42]. We did not observe a significantly prolonged survival in AML-M0 patients who received HSCT in this study. The small number of patients who received HSCT and the delayed timing for HSCT in our cohort may effectuate the lack of significant benefit of HSCT in our patients. Complete gene mutation profiling for AML-M0 patients at the diagnosis may further help in identifying biologic risk factors and potential therapeutic targets. The relevance of gene mutations in predicting therapeutic response of myeloid malignancies to targeted agents has not been well established. Flt3 antagonists combined with chemotherapy or hypomethylating agents have been reported to have impact on response rates in a phase II study [43]. Potential links between gene mutations and the treatment responses to hypomethylating agents have been reported in a few small series. Itzykson et al. reported that patients with mutated TET2 and favorable cytogenetics had a higher response rate to azacitidine compared to patients with wild-type TET2 in myelodysplastic syndromes and low blast count AML [44]. Metzeler et al. reported a higher complete remission rate in AML patients with DNMT3A mutations treated with decitabine [45]. TET2 and/or DNMT3A mutations were found to be independent response predictor to methyltransferase inhibitors in patients with myelodysplastic syndrome or its related disorders [46]. However, these results have not been confirmed in randomized clinical trials. IDH also serves as a potential therapeutic target in AML. Chaturvedi et al. recently reported that a small molecular inhibitor targeting mutant IDH1 induced apoptosis and decreased colony formation in methylcellulose of IDH1-mutant human primary bone marrow cells [47]. Our study reveals the complex and heterogeneous molecular aberrations in AML-M0. As coexistence of gene mutations occurs frequently, combined therapy through multiple targeting might be the reasonable approach in future studies.
In summary, we analyzed a broad spectrum of known mutated genes involved in myeloid neoplasms from 67 patients with de novo AML-M0 at diagnosis. To the best of our knowledge, this is the most comprehensive study regarding the gene mutation patterns in AML-M0. We found that AML-M0 was characterized by high frequency of FLT3-ITD, RUNX1, and IDH mutations. In addition to the signaling pathway, we further demonstrated that AML-M0 was frequently associated with mutations of epigenetic regulator genes, occurring in more than 40% of patients. Other than old age, RUNX1 mutation was associated with a trend of inferior survival by multivariate analysis, while FLT3, IDH, and other gene mutations did not have impact on the outcome. Cooperation of multiple mutations in different classes of genes was common. Our findings suggested that AML-M0 is a complex and heterogeneous subtype of AML in terms of molecular aberrations. The high frequency of gene mutations in epigenetic modifiers implies that epigenetic deregulation, which frequently cooperates with other gene mutations, may play an important role in the pathogenesis of AML-M0. Further studies on how epigenetic regulator mutations interact with mutated genes affecting cell proliferation and/or differentiation in AML-M0 are warranted. Our findings also suggest the potential implication of combined therapies with targeted agents in this rare and unfavorable subtype of AML.
Acknowledgments
This work was supported by a grant from the National Health Research Institute (Miaoli,Taiwan; NHRI-EX96-9434SI), a grant from the National Science Council (Taipei, Taiwan; NSC100-2314-B-182-023-MY3), a grant from the Department of Health (Taipei, Taiwan; DOH100-TD-C-111-006), grants from Mackay Memorial Hospital (Taipei, Taiwan; MMH-E-99009 and MMH-E-100009), and grants from Chang Gung Memorial Hospital (Taipei, Taiwan; OMRPG380031 and OMRPG3C0021). The authors declare that they have no competing interest. The authors also thank Ting-Yu Huang and Yu-Feng Wang for their secretarial assistance and Judy Sheu for her kind help in English editing.
Footnotes
This article refers to supplementary materials, which are designated by Tables W1–W4 and Figures W1–W3 and are available online at www.neoplasia.com.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.06.002.
Appendix A. Supplementary Data
The following are the supplementary data related to this article.
Supplementary Materials.
