

Abstract
NF-κB1 p105 forms a high-affinity, stoichiometric interaction with TPL-2, a MEK kinase essential for TLR4 activation of the ERK mitogen-activated protein kinase cascade in lipopolysaccharide (LPS)-stimulated macrophages. Interaction with p105 is required to maintain TPL-2 metabolic stability and also negatively regulates TPL-2 MEK kinase activity. Here, affinity purification identified A20-binding inhibitor of NF-κB 2 (ABIN-2) as a novel p105-associated protein. Cotransfection experiments demonstrated that ABIN-2 could interact with TPL-2 in addition to p105 but preferentially formed a ternary complex with both proteins. Consistently, in unstimulated bone marrow-derived macrophages (BMDMs), a substantial fraction of endogenous ABIN-2 was associated with both p105 and TPL-2. Although the majority of TPL-2 in these cells was complexed with ABIN-2, the pool of TPL-2 which could activate MEK after LPS stimulation was not, and LPS activation of TPL-2 was found to correlate with its release from ABIN-2. Depletion of ABIN-2 by RNA interference dramatically reduced steady-state levels of TPL-2 protein without affecting levels of TPL-2 mRNA or p105 protein. In addition, ABIN-2 increased the half-life of cotransfected TPL-2. Thus, optimal TPL-2 stability in vivo requires interaction with ABIN-2 as well as p105. Together, these data raise the possibility that ABIN-2 functions in the TLR4 signaling pathway which regulates TPL-2 activation.
NF-κB transcription factors play an essential role in immune and inflammatory responses (27). In unstimulated cells, NF-κB dimers are inactive due to their retention in the cytoplasm by associated IκB proteins (11). In response to stimulation with agonists, such as tumor necrosis factor alpha (TNF-α) and bacterial lipopolysaccharide (LPS), IκBs are degraded by the proteasome. This degradation releases associated NF-κB subunits, allowing them to translocate into the nucleus and modulate gene expression.
NF-κB1 functions both as an IκB in its longer precursor form, p105, and as an active transcription factor, p50, after proteolytic processing by the proteasome, which removes the IκB-like C-terminal half of p105. Processing to p50 occurs in a constitutive, unregulated fashion (18). However, following cellular stimulation with ligands, such as TNF-α, two serines in the p105 PEST domain are rapidly phosphorylated by the IκB kinase (IKK) complex, triggering complete p105 degradation with little effect on processing to p50 (12, 19, 24). This degradation results in the release of associated p50 and other NF-κB subunits, which can then translocate into the nucleus. Analysis of knockout mice that lack the C-terminal (IκB-like) half of p105 while still expressing p50 has suggested an essential role for p105 in the regulation of the nuclear translocation of p50 homodimers (16).
In addition to NF-κB activation, LPS triggering of proinflammatory cytokine gene expression involves the activation of the signaling pathways that regulate all of the major mitogen-activated protein (MAP) kinase (MAP K) subtypes (extracellular signal-regulated kinases 1 and 2 [ERK-1/2], Jun amino-terminal kinases [JNK], and p38 [15, 33]). MAP K activation involves three-tiered kinase cascades in which MAP Ks are activated by MAP K kinases (MAP 2-K), which in turn are activated by MAP K kinase kinases (MAP 3-K) (6). In macrophages, LPS activation of MEK-1/2, the MAP 2-Ks which phosphorylate and activate ERK-1/2, is mediated by the MAP 3-kinase TPL-2 (9). Recent research has revealed an unexpected function for NF-κB1 p105 in regulating this signaling pathway (31). The C-terminal half of p105 forms a high-affinity, stoichiometric association with TPL-2 (2, 3). Interaction with p105 is required for stabilization of TPL-2 protein and also inhibits TPL-2 MEK kinase activity by preventing its access to MEK (2, 31). Thus, in addition to its roles as a precursor for p50 and an IκB, p105 functions as a negative regulator of TPL-2.
In order to more fully understand the regulation and function of the TPL-2/p105 complex, it is essential to identify any additional proteins with which it associates. In the present study, affinity purification and peptide mass fingerprinting revealed A20-binding inhibitor of NF-κB 2 (ABIN-2) (29) as a novel p105-associated protein. A20 is an inhibitor of NF-κB activation, and overexpression experiments have suggested that ABIN-2 may be a downstream effector of A20 which mediates this inhibitory function (4, 29). Evidence that the majority of ABIN-2 in macrophages forms a ternary complex with TPL-2 and p105 and that ABIN-2 is required for stable TPL-2 protein expression is presented here. The potential role of ABIN-2 in the regulation of TPL-2 function is discussed.
MATERIALS AND METHODS
cDNA constructs.
For transient-transfection experiments in 293 cells, all hemagglutinin (HA) epitope-tagged NF-κB1 p105 (HA-p105) cDNAs were cloned into the pcDNA3 vector (Invitrogen). Deletion and point mutant versions of HA-p105 and HA-p50 have been described previously (1, 2, 24). For stable transfection of HeLa S3 cells, HA-p105(S927A) was subcloned in the pMX-1 vector (Ingenius). Myc epitope-tagged p105 (Myc-p105) was generated by PCR and verified by DNA sequencing. Myc-tagged and untagged versions of TPL-2, kinase-inactive TPL-2(D270A) and TPL-2ΔC have been described previously (3). Myc-A20 was kindly provided by Nancy Raab-Traub (University of North Carolina) (10).
Human ABIN-2 cDNA (Image clone 4287014) was obtained from the United Kingdom Human Genome Mapping Project Resource Centre (Cambridge, United Kingdom). Wild-type ABIN-2 was FLAG tagged on its C terminus (ABIN-2-FL) using PCR and cloned into the pcDNA3 vector (Invitrogen). PCR was also used to generate the following panel of ABIN-2 deletion mutants subcloned into pGEX-2T (Amersham Biosciences): GST-ABIN-21-429, GST-ABIN-21-89, GST-ABIN-21-108, GST-ABIN-21-129, GST-ABIN-21-193, GST-ABIN-21-250, GST-ABIN-290-250, GST-ABIN-2130-250, GST-ABIN-2194-250, and GST-ABIN-2251-429. All constructs were verified by DNA sequencing.
Recombinant proteins, peptides and antibodies.
Glutathione S-transferase (GST)-ABIN-2 fusion proteins were expressed at 30°C in Escherichia coli BL21(DE3) and purified by affinity chromatography on glutathione-Sepharose 4B (Amersham Biosciences). The purity of GST fusion proteins was estimated to be >90% by Coomassie brilliant blue (Novex) staining after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). GST-p105497-968 fusion protein has been described previously (2). GST-MEK1, GST-MEK1(K207A), and GST-ERK fusion proteins were kindly provided by Richard Marais (Cancer Research-U.K., London, United Kingdom).
Antibodies to HA, Myc, and FLAG epitope tags have been described previously (1). Anti-human p105C, anti-murine p105C, anti-human p105C, and anti-murine p105N antibodies have also been described previously (7, 24). The 70-mer anti-TPL-2 antibody was raised in rabbits against a synthetic peptide corresponding to the C-terminal 70 amino acids of rat TPL-2 coupled to keyhole limpet hemocyanin (Pierce) and was used for immunoprecipitation of endogenous TPL-2 protein. A commercial anti-TPL-2 antibody (M20; Santa Cruz) was used to detect TPL-2 in Western blots. Anti-ABIN-2 antibody was raised in rabbits against GST-ABIN-2251-429 fusion protein. Endogenous p100 was detected using a commercial anti-p100 antibody (UBI 05-361). Anti-MEK-1/2 and anti-phospho(S217/S221)-MEK-1/2 (anti-phospho-MEK-1/2) antibodies were purchased from Cell Signaling Technology. Tubulin was detected with the TAT-1 anti-α-tubulin MAb (kindly provided by Keith Gull, University of Manchester, Manchester, United Kingdom) and actin using a commercial anti-actin monoclonal antibody (MAb) (Sigma). Tubulin and actin were used as loading controls for Western blots of cell lysates.
3x-HA peptide was synthesized by Pete Fletcher (Protein Structure, National Institute for Medical Research) and consisted of the sequence to which the 12CA5 anti-HA MAb was raised (YPYDVPDYA) triplicated, with three glycine residue spacers between each HA sequence. This was significantly more efficient for elution of affinity-purified protein from 12CA5 MAb than a single-copy peptide (data not shown).
Cell culture.
HeLa S3 and 293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (50 U/ml) (complete DMEM). Cells were maintained in a rapid growth phase prior to use in experiments.
For stable transfection of HeLa S3 cells, 7 × 105 cells were plated in a 60-mm-diameter dish (Nunc), and after 18 h in culture, the cells were transfected using a standard calcium phosphate method with 5 μg of pMX-1 HA-p105(S927A). Transfected cells were cultured for a further 48 h and then selected for neomycin resistance with 1 mg of G418 (Invitrogen) per ml. After 3 to 4 weeks, 48 clones were picked manually and then expanded. Five of these clones tested positive for expression of HA-p105(S927A) protein by Western blotting. Clone C3.25, which expressed relatively high levels of HA-p105(S927A) protein, was selected for preparative experiments. A clone transfected with the empty pMX-1 vector was used as a control.
Bone marrow-derived macrophages (BMDMs) were prepared as described previously (30). Briefly, BALB/c bone marrow cells were plated at 2 × 106 cells/ml in 10 ml of complete BMDM medium (RPMI 1640 [Invitrogen] plus 10% fetal bovine serum [FBS] and antibiotics supplemented with 10% L-cell conditioned medium) in 25-cm2 tissue culture flasks (Nunc). After 24 h, nonadherent cells were then transferred to a 80-cm2 tissue culture flask (Nunc), and another 10 ml of complete BMDM medium was added. Flasks were then incubated for 3 days at 37°C, at which time another 10 ml of complete BMDM medium was added. After a total of 7 days of culture, adherent macrophages were harvested, replated, and cultured in RMPI 1640 medium plus 0.5% FBS and antibiotics for a further 24 h before use in experiments. More than 95% of the resulting cell populations were macrophages as judged by flow cytometric analysis (data not shown).
nf-κb1−/− mice (26) were obtained from the Jackson Laboratory. BMDMs from these knockout mice and heterozygous littermates (6 to 10 weeks old) were prepared as described above.
Affinity purification of HA-p105(S927A).
Cell pellets (20 ml) of C3.25 HA-p105(S927A) and EV HeLa S3 cells were prepared by centrifugation from large-scale suspension cell cultures (20 liters). Cells were lysed in 10 volumes of ice-cold buffer A (1% Nonidet P-40 [NP-40], 50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 20 mM NaF, 1 mM Na3VO4, 1 mM Na4P2O7 plus a mixture of protease inhibitors [Roche Molecular Biochemicals]). All subsequent steps were performed at 4°C. Lysates were centrifuged at 20,000 × g for 30 min and passed through a glass-fiber filter (catalog no. 189-2000; Nalgene) to remove lipids. Filtered lysates were recentrifuged at 100,000 × g for a further 30 min and RQI RNase-free DNase (catalog no. M610A; Promega) (1/1,000 stock) was added.
Lysates were precleared of nonspecific binding proteins first by two sequential batch incubations (overnight and 2 h) with 1-ml aliquots of protein A-Sepharose beads (Amersham Biosciences). Lysates were precleared for another 3 h by batch incubation with 0.5 ml of protein A-Sepharose coupled to 3.5 mg of purified mouse immunoglobulin G (IgG) (technical grade; Sigma). Lysates were finally precleared by passing under gravity through 5 ml of protein A-Sepharose packed in a disposable column (catalog no. 732-1010; Bio-Rad).
HA-p105(927A) protein was affinity purified by incubating precleared lysate overnight with 0.5 ml of protein A-Sepharose beads coupled to 3.5 mg of 12CA5 anti-HA MAb. The suspension of the 12CA5 beads in lysate was then transferred to a disposable column (catalog no. 732-1010; Bio-Rad) and washed with 100 column volumes of buffer A (flow rate, <1 ml/min). Washed beads were transferred to siliconized tubes (0.5 ml; Bioquote), which were used in all subsequent steps, and excess buffer was removed. To elute bound protein, 250 μl of peptide elution buffer (3x-HA peptide dissolved in a solution consisting of 50 mM Tris [pH 7.5], 150 mM NaCl, and 0.05% NP-40) was added, and beads were incubated for 15 min with rotation. Eluate was removed from centrifuged beads and transferred to new tubes. This process was repeated five times, and eluates were combined (total volume, 1.5 ml), snap-frozen, and stored at −70°C.
One-third of the 3x-HA peptide eluate (0.5 ml) was diluted 1/2 in buffer A and precleared twice (overnight and for 1 h) by incubation with 25 μl of rabbit IgG (Sigma) coupled to protein A-Sepharose beads (0.5 mg/ml). Eluted HA-p105(S927A) protein was then reimmunoprecipitated by incubation for 5 h with 40 μl of anti-p105C antibody IgG coupled to protein A-Sepharose (0.35 mg/ml). Beads were washed five times with buffer A and once with water, and bound protein was eluted with 50 μl of 0.1 M glycine (pH 3.0)-0.05% NP-40. Low-pH elution was repeated five times, and eluates were combined and neutralized with 1/10 volume of 1 M Tris (pH 8.0). A portion (1/200) of the eluate (370 μl), containing purified HA-p105(S927A) and associated proteins, was resolved by SDS-PAGE (10% acrylamide), and isolated proteins were revealed by silver staining (14). The remainder of the eluate was snap-frozen and stored at −70°C until processing for mass spectroscopic analysis.
Mass spectroscopic analysis.
Affinity-purified eluate (270 μl) containing HA-p105(S927A) was concentrated to 30 μl using a Microcon YM-10 (Millipore) and then mixed with 7.5 μl of 4× sample buffer. Isolated proteins were resolved by SDS-PAGE (10% acrylamide) and revealed by staining with colloidal Coomassie brilliant blue (Novex). The stained gel was not scanned prior to excision of protein bands to minimize handling and potential introduction of contaminating keratins. Excised protein bands were reduced with 20 mM dithiothreitol and alkylated with 5 mM iodoacetamide. Bands were then dried and reswollen in 5 mM ammonium bicarbonate containing 2 ng of trypsin (modified sequencing grade; Promega) per μl. After overnight digestion at 32°C, the supernatant was acidified by addition of 1/10 volume of trifluoroacetic acid.
Peptide mass fingerprinting was performed using a Reflex III matrix-assisted laser desorption ionization (MALDI)-time-of-flight mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a nitrogen laser and a Scout-384 probe to obtain positive-ion mass spectra. A portion (0.4 μl) of digestion supernatant was analyzed after desalting with water on the matrix surface. Peptide mass fingerprints were searched against the nonredundant protein database at the National Center for Biotechnology Information (NCBI) using the MASCOT program.
Protein analyses.
To analyze interactions with endogenous ABIN-2 protein, HeLa S3 cells were plated at 5 × 106 cells per 60-mm-diameter dish (Nunc) and cultured overnight. Cells were then washed in phosphate-buffered saline and lysed in 1 ml of buffer A. Lysates were cleared of particulate matter by centrifugation at 100,000 × g for 10 min. Immunoprecipitation and Western blotting of proteins were performed as described previously (17). However, two additional steps were taken to minimize detection of Ig heavy chain in immunoprecipitates, which comigrates with both ABIN-2 and TPL-2. First, all antibodies were covalently coupled to protein A-Sepharose using dimethylpimelimidate (25), and immunoprecipitated protein was eluted using a low-pH buffer (0.2 M glycine [pH 2.5], 0.05% NP-40) (50 μl). Eluate was neutralized by addition of 10 μl of 1 M Tris (pH 8) and then mixed with an equal volume of 2× SDS-PAGE sample buffer. Second, Western blot membranes were blocked with protein A (5 μg/ml; Sigma) prior to probing with primary rabbit antibody. Bound antibody was then revealed with protein A coupled to horseradish peroxidase (Amersham Biosciences) and enhanced chemiluminescence (Amersham Biosciences).
293 cells (3 × 105 cells per 60-mm-diameter Nunc dish) were transiently transfected using Lipofectamine (Invitrogen) and cultured for a total of 48 h, as described previously (2). For protein association experiments, cell lysates were prepared using 1% NP-40 buffer A. Immunoprecipitation and Western blotting were performed as described previously (17). In pulse-chase experiments, cells were washed in phosphate-buffered saline after 24 h of culture and then incubated for 45 min in methionine-cysteine-free minimal Eagle medium (Sigma) plus 0.5% FBS. Cells were pulse-labeled with 2.65 MBq of [35S]methionine-[35S]cysteine (Pro-Mix; Amersham Biosciences) for 30 min and chased for the indicated times in DMEM plus 2% fetal calf serum. Lysis was performed using buffer A supplemented with 0.5% deoxycholate and 0.1% SDS (radioimmunoprecipitation assay [RIPA] buffer). TPL-2 was isolated using 70-mer anti-TPL-2 antibody, and labeled bands were revealed by autoradiography after SDS-PAGE (10% acrylamide).
BMDMs were plated in 60-mm-diameter dishes (3 × 106 cells; Nunc) and cultured for 18 h prior to lysis in 1% NP-40 buffer A. Dishes with a diameter of 35 mm (106 cells; Nunc) were used in experiments in which cells were stimulated with LPS (1 μg/ml) (Salmonella minnesota; Alexis Biochemicals). Where indicated, cells were preincubated for 30 min with MG132 proteasome inhibitor (40 μM; Biomol) or dimethyl sulfoxide vehicle control prior to LPS stimulation. Proteins were removed from lysates by immunoprecipitation with preclearing antibody or preimmune rabbit IgG as a control, both covalently coupled to protein A-Sepharose. In some experiments, precleared lysates were reimmunoprecipitated overnight with the indicated specific antibody. Lysates and reimmunoprecipitated proteins were resolved by SDS-PAGE (10% acrylamide) and Western blotted.
For pulldown assays with GST-ABIN-2 fusion proteins, 2 μg of recombinant protein was added to ultracentrifuged lysate of transfected 293 cells. For some of these experiments, 1% Brij 58 was used as the detergent component of buffer A used for lysis, rather than 1% NP-40, as indicated in the figure legends. Lysates were incubated overnight with mixing, and fusion proteins were affinity isolated by addition of 10 μl of glutathione-Sepharose 4B beads (Amersham Biosciences) and incubation for a further 30 min. Beads were then washed extensively in buffer A (1% NP-40 or 1% Brij 58, as appropriate), and isolated protein was analyzed by Western blotting.
To investigate whether ABIN-2 could bind to the C-terminal half of p105, ABIN-2-FL was synthesized from its expression vector and labeled with [35S]methionine (Amersham Bioscience) by cell-free translation (25-μl reaction volume; Promega TNF-coupled rabbit reticulocyte system). Translated protein was diluted in 1 ml of 1% NP-40 buffer A and then incubated with 5 μg of GST-p105497-968 fusion protein bound to glutathione-Sepharose 4B (Amersham Bioscience). After an overnight incubation at 4°C, beads were extensively washed with buffer A, and bound 35S-labeled protein was visualized by autoradiography after SDS-PAGE (10% acrylamide).
To demonstrate binding of ABIN-2 to the isolated TPL-2 C terminus, 30 μg of biotinylated TPL-2398-467 peptide (2) was incubated for 2 h with lysates (1% NP-40 buffer A) of 293 cells transfected with a plasmid encoding ABIN-2-FL. TPL-2 peptide was captured on streptavidin-agarose beads (Sigma), which were then washed extensively with buffer A. Isolated protein was resolved by SDS-PAGE (10% acrylamide) and Western blotted.
RNA interference.
RNA interference was used to deplete HeLa S3 and 293 cells of endogenous ABIN-2. Small interfering RNAs (siRNAs) were synthesized by Xeragon, Inc. The sequences of the ABIN-2 siRNAs used were (sense) GUAUUUGGCCGCCGACGCAd(TT) and (antisense) UGCGUCGGCGGCCAAAUACd(TT). Commercial control siRNAs (catalog no. 1022076; Xeragon, Inc.) were used to confirm the specificity of the effects of ABIN-2 siRNAs.
For gene knockdown experiments, HeLa S3 cells (5 × 105) or 293 cells (2 × 105) were plated in 60-mm-diameter dish (Nunc) and cultured for 12 to 16 h in complete DMEM medium without antibiotics. Cells were transfected with siRNAs (0.4 nmol per well) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 24 h of culture, cells were retransfected with siRNAs and then recultured for another 48 h. Protein expression was analyzed by Western blotting of cell lysates. Semiquantitative reverse transcription-PCR (RT-PCR) of TPL-2 mRNA was performed by utilizing the QIAGEN OneStep RT-PCR kit. Total RNA was isolated from cells using the QIAGEN RNeasy kit. The TPL-2 primer pairs used were the (5′)-primer (5′-ACGCTAGTCGACTCACCTGTACGTCAGCTTCCACGG-3′) and (3′)-primer (5′-GCCCAGGGGATCCGAATGGAGTACATGAGCACCG-3′). The 18S rRNA loading control oligonucleotides used were 5′)-primer (5′-GGCGGCTTGGTGACTCTAGATA-3′) and (3′)-primer (5′-GCTCGGGCCTGCTTTGAACAC-3′).
Semiquantitative RT-PCR was also used to quantify ABIN-2 mRNA in wild-type and NF-κB1-deficient 3T3 fibroblasts using the methodology described above and the following pair of primers: (5′)-primer (5′-CCATGTCGTCTGGGGACGCAA-3′) and (3′) primer (5′-TGGCAGCACTCAGACAGGTGC-3′).
MEK kinase assays.
To assay MEK kinase activity of endogenous TPL-2, BMDMs (8 × 106) were plated in 90-mm-diameter dishes (Nunc). After 18 h in culture, cells were stimulated with LPS for 15 min and then lysed in kinase lysis buffer (buffer A containing 0.5% NP-40, 5 mM sodium β-glycerophosphate, and 0.1% 2-mercaptoethanol). Lysates were immunoprecipitated for 4 h with anti-TPL-2 antibody coupled to protein A-Sepharose beads, and the beads were washed four times in kinase lysis buffer and twice in kinase buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 5 mM β-glycerophosphate, 2 mM dithiothreitol, 0.1 mM Na3VO4, 10 mM MgCl2, 1 mM EGTA, 0.03% Brij 35). The beads were then resuspended in 25 μl of kinase buffer supplemented with 1 mM ATP, 6.5 μg of GST-MEK per ml, and 100 μg of GST-ERK per ml and incubated for 30 min at room temperature. After centrifugation, 2 μl of the supernatant was added to 48 μl of kinase buffer containing 0.33 mg of myelin basic protein (MBP) (Sigma) per ml, 0.1 mM ATP, and 2.5 μCi of [γ-32P]ATP (Amersham Biosciences) and incubated at room temperature for 10 min. The assay was terminated by adding 50 μl of 2× SDS-PAGE sample buffer, and labeled MBP was revealed by autoradiography after SDS-PAGE (12.5% acrylamide). Immunoprecipitated protein was eluted from anti-TPL-2 antibody beads with 0.2 M glycine (pH 2.5) and resolved by SDS-PAGE (10% acrylamide) and Western blotted.
To test the effect of ABIN-2-FL coexpression on TPL-2 MEK kinase activity, Myc-TPL-2 was isolated by immunoprecipitation from lysates of cotransfected 293 cells as described above. After the beads were washed, they were resuspended in 50 μl of kinase buffer containing 1 mM ATP plus 1 μg of kinase-inactive GST-MEK1(K207A) and incubated at 30 min at room temperature. The supernatant was removed, mixed with an equal volume of 2× SDS-PAGE sample buffer, and Western blotted after SDS-PAGE (10% acrylamide). MEK phosphorylation was determined by probing blots with anti-phospho-MEK-1/2 antibody. Immunoprecipitated Myc-TPL-2 was eluted with SDS-PAGE sample buffer from the remaining anti-Myc MAb beads and quantified by Western blotting.
RESULTS
Affinity purification of HA-p105(S927A).
To more fully understand the function and regulation of NF-κB1 p105, affinity purification was used to identify novel p105-associated proteins. To do this, HeLa S3 cells were stably transfected with a vector encoding HA-p105(S927A) and the C3.25 clone was selected. The C3.25 clone expressed relatively high levels of the transfected protein. HA-p105(S927A) contains a mutation of a critical serine residue in the p105 PEST region phosphorylated by the IKK complex and is thus resistant to signal-induced proteolysis (24). Higher levels of HA-p105(S927A) were obtained than wild-type HA-p105 (data not shown), presumably due to proteolysis of the latter protein triggered by constitutive IKK activity in HeLa S3 cells.
A two-step, sequential affinity purification methodology was used to isolate HA-p105(S927A) and associated proteins from a large-scale suspension culture of C3.25 cells. Protein was first isolated from cell lysates using an anti-HA MAb covalently linked to protein A-Sepharose beads. Bound protein was specifically eluted by incubation of washed beads with a 3x-HA peptide and then reimmunoprecipitated using an antibody directed against the C terminus of p105. Reimmunoprecipitated protein was eluted from beads using a low-pH buffer, and 1/200 of the protein was resolved by SDS-PAGE. Silver staining revealed a number of bands which were isolated from lysates of C3.25 cells but not from an equivalent number of control cells stably transfected with EV (Fig. 1A). Western blotting demonstrated that the major bands at approximately 100 and 50 kDa comigrated with HA-p105 and HA-p50, respectively (data not shown).
FIG. 1.

ABIN-2 copurifies with NF-κB1 p105. (A) Protein was purified from lysate of HeLa S3 cells stably transfected with HA-p105(S927A) or empty vector (EV) by sequential affinity purification using anti-HA MAb and anti-p105C antibody. Purified protein was resolved by SDS-PAGE (10% acrylamide) and revealed by silver staining. The positions of the identified proteins are shown. For mass spectroscopic analysis, the indicated 100-kDa regions (bands 1 and 2) and 50-kDa regions (bands 3 to 8) were excised as a series of adjacent slices numbered from high to low molecular weight from a replicate SDS-polyacrylamide gel (10% acrylamide) stained with colloidal Coomassie brilliant blue (not shown). Proteins in isolated bands were identified by MALDI mass spectroscopy of tryptic digests. (B) ABIN-2 amino acid sequence showing the deduced portions of peptides (in bold type) identified by MALDI mass spectroscopic analysis of band 7. Peptide coverage corresponded to 56% of the ABIN-2 amino acid sequence. (C) Lysates of HeLa S3 cells were immunoprecipitated with the indicated specific antibodies (IP Ab) or preimmune (PI) IgG. Isolated proteins were resolved by SDS-PAGE (10% acrylamide) and Western blotting. The specificity of blotting and immunoprecipitating antibodies is denoted as α (anti) followed by the name of the recognized antigen.
To identify isolated proteins, a fraction of the remaining eluted protein was concentrated and then resolved by SDS-PAGE (10% acrylamide). Only the major bands of approximately 100 and 50 kDa were sufficiently abundant to be visualized by colloidal Coomassie blue staining, permitting further analysis (data not shown). The 100-kDa region was excised in two adjacent slices (bands 1 and 2), whereas the 50-kDa region was excised in five adjacent slices (bands 3 to 8). Isolated protein bands were subjected to in-gel digestion, and aliquots of the digest supernatants were analyzed by MALDI mass spectroscopy. Masses of the resulting protonated peptides were used to search the NCBI nonredundant database. Band 1 was identified as NF-κB1 p105, whereas band 2 was found to contain both NF-κB1 p105 and NF-κB2 p100. NF-κB1 p50 was identified as the major component of bands 3 and 4. Bands 5 to 8 were found to contain both NF-κB1 p50 and ABIN-2 (29), which had not previously been linked to p105. These data indicate that NF-κB1 p50/HA-p50, NF-κB2 p100, and ABIN-2 copurify with HA-p105(S927A) and suggest that ABIN-2 is a novel p105-associated protein.
ABIN-2 specifically associates with both p105 and TPL-2.
Having identified ABIN-2 as a protein that copurifies with stably overexpressed HA-p105(S927A), it was important to determine whether ABIN-2 also interacted with p105 at physiological levels of both proteins. The endogenous proteins were immunoprecipitated from lysates of HeLa S3 cells, and the isolated proteins were Western blotted. ABIN-2 specifically copurified in anti-p105C immunoprecipitates, and conversely, p105 copurified in anti-ABIN-2 immunoprecipitates (Fig. 1C). Importantly, NF-κB2 p100, which is closely related to p105, did not coimmunoprecipitate with ABIN-2 (Fig. 1C), confirming the specificity of the p105/ABIN-2 association. As previously reported (21, 22), however, p100 was found to coimmunoprecipitate with p105, consistent with its copurification with HA-p105(S927A) (Fig. 1A). Thus, endogenous ABIN-2 specifically copurifies with p105 in a complex distinct from that containing p100.
Our previous studies demonstrated that the MAP 3-kinase TPL-2 is stoichiometrically associated with p105 in HeLa cells (3). Interestingly, immunoprecipitation of HeLa S3 lysates with anti-TPL-2 antibody revealed that ABIN-2 specifically copurified with TPL-2 and, conversely, that TPL-2 was specifically copurified in anti-ABIN-2 immunoprecipitates (Fig. 1C).
Since p105 and TPL-2 are associated, the previous experiments did not distinguish whether ABIN-2 interacts directly with both p105 and TPL-2 or with only one of these proteins. To address this question, 293 cells were transiently transfected with plasmids encoding C-terminally FLAG-tagged ABIN-2 (ABIN-2-FL) and either HA-tagged p105 (HA-p105) or Myc-tagged TPL-2 (Myc-TPL-2). Immunoprecipitation from transfected cell lysates and Western blotting revealed that ABIN-2-FL specifically coimmunoprecipitated with both HA-p105 and Myc-TPL-2 (Fig. 2A and B). In contrast, similar experiments indicated that ABIN-2-FL did not associate with either HA-p50 (Fig. 2A), the processed product of p105, or HA-p100 (data not shown).
FIG. 2.

Transfected ABIN-2-FL coimmunoprecipitates with both HA-p105 and Myc-TPL-2. (A and B) 293 cells were cotransfected with vectors encoding HA-p105, HA-p50, or Myc-TPL-2; with empty vector (EV) and ABIN-2-FL; or with EV alone as indicated. Anti-HA (α-HA), anti-Myc, and anti-FL immunoprecipitates (Ips) and cell lysates were Western blotted with the indicated antibodies. (C) 293 cells were cotransfected with vectors encoding HA-p105 or with EV and ABIN-2-FL. Cell lysates were cleared of endogenous TPL-2 by immunoprecipitation with anti-TPL-2 antibody and then reimmunoprecipitated with anti-HA MAb. Immunoprecipitates and lysates were Western blotted with the indicated antibodies. (D) 293 cells were cotransfected with the indicated expression vectors. Cell lysates were cleared of endogenous p105 by immunoprecipitation with anti-p105C antibody and then reimmunoprecipitated with anti-FL MAb to isolate ABIN-2-FL and associated Myc-TPL-2. Immunoprecipitates and lysates were Western blotted with the indicated antibodies.
It was possible that ABIN-2-FL complexing with HA-p105 was mediated via the association of endogenous TPL-2 with HA-p105. Therefore, TPL-2 was removed from lysates of 293 cells cotransfected with vectors encoding ABIN-2-FL and HA-p105 by sequential immunoprecipitation with anti-TPL-2 antibody. Removal of endogenous TPL-2 from 293 cell lysates did not reduce the level of ABIN-2-FL which coimmunoprecipitated with HA-p105 (Fig. 2C). Similarly, immunodepletion of endogenous p105 with anti-p105C antibody did not alter binding of ABIN-2-FL to Myc-TPL-2 (Fig. 2D). Thus, ABIN-2-FL can interact independently with HA-p105 and Myc-TPL-2.
HA-p105 increases the solubility of coexpressed ABIN-2-FL in NP-40 lysis buffer.
In the course of the previous experiments, it was noticed that the steady-state levels of transfected ABIN-2-FL detected by Western blot analysis of 1% NP-40 extracts were dramatically increased by coexpression with HA-p105 (Fig. 2A and 3A). By comparison with lysis using a more stringent buffer containing ionic detergents (RIPA buffer) and quantitation of ABIN-2-FL transcription by semiquantitative RT-PCR (Fig. 3A), it was apparent that this difference was due to increased extraction of ABIN-2-FL with 1% NP-40 buffer when ABIN-2-FL was coexpressed with HA-p105, rather than increased production of ABIN-2-FL protein. Subcellular fractionation experiments indicated that this was not due to alteration in the localization of ABIN-2-FL (data not shown). Rather, the data suggest that HA-p105 binding increases ABIN-2-FL solubility.
FIG. 3.

ABIN-2 preferentially interacts with a p105/TPL-2 complex. (A and B) Duplicate cultures of 293 cells were cotransfected with vectors encoding ABIN2-FL and HA-p105 or Myc-TPL-2 or with EV. Cell lysates were prepared from each duplicate culture set using either buffer A (1% NP-40) or RIPA buffer, as indicated. Lysates were resolved by SDS-PAGE (10% acrylamide) and Western blotting (top blots). HA-p105 and Myc-TPL-2 mRNA levels in total RNA were assayed by semiquantitative RT-PCR (bottom blots). The 18S rRNA amplicon was used as an internal control. (C) 293 cells were cotransfected with vectors encoding HA-p105 and TPL-2 individually or together. Transfected proteins were affinity purified from cell lysates, prepared in 1% NP-40 buffer A, using GST-ABIN-21-429 fusion protein coupled to glutathione-Sepharose. Isolated proteins were resolved by SDS-PAGE (10% acrylamide) and Western blotting. α, anti.
Steady-state levels of ABIN-2-FL were also dramatically increased by coexpression with Myc-TPL-2 (Fig. 2B and 3B). However, this increase was evident after cell extraction with 1% NP-40 buffer A or RIPA buffer, and semiquantitative PCR indicated that this was due to Myc-TPL-2 inducing the production of higher levels of ABIN-2-FL mRNA (Fig. 3B). Kinase-inactive Myc-TPL-2(D270A), which could bind to ABIN-2 (see Fig. 5D), had little effect on steady-state levels of coexpressed ABIN-2-FL protein detected in cell lysates (data not shown).
FIG. 5.

Mapping interacting regions for ABIN-2 on p105 and TPL-2. (A) Schematic diagram of HA-p105 mutants. The relative positions of the Rel homology domain (RHD), ankyrin repeats (ANK), death domain (DD), and PEST region are shown. The N and C termini of the wild-type (WT) HA-p105 protein (amino acids 1 to 968) are indicated. (B) 293 cells were transfected with vectors encoding wild-type (WT) and mutant forms of HA-p105. Cell lysates, prepared using 1% Brij 58 buffer A, were incubated with GST-ABIN-21-429 fusion protein or GST (control) coupled to glutathione-Sepharose beads. Affinity-purified protein was resolved by SDS-PAGE (10% acrylamide) and Western blotting. α, anti. (C) GST-p105497-968 fusion protein and GST (control) were coupled to glutathione-Sepharose beads and used to affinity purify ABIN-2-FL translated and labeled with [35S]methionine in vitro. Isolated protein was detected by autoradiography of SDS-8% acrylamide gels. (D) 293 cells were transfected with vectors encoding wild-type and mutant forms of Myc-TPL-2. GST-ABIN-21-429 was used as an affinity ligand to isolate protein from cell lysates prepared with 1% NP-40 buffer A. Isolated protein was resolved by SDS-PAGE (10% acrylamide) and Western blotting. (E) TPL-2398-467 peptide coupled to streptavidin-agarose beads was used as an affinity ligand to isolate ABIN-2-FL from lysates of transfected 293 cells. Bound protein was resolved by SDS-PAGE (10% acrylamide) and Western blotting.
ABIN-2 forms a ternary complex with p105 and TPL-2.
In HeLa cells, the majority of TPL-2 is complexed with p105 (3). To determine whether ABIN-2 can interact with a p105/TPL-2 complex rather than p105 or TPL-2 alone, GST-ABIN-2 fusion protein was used as an affinity ligand to isolate HA-p105 and TPL-2 from lysates of transiently transfected 293 cells. When expressed individually, GST-ABIN-2 interacted with TPL-2 but not HA-p105 (Fig. 3C). However, to allow the formation of a TPL-2/HA-p105 complex in vivo, coexpression of TPL-2 and HA-p105 facilitated GST-ABIN-2 pulldown of HA-p105, and the level of Myc-TPL-2 isolated was significantly increased. These data indicate that GST-ABIN-2 preferentially forms a ternary complex with HA-p105 and TPL-2.
Although GST-ABIN-2 did not form stable complexes with HA-p105 (Fig. 3C) or Myc-p105 (data not shown) when extracted from cells using 1% NP-40 detergent, interaction was clearly detected with HA-p105 when cell lysates were prepared using Brij 58, a milder detergent (see Fig. 5B). In contrast, TPL-2 or Myc-TPL-2 bound to GST-ABIN-2 after extraction with either detergent (Fig. 3C and 6B). These data suggest that the affinity of GST-ABIN-2 for TPL-2 is greater than that for HA-p105.
FIG. 6.

Mapping regions of ABIN-2 which interact with p105 and TPL-2. (A) Schematic diagram of recombinant GST-ABIN-2 fusion proteins. The positions of the ABIN homology domain (AHD) and the binding regions for TPL-2, p105, and A20 are shown. The N and C termini of the wild-type (WT) GST-ABIN-2 protein (amino acids 1 to 420) are indicated. (B and C) 293 cells were transfected with vectors encoding Myc-p105, Myc-TPL-2, or Myc-A20. Cell lysates were prepared using 1% Brij 58 buffer A and incubated with the indicated GST-ABIN-2 fusion proteins or GST (control) coupled to glutathione-Sepharose 4B. Affinity-purified protein was resolved by SDS-PAGE (10% acrylamide) and Western blotting. α, anti.
The majority of endogenous ABIN-2 is associated with TPL-2/p105 complexes.
In macrophages, TPL-2 is essential for LPS activation of the MEK/ERK MAP K pathway (9) and must interact with p105 to maintain its steady-state expression (31). To determine whether ABIN-2 is associated with TPL-2 and p105 in this physiologically relevant cell type, lysates were prepared from BMDMs. Immunoprecipitation and Western blot analysis confirmed that both p105 and TPL-2 copurified with ABIN-2 in these cells (Fig. 4A).
FIG. 4.

The majority of endogenous ABIN-2 forms a ternary complex with p105 and TPL-2. (A) BMDM lysate was immunoprecipitated with anti-ABIN-2 antibody (α-ABIN-2) (Ip Ab) or control preimmune rabbit IgG (PI). Isolated protein was resolved by SDS-PAGE (10% acrylamide) and Western blotting. (B to D) ABIN-2, p105, and TPL-2 were individually removed from lysates of BMDMs by immunoprecipitation with specific antibodies. Control preclearing was performed using preimmune rabbit IgG (PI). Precleared lysates were then reimmunoprecipitated (Re-Ip) with the indicated specific antibodies. Reimmunoprecipitated protein (B and D) and precleared cell lysate (C) was resolved by SDS-PAGE (10% acrylamide) and Western blotting.
Next, it was investigated whether an ABIN-2/TPL-2/p105 ternary complex exists in BMDMs. To do this, endogenous ABIN-2 was depleted from BMDM lysates by serial immunoprecipitation with anti-ABIN-2 antibody. ABIN-2 immunodepletion removed the majority of TPL-2 detected directly in cell lysates or after reimmunoprecipitation with anti-TPL-2 or anti-p105 antibodies (Fig. 4B and C). Immunodepletion of p105 using anti-p105C antibody also removed a substantial fraction of ABIN-2 (Fig. 4C) and cleared TPL-2 from lysates, similar to the results of earlier experiments with HeLa cells (3). Together, these data imply that TPL-2 is present in a complex with both p105 and ABIN-2 in BMDMs. Consistent with this conclusion, anti-TPL-2 antibody immunodepletion removed only a small fraction of total p105 from cell lysates (Fig. 4C) but the majority of ABIN-2-associated p105 (Fig. 4D). Significantly, immunodepletion of TPL-2 removed a substantial fraction of total ABIN-2 detected directly in lysates (Fig. 4 C) or after reimmunoprecipitation with anti-ABIN-2 antibody (Fig. 4D). Thus, the majority of ABIN-2 is associated with p105/TPL-2 complexes and the majority of TPL-2 is associated with ABIN-2 in macrophages. However, only a small fraction of total cellular p105 participates in these ternary complexes (Fig. 4C).
Mapping the regions involved in interaction of ABIN-2 with p105 and TPL-2.
To further characterize the association between ABIN-2 and the p105/TPL-2 complex, the interacting regions of each protein were mapped. To analyze the interaction of p105 with ABIN-2, 293 cells were transiently transfected with vectors encoding a panel of deletion and point mutants of HA-p105 (Fig. 5A) (2). Lysates prepared using buffer A containing 1% Brij 58 detergent were then incubated with GST-ABIN-21-429 fusion protein in a pulldown assay. Western blotting of isolated proteins demonstrated binding of GST-ABIN-21-429 to wild-type HA-p105 (Fig. 5B). Binding was significantly decreased by deletion of the PEST region (HA-p1051-892) and completely lost by further deletion to remove the death domain (DD) (HA-p1051-801). Binding was also eliminated by internal deletion of the DD (HA-p105ΔDD) or functional inactivation of the DD by point mutation (HA-p105L841A) (2). Deletion of the region N terminal to the ankyrin repeats that binds to the TPL-2 C terminus (HA-p105Δ497-538) also slightly reduced binding to GST-ABIN-2. Thus, the p105 DD is essential for ABIN-2 binding to p105, but optimal association also requires the PEST region and, to a lesser extent, p105 residues 497 to 538. Previous experiments have indicated that maximal interaction of TPL-2 with p105 requires the p105 DD and p105 residues 497 to 538 but does not involve the p105 PEST region (2). Thus, ABIN-2 interacts with regions of p105 similar to those of TPL-2 but not identical. A pulldown experiment with GST-p105497-968 protein (2) confirmed that the isolated C-terminal half of p105 was sufficient for binding to ABIN-2-FL (Fig. 5C).
Activation of the oncogenic potential of TPL-2 requires deletion of its C terminus (5). In an earlier study, it was demonstrated that the TPL-2 C terminus forms a high-affinity interaction with a region N terminal to the ankyrin repeats of p105 (2). To determine whether the TPL-2 C terminus is also involved in interaction with ABIN-2, 293 cells were transfected with vectors encoding Myc-TPL-2 and Myc-TPL-2ΔC. Pulldown assays revealed that the TPL-2 C terminus was required for interaction with GST-ABIN-21-429 (Fig. 5D). The degree of binding of Myc-TPL-2(D270A) to GST-ABIN-21-429 was similar to that of wild-type Myc-TPL-2, indicating that its kinase activity is not required for TPL-2/ABIN-2 interaction. In contrast, previous experiments have indicated that the D270A mutation significantly decreases the interaction of TPL-2 with p105 (2). A pulldown assay with biotinylated TPL-2398-467 peptide (2) coupled to streptavidin-agarose beads demonstrated that the isolated TPL-2 C terminus is sufficient for binding to ABIN-2-FL (Fig. 5E).
A fragment comprising amino acids 251 to 429 of ABIN-2 contains the A20-binding and NF-κB inhibitory functions of ABIN-2 (29). To determine whether p105 and TPL-2 interact with the same region of ABIN-2, GST-ABIN-21-250 and GST-ABIN-2251-429 fusion proteins were assayed for their ability to interact with Myc-p105 or Myc-TPL-2 in pulldown assays. Both Myc-p105 and Myc-TPL-2 bound to GST-ABIN-21-250 but not GST-ABIN-2251-429 (Fig. 6B). In contrast, Myc-A20 interacted with GST-ABIN-2251-429 but not GST-ABIN-21-250 (Fig. 6B), consistent with previously published results (29). Thus, p105 and TPL-2 interact with a region of ABIN-2 distinct from A20.
To more finely map the region of the ABIN-2 N-terminal half involved in interaction with Myc-TPL-2 and Myc-p105, additional GST-ABIN-2 fusion proteins were generated (Fig. 6A). Pulldown experiments revealed that Myc-TPL-2 bound to a region containing amino acids 194 to 250 of ABIN-2 (Fig. 6C). Homology searches did not reveal any close similarity between this region and other proteins in the database. In contrast, Myc-p105 interacted only with the entire fragment of ABIN-2 (amino acids 1 to 250) (Fig. 6C). These data indicate that the interactions of p105 and TPL-2 with ABIN-2 are distinct, consistent with the earlier conclusion that p105 and TPL-2 can bind to ABIN-2 independently (Fig. 3A and B).
ABIN-2 is required to maintain the metabolic stability of TPL-2 protein.
Previous studies have indicated that p105 binding to TPL-2 is required to stabilize TPL-2 protein and maintain its steady-state levels in both macrophages and fibroblasts (2, 31). Since TPL-2 is present in a ternary complex with p105 and ABIN-2 in cells, it was of interest to determine whether TPL-2 protein stability was also influenced by ABIN-2 binding. To investigate this, siRNA-mediated gene suppression was used to deplete endogenous ABIN-2 expression in HeLa S3 cells. Western blotting of cell lysates confirmed that ABIN-2 siRNA, but not an irrelevant control siRNA, significantly reduced steady-state levels of ABIN-2 protein (Fig. 7A, top blots). TPL-2 protein levels were also strikingly reduced by ABIN-2 siRNA treatment. ABIN-2 depletion by RNA interference in 293 cells similarly reduced steady-state levels of TPL-2 protein (Fig. 7B, top blots). Semiquantitative RT-PCR demonstrated that ABIN-2 depletion did not alter steady-state levels of TPL-2 mRNA in HeLa cells (Fig. 7A, bottom blots) or 293 cells (Fig. 7B, bottom blots), suggesting that TPL-2 protein levels were downregulated posttranscriptionally. ABIN-2 depletion did not affect steady-state p105 levels in either HeLa (Fig. 7A) or 293 cells (Fig. 7B). In addition, the p105/p50 ratio was not affected by ABIN-2 knockdown (Fig. 7C), suggesting that ABIN-2 is not required for constitutive processing of p105 to p50.
FIG. 7.

Depletion of ABIN-2 by RNA interference dramatically reduces steady-state levels of TPL-2. (A and B) ABIN-2 expression in HeLa S3 cells (A) and 293 cells (B) was decreased by siRNA treatment (−ABIN-2). Control cells were treated with an irrelevant siRNA oligonucleotide pair (+ABIN-2). Cell lysates were resolved by SDS-PAGE (10% acrylamide) and Western blotting (top blots). TPL-2 mRNA levels in total RNA were assayed by semiquantitative RT-PCR (bottom blots). The 18S rRNA amplicon was used as an internal control. α, anti. (C) ABIN-2 was depleted by RNA interference in HeLa and 293 cells as described above for panels A and B. Expression of p105, p50, or ABIN-2 was determined by Western blotting of cell lysates. The position of the ABIN-2 band in the Western blot of HeLa cell lysates is indicated by an asterisk. (D) 293 cells were cotransfected with vectors encoding TPL-2 and ABIN-2-FL or with EV. Lysates were resolved by SDS-PAGE (10% acrylamide) and Western blotting (top blots). In duplicate cultures, transfected TPL-2 mRNA levels were determined by semiquantitative RT-PCR (bottom blots). 18S rRNA was used as an internal control. (E) 293 cells were cotransfected with expression vectors encoding TPL-2 and ABIN-2-FL or with EV. After 24 h, cells were metabolically pulse-labeled with [35S]methionine-[35S]cysteine (30 min) and then chased for the times indicated. Anti-TPL-2 immunoprecipitates were resolved by SDS-PAGE (8% acrylamide) and visualized by fluorography. Amounts of immunoprecipitated labeled TPL-2 were quantified by densitometry (n = 3).
The low level of TPL-2 expression in both HeLa and 293 cells prevented its detection after metabolic labeling with [35S]methionine and [35S]cysteine. Therefore, it was not possible to determine by pulse-chase metabolic labeling whether ABIN-2 knockdown increased TPL-2 turnover. As an alternative approach to investigate whether ABIN-2 binding modulates TPL-2 stability, the effect of ABIN-2-FL binding to TPL-2 was investigated by transient transfection of 293 cells. ABIN-2-FL coexpression significantly increased steady-state levels of cotransfected TPL-2 protein over those of cells cotransfected with EV (Fig. 7D, top blots). However, semiquantitative RT-PCR revealed that ABIN-2-FL did not alter the levels of cotransfected TPL-2 mRNA (Fig. 7D, bottom blots). These data suggest that ABIN-2-FL binding stabilizes TPL-2 protein. Consistent with this conclusion, pulse-chase metabolic labeling experiments revealed that ABIN-2-FL increased the half-life of cotransfected TPL-2 (Fig. 7E). Together, the data in this section indicate that TPL-2 must interact with ABIN-2 to maintain TPL-2 metabolic stability.
Steady-state levels of ABIN-2 protein are substantially reduced in NF-κB1-deficient cells.
TPL-2 protein levels are severely reduced in NF-κB1-deficient cells, as p105 binding is required to maintain the metabolic stability of TPL-2 protein (2, 31). Since the majority of cellular ABIN-2 is associated with p105, the effect of p105 deficiency on steady-state levels of ABIN-2 was investigated. To do this, ABIN-2 was immunoprecipitated from lysates of wild-type (nf-κb1+/+) and nf-κb1−/− 3T3 fibroblasts. Western blotting revealed that ABIN-2 was not detected in the p105-deficient cells, although it was clearly present in wild-type cells (Fig. 8A). However, semiquantitative RT-PCR indicated that ABIN-2 mRNA levels were similar in both cell lines (Fig. 8B). ABIN-2 protein levels were also severely reduced in primary nf-κb1−/− BMDMs compared with nf-κb1+/+ control cells (Fig. 8C). Thus, expression of NF-κB1 p105 (or p50) is required to maintain steady-state levels of endogenous ABIN-2 protein. Consequently, lack of TPL-2 expression in nf-κb1−/− cells is likely to be caused by deficiency of both p105 and ABIN-2 (2, 31).
FIG. 8.

nf-κb1−/− cells have dramatically reduced levels of ABIN-2 protein. (A and C) Lysates from nf-κb1−/− and wild-type (nf-κb1+/+) 3T3 fibroblasts (A) and BMDMs (C) were immunoprecipitated with the indicated antibodies. Immunoprecipitates (Ip) and cell lysates were resolved by SDS-PAGE (10% acrylamide) and Western blotting. α, anti. (B) ABIN-2 mRNA levels in total RNA isolated from nf-κb1−/− and wild-type 3T3 fibroblasts were assayed by semiquantitative PCR. The 18S rRNA amplicon was used as an internal control.
ABIN-2 is not associated with active TPL-2 in LPS-stimulated BMDMs.
The observation that the majority of TPL-2 in BMDMs is associated with ABIN-2 (Fig. 4B and C) suggested that ABIN-2 might function in the TLR4/TPL-2/MEK/ERK signaling pathway (9). It has previously been shown that LPS induces the proteolysis of p105 and the long form of TPL-2 (M1-TPL-2) (8, 31). In initial experiments, the effect of LPS stimulation on ABIN-2 stability was determined in BMDMs. Stimulation with LPS for 60 min induced degradation of a substantial fraction of ABIN-2 (Fig. 9A). LPS activation of endogenous MEK phosphorylation preceded degradation of ABIN-2 (Fig. 9A), suggesting that ABIN-2 proteolysis may be involved in downregulation of the signaling pathway. LPS also induced proteolysis of M1-TPL-2 and p105, as expected (8, 31). Pretreatment of cells with proteasome inhibitor MG132 blocked LPS-stimulated proteolysis of ABIN-2, M1-TPL-2, and p105 (Fig. 9B). Thus, each of the components of the ABIN-2/TPL-2/p105 ternary complex is proteolyzed by the proteasome after LPS stimulation of BMDMs.
FIG. 9.

ABIN-2 is not associated with active TPL-2 in LPS-stimulated BMDMs. (A and B) BMDMs from wild-type BALB/C mice were stimulated for the indicated times with LPS or left unstimulated. In panel B, BMDMs were preincubated with MG132 or vehicle control for 30 min prior to stimulation with LPS. Lysates were resolved by SDS-PAGE (10% acrylamide) and Western blotting. α, anti. (C) BMDMs were stimulated for 15 min with LPS or left unstimulated. Lysates were immunoprecipitated (Ip) with the indicated antibodies, and associated MEK kinase activity was determined by coupled MEK/ERK kinase assay (KA). Labeled MBP was visualized by autoradiography after SDS-PAGE (10% acrylamide). Levels of immunoprecipitated proteins were determined by Western blotting. The LPS-induced mobility shift of M1-TPL-2 is due to phosphorylation (data not shown). (D) BMDM lysates were depleted of ABIN-2 by immunoprecipitation with anti-ABIN-2 antibody. ABIN-2 precleared lysates and control untreated lysates were then Western blotted. (E) 293 cells were cotransfected with vectors encoding Myc-TPL-2 and ABIN-2-FL or with EV. The MEK kinase activity of immunoprecipitated Myc-TPL-2 was determined using GST-MEK1(K207A) protein as a substrate. Phosphorylation was assayed by Western blotting of reaction mixtures and probing with anti-phospho-MEK-1/2 antibody (α-P-MEK). Western blotting of anti-Myc immunoprecipitates demonstrated that similar amounts of TPL-2 were assayed in each reaction mixture.
LPS stimulation activates the MEK kinase activity of TPL-2 in BMDMs (31), consistent with its essential role in inducing MEK phosphorylation in these cells (9). To investigate whether ABIN-2 is associated with active TPL-2, ABIN-2 was immunoprecipitated from LPS-stimulated BMDMs and the MEK kinase activity of associated TPL-2 determined in a coupled MEK/ERK kinase assay (23). Although large amounts of TPL-2 were present in anti-ABIN-2 antibody immunoprecipitates, no associated MEK kinase was detected with or without LPS stimulation (Fig. 9C). In contrast, substantial MEK kinase activity was evident in anti-TPL-2 immunoprecipitates from lysates of LPS-stimulated cells, as expected (31). Thus, ABIN-2 is not associated with the active pool of TPL-2, suggesting that TPL-2 might dissociate from ABIN-2 after LPS stimulation. To investigate this possibility, lysates of BMDMs were precleared of ABIN-2 by immunoprecipitation and then Western blotted for TPL-2. Similar to previous results (Fig. 4C), minimal ABIN-2-free TPL-2 was detected in unstimulated cells (Fig. 9D). However, LPS stimulation induced a substantial increase of both long and short forms of TPL-2 in the ABIN-2-depleted lysate. Thus, LPS-stimulated activation of TPL-2 (Fig. 9C) correlates with its release from ABIN-2.
No MEK kinase activity was detected in anti-p105 immunoprecipitates from LPS-stimulated BMDM lysates (Fig. 9C), consistent with published data showing that p105 functions as an inhibitor of TPL-2 (2, 31). To determine whether ABIN-2 might also inhibit TPL-2 MEK kinase activity, Myc-TPL-2 was transiently coexpressed with ABIN-2-FL in 293 cells and then isolated by immunoprecipitation with anti-Myc MAb. Myc-TPL-2 MEK kinase activity was not affected by coexpression with ABIN-2-FL (Fig. 9E), although ABIN-2-FL was clearly associated with Myc-TPL-2. In contrast, HA-p105 coexpression dramatically inhibited Myc-TPL-2 activity, as reported previously (2). Thus, ABIN-2 does not appear to function as an inhibitor of TPL-2 MEK kinase activity.
DISCUSSION
This study identifies ABIN-2 as a protein which interacts with both NF-κB1 p105 and TPL-2. In BMDMs, the majority of ABIN-2 and TPL-2 is shown to be associated with p105 in a ternary complex (Fig. 4). ABIN-2 is demonstrated to be essential to maintain steady-state protein levels of TPL-2 but not p105 (Fig. 7A and B). Furthermore, NF-κB1 p105 (p50) is required to maintain protein levels of both ABIN-2 (Fig. 8) and TPL-2 (2, 31). Thus, in unstimulated cells, ABIN-2 and TPL-2 appear to be confined to a ternary complex with p105 and are not present in isolation.
Binding to p105 dramatically increases the solubility of ABIN-2 (Fig. 3A), suggesting that correct folding of ABIN-2 may require its association with p105. In addition, maintenance of steady-state levels of ABIN-2 protein, but not mRNA, require p105 (or p50) expression (Fig. 8). While these latter data do not rule out a role for TPL-2 in controlling ABIN-2 stability (2, 31), they nevertheless demonstrate a close relationship between ABIN-2 and NF-κB1 p105. These data also raise the possibility that the phenotype of nf-κb1−/− mice (26) may result not only from the lack of p105 or p50, but also from deficiency of both TPL-2 (2, 31) and ABIN-2 proteins.
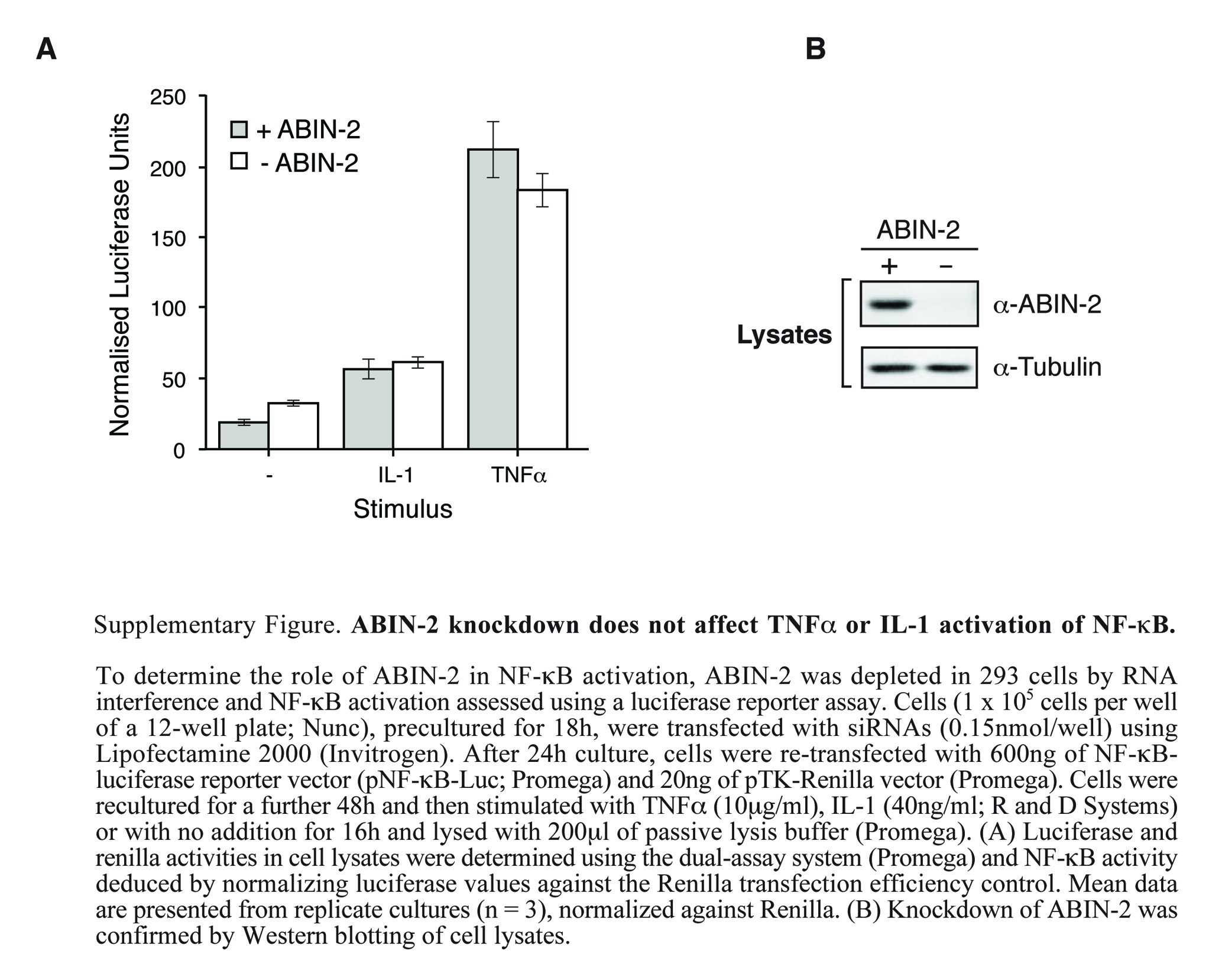
ABIN-2 was originally isolated in a two-hybrid screen using the zinc finger protein A20 as a bait (29). A20 is a primary response gene that is induced by a number of stimuli which activate NF-κB, including TNF-α, interleukin 1, and LPS (4). Analysis of A20-deficient mice has indicated that A20 is required for termination of TNF-α-induced NF-κB activation and for blockade of TNF-α-mediated apoptosis (20). Overexpression of ABIN-2 in 293 cells inhibits NF-κB activation by TNF-α, and it has been suggested that ABIN-2 may contribute to the NF-κB inhibitory function of A20 (29). Surprisingly, however, ABIN-2 knockdown by RNA interference does not affect TNF-α or interleukin 1 activation of an NF-κB reporter gene in 293 cells (see Fig. S1 in the supplemental material). Therefore, it is unclear whether ABIN-2 acts physiologically to inhibit NF-κB activation.
TPL-2 is the critical MEK kinase required for TLR4 stimulation of the MEK/ERK MAP K pathway in LPS-stimulated BMDMs (9). In resting BMDMs, TPL-2 is in a complex with p105, which inhibits TPL-2 MEK kinase activity (31). Consequently, it has been proposed that LPS may activate TPL-2 by promoting its release from p105 (31). The majority of TPL-2 in these cells is also complexed with ABIN-2 (Fig. 4). Together with the observed requirement for ABIN-2 to maintain steady-state levels of TPL-2 protein in both HeLa and 293 cells (Fig. 7A and B), this strongly suggests that ABIN-2 is involved in the regulation of TPL-2 function. However, ABIN-2 is not associated with the pool of TPL-2 that activates MEK in LPS-stimulated BMDMs (Fig. 9C), and LPS-induced TPL-2 activation correlates with its release from ABIN-2 (Fig. 9D). The importance of TPL-2 release from ABIN-2 is unclear, but ABIN-2 does not appear to function as an inhibitor of TPL-2 MEK kinase activity (Fig. 9E) similar to p105 (2, 31). Together, these data raise the possibility that ABIN-2 might function upstream of TPL-2 in the TLR4 signaling pathway that regulates TPL-2 activation.
ABIN-2 contains a region (residues 298 to 320) (Fig. 6A), distinct from the TPL-2/p105 binding regions (Fig. 6B and C), which has homology with regions in both ABIN-1 and the noncatalytic component of the IKK complex, NEMO (13). These homologous regions have been termed ABIN homology domains (AHDs). Interestingly, the AHD of NEMO overlaps with a region required for its oligomerization, which is essential for NEMO to couple activating signals from TLR4 to the IKK complex (28). Given this similarity with NEMO, it is possible that ABIN-2 may have an analogous function, perhaps acting as an adapter which couples TPL-2 to signaling pathways downstream of TLR4.
A20 binds to a region of ABIN-2 that is distinct from regions bound by TPL-2 and p105 (Fig. 6). Since LPS triggers upregulation of A20 (4), this raises the possibility that A20 may be recruited to the ABIN-2/TPL-2/p105 complex in LPS-stimulated macrophages. Thus, A20 could potentially regulate TLR4 activation of the TPL-2/MEK/ERK MAP K signaling pathway in these cells. In an analogous fashion, the NF-κB inhibitory function of A20, which is mediated upstream of IKK (20), is thought to involve its direct recruitment to the IKK complex via NEMO (32).
In future research, analysis of ABIN-2 knockout mice will be important to establish the physiological function of ABIN-2 in LPS signaling. However, the results of the present study indicate that interpretation of such experiments will be complicated by the requirement for ABIN-2 expression to maintain steady-state levels of TPL-2 protein.
Supplementary Material
Acknowledgments
We thank David Baltimore, Alexander Hoffmann, Alain Israel, and Phillip Tsichlis for reagents used in this study. TNF-α, which was produced by Genentech, Inc. (San Francisco, Calif.), was obtained from the Centralised Facility for AIDS Reagents (supported by EU Programme EVA/MRC and the United Kingdom Medical Research Council). We are grateful to Lee Johnston for critical reading of the manuscript, to the Large Scale Lab for culture of C3.25 cells, and to other members of the Ley laboratory for their support during the course of this work.
This study was supported in part by the United Kingdom Medical Research Council and the Arthritis Research Campaign project grants L0536 (V.L. and S.J.W.) and L0549 (A.S.).
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Beinke, S., M. P. Belich, and S. C. Ley. 2002. The death domain of NF-κB1 p105 is essential for signal-induced p105 proteolysis. J. Biol. Chem. 277:24162-24168. [DOI] [PubMed] [Google Scholar]
- 2.Beinke, S., J. Deka, V. Lang, M. P. Belich, P. A. Walker, S. Howell, S. J. Smerdon, S. J. Gamblin, and S. C. Ley. 2003. NF-κB p105 negatively regulates TPL-2 MEK kinase activity. Mol. Cell. Biol. 23:4739-4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belich, M. P., A. Salmeron, L. H. Johnston, and S. C. Ley. 1999. TPL-2 kinase regulates the proteolysis of the NF-κB inhibitory protein NF-κB1 p105. Nature 397:363-368. [DOI] [PubMed] [Google Scholar]
- 4.Beyaert, R., K. Heyninck, and S. van Huffel. 2000. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-κB-dependent gene expression and apoptosis. Biochem. Pharmacol. 60:1143-1151. [DOI] [PubMed] [Google Scholar]
- 5.Ceci, J. D., C. P. Patriotis, C. Tsatsanis, A. M. Makris, R. Kovatch, D. A. Swing, N. A. Jenkins, P. N. Tsichlis, and N. G. Copeland. 1997. TPL-2 is an oncogenic kinase that is activated by carboxy-terminal truncation. Genes Dev. 11:688-700. [DOI] [PubMed] [Google Scholar]
- 6.Chang, L., and M. Karin. 2001. Mammalian MAP kinase signalling cascades. Nature 410:37-40. [DOI] [PubMed] [Google Scholar]
- 7.Coope, H. J., P. G. P. Atkinson, B. Huhse, M. P. Belich, J. Janzen, M. Holman, G. G. B. Klaus, L. H. Johnston, and S. C. Ley. 2002. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 21:5375-5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donald, R., D. W. Ballard, and J. Hawiger. 1995. Proteolytic processing of NF-κB/IκB in human monocytes. J. Biol. Chem. 270:9-12. [DOI] [PubMed] [Google Scholar]
- 9.Dumitru, C. D., J. D. Ceci, C. Tsatsanis, D. Kontoyiannis, K. Stamatakis, J.-H. Lin, C. Patriotis, N. A. Jenkins, N. G. Copeland, G. Kollias, and P. N. Tsichlis. 2000. TNFα induction by LPS is regulated posttranscriptionally via a TPL2/ERK-dependent pathway. Cell 103:1071-1083. [DOI] [PubMed] [Google Scholar]
- 10.Fries, K. L., W. E. Miller, and N. Raab-Traub. 1999. The A20 protein interacts with the Epstein-Barr virus latent membrane protein 1 (LMP1) and alters the LMP1/TRAF1/TRADD complex. Virology 264:159-166. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh, S., M. J. May, and E. B. Kopp. 1998. NF-κB and Rel proteins: evolutionary conserved mediators of immune responses. Annu. Rev. Immunol. 16:225-260. [DOI] [PubMed] [Google Scholar]
- 12.Heissmeyer, V., D. Krappmann, E. N. Hatada, and C. Scheidereit. 2001. Shared pathways of IκB kinase-induced SCFβTrCP-mediated ubiquitination and degradation for the NF-κB precursor p105 and IκBa. Mol. Cell. Biol. 21:1024-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heyninck, K., M. M. Kreike, and R. Beyaert. 2003. Structure-function analysis of the A20-binding inhibitor of NF-κB activation, ABIN-1. FEBS Lett. 536:135-140. [DOI] [PubMed] [Google Scholar]
- 14.Hochstrasser, D. F., and C. R. Merril. 1988. ′Catalysts' for polyacrylamide gel polymerization and detection of proteins by silver staining. Appl. Theor. Electrophor. 1:35-40. [PubMed] [Google Scholar]
- 15.Holloway, A. F., S. Rao, and M. F. Shannon. 2001. Regulation of cytokine gene transcription in the immune system. Mol. Immunol. 38:567-580. [DOI] [PubMed] [Google Scholar]
- 16.Ishikawa, H., E. Claudio, D. Dambach, C. Raventos-Suarez, C. Ryan, and R. Bravo. 1998. Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-κB1) but expressing p50. J. Exp. Med. 187:985-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabouridis, P. S., A. I. Magee, and S. C. Ley. 1997. S-acylation of LCK protein tyrosine kinase is essential for its signalling function in T lymphocytes. EMBO J. 16:4983-4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18:621-663. [DOI] [PubMed] [Google Scholar]
- 19.Lang, V., J. Janzen, G. Z. Fischer, Y. Soneji, S. Beinke, A. Salmeron, H. Allen, R. T. Hay, Y. Ben-Neriah, and S. C. Ley. 2003. βTrCP-mediated proteolysis of NF-κB1 p105 requires phosphorylation of p105 serines 927 and 932. Mol. Cell. Biol. 23:402-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee, E. G., D. L. Boone, S. Chai, S. L. Libby, M. Chien, J. P. Lodolce, and A. Ma. 2000. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 289:2350-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercurio, F., J. A. DiDonato, C. Rosette, and M. Karin. 1993. p105 and p98 precursor proteins play an active role in NF-κB-mediated signal transduction. Genes Dev. 7:705-718. [DOI] [PubMed] [Google Scholar]
- 22.Rice, N. R., M. L. MacKichan, and A. Israel. 1992. The precursor of NF-κB p50 has IκB-like functions. Cell 71:243-253. [DOI] [PubMed] [Google Scholar]
- 23.Salmeron, A., T. B. Ahmad, G. W. Carlile, D. Pappin, R. P. Narsimhan, and S. C. Ley. 1996. Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 15:817-826. [PMC free article] [PubMed] [Google Scholar]
- 24.Salmeron, A., J. Janzen, Y. Soneji, N. Bump, J. Kamens, H. Allen, and S. C. Ley. 2001. Direct phosphorylation of NF-κB p105 by the IκB kinase complex on serine 927 is essential for signal-induced p105 proteolysis. J. Biol. Chem. 276:22215-22222. [DOI] [PubMed] [Google Scholar]
- 25.Schneider, C., R. A. Newman, D. R. Sutherland, U. Asser, and M. F. Greaves. 1982. A one-step purification of membrane proteins using a high efficiency immunomatrix. J. Biol. Chem. 257:10766-10769. [PubMed] [Google Scholar]
- 26.Sha, W. C., H.-C. Liou, E. I. Tuomanen, and D. Baltimore. 1995. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell 80:321-330. [DOI] [PubMed] [Google Scholar]
- 27.Takeda, K., and S. Akira. 2004. TLR signaling pathways. Semin. Immunol. 16:3-9. [DOI] [PubMed] [Google Scholar]
- 28.Tegethoff, S., J. Behlke, and C. Scheidereit. 2003. Tetrameric oligomerization of IκB kinase γ (IKKγ) is obligatory for IKK complex activity and NF-κB activation. Mol. Cell. Biol. 23:2029-2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Huffel, S., F. Delaei, K. Heyninck, D. de Valck, and R. Beyaert. 2001. Identification of a novel A20-binding inhibitor of nuclear factor-κB activation termed ABIN-2. J. Biol. Chem. 276:30216-30223. [DOI] [PubMed] [Google Scholar]
- 30.Warren, M. K., and S. N. Vogel. 1985. Bone marrow-derived macrophages: development and regulation of differentiation markers by colony-stimulating factor and interferons. J. Immunol. 134:982-989. [PubMed] [Google Scholar]
- 31.Waterfield, M. R., M. Zhang, L. P. Norman, and S.-C. Sun. 2003. NF-κB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the TPL-2 kinase. Mol. Cell 11:685-694. [DOI] [PubMed] [Google Scholar]
- 32.Zhang, S. Q., A. Kovalenko, G. Cantarella, and D. Wallach. 2000. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKγ) upon receptor stimulation. Immunity 12:301-311. [DOI] [PubMed] [Google Scholar]
- 33.Zhu, W., J. S. Downey, J. Gu, F. D. Padova, H. Gram, and J. Han. 2000. Regulation of TNF expression by multiple mitogen-activated protein kinase pathways. J. Immunol. 164:6349-6358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}