

Abstract
Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations and epidermal growth factor receptor (EGFR) family signaling are drivers of tumorigenesis in pancreatic ductal adenocarcinoma (PDAC). Previous studies have demonstrated that combinatorial treatment of PDAC xenografts with the mitogen-activated protein kinase–extracellular-signal-regulated kinase (ERK) kinase1/2 (MEK1/2) inhibitor trametinib and the dual EGFR/human epidermal growth factor receptor 2 (HER2) inhibitor lapatinib provided more effective inhibition than either treatment alone. In this study, we have used the therapeutic antibodies, panitumumab (specific for EGFR) and trastuzumab (specific for HER2), to probe the role of EGFR and HER2 signaling in the proliferation of patient-derived xenograft (PDX) tumors. We show that dual anti-EGFR and anti-HER2 therapy significantly augmented the growth inhibitory effects of the MEK1/2 inhibitor trametinib in three different PDX tumors. While significant growth inhibition was observed in both KRAS mutant xenograft groups receiving trametinib and dual antibody therapy (tumors 366 and 608), tumor regression was observed in the KRAS wild-type xenografts (tumor 738) treated in the same manner. Dual antibody therapy in conjunction with trametinib was equally or more effective at inhibiting tumor growth and with lower apparent toxicity than trametinib plus lapatinib. Together, these studies provide further support for a role for EGFR and HER2 in pancreatic cancer proliferation and underscore the importance of therapeutic intervention in both the KRAS–rapidly accelerated fibrosarcoma kinase (RAF)–MEK–ERK and EGFR-HER2 pathways to achieve maximal therapeutic efficacy in patients.
Abbreviations: EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FDA, Food and Drug Administration; HER2, human epidermal growth factor receptor 2; KRAS, Kirsten rat sarcoma viral oncogene homolog; MEK1/2, mitogen-activated protein kinase–ERK kinase1/2; PDAC, pancreatic ductal adenocarcinoma; PDX, patient-derived xenograft; pEGFR, phospho-EGFR; pHER2, phospho-HER2; pJNK, phospho–c-Jun N-terminal kinase; pRTK, phospho–receptor tyrosine kinase; RAF, rapidly accelerated fibrosarcoma kinase; Tra + P + T, trametinib plus panitumumab plus trastuzumab
Introduction
Pancreatic cancer is the 10th most common US cancer and the 4th leading cause of cancer death in the United States. The 5-year survival from this disease has barely improved from 2% to 6% in the last 40 years [1]. These poor outcomes coupled with a projected increase in disease incidence of 55% in the next 20 years highlight the pressing need for improved systemic therapies for this disease [2].
Central to the failure of existing treatment strategies is the marked genetic heterogeneity and resultant molecular signaling complexity observed in pancreatic cancers [3], [4], [5]. Despite this diversity, a conserved sequence of acquired genetic alterations is observed in a majority of pancreatic ductal epithelial cells during their malignant transformation to adenocarcinoma. Activating mutations of the Kirsten rat sarcoma viral oncogene homolog (KRAS) have been reported in 75% to 95% of pancreatic ductal adenocarcinomas (PDAC), and the importance of this oncogene in pancreatic tumorigenesis has been demonstrated in genetically engineered mouse models [6], [7], [8], [9], [10], [11]. Developing strategies to target RAS pathway signaling at the preclinical and clinical levels has been emphasized as a high priority in pancreatic cancer research [12]. Small molecule inhibitors of both rapidly accelerated fibrosarcoma kinase (RAF) and mitogen-activated protein kinase (MAPK)–extracellular-signal-regulated (ERK) kinase1/2 (MEK1/2) within the RAS-RAF-MEK-ERK signaling cascade are in clinical development with promising early results in mutant RAS/RAF-driven tumors [13], [14].
The epidermal growth factor receptor (EGFR) families have also been identified as promising targets for PDAC treatment. The observation that 40% to 70% of PDAC tumors overexpress EGFR led to the testing and US Food and Drug Administration (FDA) approval of erlotinib, an EGFR-specific tyrosine kinase inhibitor, in combination with gemcitabine for the treatment of patients with advanced disease [15], [16]. Additionally, human epidermal growth factor receptor 2 (HER2) has been identified as overexpressed in approximately 20% of PDAC tumors [17]. Its overexpression has been associated with worse patient outcomes, and anti-HER2 therapy has exhibited therapeutic synergism with anti-EGFR agents in pancreatic xenografts expressing moderate and even low levels of HER2 [18], [19], [20].
Despite promising early results with RAS pathway inhibitors and anti-EGFR family therapy, the genetic heterogeneity, signaling redundancy, and plasticity of pancreatic tumor cells suggest that monotherapy or single pathway treatment strategies are unlikely to result in significant and durable responses [5]. Importantly, preclinical studies evaluating combination therapy with EGFR and RAS pathway inhibitors in pancreatic cancer xenografts have shown promising results [21], [22], [23].
To develop and test rational approaches to therapy for PDAC, we have established a patient-derived xenograft (PDX) model of pancreatic cancer with orthotopic implantation of tumors into immunocompromised mice. Genetic and molecular profiling of the initial 15 PDXs in this tumor bank revealed a high frequency of tumors with KRAS mutations and activated EGFR and a smaller cohort with activated HER2 [24]. In this preclinical model, we evaluated combination therapy with trametinib (GSK1120212), a selective allosteric inhibitor of MEK1/2 [16], [25], [26], plus lapatinib, an inhibitor of both EGFR and HER2 receptor tyrosine kinase (RTK) activity [27], [28], [29]. We reported that trametinib-mediated tumor growth inhibition was significantly enhanced by concomitant lapatinib therapy in four of five patient-derived tumors assessed [23].
While this combination therapy was highly efficacious and considered for a clinical trial, there were concerns about potential patient toxicity as a recent phase I/Ib study evaluating trametinib in combination with erlotinib in patients with non–small cell lung and pancreatic cancers reported treatment-limiting gastrointestinal toxicity [30]. We therefore sought to identify alternate agents targeting EGFR and HER2 to use in combination with trametinib.
In this study, we have used two well-studied therapeutic antibodies, panitumumab (specific for EGFR) and trastuzumab (specific for HER2), to probe the role of EGFR and HER2 signaling in the proliferation of PDX tumors bearing mutant and wild-type KRAS alleles. We show that dual anti-EGFR and anti-HER2 therapy significantly augmented the growth inhibitory effects of the MEK1/2 inhibitor trametinib in different PDX tumors. Particularly noteworthy was the observation that two different tumors bearing wild-type KRAS alleles were particularly sensitive to trametinib plus dual antibody therapy exhibiting significant tumor regression. In vitro and in vivo studies confirmed that treatment with panitumumab or trastuzumab effectively inhibited the epidermal growth factor (EGF)–dependent autophosphorylation of EGFR and HER2, respectively. These studies using PDX tumors support the role for EGFR and HER2 in pancreatic cancer proliferation and underscore the importance of therapeutic intervention in both the KRAS-RAF-MEK-ERK and EGFR-HER2 pathways to achieve maximal therapeutic efficacy in vivo.
Materials and Methods
Orthotopic PDXs and Cell Lines
PDAC cell line and tumor samples MAD 08-608, 08-738, 09-366, and 10-215 (T608, T738, T366, and T215, respectively) were generated from fresh human tumor specimens collected with the approval of the University of Virginia’s Institutional Review Board and Animal Care and Use Committee following informed consent from each patient as previously described [23], [31]. Six- to 8-week-old male athymic nude mice were used for all in vivo experiments. For in vitro experiments, cells were maintained in Roswell Park Memorial Institute 1640 medium containing 10% FBS and 1% penicillin/streptomycin and cultured in a humidified (37 °C, 5% CO2) incubator. Fresh cell aliquots were thawed, propagated, and used for experiments every 6 months. Cell lines were authenticated in 2010 by the University of Virginia Biomedical Research Facility as previously described [23].
MEK1/2 Inhibitor, EGFR/HER2 Inhibitor, and Antibodies
Trametinib (GSK1120212), a selective allosteric inhibitor of MEK1/2 [16], [25], [26], and lapatinib, a dual tyrosine kinase inhibitor of EGFR and HER2, were kindly provided by GlaxoSmithKline (Brentford, United Kingdom). Panitumumab (Amgen, Thousand Oaks, CA) is a fully humanized, anti-EGFR monoclonal antibody that is FDA approved for the treatment of metastatic colorectal cancer with disease progression on standard therapy [32]. Trastuzumab (Genentech, San Francisco, CA) is a humanized anti-HER2 monoclonal antibody that is FDA approved for the treatment of HER2 overexpressing breast cancer [33]. Pertuzumab (Roche, Basel, Switzerland) is a humanized anti-HER2 monoclonal antibody that inhibits HER2 dimerization and has improved survival for patients with metastatic breast cancer in combination with trastuzumab and docetaxel [34]. The University of Virginia Medical Center’s Investigational Drugs Pharmacy kindly provided panitumumab, trastuzumab, and pertuzumab.
In Vitro Molecular Response Assays
To assess the molecular responses to EGF stimulation, tumor 366 cells were plated and allowed to adhere overnight in a six-well plate in regular culture conditions. Cells were then starved in serum-free media for 4 hours before the addition of drug combinations (panitumumab, 5 μg/ml; trastuzumab, 20 μg/ml; trametinib, 10 nM) or phosphate-buffered saline (PBS)/DMSO control. One cohort of cells remained in 10% FBS-containing media as a control population. After 1 hour of drug treatment, one cohort of cells was stimulated with 100 ng/ml human EGF or PBS control. Thirty minutes later, samples were lysed and Western blot performed with previously described techniques and antibodies [23].
Tumor Xenografts and Treatment
Tumor pieces (~ 50 mg) were orthotopically implanted onto the pancreata of 6- to 8-week-old male athymic nude mice. Tumors were allowed to grow for 3 to 4 weeks to a volume of 100 to 500 mm3 as assessed by volumetric magnetic resonance imaging (MRI), at which point drug treatment commenced. Mice were treated with either vehicle (0.5% hydroxypropyl methylcellulose in 0.1% Tween 80) or a combination of trametinib, panitumumab, and/or trastuzumab as indicated in the text and figures. Dosing was given as follows: trametinib (0.3 mg/kg orally, once daily), panitumumab (200 μg, intraperitoneal injection, twice weekly), trastuzumab (500 μg, intraperitoneal injection, twice weekly), pertuzumab (200 μg, intraperitoneal injection, twice weekly). In all experiments, volumetric MRI was used to assess changes in tumor volume at 7-day intervals while on drug treatment, as previously described [23]. Mice were then sacrificed, tumors were completely excised, weighed, and measured by calipers, and mice were examined for the presence of metastatic disease. To determine the therapeutic efficacy of drug combinations, an MRI was obtained just before the start of treatment to establish an index tumor volume for each mouse. Subsequent interval MRI studies were used to assess the change in tumor volume while on treatment. The interval tumor volumes were divided by the index tumor volume to calculate the relative change in tumor volume (fold change) for each tumor. Linear regression was used to model a line of best fit for the tumor fold change data plotted relative to time. The slope of that line of best fit served as an estimate of the tumor growth rate for each treatment group expressed as the fold change per week on treatment. Upon completion of the experiment, pieces of tumor were placed in Allprotect (Qiagen, Valencia, CA), snap-frozen in liquid nitrogen, or fixed in formalin for histologic analysis.
Phospho-RTK and Phospho-MAPK Array Analyses
The activation status of 42 RTKs and 26 protein kinases including 9 MAPKs was assessed using the Proteome Profiler human phospho-RTK (pRTK) array kit and human phospho-MAPK array kit (R&D Systems, Minneapolis, MN), as previously described [23].
Statistical Analysis and Image Acquisition
The tumor volume of individual mice during treatment was divided by starting volume to calculate relative change in tumor volume, and linear regression was used to model tumor growth rate. Student’s t test was used to compare continuous variables. All group comparisons were unpaired. Continuous variables were expressed as means ± SEM. All P values reported are two-tailed, and statistical significance was indicated by P values of < .05. GraphPad Prism (Version 5.0b) software (La Jolla, CA) was used for all statistical analyses. Image acquisition and analysis for all Western blot data were performed using the Odyssey Infrared Imaging System (LI-COR, Lincoln, NE) and Image Studio V2.1 software (LI-COR) and for all array data using a Bio-Rad GS-800 calibrated densitometer (Bio-Rad, Hercules, CA) and ImageQuant TL 2005 (GE Healthcare, Piscataway, NJ) software. Blots and array images were cropped and spliced for clarity and ease of comparison using Adobe Photoshop CS4 (Adobe, San Jose, CA).
Results
Genetic and Molecular Profiles of PDX Cells and Tumors
We have previously described the results of the genetic and molecular profiling of 15 PDX tumors [24]. For the present study, we selected a cohort of four PDX tumors for which key genetic and molecular characteristics are summarized in Table 1. Two of four tumors (T366 and T608) harbor KRAS mutations (codon 12) and were derived from liver metastases. The KRAS wild-type tumors under study (T738 and T215) were derived from a primary pancreatic tumor. Phospho-array analysis demonstrated that T608 and T215 exhibited high levels of activated phospho-EGFR (pEGFR) and phospho-HER2 (pHER2) receptors, T366 exhibited readily detectable pEGFR and low-level pHER2, and T738 showed the lowest levels of both pEGFR and pHER2 [24]. No detectable pHER3 or pHER4 signals were observed for any of the tumors under study.
Table 1.
Characteristics of PDX Tumors and Cells
| Tumor | Primary or Metastatic | Differentiation⁎ | Stromal Content | Genetic Alterations† |
Relative Activation‡ |
||||
|---|---|---|---|---|---|---|---|---|---|
| KRAS | TP53 | p16 | SMAD4 | pEGFR | pHER2 | ||||
| 366 | Metastatic | Poor | Low | mut | mut | wt | mut | High | Low |
| 608 | Metastatic | Well-Mod | Low | mut | mut | wt | mut | High | High |
| 738 | Primary | Poor | High | wt | mut | wt | mut | Low | Low |
| 215 | Metastatic | Poor | High | wt | wt | wt | wt | High | High |
Poor, marked cellular atypia, solid single-cell growth; Well-Mod, mild to moderate cytologic atypia, gland forming.
mut, mutant; wt, wild type.
Phosphorylation state relative to background on R&D Systems pRTK array; Low, 1 to 2 × background; High, > 3 × background.
Panitumumab and Trastuzumab Inhibit the EGF-Dependent Activation of EGFR and HER2 and Blunt the Feedback Activation of AKT by Trametinib
To confirm the target specificity for panitumumab and trastuzumab and their respective inhibition of receptor-mediated signaling, we compared the relative pEGFR and pHER2 levels in 366 cells following stimulation with EGF for 30 minutes (Figures 1 and W1). Cells exposed to panitumumab alone or in combination with trastuzumab showed a significant reduction in ligand-activated pEGFR relative to the no antibody control (Figure 1, left panel). Trametinib therapy alone produced no significant change in pEGFR, while trastuzumab alone or in combination with trametinib produced a statistically significant increase in relative pEGFR indicating that anti-HER2 therapy absent anti-EGFR therapy promoted EGFR phosphorylation. Cells exposed to either panitumumab or trastuzumab in any combination elicited a reduction in pHER2 relative to no antibody treatment control. The greatest decrease in pHER2 was seen with dual antibody–treated cells (Figure 1, right panel). We speculate that anti-EGFR therapy likely inhibits the phosphorylation of EGFR-HER2 heterodimers and subsequent EGF-dependent HER2 signaling. Interestingly, trametinib therapy alone produced a marked increase in pHER2 consistent with a MEK-ERK–mediated feedback activation of EGFR/HER2 (see below).
Figure 1.
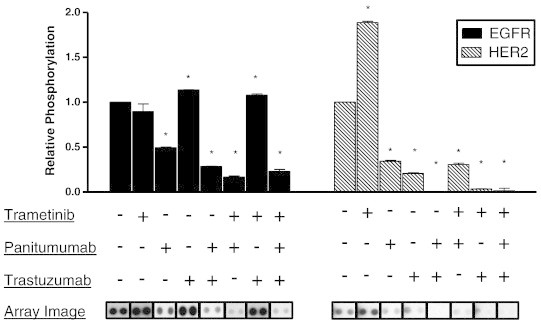
Antibody-mediated inhibition of EGFR and HER2 in response to EGF stimulation. Phospho-RTK arrays were used to assess the relative phosphorylation of EGFR (black bars) and HER2 (lined bars) from EGF-stimulated tumor 366 cells following 90 minutes of drug treatment with DMSO, panitumumab (5 μg/ml), trastuzumab (20 μg/ml), trametinib (10 nM), or the indicated combination therapy (+ indicates drug present and − indicates drug absent). Images from the pRTK arrays used to assay relative phosphorylation level are depicted below each treatment group. Images were cropped and aligned from individual arrays exposed for the same period of time for ease of comparison with vertical bars separating individual arrays. The mean pixel density of two replicates for each phospho-antibody was quantified, normalized to control, and depicted in bar graph form. Image acquisition and analysis were performed using a Bio-Rad GS-800 calibrated densitometer and ImageQuant TL 2005 software. Significant difference in mean pixel density for each treatment group compared to control is denoted as *P < .05. Figure W1 shows the complete array images.
Next, we assessed changes in intracellular MAPK signaling in extracts from EGF-stimulated cells pretreated with panitumumab, trastuzumab, or combination treatment (Figure 2). In the 366 KRAS mutant cell line, addition of EGF to serum-starved cells results in a 50% increase in pERK (as measured by the ratio of pERK/ERK). Addition of panitumumab, trastuzumab, or the combination of antibodies had only modest effects on basal or EGF-dependent phosphorylation of pERK (Figure 2). As anticipated, trametinib treatment markedly reduced pERK in both starved and EGF-stimulated conditions. Addition of either antibody or both antibodies to trametinib treatment effectively blocked the EGF-dependent activation of pERK levels. Interestingly, we observed that in the presence of trametinib, EGF stimulation produced an increase in pAKT (pS473). This trametinib-dependent compensatory response was blunted with the concomitant addition of either panitumumab or combination antibody treatment consistent with the feedback activation of the EGFR/HER2 pathway.
Figure 2.
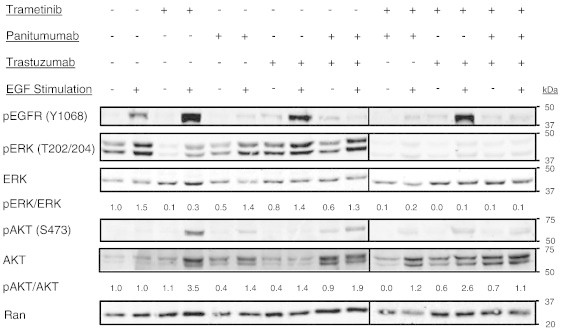
ERK and AKT signaling in a PDX-derived tumor cell line. Western blot depicting the in vitro response of tumor 366 cells following 90 minutes of drug treatment with DMSO, panitumumab (5 μg/ml), trastuzumab (20 μg/ml), trametinib (10 nM), or the indicated combination therapy (+ indicates drug present and − indicates drug absent) under EGF-stimulated or starved conditions. Starved cells were maintained in serum-free media for 4 hours before drug therapy and then given PBS after 60 minutes of drug exposure; EGF-stimulated cells (EGF +) were starved for 4 hours before drug therapy and then exposed to 100 ng/ml of human EGF after 60 minutes of drug exposure. Lysates prepared from cells exposed to each indicated treatment group were blotted for pEGFR (Y1068), pERK1/2 (T202/204), ERK, pAKT (S473), AKT, and RAN. The pERK/ERK and pAKT/AKT ratios are presented to permit quantitative comparisons between groups. Vertical bars denote the alignment of different regions of the same blot. All samples in two separate gels were obtained from one experiment and the same control was included on both gels. Image acquisition and analysis were performed using the Odyssey Infrared Imaging System and Image Studio V2.1 software.
In Vivo Response to Anti-EGFR, Anti-HER2, and MEK1/2 Inhibitor Therapy
To determine if the inhibitory antibodies, panitumumab and trastuzumab, augment the trametinib-dependent inhibition of tumor growth in vivo, serial MRI (sample images are shown in Figure 3, B, D, and F) was used to determine the rate of growth of three different orthotopically implanted PDX tumors. To assess the extent to which blockade of EGFR and HER2 augmented the inhibition of tumor growth by trametinib, mice bearing PDX tumors were initially treated with trametinib, panitumumab, or combination trametinib plus panitumumab for 2 to 4 weeks (Figures 3 and W2). During this initial treatment regimen, treatment with either trametinib or panitumumab reduced tumor volume (Figure W2) and the rate of tumor growth (Figure 3, hatched bars vs black bar) in all three PDX tumors. In the KRAS mutant tumors 366 and 608, the addition of panitumumab to trametinib therapy enhanced the inhibition of tumor growth. In T738, both trametinib and panitumumab treatment yielded effective inhibition of tumor growth with the addition of panitumumab to trametinib yielding no additional inhibition.
Figure 3.
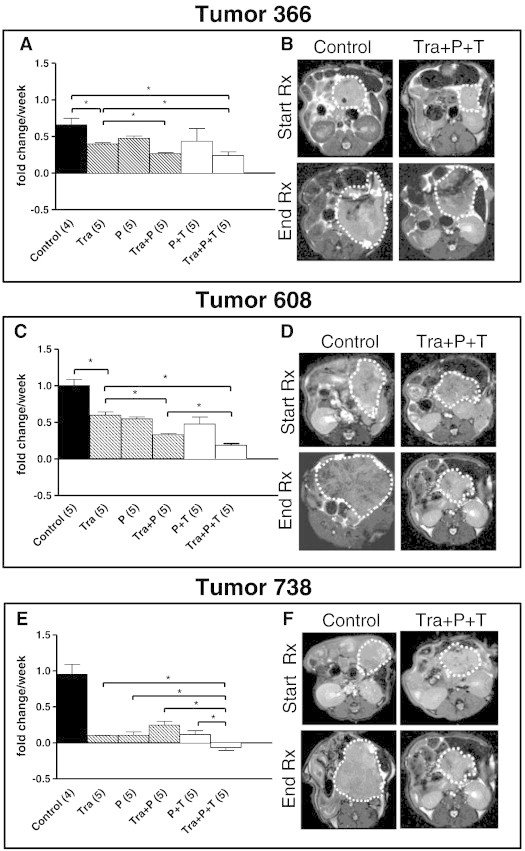
In vivo response of PDX tumors to trametinib and antibody treatment. (A, C, and E) In vivo response of three different established PDAC tumors (100-500 mm3 before starting therapy) to treatment with vehicle control (black bar), trametinib (Tra), panitumumab (P), or trametinib plus panitumumab (Tra + P) (treated mice, hatched bars). The number of mice in each treatment group is indicated in parentheses. An MRI was obtained just before the start of treatment to establish an index tumor volume for each mouse. Subsequent interval MRI studies were used to assess the change in tumor volume while on treatment (see Figure W2). To calculate the relative change in tumor volume (fold change) for each tumor, the interval tumor volumes were divided by the index tumor volume and linear regression was used to model a line of best fit for the tumor fold change data plotted relative to time. The slope of that line of best fit demonstrates the tumor growth rate for each treatment group expressed as the fold change per week on treatment. Mean fold change per week plus the standard error of the mean are displayed for each treatment group as bar graphs. Significance is denoted as *P < .05. Following the initial treatment period, a group of mice treated with trametinib was maintained on trametinib. Mice in the panitumumab (P) group were switched to panitumumab plus trastuzumab (P + T) and mice in the trametinib plus panitumumab (Tra + P) group were switched to combined treatment with panitumumab, trastuzumab, and trametinib (Tra + P + T) for an additional 2 to 4 weeks (open bars, Figure W2). (B, D, and F) Representative MRI images of control and triple therapy–treated (Tra + P + T) tumors obtained just before the onset of treatment (Start Rx) and at the conclusion of treatment just before sacrifice (End Rx). The tumors are outlined and these images depict the axial slice with the largest cross-sectional area in each tumor.
To determine whether addition of the HER2 inhibitory antibody trastuzumab would augment the effects of panitumumab, in the initial treatment groups (trametinib plus panitumumab and panitumumab alone), mice were “rolled on” to therapy with trastuzumab for an additional 2 weeks of therapy (Figures 3, A, C, and E, open bars, and W2). For T366, the addition of trastuzumab had no impact on the growth rate of tumors treated with trametinib plus panitumumab (Figure 3A). In contrast, tumors 608 and 738 showed statistically significant decreases in growth rate with the addition of anti-HER2 therapy to trametinib plus panitumumab therapy (Figure 3, C and E). In a second KRAS wild-type tumor, T215, addition of panitumumab and trastuzumab to trametinib therapy again yielded significant inhibition of tumor growth (Figure W2). These findings indicated that dual antibody therapy may prove more efficacious in some PDX tumors, particularly with tumors with higher levels of HER2 expression or with wild-type KRAS expression.
Triple Therapy Is as Effective as Trametinib plus Lapatinib Treatment
To directly compare the efficacy and tolerability of triple therapy versus trametinib plus lapatinib therapy as described previously [23], mice bearing established T608 and T738 xenografts were randomized to receive control, trametinib plus lapatinib, or trametinib plus panitumumab/trastuzumab treatment for 4 weeks (Figures 4 and W3). Whereas both tumor xenografts demonstrated significant growth inhibition with the trametinib/lapatinib or trametinib/antibody treatment regimen, T738 exhibited a pronounced and durable tumor regression (Figures 4, A and B, and W3, A and C). Mean tumor volume in T738 triple therapy–treated mice decreased by 75% without evidence of outgrowth while on therapy. In contrast, trametinib plus lapatinib–treated tumors demonstrated tumor outgrowth after 2 to 3 weeks of treatment despite an initial tumor regression (T738) or cytostatic response (T608).
Figure 4.
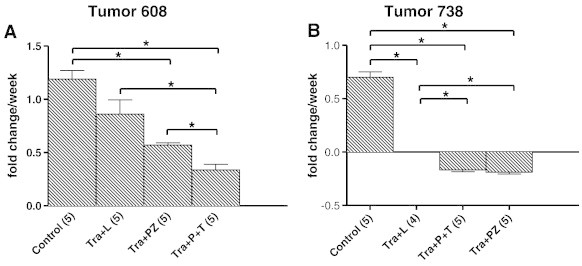
Comparison of in vivo response of PDX tumors to trametinib plus lapatinib versus trametinib plus antibody treatment. (A and B) In vivo response of two different PDAC tumors to treatment with vehicle control, trametinib plus lapatinib (Tra + L), trametinib plus pertuzumab (Tra + PZ), or trametinib plus panitumumab plus trastuzumab (Tra + P + T). Tumors were allowed to grow to a starting volume of 100 to 500 mm3 before the onset of treatment. Mice were treated with vehicle control or drug therapy for 4 weeks (the number of mice in each treatment group is indicated in parentheses). Initial tumor volume was assessed by MRI before the start of dosing and subsequent weekly MRI assessments were carried out to calculate the change in relative tumor volume (see also Figure W3). The mean relative tumor volume ± the standard error of the mean over time is plotted in line graph form for each treatment group. Linear regression was used to model a line of best fit for the tumor fold change data plotted relative to time. The slope of that line of best fit that served as an estimate of the tumor growth rate for each treatment group expressed as fold change per week on treatment was determined as described in the Materials and Methods section. The mean fold change per week ± the standard error of the mean are displayed for each treatment group as bar graphs. Significance is denoted as *P < .05.
The tolerability of each regimen was assessed by comparing the percent weight change of mice from each treatment group while on therapy (Figure W3, B and D). We observed that for T608 trametinib plus lapatinib–treated mice lost a significantly higher percentage of their body weight during treatment than triple therapy–treated mice after 2 and 3 weeks of drug exposure. A similar trend was observed early for T738 although significance was not achieved as toxicity to trametinib plus lapatinib was much less compared to T608. Collectively, these findings indicate that triple therapy is as or more effective than trametinib plus lapatinib therapy but appears to be less toxic.
Given the effectiveness of panitumumab + trastuzumab in combination with trametinib in tumors 608 and 738, we tested the activity of the HER2 dimerization inhibitor pertuzumab. Mice with established T608 and T738 xenografts were treated with trametinib in combination with pertuzumab for 4 weeks as above. As seen in Figure 4, pertuzumab in combination with trametinib significantly inhibited tumor growth relative to control in both xenografts. The rate of tumor regression was similar in T738 xenografts treated with triple therapy and trametinib plus pertuzumab underscoring the importance HER2 heterodimer activation in this tumor.
In Vivo Molecular Response to EGFR, HER2, and MEK1/2 Inhibition
To assess the signaling pathways impacted by the inhibitory antibodies used in combination with trametinib, array platforms were used to compare the changes in pEGFR and pHER2 and as well as pERK, pAKT, c-Jun N-terminal kinase, phospho–c-Jun N-terminal kinase (pJNK), and p70S6K. In all tumors, dual antibody therapy produced a significant decrease in relative pEGFR compared to untreated control tumors (Figure 5). As expected from the above experiments, trametinib treatment alone yielded a more modest decrease in relative level of pEGFR. In T608 and T738, combined trametinib and antibody treatment sustained the decrease in pEGFR. However, in T366, the relative pEGFR of triple therapy–treated tumors was not significantly different from control values. This failure to inhibit EGFR activation may have contributed to the modest growth inhibition of T366 triple therapy–treated tumors relative to tumors 608 and 738. In all tumors, pHER2 levels decreased upon antibody therapy, consistent with the expected inhibition of EGFR and HER2 signaling. Interestingly, in tumor 738, which harbors a wild-type KRAS, we observed little inhibition of pHER2 with trametinib treatment alone; however, it should be noted that the overall pHER2 signals in this tumor were low.
Figure 5.
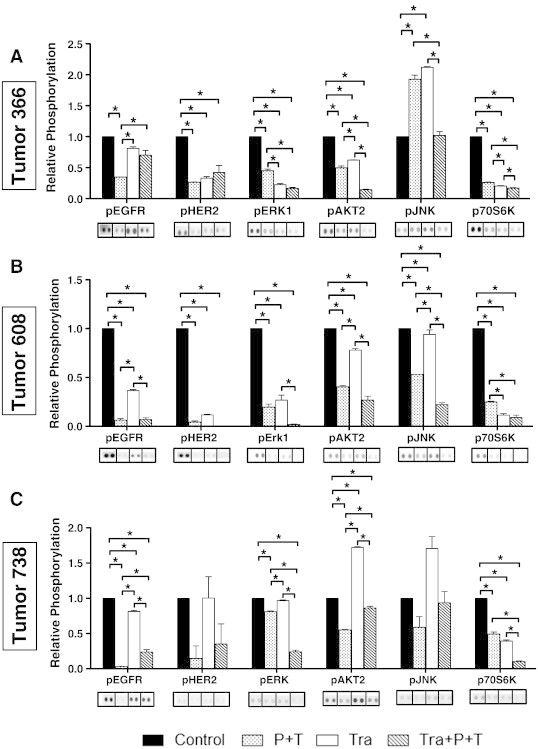
Relative phosphorylation of RTKs and MAPKs in different PDX tumors. Extracts were prepared from PDX tumors following 4 to 6 weeks of in vivo treatment with vehicle control, panitumumab plus trastuzumab (P + T), trametinib (Tra), or triple therapy (Tra + P + T) and analyzed on duplicate arrays. Bar graphs depict relative pEGFR (pan Y), pHER2 (pan Y), pERK1 (T202/Y204), pAKT2 (S474), pJNK-pan (T183/Y185, T221/Y223), and p70S6K (T421/S424) levels in treated tumor 366 (A), tumor 608 (B), and tumor 738 (C) xenografts. Array images from duplicate arrays were used to calculate the mean pixel density for each phospho-protein under each treatment condition. Images were cropped and aligned from individual arrays exposed for the same period of time for ease of comparison with vertical bars separating individual arrays. All phosphorylation values for each treatment group are presented normalized to control for each phospho-protein assessed. Differences in relative phosphorylation were compared between groups with significance denoted as *P < .05. Image acquisition and analysis were as described above in Figure 1. Figure W4 shows the complete array images.
Use of the array platforms to assess pERK, pAKT, pJNK, and p70S6K revealed two response patterns. In the case of the mutant KRAS T366 and T608, dual antibody treatment as well as trametinib treatment yielded significant decreases in pERK1 and p70S6K. The combination treatment with both antibodies and trametinib was also efficient in reducing pERK1 levels (Figures 5 and W4). These observations support the idea that EGFR and HER2 contribute significantly to activation of pERK even in the setting of a mutant KRAS. In contrast, dual antibody treatment or trametinib yielded only modest effects on pAKT and pJNK, consistent with the earlier noted possible feedback activation of the AKT pathway in response to trametinib (Figure 5, A and B). In the case of the wild-type KRAS T738, inhibition of pERK and p70S6K was most efficient in tumors treated with both antibodies and trametinib and correlates with the significant growth inhibition of this tumor observed upon triple therapy. Interestingly, in this tumor, triple therapy blunted the trametinib-induced increase in pAKT2 and pJNK, returning pAKT2 and pJNK to levels similar to those observed in control tumors (Figure 5C).
Discussion
PDX tumor models in which surgically resected human pancreatic adenocarcinomas are propagated orthotopically in the pancreas of immunocompromised mice provide an ideal system to evaluate combinatorial therapies in the context of a complex tumor microenvironment. Not surprisingly, the effects of such therapies can often differ from those observed in tissue culture models. In this study, we have used the therapeutic antibodies panitumumab (specific for EGFR) and trastuzumab (specific for HER2) to explore the role of EGFR and HER2 signaling in the proliferation of PDX tumors bearing mutant and wild-type KRAS alleles. We show that dual anti-EGFR and anti-HER2 therapy significantly augmented the growth inhibitory effects of the MEK1/2 inhibitor trametinib in three different PDX tumors. While significant growth inhibition was observed in both KRAS mutant xenograft groups receiving trametinib and dual antibody therapy (T366 and T608), tumor regression was observed in the KRAS wild-type xenografts (T738) treated in the same manner. We observed that dual antibody therapy in conjunction with trametinib was equally or more effective at inhibiting tumor growth than trametinib plus lapatinib.
A possible role for trametinib as front-line therapy for pancreatic cancer remains unclear. In the recent report of a phase 1b study of trametinib in combination with gemcitabine for advanced solid tumors, it was noted that of 10 patients with measurable pancreatic cancer, three partial responses (30%) were documented. In a randomized double-blind placebo-controlled trial of trametinib in combination with gemcitabine for patients with untreated metastatic adenocarcinoma, no improvement in overall survival, progression-free survival, or response rate in patients was observed (discussed in [35]). These studies underscore the difficulty of using single agents to inhibit the growth of KRAS-driven cancers. The results reported above provide additional evidence that concurrent blockade of EGFR, HER2, and MEK1/2 pathways may lead to more effective pancreatic tumor growth inhibition through a more complete inhibition of RAS and phosphoinositide 3-kinase pathway signaling. Importantly, combining monoclonal antibodies targeting EGFR and HER2 with a MEK inhibitor provides an alternative and perhaps better tolerated combination than lapatinib plus trametinib.
The importance of KRAS mutations in pancreas cancer is widely accepted; in contrast, the contribution of cell-surface RTKs such as EGFR and HER2 in pancreatic cancer progression is poorly understood. One or more of the members of the EGF family of receptors is expressed in a large proportion of pancreatic cancers [36], [37]. Further, studies using both mouse genetic models and human pancreatic cancer cell lines suggest that development of pancreatic adenocarcinomas is totally dependent on EGFR signaling [38]. The EGFR inhibitor erlotinib is approved for use in metastatic pancreatic cancer in combination with gemcitabine, although its overall efficacy in clinical trials of unselected patients has been minimal [39]. A recent report shows that overexpression of HER2 receptors is an independent factor for a worse patient outcome [18]. In preclinical studies, the combination of cetuximab (anti-EGFR monoclonal antibody) and trastuzumab exhibited a synergistic therapeutic effect on the growth of human pancreatic cancer cell lines and xenografts [20]. In these studies, combination therapy (cetuximab/trastuzumab) induced the stable down-regulation of EGFR and HER2 and the downstream blockade of AKT phosphorylation. In other studies, heterocombinations of monoclonal antibodies against EGFR and HER2 exhibited enhanced efficacy through a mechanism that enhanced receptor degradation [40]. These studies provide additional evidence for the importance of EGFR and HER2 in pancreatic cancer and support the approach of combining antibody therapy with targeted inhibition of signaling pathways.
As we report here, in vitro studies using cells cultured from KRAS mutant PDX tumor 366 showed that pretreatment with panitumumab or trastuzumab effectively inhibited the EGF-dependent autophosphorylation of EGFR and HER2, respectively. Interestingly, in the presence of trametinib, we observed a significant EGF-dependent stimulation of HER2 autophosphorylation, consistent with the feedback activation of this pathway. Indeed, in the presence of trametinib, EGF stimulated the phosphorylation of AKT on S473, and this phosphorylation was blunted by preincubation with panitumumab, trastuzumab, or the combination of both antibodies. These observations parallel our previous in vivo PDX studies [23] that showed a similar increase in AKT phosphorylation following trametinib treatment that was, in turn, blunted by lapatinib. We suggest that, at least in T366 cells, the increase in AKT phosphorylation of S473 may occur through the feedback activation of EGFR-HER2 heterodimers, a process that is inhibited by treatment with panitumumab and trastuzumab. It is important to note that in vivo treatment of T366 (as well as T608 and T738) with trametinib and both panitumumab and trastuzumab effectively inhibited the phosphorylation of S473. Paradoxically, in T366 cells, pretreatment with panitumumab, trastuzumab, or the combination failed to inhibit EGF-stimulated ERK phosphorylation. A recent report demonstrated that in head and neck cancers, HER2 (ERBB2), EGFR (ERBB1), and the ligand ephrinB1 (EFNB1) form a complex that enhances ERK signaling and that the antibodies cetuximab (anti-EGFR) and trastuzumab failed to block the EGF-stimulated signaling to ERK1/2 [41]. These studies underscore the complexities of heterodimeric receptor signaling and point out the need to understand more about the dynamics of EGFR family signals in pancreatic cancers.
In the studies reported here and in our previous studies [23], we observed that the KRAS wild-type T738 showed a notable sensitivity to triple therapy and to combined trametinib-lapatinib therapy. A second wild-type KRAS PDX tumor (T215) also showed increased sensitivity to trametinib/dual antibody treatment (Figure W2). While it is unclear what the oncogenic drivers are for these wild-type KRAS tumors, it indicates that trametinib therapy may be more effective in such a KRAS wild-type pancreatic cancer patient population.
In summary, the data presented here using PDX tumors support a role for EGFR and HER2 in pancreatic cancer proliferation and underscore the importance of therapeutic intervention in both the KRAS-RAF-MEK-ERK and EGFR-HER2 pathways to achieve maximal therapeutic efficacy in vivo. A clinical trial evaluating MEK inhibitor plus panitumumab and trastuzumab or MEK inhibitor plus pertuzumab should be considered in patients with pancreatic cancer.
Acknowledgements
The authors thank Clifford Cutchins for his assistance with experiments and animal care.
Footnotes
This article refers to supplementary materials, which are designated by Figures W1 to W4 and are available online at www.neoplasia.com.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.06.004.
Appendix A. Supplementary Materials
Figure S1. Antibody-mediated inhibition of EGFR and HER2 in response to EGF stimulation. Phospho-RTK arrays were used to assess the relative phosphorylation of EGFR and HER2 from EGF-stimulated tumor 366 cells following 90 minutes of drug treatment with DMSO, panitumumab (5 μg/ml; P), trastuzumab (20 μg/ml; T), trametinib (10 nM; Tra), or the indicated combination. Images of pRTKs from each treatment group are depicted. Array images following a 30-second exposure are depicted on the left and used to quantify relative pEGFR (1). A 10-minute exposure of these same arrays is used to quantify relative pHER2 (2) and is depicted by the images on the right. These arrays are presented complete without cropping for comparison and serve as the source for the cropped and spliced images depicted in Figure 1. A Bio-Rad GS 800 calibrated densitometer was used for image acquisition.
Figure S2. In vivo response of three different PDAC tumors to treatment with vehicle control, trametinib, panitumumab, or trametinib plus panitumumab. Tumors were allowed to grow to a starting volume of 100 to 500 mm3 before the onset of treatment. (A–C) Mice were then exposed to vehicle control or drug treatment as described in the Materials and Methods section for the times indicated (initial treatment). After the initial treatment period, mice treated with panitumumab (P) or trametinib and panitumumab (Tra + P) were “rolled” on to treatment with trastuzumab as indicated (plus trastuzumab). (D) Treatment of a PDAC tumor 215. Mice bearing 215 tumors were exposed to vehicle control, trametinib alone, or trametinib, panitumumab, and trastuzumab as described in the Materials and Methods section. Data presented are mean relative tumor volume ± SEM.
Figure S3. Comparison of tumor response and mouse toxicity to treatment with trametinib plus lapatinib, trametinib plus concurrent panitumumab and trastuzumab, or trametinib plus pertuzumab.
Figure S4. Relative phosphorylation of RTKs and MAPKs in different PDX tumors. Extracts were prepared from PDX tumors following 4 to 6 weeks of in vivo treatment as described in the text and exposed to R&D Systems pRTK and phospho-MAPK array platforms. pEGFR (pan Y) (1), pHER2 (panY) (2), pAKT2 (S474) (3), pERK1 (T202/204) (4), pJNK-pan (T183/Y185) (5), and p70S6K (T421/424) (6) levels in treated tumor 366(A), tumor 608(B), and tumor 738(C) xenografts are outlined for comparison between treatment groups. These arrays are presented complete without cropping for comparison and serve as the source for the cropped and spliced images depicted in Figure 5. A Bio-Rad GS800 calibrated densitometer was used for image acquisition.
References
- 1.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 2.Smith BD, Smith GL, Hurria A, Hortobagyi GN, Buchholz TA. Future of cancer incidence in the United States: burdens upon an aging, changing nation. J Clin Oncol. 2009;27:2758–2765. doi: 10.1200/JCO.2008.20.8983. [DOI] [PubMed] [Google Scholar]
- 3.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furukawa T. Molecular pathology of pancreatic cancer: implications for molecular targeting therapy. Clin Gastroenterol Hepatol. 2009;7:S35–S39. doi: 10.1016/j.cgh.2009.07.035. [DOI] [PubMed] [Google Scholar]
- 5.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 6.Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995;18:1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- 7.Shibata D, Capella G, Perucho M. Mutational activation of the c-K-ras gene in human pancreatic carcinoma. Baillieres Clin Gastroenterol. 1990;4:151–169. doi: 10.1016/0950-3528(90)90044-h. [DOI] [PubMed] [Google Scholar]
- 8.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 10.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U. Both p16Ink4a and the p19Arf-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Philip PA, Mooney M, Jaffe D, Eckhardt G, Moore M, Meropol N, Emens L, O'Reilly E, Korc M, Ellis L. Consensus report of the national cancer institute clinical trials planning meeting on pancreas cancer treatment. J Clin Oncol. 2009;27:5660–5669. doi: 10.1200/JCO.2009.21.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poulikakos PI, Solit DB. Resistance to MEK inhibitors: should we co-target upstream? Sci Signal. 2011;4:1–13. doi: 10.1126/scisignal.2001948. [pe16] [DOI] [PubMed] [Google Scholar]
- 15.Yamanaka Y, Friess H, Kobrin MS, Buchler M, Beger HG, Korc M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993;13:565–569. [PubMed] [Google Scholar]
- 16.Yamaguchi T, Yoshida T, Kurachi R, Kakegawa J, Hori Y, Nanayama T, Hayakawa K, Abe H, Takagi K, Matsuzaki Y. Identification of JTP-70902, a p15INK4b-inductive compound, as a novel MEK1/2 inhibitor. Cancer Sci. 2007;98:1809–1816. doi: 10.1111/j.1349-7006.2007.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Safran H, Steinhoff M, Mangray S, Rathore R, King TC, Chai L, Berzein K, Moore T, Iannitti D, Reiss P. Overexpression of the HER-2/neu oncogene in pancreatic adenocarcinoma. Am J Clin Oncol. 2001;24:496–499. doi: 10.1097/00000421-200110000-00016. [DOI] [PubMed] [Google Scholar]
- 18.Komoto M, Nakata B, Amano R, Yamada N, Yashiro M, Ohira M, Wakasa K, Hirakawa K. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci. 2009;100:1243–1247. doi: 10.1111/j.1349-7006.2009.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larbouret C, Robert B, Navarro-Teulon I, Thèzenas S, Ladjemi MZ, Morisseau S, Campigna E, Bibeau F, Mach JP, Pèlegrin A. In vivo therapeutic synergism of anti-epidermal growth factor receptor and anti-HER2 monoclonal antibodies against pancreatic carcinomas. Clin Cancer Res. 2007;13:3356–3362. doi: 10.1158/1078-0432.CCR-06-2302. [DOI] [PubMed] [Google Scholar]
- 20.Larbouret C, Gaborit N, Chardès T, Coelho M, Campigna E, Bascoul-Mollevi C, Mach JP, Azria D, Robert B, Pèlegrin A. In pancreatic carcinoma, dual EGFR/HER2 targeting with cetuximab/trastuzumab is more effective than treatment with trastuzumab/erlotinib or lapatinib alone: implication of receptors' down-regulation and dimers' disruption. Neoplasia. 2012;14:121–130. doi: 10.1593/neo.111602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jimeno A, Rubio-Viqueira B, Amador ML, Grunwald V, Maitra A, Iacobuzio-Donahue C, Hidalgo M. Dual mitogen-activated protein kinase and epidermal growth factor receptor inhibition in biliary and pancreatic cancer. Mol Cancer Ther. 2007;6:1079–1088. doi: 10.1158/1535-7163.MCT-06-0448. [DOI] [PubMed] [Google Scholar]
- 22.Diep CH, Munoz RM, Choudhary A, Von Hoff DD, Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011;17:2744–2756. doi: 10.1158/1078-0432.CCR-10-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT, Chopivsky ME. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia. 2013;15:143–155. doi: 10.1593/neo.121712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walters DM, Stokes JB, Adair SJ, Stelow EB, Borgman CA, Lowrey BT, Xin W, Blais EM, Lee JK, Papin JA. Clinical, molecular and genetic validation of a murine orthotopic xenograft model of pancreatic adenocarcinoma using fresh human specimens. PLoS One. 2013;8:1–10. doi: 10.1371/journal.pone.0077065. [e77065] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39:23–31. doi: 10.3892/ijo.2011.1015. [DOI] [PubMed] [Google Scholar]
- 27.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 28.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 29.Schwartzberg LS, Franco SX, Florance A, O'Rourke L, Maltzman J, Johnston S. Lapatinib plus letrozole as first-line therapy for HER-2 + hormone receptor-positive metastatic breast cancer. Oncologist. 2010;15:122–129. doi: 10.1634/theoncologist.2009-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becerra C, Infante JR, Garbo L, Gordon M, Smith D, Braiteh F, Gandara D, Jotte R, Reckamp K, Janku F. A five-arm, open-label, phase I/Ib study to assess safety and tolerability of the oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with chemotherapy or erlotinib in patients with advanced solid tumors. J Clin Oncol. 2012;30:3023. [Google Scholar]
- 31.Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, Hwang RF. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10:2135–2145. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: panitumumab (Vectibix) Oncologist. 2007;12:577–583. doi: 10.1634/theoncologist.12-5-577. [DOI] [PubMed] [Google Scholar]
- 33.Ahn ER, Wang E, Gluck S. Is the improved efficacy of trastuzumab and lapatinib combination worth the added toxicity? A discussion of current evidence, recommendations, and ethical issues regarding dual HER2-targeted therapy. Breast Cancer (Auckl) 2012;6:191–207. doi: 10.4137/BCBCR.S9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baselga J, Cortés J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kothari N, Saif MW, Kim R. First-line treatment for advanced pancreatic cancer. JOP. 2013;14:129–132. doi: 10.6092/1590-8577/1477. [DOI] [PubMed] [Google Scholar]
- 36.Lemoine NR, Hughes CM, Barton CM, Poulsom R, Jeffery RE, Kloppel G, Hall PA, Gullick WJ. The epidermal growth factor receptor in human pancreatic cancer. J Pathol. 1992;166:7–12. doi: 10.1002/path.1711660103. [DOI] [PubMed] [Google Scholar]
- 37.Kimura K, Sawada T, Komatsu M, Inoue M, Muguruma K, Nishihara T, Yamashita Y, Yamada N, Ohira M, Hirakawa K. Antitumor effect of trastuzumab for pancreatic cancer with high HER-2 expression and enhancement of effect by combined therapy with gemcitabine. Clin Cancer Res. 2006;12:4925–4932. doi: 10.1158/1078-0432.CCR-06-0544. [DOI] [PubMed] [Google Scholar]
- 38.Navas C, Hernández-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-Ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 40.Maron R, Schechter B, Mancini M, Mahlknecht G, Yarden Y, Sela M. Inhibition of pancreatic carcinoma by homo- and heterocombinations of antibodies against EGF-receptor and its kin HER2/ErbB-2. Proc Natl Acad Sci U S A. 2013;110:15389–15394. doi: 10.1073/pnas.1313857110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vermeer PD, Colbert PL, Wieking BG, Vermeer DW, Lee JH. Targeting ERBB receptors shifts their partners and triggers persistent ERK signaling through a novel ERBB/EFNB1 complex. Cancer Res. 2013;73:5787–5797. doi: 10.1158/0008-5472.CAN-13-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Antibody-mediated inhibition of EGFR and HER2 in response to EGF stimulation. Phospho-RTK arrays were used to assess the relative phosphorylation of EGFR and HER2 from EGF-stimulated tumor 366 cells following 90 minutes of drug treatment with DMSO, panitumumab (5 μg/ml; P), trastuzumab (20 μg/ml; T), trametinib (10 nM; Tra), or the indicated combination. Images of pRTKs from each treatment group are depicted. Array images following a 30-second exposure are depicted on the left and used to quantify relative pEGFR (1). A 10-minute exposure of these same arrays is used to quantify relative pHER2 (2) and is depicted by the images on the right. These arrays are presented complete without cropping for comparison and serve as the source for the cropped and spliced images depicted in Figure 1. A Bio-Rad GS 800 calibrated densitometer was used for image acquisition.
Figure S2. In vivo response of three different PDAC tumors to treatment with vehicle control, trametinib, panitumumab, or trametinib plus panitumumab. Tumors were allowed to grow to a starting volume of 100 to 500 mm3 before the onset of treatment. (A–C) Mice were then exposed to vehicle control or drug treatment as described in the Materials and Methods section for the times indicated (initial treatment). After the initial treatment period, mice treated with panitumumab (P) or trametinib and panitumumab (Tra + P) were “rolled” on to treatment with trastuzumab as indicated (plus trastuzumab). (D) Treatment of a PDAC tumor 215. Mice bearing 215 tumors were exposed to vehicle control, trametinib alone, or trametinib, panitumumab, and trastuzumab as described in the Materials and Methods section. Data presented are mean relative tumor volume ± SEM.
Figure S3. Comparison of tumor response and mouse toxicity to treatment with trametinib plus lapatinib, trametinib plus concurrent panitumumab and trastuzumab, or trametinib plus pertuzumab.
Figure S4. Relative phosphorylation of RTKs and MAPKs in different PDX tumors. Extracts were prepared from PDX tumors following 4 to 6 weeks of in vivo treatment as described in the text and exposed to R&D Systems pRTK and phospho-MAPK array platforms. pEGFR (pan Y) (1), pHER2 (panY) (2), pAKT2 (S474) (3), pERK1 (T202/204) (4), pJNK-pan (T183/Y185) (5), and p70S6K (T421/424) (6) levels in treated tumor 366(A), tumor 608(B), and tumor 738(C) xenografts are outlined for comparison between treatment groups. These arrays are presented complete without cropping for comparison and serve as the source for the cropped and spliced images depicted in Figure 5. A Bio-Rad GS800 calibrated densitometer was used for image acquisition.