References
- 1.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, Sultan C. Proposal for the recognition of minimally differentiated acute myeloid leukemia (AML-M0) Br J Haematol. 1991;78:325–329. doi: 10.1111/j.1365-2141.1991.tb04444.x. [DOI] [PubMed] [Google Scholar]
- 2.Stasi R, Delpoeta G, Venditti A, Masi M, Stipa E, Dentamaro T, Cox C, Dallapiccola B, Papa G. Analysis of treatment failure in patients with minimally differentiated acute myeloid leukemia (AML-M0) Blood. 1994;83:1619–1625. [PubMed] [Google Scholar]
- 3.Béné M-C, Bernier M, Casasnovas RO, Castoldi G, Doekharan D, Van Der Holt B, Knapp W, Lemež P, Ludwig W-D, Matutes E. Acute myeloid leukaemia M0: haematological, immunophenotypic and cytogenetic characteristics and their prognostic significance: an analysis in 241 patients. Br J Haematol. 2001;113:737–745. doi: 10.1046/j.1365-2141.2001.02801.x. [DOI] [PubMed] [Google Scholar]
- 4.Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 5.Roumier C, Eclache V, Imbert M, Davi F, MacIntyre E, Garand R, Talmant P, Lepelley P, Lai JL, Casasnovas O. M0 AML, clinical and biologic features of the disease, including AML1 gene mutations: a report of 59 cases by the Groupe Français d'Hématologie Cellulaire (GFHC) and the Groupe Français de Cytogénétique Hématologique (GFCH) Blood. 2003;101:1277–1283. doi: 10.1182/blood-2002-05-1474. [DOI] [PubMed] [Google Scholar]
- 6.Matsuno N, Osato M, Yamashita N, Yanagida M, Nanri T, Fukushima T, Motoji T, Kusumoto S, Towatari M, Suzuki R. Dual mutations in the AML1 and FLT3 genes are associated with leukemogenesis in acute myeloblastic leukemia of the M0 subtype. Leukemia. 2003;17:2492–2499. doi: 10.1038/sj.leu.2403160. [DOI] [PubMed] [Google Scholar]
- 7.Roumier C, Lejeune-Dumoulin S, Renneville A, Goethgeluck AS, Philippe N, Fenaux P, Preudhomme C. Cooperation of activating Ras/rtk signal transduction pathway mutations and inactivating myeloid differentiation gene mutations in M0 AML: a study of 45 patients. Leukemia. 2006;20:433–436. doi: 10.1038/sj.leu.2404097. [DOI] [PubMed] [Google Scholar]
- 8.Silva F.P., Almeida I, Morolli B, Brouwer-Mandema G, Wessels H, Vossen R, Vrieling H, Marijt EWA, Valk PJM, Kluin-Nelemans HC. Genome wide molecular analysis of minimally differentiated acute myeloid leukemia. Haematologica. 2009;94:1546–1554. doi: 10.3324/haematol.2009.009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dash A, Gilliland DG. Molecular genetics of acute myeloid leukaemia. Best Pract Res Clin Haematol. 2001;14:49–64. doi: 10.1053/beha.2000.0115. [DOI] [PubMed] [Google Scholar]
- 10.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116:4086–4094. doi: 10.1182/blood-2010-05-283291. [DOI] [PubMed] [Google Scholar]
- 12.Boissel N, Nibourel O, Renneville A, Gardin C, Reman O, Contentin N, Bordessoule D, Pautas C, de Revel T, Quesnel B. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association Group. J Clin Oncol. 2010;28:3717–3723. doi: 10.1200/JCO.2010.28.2285. [DOI] [PubMed] [Google Scholar]
- 13.Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, Curfman J, Holland KB, Schwind S, Whitman SP. TET2 mutations improve the New European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shih LY, Huang CF, Wang PN, Wu JH, Lin TL, Dunn P, Kuo MC. Acquisition of FLT3 or N-Ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia. 2004;18:466–475. doi: 10.1038/sj.leu.2403274. [DOI] [PubMed] [Google Scholar]
- 15.Shih LY, Huang CF, Wu JH, Wang PN, Lin TL, Dunn P, Chou MC, Kuo MC, Tang CC. Heterogeneous patterns of FLT3 Asp835 mutations in relapsed de novo acute myeloid leukemia: a comparative analysis of 120 paired diagnostic and relapse bone marrow samples. Clin Cancer Res. 2004;10:1326–1332. doi: 10.1158/1078-0432.ccr-0835-03. [DOI] [PubMed] [Google Scholar]
- 16.Shih LY, Liang DC, Huang CF, Chang YT, Lai CL, Lin TH, Yang CP, Hung IJ, Liu HC, Jaing TH. Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia. 2008;22:303–307. doi: 10.1038/sj.leu.2404995. [DOI] [PubMed] [Google Scholar]
- 17.Liang DC, Shih LY, Fu JF, Li HY, Wang HI, Hung IJ, Yang CP, Jaing TH, Chen SH, Liu HC. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer. 2006;106:950–956. doi: 10.1002/cncr.21687. [DOI] [PubMed] [Google Scholar]
- 18.Kuo MC, Liang DC, Huang CF, Shih YS, Wu JH, Lin TL, Shih LY. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426–1431. doi: 10.1038/leu.2009.48. [DOI] [PubMed] [Google Scholar]
- 19.Shih LY, Huang CF, Lin TL, Wu JH, Wang PN, Dunn P, Kuo MC, Tang TC. Heterogeneous patterns of CEBPα mutation status in the progression of myelodysplastic syndrome and chronic myelomonocytic leukemia to acute myelogenous leukemia. Clin Cancer Res. 2005;11:1821–1826. doi: 10.1158/1078-0432.CCR-04-1932. [DOI] [PubMed] [Google Scholar]
- 20.Shih LY, Liang DC, Fu JF, Wu JH, Wang PN, Lin TL, Dunn P, Kuo MC, Tang TC, Lin TH. Characterization of fusion partner genes in 114 patients with de novo acute myeloid leukemia and MLL rearrangement. Leukemia. 2006;20:218–223. doi: 10.1038/sj.leu.2404024. [DOI] [PubMed] [Google Scholar]
- 21.Liang DC, Liu HC, Yang CP, Jaing TH, Hung IJ, Yeh TC, Chen SH, Hou JY, Huang YJ, Shih YS. Cooperating gene mutations in childhood acute myeloid leukemia with special reference on mutations of ASXL1, TET2, IDH1, IDH2, and DNMT3A. Blood. 2013;121:2988–2995. doi: 10.1182/blood-2012-06-436782. [DOI] [PubMed] [Google Scholar]
- 22.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19:1405–1413. doi: 10.1200/JCO.2001.19.5.1405. [DOI] [PubMed] [Google Scholar]
- 23.King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms' tumor gene WT1 in leukemias. Blood. 1996;87:2171–2179. [PubMed] [Google Scholar]
- 24.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 25.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 26.Liang DC, Chang TT, Lin KH, Lin DT, Lu MY, Chen SH, Liu HC, Lin MT, Lee MT, Shu SG. Improved treatment results for childhood acute myeloid leukemia in Taiwan. Leukemia. 2006;20:136–141. doi: 10.1038/sj.leu.2403979. [DOI] [PubMed] [Google Scholar]
- 27.Dicker F, Haferlach C, Kern W, Haferlach T, Schnittger S. Trisomy 13 is strongly associated with AML1/RUNX1 mutations and increased FLT3 expression in acute myeloid leukemia. Blood. 2007;110:1308–1316. doi: 10.1182/blood-2007-02-072595. [DOI] [PubMed] [Google Scholar]
- 28.Silva F.P., Lind A, Brouwer-Mandema G, Valk P.J., Giphart-Gassler M. Trisomy 13 correlates with RUNX1 mutation and increased FLT3 expression in AML-M0 patients. Haematologica. 2007;92:1123–1126. doi: 10.3324/haematol.11296. [DOI] [PubMed] [Google Scholar]
- 29.Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, Haferlach C, Haferlach T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117:2348–2357. doi: 10.1182/blood-2009-11-255976. [DOI] [PubMed] [Google Scholar]
- 30.Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, Asou N, Kuriyama K, Jinnai I, Shimazaki C. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- 31.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 32.Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, Späth D, Kayser S, Zucknick M, Götze K. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 33.Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, Huang CF, Lee FY, Liu MC, Yao M. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114:5352–5361. doi: 10.1182/blood-2009-05-223784. [DOI] [PubMed] [Google Scholar]
- 34.Döhner K, Schlenk RF, Habdank M, Scholl C, Rücker FG, Corbacioglu A, Bullinger L, Fröhling S, Döhner H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–3746. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 35.Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, Haferlach T, Hiddemann W, Falini B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–3739. doi: 10.1182/blood-2005-06-2248. [DOI] [PubMed] [Google Scholar]
- 36.Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. 2011;25:246–253. doi: 10.1038/leu.2010.267. [DOI] [PubMed] [Google Scholar]
- 38.Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, Gale RE. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. 2011;118:409–412. doi: 10.1182/blood-2010-12-322479. [DOI] [PubMed] [Google Scholar]
- 39.Boissel N, Nibourel O, Renneville A, Huchette P, Dombret H, Preudhomme C. Differential prognosis impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. 2011;117:3696–3697. doi: 10.1182/blood-2010-11-320937. [DOI] [PubMed] [Google Scholar]
- 40.Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P, Preudhomme C. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008;22:915–931. doi: 10.1038/leu.2008.19. [DOI] [PubMed] [Google Scholar]
- 41.Cascavilla N, Melillo L, D'Arena G, Greco MM, Carella AM, Sajeva MR, Perla G, Matera R, Minervini MM, Carotenuto M. Minimally differentiated acute myeloid leukemia (AML M0): clinico-biological findings of 29 cases. Leuk Lymphoma. 2000;37:105–113. doi: 10.3109/10428190009057633. [DOI] [PubMed] [Google Scholar]
- 42.Kuzmanovic M, Rasovic N, Bunjevacki G, Scekic-Guc M, Bunjevacki V. Minimally differentiated acute myeloid leukemia (AML-M0) in children: a single center experience. Med Pediatr Oncol. 2000;34:364–365. doi: 10.1002/(sici)1096-911x(200005)34:5<364::aid-mpo11>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 43.Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, Pierce S, Daver N, Garcia-Manero G, Faderl S. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121:4655–4662. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, Quesnel B, Vey N, Gelsi-Boyer V, Raynaud S. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–1152. doi: 10.1038/leu.2011.71. [DOI] [PubMed] [Google Scholar]
- 45.Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, Blum W, Marcucci G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26:1106–1107. doi: 10.1038/leu.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, Sugimoto Y, Szpurka H, Makishima H, O'Keefe CL. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28:78–87. doi: 10.1038/leu.2013.269. [DOI] [PubMed] [Google Scholar]
- 47.Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Görlich K, Wichmann M, Schwarzer A, Preller M, Thol F. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013;122:2877–2887. doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials.