

Abstract
As glioma cells infiltrate the brain they become associated with various microanatomic brain structures such as blood vessels, white matter tracts, and brain parenchyma. How these distinct invasion patterns coordinate tumor growth and influence clinical outcomes remain poorly understood. We have investigated how perivascular growth affects glioma growth patterning and response to antiangiogenic therapy within the highly vascularized brain. Orthotopically implanted rodent and human glioma cells are shown to commonly invade and proliferate within brain perivascular space. This form of brain tumor growth and invasion is also shown to characterize de novo generated endogenous mouse brain tumors, biopsies of primary human glioblastoma (GBM), and peripheral cancer metastasis to the human brain. Perivascularly invading brain tumors become vascularized by normal brain microvessels as individual glioma cells use perivascular space as a conduit for tumor invasion. Agent-based computational modeling recapitulated biological perivascular glioma growth without the need for neoangiogenesis. We tested the requirement for neoangiogenesis in perivascular glioma by treating animals with angiogenesis inhibitors bevacizumab and DC101. These inhibitors induced the expected vessel normalization, yet failed to reduce tumor growth or improve survival of mice bearing orthotopic or endogenous gliomas while exacerbating brain tumor invasion. Our results provide compelling experimental evidence in support of the recently described failure of clinically used antiangiogenics to extend the overall survival of human GBM patients.
“Et pour arriver a une meilleure compréhension biologique des gliomes, l’analyse des permiers débuts des processus gliomateux nous semble, pour le moment, plus importante que celle des manifestations tardives.”
“La croissance périvasculaire joue un grand role, dans beaucoup de gliomes, comme phénomène de croissance primitive.”
“And to achieve a better biological understanding of gliomas, we believe that the analysis of the earliest stages of glioma formation, for the moment, are more important than the study of late manifestations.”
“Perivascular growth plays an important role in many gliomas as a phenomenon of early growth.”
Hans Joachim Scherer, 1937
Discovery is to see what everybody else has seen, and to think what nobody else has thought.
Albert Szent-Gyorgyi
Introduction
Gliomas are brain cancers arising from transformed glial cells. Of all gliomas, World Health Organization grade IV glioblastoma (GBM) is the most prevalent and aggressive [1]. The diffusely invasive nature of GBM precludes its complete surgical resection, which inevitably leads to tumor recurrence and patient death within 15 to 21 months [2].
In the 1930s, Hans Joachim Scherer classified the pathology of malignant glioma growth and invasion by examining several hundred post-mortem brains of GBM patients. Scherer discovered that malignant glioma cells are preferentially associated with four distinct microanatomic structures: white matter fiber bundles, perineuronal spaces, the subpial space, and brain microvessels [3], [4]. Collectively, these infiltrative tumor satellites have become known as the “secondary structures of Scherer”. Despite his histopathologic observations, Scherer’s work was unable to experimentally test the causes and biologic consequences of these various forms of glioma growth and invasion. More recently, others have demonstrated the occurrence of perivascular invasion in multiple experimental brain tumor models [5], [6], [7], and it has been shown that perivascular invasion increases in vascular endothelial growth factor (VEGF)–deficient glioma cells [8], [9] and brain tumor xenografts treated with anti-VEGF blocking antibodies [10], [11], [12], [13]. Clinical GBMs resistant to bevacizumab therapy also demonstrate the tendency toward perivascular invasion [14]. A better understanding of the cellular and molecular mechanisms governing the behavior of glioma growth and invasion will be required for the development of effective anti-GBM therapies.
How the behavior of individual glioma cells determines clinically relevant macroscopic tumor growth remains poorly understood. We demonstrate that orthotopic glioma cells (and glioma stem cells) from mice, rats, and humans grow perivascularly throughout tumor progression (i.e., from earliest stages to death of the host). We also observe perivascular growth in human biopsies of primary GBM, genetically induced endogenous mouse gliomas, and peripheral metastasis to the human brain. Perivascular growth causes such tumors to become autovascularized as they continuously grow along preexisting normal brain microvessels.
Agent-based computational modeling predicted that the perivascular growth pattern was independent of neoangiogenesis. We tested this prediction experimentally by blocking angiogenic signaling using antibodies targeting the VEGF-A signaling axis. VEGF-A inhibitors failed to curb progressive tumor growth and extend median survival in multiple glioma models. Our data provide a novel interpretation and understanding of perivascular brain tumor growth and invasion as a VEGF-independent mechanism of tumor vascularization, which helps explain why neoangiogenesis is dispensable for brain tumor progression and why clinical antiangiogenics fail to meaningfully extend patient survival in GBM. We propose that novel therapeutics ought to specifically target normal brain blood vessels used by perivascularly invading glioma cells to sustain progressive tumor growth.
Materials and Methods
Malignant Glioma Cell Lines and Patient-Derived GBM Samples
The mouse GL26 glioma cell line was obtained from the National Cancer Institute (Bethesda, MD). The rat CNS-1 glioma cell line was kindly provided by William Hickey (Department of Pathology, Dartmouth Medical Center, Lebanon, NH). HF2303 primary human GBM cancer stem cells were provided by Dr Tom Mikkelsen, MD (Department of Neurology, Henry Ford Hospital, Detroit, MI). The human U251 glioma cell line was provided by the National Institutes of Health (Bethesda, MD) cell bank. The human brain biopsy of mammary carcinoma metastasis was provided by Dr. W.S. Nichols, and human GBM clinical biopsies were obtained from neurosurgical patients at the University of Michigan Health System after informed patient consent (Institutional Review Board No. HUM00024610).
Animal Strains
Six- to 7-week-old C57BL/6J, Rag1tm1Mom/J, and Rag1tm1MomTg(TIE2GFP)287Sato/J mice were purchased from Jackson Laboratory (Bar Harbor, ME). RA/EGxdelCre mice were kindly provided by Angelika Bierhaus of the Department of Internal Medicine I, University of Heidelberg (Heidelberg, Germany). LEW/SsNHsd Lewis rats (200-240 g) were purchased commercially from Harlan Laboratories (Indianapolis, IN). All animal experiments were conducted in accordance with procedures approved by the University Committee on Use and Care of Animals and conformed to the policies and procedures of the Unit for Laboratory Animal Medicine at the University of Michigan.
Antibodies
The following primary antibodies were used: rat monoclonal anti-CD31/Platelet Endothelial Cell Adhesion Molecule-1 (clone: MEC13.3), Catalog No. 550274, BD Pharmingen (San Jose, CA); rabbit polyclonal anti-laminin, Catalog No. L9393, Sigma-Aldrich (St. Louis, MO); mouse monoclonal anti-human nestin (clone: 10C2), Catalog No. MAB5326, Millipore (Billerica, MA); mouse monoclonal anti–α-smooth muscle actin (SMA; clone: 1A4), Catalog No. M0851, Dako (Carpenteria, CA); humanized mouse monoclonal anti–VEGF-A (bevacizumab, Avastin), NDC: 50242-060-01, Genentech (San Francisco, CA); non-specific IgGs, Equitech-Bio Inc. (Kerrville, Tx).
Generation of De Novo Mouse GBM Using the Sleeping Beauty Transposase System
De novo GBMs were induced in mice using the Sleeping Beauty transposase system as previously described [15]. Plasmids encoding p53 shRNA and oncogenic NRAS, each flanked by Inverted Repeat/Direct Repeat sequences, were co-injected into the lateral ventricle of neonatal C57BL/6J mice along with a plasmid encoding the Sleeping Beauty transposase at a ratio of (1:1:1). Tumor formation was monitored by bioluminescence imaging due to luciferase expression by the Sleeping Beauty plasmid. Mice developing signs of tumor burden were euthanized, and brains were harvested and processed for immunohistochemical evaluation.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Data are reported as mean ± SEM and were examined with the statistical tests specified in each figure legend. Values were considered significant at the P ≤ .05 level.
Results
Orthotopic Glioma Growth Is Patterned by Preexisting Brain Microvasculature
Since human malignant glioma is known to associate with distinct microanatomic brain structures, we initially sought to characterize the routes of migration used by experimental GBM cell lines from the earliest time points following their intracranial implantation until moribundity. To facilitate tumor cell visualization at single cell resolution, we genetically modified mouse (GL26) and rat (CNS-1) glioma cell lines to express mCitrine fluorescent protein, a bright and photostable mutant of yellow fluorescent protein [16]. The resultant fluorescently modified cell lines were referred to as GL26-Cit and CNS-1-Cit, respectively.
Detailed time-course analyses of intracranial GL26-Cit glioma growth from 15 minutes to 120 hours post-implantation (hpi) into the right striatum of 6- to 7-week-old syngeneic RAG1−/− mice revealed three distinct phases of early glioma growth over nine time points analyzed (n = 54; six mice per time point) (Figure 1, A and B). Tumors < 24 hpi were characterized by round glioma cells confined to the site of initial tumor implantation (phase I). Tumor morphology then shifted after 24 hours displaying chains of elongated glioma cells radiating away from the site of initial tumor implantation that assumed a branch-like structure (phase II). Tumor centers became increasingly dense with glioma cells > 96 hours in vivo, resulting in a progressive widening of the tumor mass while maintaining an invasive pattern at its margins (phase III).
Figure 1.
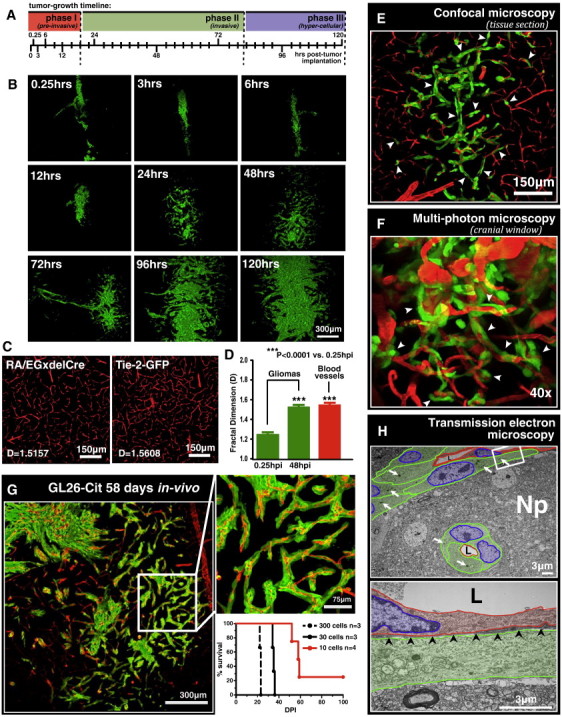
GL26-Cit glioma uses preexisting brain microvasculature as a scaffold for brain tumor invasion. (A) Tumor growth timeline indicating the time points analyzed over the first 120 hours of intracranial GL26-Cit glioma growth. Colored bars represent time frames corresponding to the three distinct phases of early intracranial GL26-Cit glioma growth. (B) Representative fluorescence scanning confocal micrographs of GL26-Cit glioma in the RAG1−/− mouse striatum imaged at the nine predetermined time points indicated in A. (C) Representative fluorescence scanning confocal micrographs of mouse brain microvessels from RA/EGxdelCre (left) and Rag1tm1MomTg(TIE2GFP)287Sato/J (i.e., Tie-2-GFP) (right) mice used to determine the fractal dimension of normal mouse brain microvasculature. Fractal dimension values (D values) for each micrograph are shown. (D) Average D values of gliomas from RAG1−/− mice at 0.25 and 48 hpi (n = 6 mice per group) compared to the average D value of brain microvasculature from the two GFP+ mouse strains represented in C (n = 8; four mice per strain with five distinct striatal regions imaged per mouse). ***P < .0001 versus tumors 0.25 hpi; one-way analysis of variance followed by Tukey post-test. Data represent the mean ± SEM. (E) Fluorescence scanning confocal micrograph of GL26-Cit glioma 48 hpi into the RA/EGxdelCre mouse striatum. GFP+ mouse brain microvessels have been pseudocolored red. White arrowheads point to several examples of microvasculature-associated glioma invasion. (F) Intravital multiphoton micrograph of GL26-Cit glioma imaged through an intracranial window 120 hpi into the somatosensory cortex of the RAG1−/− mouse brain (imaging depth = 129 μm). Mouse brain microvasculature was labeled intraluminally by tail-vein injection of rhodamine B isothiocyanate–conjugated dextran (mw = 70 kDa) before imaging. White arrowheads point to numerous examples of microvasculature-associated tumor invasion. (G) GL26-Cit after 58 days post-tumor implantation into the RAG1−/− mouse brain shown at low (left) and high (right) magnification. Kaplan-Meier survival analysis (lower right) demonstrates that initial injections of low numbers of glioma cells extend animal survival to study late-stage tumor invasion. (H) TEM at low power (top image) showing longitudinal and transverse capillary segments (pseudocolored red) enveloped by perivascularly invading GL26-Cit glioma cells (cytoplasm, green; nuclei, blue) 48 hpi in the RAG1−/− mouse brain. Tumor cells displace the immediately surrounding neuropil as they enter the perivascular space. White arrows indicate USPIOs used to label tumor cells in vitro before implantation. Higher magnification image of the area outlined by the white box is shown below. Black arrowheads in lower micrograph point to the vascular basement membrane covering the adluminal surface of the vascular endothelium. L, vessel lumen.
The growth pattern of phase II (invasive) gliomas was reminiscent of branching (i.e., bifurcating) blood vessel architecture. To test whether GL26-Cit glioma uses preexisting brain microvasculature as a microanatomic scaffold for tissue invasion, we performed fractal dimension analysis on fluorescence scanning confocal micrographs of these tumors using the box-counting algorithm of ImageJ quantitative analytical software (National Institutes of Health). This allowed us to obtain a quantitative description of tumor morphology. Tumor fractal dimension values (D values) were compared to those obtained from tumor-naïve microvasculature of two distinct transgenic mouse strains (RA/EGxdelCre and Rag1tm1MomTg(TIE2GFP)287Sato/J), both of which express green fluorescent protein (GFP) in brain endothelium [17], [18] (Figure 1C). Our analysis indicated that invasive gliomas (48 hpi) displayed an average D value of 1.532 ± 0.015, directly overlapping the average D value obtained from tumor-naïve mouse brain microvasculature (1.567 ± 0.024). However, preinvasive gliomas (0.25 hpi) had significantly lower average D values (1.230 ± 0.010) (Figure 1D). The overlap in the fractal dimension of invasive gliomas with that of tumor-naïve brain microvessels indicated that GL26-Cit assumes microvasculature-like morphology at the onset of tumor invasion. This implicated the use of preexisting brain microvessels as a scaffold for GL26-Cit glioma invasion as previously observed in mouse GL261 and rat C6 gliomas [5], [6], [7].
To substantiate our hypothesis, we implanted GL26-Cit glioma cells directly into the brain of RA/EGxdelCre mice. Fluorescence scanning confocal microscopy was used to analyze tumor morphology in brain tissue sections corresponding to the aforementioned nine time points (n = 54; six mice per time point). Not only did we find direct evidence for GL26-Cit cells using preexisting mouse brain microvessels as a scaffold for tumor invasion (Figure 1E), but we were also unable to find even a single example of these cells using any other known route of glioma invasion throughout the 324 brain tissue sections analyzed among 54 mice. Transcardial injections of fluorophore-conjugated lysine-fixable dextrans and immunohistochemistry with blood vessel–specific antibodies (i.e., CD31, α-SMA, and laminin) were used as alternative methods to label the mouse brain microvessels (Figure S1). Both methods corroborated the presence of microvasculature-associated growth and invasion. We conclude that a strict association with preexisting brain microvasculature characterizes the intracranial invasion of GL26-Cit glioma.
To further examine GL26-Cit perivascular invasion, we performed intravital imaging with multiphoton laser scanning microscopy to examine brain tumor growth and invasion in real time through cranial windows installed over the somatosensory cortex of living RAG1−/− mice. Phase II brain tumors (i.e., 48 hpi) were imaged for up to 24 hours in mice maintained under anesthesia with inhaled isoflurane (n = 10). Mouse brain microvessels were visualized by intravenous tail-vein injections of rhodamine B isothiocyanate–conjugated dextrans (mw = 70 kDa) before fitting the animal on the microscope stage. Imaging experiments revealed the dynamics of early microvasculature-associated GL26-Cit invasion in real time at single-cell resolution, further demonstrating that GL26-Cit glioma invasion is guided by preexisting brain microvasculature (Figure 1F and Movie S1).
We next asked whether perivascular invasion occurs throughout the entire course of brain tumor progression. To assess this, we implanted low numbers of GL26-Cit cells (from 300 to as few as 10) into the striatum of RAG1−/− mice. Implantations of approximately 10 GL26-Cit cells extended median survival to nearly 60 days from a median survival of only 21 days at a dose of 300 cells (Figure 1G). Histologic analysis of GL26-Cit glioma during end-stage disease [i.e., 58 days post-implantation (dpi)] confirmed the presence of continuous perivascular invasion (Figure 1G).
Identification of the Microanatomic Niche Occupied by Perivascularly Invading Glioma Cells
To determine the microanatomic localization of perivascular glioma cells, we labeled GL26-Cit cells in vitro with electron-dense ultrasmall particles of iron oxide (USPIOs) and visualized these cells using transmission electron microscopy (TEM). TEM samples bearing gliomas corresponding to phase II (invasive) tumor growth (n = 6) were chosen to capture perivascular migration. TEM analysis consistently revealed invasive glioma cells in juxtaposition to the vascular basement membrane covering the abluminal surface of brain microvessels (Figures 1H and S2). GL26-Cit tumor cells displaced normal brain tissue adjacent to the adluminal microvessel surface as they entered and migrated throughout the perivascular space, a potential space facilitating bulk diffusion of interstitial fluid throughout the brain [19], [20], [21]. The process of iterative perivascular glioma growth and invasion is illustrated in Figure 2 (A and B).
Figure 2.
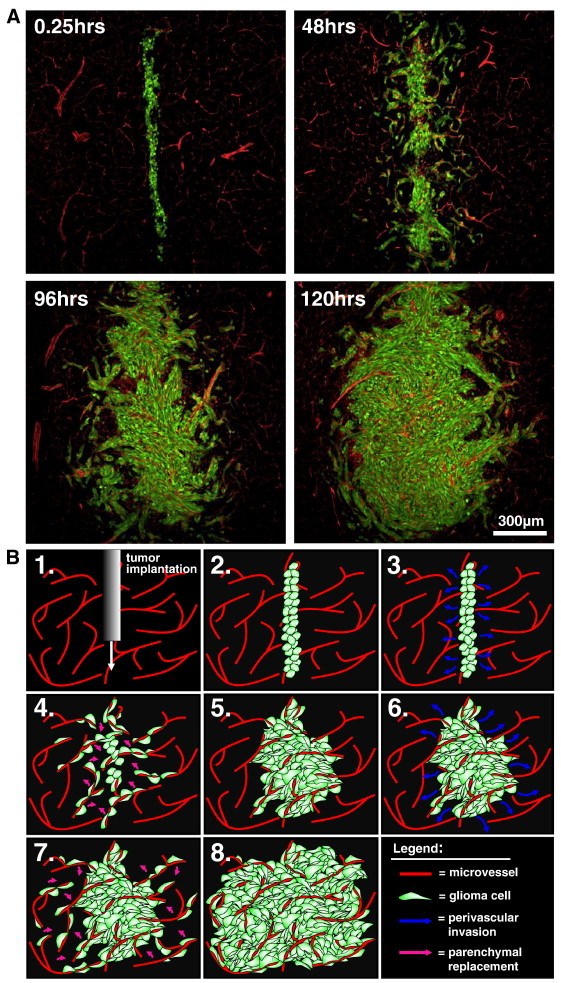
Perivascular invasion is an iterative growth process. (A) Scanning fluorescence confocal micrographs of GL26-Cit glioma cells from 0.25 to 120 hours in the RA/EGxdelCre mouse brain. Glioma cells are shown in green; brain microvessels are shown in red. (B) Schematic representation of the process of perivascular glioma growth and invasion beginning immediately post-tumor implantation (Step 1-2). Implanted glioma cells initially infiltrate throughout the perivascular space (Step 3-4). Cell division causes normal brain parenchyma present between adjacent microvessels to be displaced by neoplastic tissue, leaving normal brain microvessels within the tumor core (Step 4-5). Iterations of further perivascular invasion and cell division produce well-vascularized brain tumors throughout tumor growth (Steps 6-8).
Brain Tumors of Diverse Species and Cellular Origin Exhibit Perivascular Invasion
We next evaluated whether perivascular invasion characterized brain tumors of species other than mouse. Indeed, both rat CNS-1-Cit glioma cells implanted into the striatum of syngeneic Lewis rats (n = 6; 3 rats per time point) and human U251 glioma cells implanted into the striatum of NU/J (i.e., nude) mice [22] (n = 5) used brain microvasculature as a scaffold for tumor invasion (Figure 3, A and B).
Figure 3.
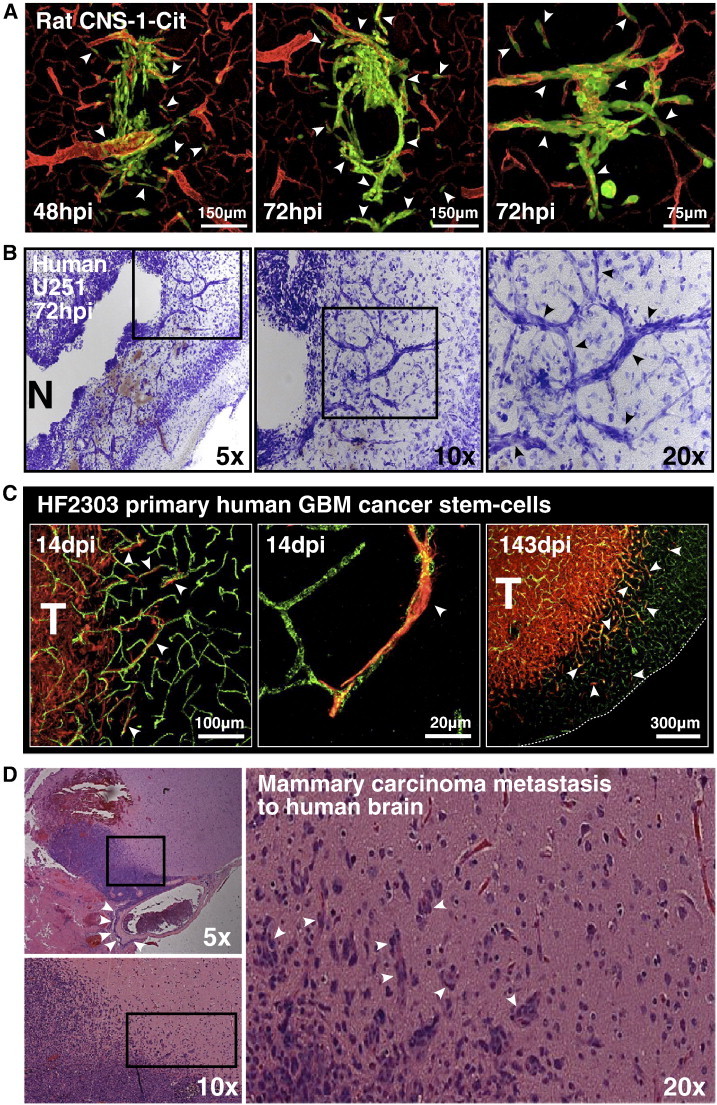
Brain tumors of diverse species and cellular origin exhibit perivascular invasion. (A) Fluorescence scanning confocal micrographs of fluorescently modified CNS-1-Cit rat glioma cells in the brain of syngeneic Lewis rats after 48 hpi (left image) and 72 hpi (right two images). Brain microvasculature was immunolabeled with anti-laminin antibodies, a marker of the vascular basement membrane. White arrowheads point to distinct examples of microvasculature-associated tumor invasion. (B) Nissl staining of human U251 glioma cells in NU/J mice 72 hpi. Black boxes in each brightfield micrograph outline the field of view shown to the right at increasing magnification. Black arrows in the far right image point to tumor cell bodies heavily stained with Nissl that assume vascular morphology as they migrate away from the main tumor mass. N, tumor necrosis. (C) Fluorescence scanning confocal micrographs of HF2303 primary human GBM cancer stem cells 14 and 143 dpi into the RAG1−/− mouse striatum. Primary human GBM cancer stem cells are immunoreactive for the neural stem cell marker nestin. Brain microvessels have been labeled with anti-CD31 antibodies. White arrowheads indicate several examples of microvasculature-associated tumor invasion. T, bulk tumor mass. (D) Tissue biopsy from a human patient bearing mammary carcinoma metastasis to the brain. The metastatic lesion (purple) seen at low magnification (top left panel) can be seen adjacent to a large superficial blood vessel heavily infiltrated with perivascular tumor cells (white arrowheads). Higher magnification (bottom left panel) of the area outlined by the black box in the panel above reveals the invasive margin of the metastatic lesion. Largest panel to the right shows the invasive margin outlined by the black box in the bottom left image at × 20 magnification. White arrowheads point to several examples of invasive cancer cells (purple) surrounding brain microvessels (seen in red due to the presence of intraluminal red blood cells).
Transformed glial cells with stem cell-like properties are thought to be important contributors to clinical brain tumor initiation and progression [23]. We therefore wished to see if perivascular invasion characterized the behavior of bona fide GBM cancer stem cells. To test this, we used a patient-derived sample of primary human GBM cancer stem cells, referred to as HF2303 [24]. These cells form neurospheres when grown under serum-free conditions in the presence of human epidermal growth factor and human basic fibroblast growth factor. HF2303 cells have previously been shown to 1) express the neural stem cell markers Sox2, Musashi-1, and nestin; 2) phenocopy the original patient-derived tumor when orthotopically xenografted into immunocompromised rodents; 3) differentiate into both glial and mesenchymal cell lineages when grown intracranially [24]. HF2303 cells implanted into the striatum of RAG1−/− mice (n = 15) overtly demonstrated perivascular-associated invasion that persisted to the moribund state ~ 143 dpi (Figure 3C). This result indicated that perivascular invasion may underlie the invasion of clinical GBM cancer stem–like cells. We conclude that perivascular invasion is a common mechanism of orthotopic GBM invasion.
Brain microvasculature has been proposed as the “soil” in Paget’s “seed and soil” hypothesis of metastasis formation [25], [26]. We therefore wished to determine whether perivascular invasion also characterizes peripheral metastasis to the brain. Histologic examination of hematoxylin and eosin–stained tissue biopsies from a human patient with mammary carcinoma metastasis to the brain revealed extensive perivascular invasion at the infiltrative tumor margin (Figure 3D). This finding corroborates the notion of brain microvasculature as “soil” in brain metastasis and demonstrates that even peripheral cancers preferentially use preexisting microvessels as a scaffold for tissue invasion upon entering the brain.
De Novo Endogenous Mouse GBM and Clinical GBM Biopsies Exhibit Perivascular Invasion
We next wished to test whether perivascular invasion also occurs in endogenous brain tumors. To test this, we generated de novo mouse GBMs by co-injecting plasmids encoding p53 shRNA and oncogenic NRAS, each flanked by IR/DR sequences, along with a plasmid encoding the Sleeping Beauty transposase at a ratio of 1:1:1 into the lateral ventricle of neonatal C57BL/6J mice. Resultant GBMs were positive for the neural precursor marker, nestin, which was used to identify tumor cells. Co-immunolabeling with anti-nestin and anti–Von Willebrand factor antibodies was used to test the association of endogenous GBM cells with brain microvessels. Invasive nestin+ tumor cells at the infiltrative tumor margins were extensively associated with preexisting microvasculature (Figure 4A). We conclude that perivascular invasion characterizes the invasion of tumor cells arising de novo from brain-resident cells and also rules out the notion that perivascular invasion is an artifact of orthotopic brain tumor implantation.
Figure 4.
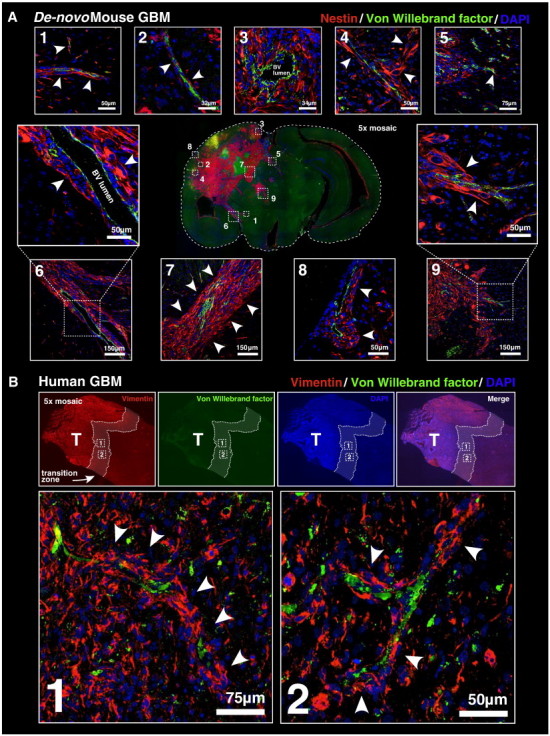
De novo mouse GBM and clinical GBM biopsies exhibit perivascular invasion. (A) De novo mouse GBM generated using the Sleeping Beauty transposase system at 60 days post-plasmid injection. At the center, a 5 × mosaic epifluorescence micrograph of a coronal mouse brain tissue section is shown. Tumor cells have been labeled using anti-nestin antibodies. Brain microvasculature has been labeled with anti–Von Willebrand factor antibodies. 4',-diamidino-2-phenylindole (DAPI) has been used as a counterstain (blue). Numbered white boxes within the central epifluorescence micrograph correspond to respectively numbered higher magnification fluorescence scanning confocal micrographs surrounding it. Several examples of perivascular tissue invasion are indicated by white arrowheads. (B) Human GBM clinical biopsy. A 5 × mosaic epifluorescence micrograph of a tissue biopsy immunolabeled with anti-vimentin antibodies, anti–Von Willebrand factor antibodies, and DAPI (blue) as a counterstain is shown. The zone of transition between tumor tissue (left) and normal brain (right) lies within the white dashed lines overlying each fluorescence channel. Panels below the high-magnification fluorescence scanning confocal micrographs correspond to the dashed white boxes from the epifluorescence images above. White arrowheads in each confocal micrograph point to examples of perivascular tissue invasion.
The clinical relevance of perivascular invasion rests upon whether this phenomenon also occurs in human GBM. We therefore wished to test whether human GBM clinical biopsies would also reveal evidence of perivascular invasion. Human GBM patient biopsies were examined by a clinical neuropathologist blinded to the location of each sample. Biopsies determined to contain both glioma and normal tissue were assumed to represent the zone of transition between these two tissue types (i.e., the infiltrative tumor margin). These biopsies were then chosen for immunohistochemical analysis in our laboratory. Biopsy tissue contained cells immunoreactive for Vimentin (a marker of brain progenitor cells) and Von Willebrand factor (a marker of blood vessels). Co-immunolabeling with these two antibodies revealed the presence of perivascular-associated Vimentin+ tumor cells at the transition zone between neoplastic and normal tissues (Figure 4B). This important finding substantiates perivascular tissue invasion as a clinically relevant mechanism of human malignant brain tumor growth.
Simulating Perivascular Brain Tumor Growth and Invasion
We constructed an agent-based computational model to identify the minimum number of biologic parameters required for the emergence of a perivascular pattern of brain tumor invasion. An agent-based model was chosen for its ability to simultaneously simulate the operations and interactions of multiple agents (i.e., individual glioma cells) with the underlying assumption that simple behavioral rules can generate complex behavioral patterns, leading to emergent phenomena [27]. For our purposes, we were interested in applying an agent-based model to simulate intracranial glioma behavior at the single-cell level under the assumption that glioma cells exhibit a predilection for preexisting brain microvasculature, as demonstrated by our empirical observations.
The model describes individual glioma cells by a position , a velocity , and an orientation vector , whose angle is denoted by θi [i.e., ] on a two-dimensional blood vessel domain taken from real mouse brain microvessel data, which is described by a density distribution g that remains static over time (i.e., blood vessel density and position do not change over the course of the simulation). We model the displacement of each cell by a correlated random walk [28], where the transition probabilities for a single cell only depend on its last position and direction of motion. However, the displacement of glioma cells is influenced by the presence of nearby blood vessels. The motion of an individual glioma cell is fully described by the following differential equation:
| (1) |
where the migration speed of the each glioma cell is a constant (i.e., ) and is only dependent on the glioma cell’s association with a blood vessel (BV) according to the following rule:
| (2) |
Glioma cells contacting a blood vessel move slower due to adhesion effects, thus we take C1 > C0. The predilection of glioma cells for blood vessels is expressed by the velocity field , where the gradient indicates the direction of largest increase of blood vessels. This velocity field attracts the glioma cells toward nearby blood vessels. We further denote the projection operator by:
| (3) |
This projection ensures that the blood vessel attractive force only changes the direction, not the speed, of the glioma cell’s migration. Finally, the term dBt signifies the stochastic noise in the system by modeling the random perturbations exerted on glioma cells. The parameter σ in Equation (1) controls the intensity of this perturbation. Upon contacting a blood vessel (i.e., ), glioma cells divide at a rate μb and reduce their migration speed due to adhesion effects. The daughter cell created upon cell division then begins its own independent correlated random walk. Glioma cells not in contact with a blood vessel (i.e., ) eventually die at a rate μd, where the life expectancy of an individual glioma cell is defined as 1/μd. The key elements of the model, and its comparison to in vivo glioma growth, are summarized in Figure 5 (A and B). To implement the model numerically, we discretize the equation using the Euler method [29]. By optimizing only few parameters such as cell migration speed, strength of attraction to blood vessels, and cell birth and death rate, we have non-trivially recapitulated the three distinct phases of early malignant glioma growth shown empirically through our experimental data (Figure 5C and Movie S2). Further details on the agent-based computational model can be found in the Supplementary Materials and Methods section (Figure S3, Figure S4).
Figure 5.
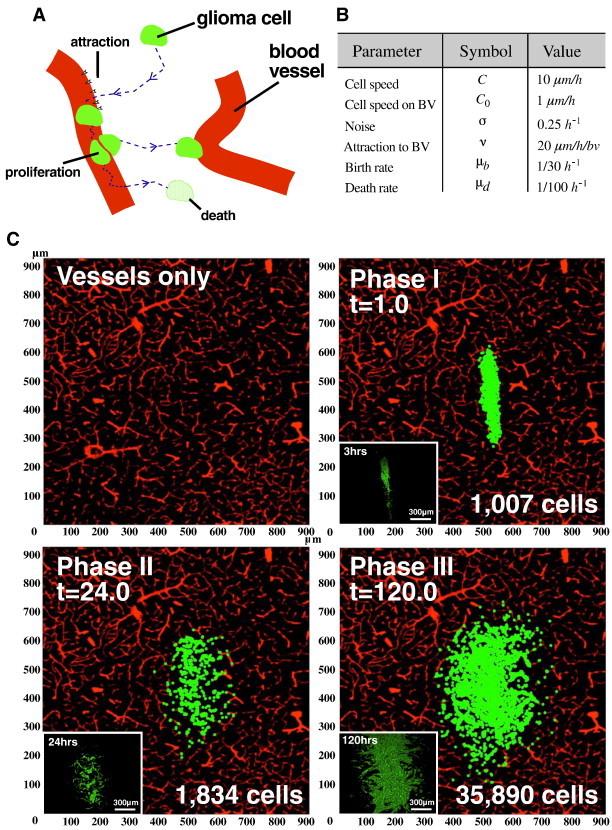
In silico computational modeling mimics perivascular brain tumor growth. (A) Schematic representation of an agent-based computational model describing perivascular brain tumor growth and invasion. Individual glioma cells move toward nearby blood vessels under the influence of a velocity field that mimics the predilection of glioma cells for blood vessels (an underlying assumption of the model). Cells move by a correlated random walk with constant speed toward nearby blood vessels. Once tumor cells make contact with a vessel, they slow their migration speed and proliferate at a rate μb. Tumor cells not associated with a blood vessel eventually undergo cell death at a rate μd and have an average life expectancy of 1/μd. BV, blood vessel. (B) A table summarizing the parameters used in the agent-based model. The respective symbols and optimal values are indicated for each parameter. (C) Representative snapshots of simulated brain tumors corresponding to the three phases of early perivascular tumor growth. The microvasculature domain (vessels only) has been taken from a fluorescence scanning confocal micrograph of RA/EGxdelCre mouse brain microvessels. The simulated tumor initially consists of 1 × 103 cells centered with the microvasculature domain at t = 0, simulating preinvasive phase I glioma soon after implantation (top right panel). Tumor cells then begin to migrate toward nearby microvessels resulting in a strong correlation between the density of tumor cells and blood vessels, causing the characteristic branch-like morphology of phase II glioma (bottom left panel). Tumor cells making contact with a blood vessel rapidly proliferate and fill the gaps between adjacent blood vessels as the total number of cells grows exponentially. As a result, the tumor center widens and becomes hypercellular while maintaining extensive microvascular contact at the tumor borders in a fashion that mimics phase III glioma (bottom right panel).
Perivascular Glioma Invasion Is a VEGF-Independent Mechanism of Tumor Vascularization
The growth pattern obtained from our in silico simulation using preexisting mouse brain microvasculature as a domain in which simulated glioma cells interact and evolve over time bore a striking resemblance to experimental data. Since our in silico simulation did not implement a parameter controlling tumor neoangiogenesis (i.e., the blood vessel domain remains static over the course of the simulation), the model predicted that perivascular invasion is independent of neoangiogenesis to sustain progressive tumor growth. We began to test this prediction by treating RAG1−/− mice bearing GL26-Cit tumors over a 144-hour time period (n = 30; two groups; five time points; three mice per group per time point) with bevacizumab, a neutralizing monoclonal antibody against VEGF-A [30], at a dose of 10 mg/kg delivered intraperitoneally (i.p.) twice weekly as previously described [31] or with non-specific control IgGs at an equivalent molar dose, route, and schedule serving as a negative control. Tumors corresponding to 24, 48, 96, 120, and 144 hours post-tumor implantation revealed no significant differences in overall tumor size or morphology between treatment groups at matched time points (Figure 6, A–C). Bevacizumab’s presence in the brain was confirmed by probing mouse brain tissue sections with secondary antibodies against the humanized Fc region of bevacizumab (Figure S5). This analysis revealed large amounts of bevacizumab antibody throughout the entirety of the brain tumor and at remote distances throughout the surrounding normal brain tissue. Evidence of bevacizumab’s biologic efficacy in stabilizing tumor-associated blood vessels (i.e., vessel normalization) was demonstrated by using antibodies against the vascular marker CD31. Tumor blood vessel integrity was preserved in bevacizumab-treated mice while being disrupted in the tumors of mice treated with control IgG antibodies (Figure 6D). These results confirmed the vasculature “normalizing” effect of bevacizumab, in which VEGF inhibitors stabilize normal vascular architecture, thus increasing vascular perfusion and reducing vasogenic edema [32], [33], [34].
Figure 6.
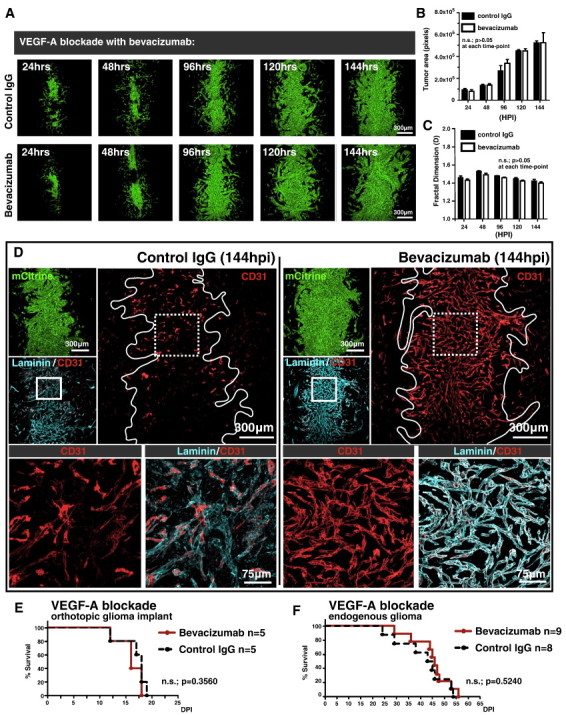
Perivascular glioma invasion obviates the requirement for tumor neoangiogenesis to support continual intracranial growth. (A) Representative fluorescence scanning confocal micrographs of GL26-Cit glioma over the initial 144 hpi in the RAG1−/− mouse brain. Mice were treated with non-specific control IgG (top row) or the VEGF-A blocking antibody bevacizumab (bottom row). (B and C) Quantification of time point–matched tumors (n = 30; three tumors per group per time point) reveals no significant difference in overall tumor size as measured by the average tumor area in pixels (B) or morphology as measured by tumor fractal dimension (C) between control IgG– and bevacizumab-treated mice over the 144-hour analysis period. The P value between treatment groups was > .05 at each time point analyzed by two-way analysis of variance followed by Bonferroni post-tests. (D) Immunohistochemistry on GL26-Cit bearing mouse brain tissue sections labeled with the vascular markers CD31 and laminin (cyan) after 144 hours of treatment with control IgG (left) or bevacizumab (right). Solid white tumor outlines in the CD31 channels circumscribe tumor-associated microvasculature and reveal the vessel “normalization” effect in the bevacizumab treatment group. Dashed white boxes outline the area imaged at higher magnification in the two images below each treatment group, further revealing the anatomic detail of vessel preservation in the bevacizumab-treated group. (E) Kaplan-Meier survival analysis of RAG1−/− mice bearing GL26-Cit glioma treated with bevacizumab (n = 5) or control IgG (n = 5). Mantel-Cox log-rank test detected no significant survival difference between the bevacizumab and control IgG treatment groups (P = .3560). (F) Kaplan-Meier survival analysis of RAG1−/− mice bearing endogenous brain tumors generated de novo using the Sleeping Beauty transposase system treated with bevacizumab (n = 9) or control IgG (n = 8). Mantel-Cox log-rank test detected no significant survival difference between the bevacizumab and control IgG treatment groups (P = .5240).
We next tested the effect of bevacizumab on the survival of mice bearing perivascularly invading gliomas. Kaplan-Meier survival analysis was performed on RAG1−/− mice treated with either bevacizumab or control IgG. Bevacizumab treatment failed to significantly extend the overall survival of mice bearing orthotopically implanted GL26-Cit mouse glioma (Figure 6E) or mice bearing oncogene-induced endogenous mouse brain tumors (Figure 6F).
We also used DC101, a monoclonal blocking antibody targeting the mouse Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) (flk-1/kdr) [35] as an alternative VEGF inhibitor at a dose of 40 mg/kg delivered i.p. twice weekly as previously described [36], [37] and compared its effect to control IgG (n = 30; five time points; three mice per group per time point). Treatment with DC101 also revealed no significant differences in overall GL26-Cit tumor size or morphology compared to control IgG treatment at matched time points over the 144-hour analysis period (Figure 7A). Kaplan-Meier survival analysis performed on RAG1−/− mice treated with either DC101 or control IgG (n = 10; 5 mice per treatment group) confirmed the failure of anti-VEGF therapy to significantly improve the overall survival of mice bearing perivascularly invading glioma (Figure 7B). Further experiments using DC101 as an adjuvant therapy in combination with an efficacious anti-glioma gene therapeutic strategy [38], [39] against GL26-bearing C57BL/6J mice revealed that DC101 also fails to provide additional survival benefit compared to gene therapy plus control IgG, or saline treatment alone, either before, or after, the administration of gene therapy (n = 30; 5 mice per treatment group) (Figure 7, C and D).
Figure 7.
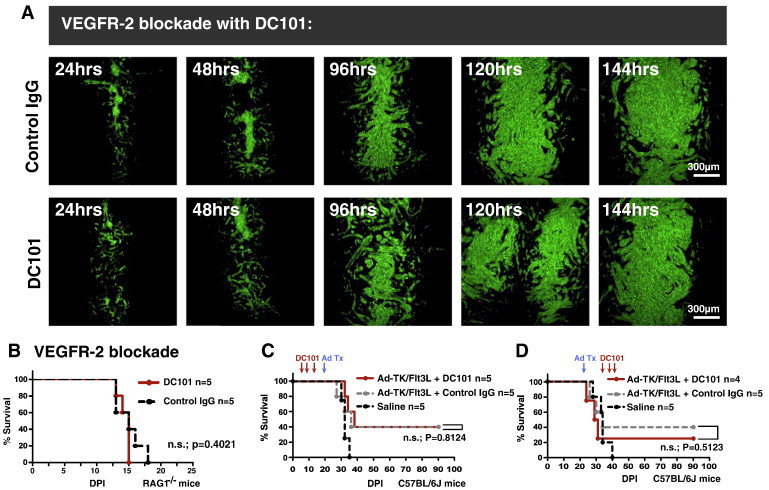
VEGFR-2 blockade with DC101 fails to inhibit tumor growth over 144 hours in vivo or improve survival alone, or as adjuvant therapy. (A) Representative fluorescence scanning confocal micrographs of GL26-Cit tumors in RAG1−/− mice treated with control IgG (top row) or DC101 (bottom row) at a dose of 40 mg/kg delivered i.p. twice weekly over 144 hours (n = 30; three mice per time point per treatment group). No discernable difference in tumor size or morphology is noted at each time point analyzed. (B) Kaplan-Meier survival analysis comparing C57BL/6J mice bearing GL26 gliomas treated with DC101 or control IgG administered three times (red arrows) before (B), or after (C), treatment with Ad-TK/Ad-Flt3L adenovirally mediated cytotoxic gene therapy. Saline treatment alone was used as a negative control. Mantel-Cox log-rank test detected no significant survival difference between Ad-TK/Ad-Flt3L plus DC101 and Ad-TK/Ad-Flt3L plus control IgG treatment either before (P = .8124) or after (P = .5123) treatment with Ad-TK/Ad-Flt3L.
Conflicting evidence over whether bevacizumab can block the activity of murine VEGF-A [40], [41], [42], [43] prompted us to treat RAG1−/− mice bearing primary human HF2303 GBM stem cells with the drug at the aforementioned dose, route, and schedule. Even after 54 total doses, given over 162 days of tumor growth, bevacizumab failed to significantly extend overall survival compared to treatment with control IgGs (Figure 8A). The brains of moribund mice were removed and photographed to document gross brain morphology before sectioning. While the brains of non-specific IgG-treated control mice exhibited extensive microvascular hemorrhage, evidence of microvascular hemorrhage was completely absent in mouse brains treated with bevacizumab (Figure 8B). Immunohistochemistry with human-specific anti-nestin and anti-CD31 antibodies revealed normalized tumor microvasculature deep within tumor tissue in bevacizumab-treated mice, as seen previously in bevacizumab-treated mouse GL26-Cit tumors. Conversely, control IgG–treated brain tumors revealed fragmented microvessels lacking mature microvascular architecture (Figure 8C). Treatment with bevacizumab also exacerbated tissue invasion (Figures 8C, Figure S6, Figure S7). Control IgG–treated tumors grew in a nodular fashion and appeared to compress the contralateral hemisphere, while bevacizumab-treated brain tumors exhibited a more diffuse growth pattern, crossing over into the contralateral striatum without evidence of compression. These results are compatible with the beneficial effects of bevacizumab in human GBM patients (i.e., reduced intracerebral edema and improved brain function).
Figure 8.
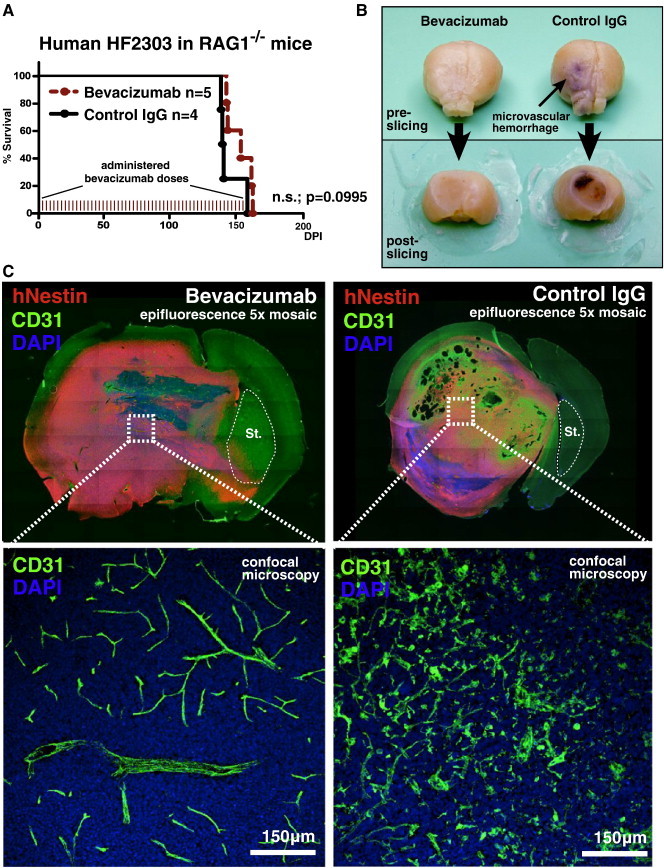
Bevacizumab does not significantly improve the survival of RAG1−/− mice bearing HF2303 primary human GBM stem cells. (A) Kaplan-Meier survival analysis of RAG1−/− mice bearing human HF2303 GBM stem cells treated with bevacizumab or non-specific control IgG. Mantel-Cox log-rank test detected no significant survival difference between the bevacizumab and control IgG treatment groups (P = .0995). Each tick denotes 1 of the 54 total doses of bevacizumab administered throughout the study. n.s., not significant. (B) Representative photographs of gross brain morphology from moribund RAG1−/− mice treated with bevacizumab (left) or control IgG (right) both before (top images) and after (bottom images) sectioning. Note the extensive microvascular hemorrhage associated with tumor neoangiogenesis in the control IgG treatment group, which is completely absent in bevacizumab-treated brain tumors. (C) 5 × mosaic epifluorescence micrographs of coronal brain tissue sections from the mouse brains shown in B. Sections have been immunolabeled with human-specific anti-nestin antibodies to label tumor cells, anti-CD31 antibodies to label brain microvasculature, and DAPI as a counterstain to label nuclei (blue). Note that the contralateral striatum in the control IgG treatment group (denoted as St.; dashed white outlines) appears compressed, presumably due to nodular tumor growth while bevacizumab treatment induces massive invasion into the contralateral hemisphere. Dashed white boxes in the respective images correspond to high-magnification fluorescence scanning confocal micrographs shown below. Normalized tumor vascular is specifically seen in bevacizumab-treated human HF2303 tumors, while control IgG–treated brain tumors are characterized by extensive microvessel fragmentation.
The phenomenon of perivascular glioma invasion, coupled with ultrastructural data indicating that glioma cells displaced normal brain tissue as they enter brain perivascular space, prompted us to quantify the average intervessel distance in the brain and the average size of glioma cells in situ so that we could estimate the number of glioma cells required to occupy the space between neighboring microvessels. To do so, we measured average intervessel distance in the striatum using confocal micrographs of brain tissue sections from immunolabeled C57BL/6J or GFP+ endothelium from RA/EGxdelCre mice using Leica analytical software (Figure 9, A and B), which both reveal calculated average intervessel distances (AIVDcalc) of 56 μm in two dimensions. Micrographs of perivascularly invading GL26-Cit glioma cells over the first 120 hours in vivo were used to calculate an average tumor cell diameter (ACDcalc) of 21 μm (Figure 9C), indicating that as few as three glioma cells (or 56 μm/21 μm) are required to span the distance between adjacent microvessels. Furthermore, a sphere of mouse brain tissue 1 mm3 is densely filled with brain microvessels (Figure 9D), thus perivascular invasion and high brain microvascular network density promote well-vascularized tumors early in development. On the basis of these quantitative results of brain microvessel density and the insights gained from our experimental and computational modeling of perivascular brain tumor growth, we propose a model of iterative, neoangiogenesis-independent, perivascular brain tumor growth and invasion referred to as “autovascularization” (Figure 10).
Figure 9.
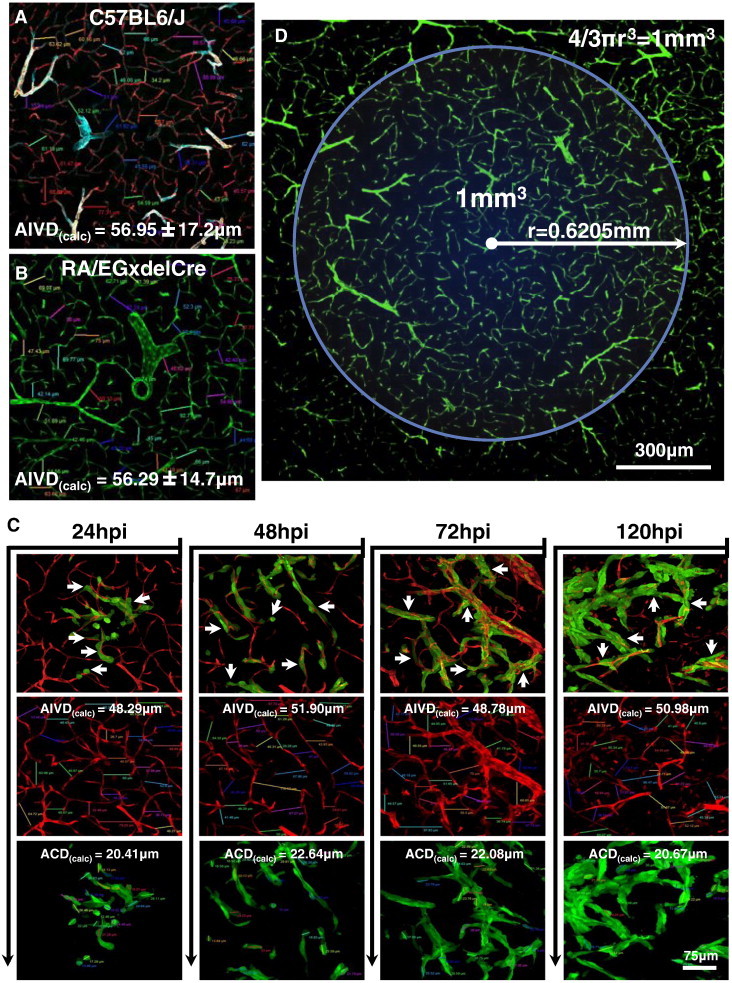
Quantification of brain microvascular network density. (A) Microvessels within the C57BL/6J tumor-naïve striatum were immunolabeled with anti-CD31, anti-laminin (cyan), and anti–α-SMA vascular markers and display a calculated average intervessel distance (AIVDcalc) of 56.95 ± 17.2 μm. (B) Microvessels within the RA/EGxdelCre tumor-naïve striatum showing an AIVDcalc of 56.29 ± 14.7 μm, in good agreement with the values obtained in A, indicating a low degree of variability in this parameter between different mouse strains. (C) Analysis of invasive GL26-Cit glioma cells at various time points at the infiltrative tumor margin within the RA/EGxdelCre mouse striatum (microvessels shown in red). Composite images (top row) show close vascular contacts made by individual tumor cells at each time point analyzed (white arrows). Individual fluorescence channels (middle and bottom rows) show AIVDcalc and calculated average tumor cell diameters (ACDcalc) for the respective micrographs. Overall averages for AIVDcalc and ACDcalc were determined to be 49.99 and 21.45 μm, respectively. (D) Low-magnification fluorescence scanning confocal micrograph of striatal microvasculature in the tumor-naïve RA/EGxdelCre brain. The blue circle illustrates the large number of brain microvessels present within a sphere of 1 mm3, a volume below which many solid tumors are thought togrow avascularly (here, we assume that microvessel density does not change significantly ± 1 mm in the Z direction). The radius of the circle has been calculated by solving for in the equation for the volume of a sphere measuring 1 mm3 (upper right corner). The white arrow indicates the radius (r = 0.6205 mm) of the 1-mm3 sphere. The combination of high microvessel density and perivascular tumor invasion is predicted to preclude an avascular phase of brain tumor growth.
Figure 10.
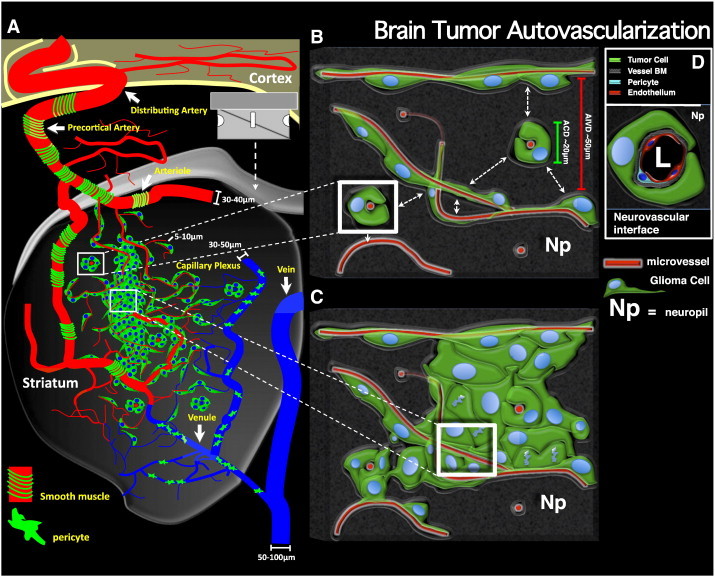
Brain tumor autovascularization. (A) Schematic representation of a coronally sectioned mouse brain revealing perivascular glioma growth and invasion within the striatum. Individual glioma cells are shown in green with blue nuclei. The tumor mass is vascularized with preexisting brain microvessels due to invasive glioma cells entering perivascular channels at the tumor border. Brain microvasculature is classified as arterial or venule (blue). Arterials are further characterized by the presence of smooth muscle (green bands) around larger diameter vessels, while the venous system is covered by patchwork pericytes (green amoeboid cells) as observed empirically using vascular-specific antibodies in situ (see Figure S7). No predilection for invasive glioma cells to migrate in association with arterials versus venules is observed. (B and C) Detailed views of invasive (B) and central (C) portions of perivascular brain tumors. White boxes shown in A correspond to the respective regions in B and C. (B) Illustration of brain perivascular potential space being infiltrated by invasive glioma cells. Adjacent microvessels have an average intervessel distance (AIVD) of ~ 50 μm (red scale bar), while the average tumor cell diameter (ACD) measures ~ 20 μm (green scale bar) as calculated empirically. Dashed white arrows represent the small intervening distance separating adjacent microvessels at the level of the capillary plexus. (C) Illustration of glioma cell division within the perivascular space. Glioma cell division within the perivascular space causes the displacement of normal brain tissue (i.e., neuropil, NP) between adjacent microvessels. The intervening space is then replaced by tumor cells while preexisting brain microvessels simultaneously become trapped within the growing tumor mass. Iterations of tumor cell invasion and division within the perivascular space causes further engulfment of preexisting brain microvessels, leading well-vascularized tumors early in tumor development through a neoangiogenesis-independent process we refer to as “autovascularization”. (D) Details of the microanatomic arrangement at the tumor-vessel interface based on empirical observations.
Discussion
Our investigations have elucidated how the preference of rodent malignant glioma cells and human glioma stem cells to migrate within brain perivascular space allows glioma cells to achieve diffuse brain infiltration and the formation of lethal macroscopic tumors without the need for new blood vessel formation. However, earlier literature has proposed that nascent tumors form without direct access to blood vessels and only grow to a limited volume (1 mm3 or 1 × 106 cells) before necessitating access to VEGF-dependent neovascularization to rescue tumors from dormancy [5], [7], [44], [45]. Our work shows that avascular tumor growth is impossible in the context of the brain, as a sphere of brain tissue 1 mm3 is already densely filled with brain microvessels. This quantitative analysis also suggests that only three to six glioma cells will suffice to fill the space between two adjacent microvessels by displacing normal brain parenchyma as they invade and divide within the perivascular space. It is therefore apparent that at no time during malignant brain tumor progression is the tumor mass devoid of a blood supply, effectively precluding an avascular phase of intracranial tumor growth. Indeed, microvessel density in GBM is comparable to the surrounding normal brain [46] supporting the notion that human glioma progression may proceed by autovascularization.
Perivascular invasion causes spontaneous tumor vascularization in a VEGF-independent manner by engulfing preexisting brain microvessels and replacing normal brain parenchyma over the course of tumor progression (see Figure 10). We propose the term “autovascularization” to describe this form of continuous brain tumor growth and invasion. The VEGF independence of glioma autovascularization is supported by our data showing that the use of VEGF inhibitors bevacizumab and DC101 fail to curb tumor progression or improve median survival in mice bearing human or mouse malignant glioma.
The brain perivascular compartment is a potential space between the vascular basement membrane and the immediately surrounding glia limitans, derived from [19] astrocytes which impart blood–brain barrier characteristics to the vascular endothelium. Because brain perivascular space serves as the point of entry for oxygen, glucose, and other nutrients into the brain, it is ideally suited to support the high metabolic demand of rapidly proliferating tumor cells. Perivascular tumor cells therefore benefit from the ample amounts of glucose entering the brain for use in the glycolytic pathway, the preferred metabolic pathway of transformed cells according to the Warburg effect [47]. Indeed, it has recently been shown that anaerobic glycolysis is favored by brain tumor initiating cells, cells thought responsible for initiating and maintaining progressive tumor growth in human glioma [48]. The basement membrane covering the adluminal microvessel surface is rich in extracellular matrix proteins such as laminin, fibronectin, and vitronectin. These highly glycosylated extracellular matrix proteins are binding partners in integrin-mediated cellular adhesion, promote cellular migration, and activate pro-survival signaling pathways [49]. For these reasons, the preference of invasive glioma cells for brain perivascular space may serve to 1) provide an oxygen, glucose, and nutrient-rich microenvironment ideal for the survival of highly metabolic neoplastic tissue and 2) promote cell adhesion/migration and stimulate pro-survival pathways in cancer cells.
Though the literature regarding the inhibitory effect of bevacizumab against mouse VEGF-A (mVEGF-A) is contradictory, the original developers of A.4.6.1, a mouse anti-VEGF monoclonal antibody subsequently humanized and branded as bevacizumab (Avastin) [50], maintain that the antibody has low affinity for mVEGF-A. However, various research groups have shown the biologic efficacy of bevacizumab against mVEGF-A [41], [42], [43], and our own results showing brain tumor vessel normalization are all consistent with the claim that bevacizumab indeed blocks mVEGF-A. A dose of 10 mg/kg delivered i.p. every 3 days normalized tumor-associated mouse brain microvessel architecture in the murine GL26-Cit glioma model. This effect was comparable to that demonstrated by bevacizumab treatment of HF2303 cells, primary human GBM cancer stem cells, suggesting that bevacizumab in fact exerts a biologic effect on both human- and mouse-derived VEGF-A in vivo.
Blood–brain barrier compromise leads to intracerebral vasogenic edema and neurologic symptoms in GBM [51]. VEGF inhibitors, such as bevacizumab, are therefore thought to improve quality of life by stabilizing normal brain microvessel structure and reducing brain edema [32], [33]. Our experimental data with HF2303 human glioma-derived stem cells treated with bevacizumab demonstrate the normalizing effect on tumor-associated microvasculature in a model of primary human GBM stem cells; these experiments also demonstrate the substantial increase in the invasion of human glioma stem cells caused by bevacizumab [11], [52]. We therefore conclude that bevacizumab-mediated symptom reduction likely involves 1) a reduction in intracerebral vasogenic edema by stabilizing tumor-associated brain microvessels and 2) a lessening of intracerebral compression of eloquent brain regions by exacerbating diffuse tumor invasion. This suggests that VEGF inhibitors, despite potential beneficial effects, may be deleterious to patients by promoting widespread tumor invasion. The recently reported failure of bevacizumab to prolong patient life in randomized human glioma clinical trials [53], [54] is compatible with our results. Indeed, our data provide a basic biological framework potentially underlying these clinical results. It remains to be examined whether increased invasion caused by bevacizumab is dependent on basal levels of tumor-derived VEGF secretion or the intrinsic tendency for glioma cells to invade perivascularly [12], [55], [56].
We have not overlooked the fact that malignant glioma cells may use other growth pathways in addition to, or in lieu of, the perivascular space. However, we suspect that all forms of malignant glioma invasion may preclude the requirement for new blood vessel formation to support tumor progression. In the future, we aim to further examine and catalogue the diverse invasion patterns of malignant glioma cells. By identifying the various ways in which malignant glioma cells invade the brain, combinatorial therapeutics that block multiple migration pathways in order to effectively treat human malignant brain tumors may be developed. We propose that new therapies directly targeting the perivascular space and normal brain blood vessels that sustain glioma growth and invasion may prove efficacious targets in the treatment of malignant glioma by curbing diffuse tumor infiltration throughout the interconnected microvessel network. Cytotoxic therapies targeting tumor-associated microvessels may also be effective against perivascularly invading gliomas by eradicating preexistent brain microvessels already engulfed by perivascularly invading glioma cells. We conclude that malignant brain tumor autovascularization is a clinically relevant mechanism of VEGF-independent tumor vascularization whose therapeutic targeting is predicted to significantly impact clinical neurooncology.
The following are the supplementary data related to this article.
Alternative methods for labeling brain microvasculature. (A) Fluorescence confocal microscopy of GL26-Cit glioma (left image) in the RAG1−/− mouse striatum (72 hpi). Blood vessels (center image) were labeled by transcardial delivery of lysine-fixable tetramethylrhodamine-conjugated dextrans immediately before animal perfusion with heparinized Tyrode’s solution and 4% PFA. Merged image (right image) reveals tumor cells surrounding dextran-perfused microvessels. (B) Confocal micrographs of GL26-Cit tumors. Blood vessels were labeled with anti-CD31 (endothelium; magenta), anti-laminin (vascular basement membrane; cyan), and anti–α-SMA (SMA/pericytes; red) primary antibodies. White arrows show vascular contacts and perivascular elongations made by glioma cells.
Labeling of glioma cells with USPIO nanoparticles permits the identification of tumor cells in the mouse brain at the ultrastructural level. (A) Cultured GL26-Cit glioma cells after overnight incubation with rhodamine B–conjugated USPIO. Phase contrast (top left) and fluorescence (top right) images are shown. Lower images show individual Gl26-Cit cells in suspension after labeling: glioma cells (left), rhodamine B–conjugated USPIOs (center), and merged image (right). Note the presence of USPIOs in all tumor cells. (B–G) TEM micrographs demonstrating the presence of USPIOs in GL26-Cit cells in the mouse brain. Note also the direct vascular apposition between all tumor cells (pseudocolored green; tumor nuclei pseudocolored blue) and capillary-sized microvessels (pseudocolored red). White arrows identify electron-dense USPIO nanoparticles within tumor cell cytoplasm. Np, neuropil, L, blood vessel lumen. Panels F and G are areas outlined by the white boxes in E shown at higher magnification to clearly illustrate parenchymal displacement due to invading tumor cells.
Exponential growth fitting. The number of tumor cells in the model grows exponentially. We plot the number of glioma cells as a function of time (in hours) since the start of the simulation (blue curve) and compare this result with an exponential fitting (green curve) to show that the simulation exhibits an exact fit with exponential growth, thus demonstrating that the simulated tumor grows exponentially with time, as would be biologically expected. (A) Glioma cell number on a linear scale. (B) Glioma cell number on a log-transformed scale.
Tumor cell density distribution. The density set of tumor cells greater than the threshold h = 0.5 cell per μm2 is shown as a color map (A) or as a contour outline (B). After an initially rapid spread (0 ≤ t ≤ 60 hours), glioma cells adhere to nearby blood vessels, which reduces their migration speed. Both the x and y axes are given in units of μm. The proportion of tumor cells on blood vessels therefore increases dramatically over the first 8 hours to reach a constant value of approximately 96% by t = 60 hours. (C) The proportion of glioma cells on a blood vessel over time on a linear scale. (D) The proportion of glioma cells on a blood vessel over time on a log-transformed scale to demonstrate the rapid initial accumulation of glioma cells on blood vessels over the first 8 hours.
In situ detection of bevacizumab within the mouse brain. Immunohistochemistry on brain tissue from RAG1−/− mice treated with control IgG (left) or bevacizumab (right) at 120 hpi using Alexa Fluor 546–conjugated goat anti-human IgG (H + L) secondary antibodies. Bevacizumab was detected due to the presence of its humanized Fc region, an epitope absent from control IgG, which allows for the specific recognition of bevacizumab in mouse brain tissue. Bevacizumab (cyan) was found throughout the tumor and the surrounding normal brain tissue of mice treated with the antibody (left), while no staining was seen in the brains of control IgG–treated mice (right). White boxes outline the areas shown below at high magnification at a single point in the Z axis. These micrographs reveal large bevacizumab aggregates between adjacent tumor cells within the tumor center in mice treated with the drug.
Bevacizumab increases the invasion of HF2303 primary human GBM stem cells; 5 × mosaic epifluorescence micrographs of bevacizumab- (left) or control IgG– (right) treated HF2303 brain tumors immunolabeled with human-specific nestin (hNestin) antibodies in the RAG1−/− mouse brain at the moribund state. Corresponding high-magnification scanning fluorescence confocal micrographs show hNestin and CD31 to reveal vasculature-associated tissue invasion. Bevacizumab treatment was associated with highly diffuse tissue invasion crossing into the contralateral striatum, while control IgG–treated tumors failed to diffusely invade, grew in a more nodular fashion, and appeared to compress the contralateral striatum. Control IgG–treated tumors also contain fragmented microvessels, while bevacizumab-treated tumors contain well-preserved microvessels both inside and outside of the tumor mass.
α-SMA morphology and vessel diameter distinguish vessel-type within the CNS. (A) Tumor-naïve C57BL/6J brain tissue sections were immunolabeled with vessel-specific markers: CD31 (endothelium), α-SMA (pericytes/smooth muscle cells), and laminin (LAM) (vessel basement membrane). 4',6-diamidino-2-phenylindole (DAPI) was used as a nuclear counterstain. Representative fluorescence scanning confocal micrographs are shown demonstrating the distinctive morphologic difference between arterioles, capillaries, and venules (top to bottom) within the mouse brain. Each fluorescence channel has been shown on its own to highlight the morphologic characteristics of these three vessel classifications. Merged images are shown with (right) and without (left) DAPI counterstain. SMA morphology is indicative of vessel class. Closely wound spiraling of SMA characterizes arterioles, while the presence of single SMA+ cells (i.e., pericytes) characterizes capillaries. Less structured and sparser SMA arrangement is associated with venules (white arrowheads). Average vessel diameters have also been included in this classification. (B) Flow chart further classifying microvessel types based on approximate vessel diameter. (D) High-magnification fluorescence scanning confocal micrograph of a C57BL/6J mouse brain arteriole showing the microanatomic arrangement of the vascular markers LAM, SMA, and CD31. L, vessel lumen.
Real-time intravital multiphoton laser scanning microscopy of perivascular GL26-Cit glioma cell migration. GL26-Cit glioma cells were imaged at the infiltrative tumor margin for ~ 2.5 hours beginning 24 hpi. Note the exclusive migration of glioma cells along the capillary plexus and the interactions between glioma cells and brain microvessels.
Agent-based model simulating perivascular growth and invasion. The model begins by simulating a tight group of 1 × 103 tumor cells implanted into the center of the microvessel domain at t = 0. The model is then allowed to evolve over time for 240 hours. Note the gradual transition of the simulated brain tumor through the three distinctive phases of early perivascular growth and invasion empirically described in Figure 1.
Supplemental Materials and Methods
Acknowledgments
We thank Roger Tsien (Department of Pharmacology, University of California San Diego) for kindly donating the pCI-neo-mCitrine expression vector. Plasmids encoding the Sleeping Beauty transposase/luciferase (pT2C-LucPGK-SB100X), shp53 (pT2-shp53-GFP4), and NRAS (pT2CAG-NRASV12) were generously provided by the laboratory of John Ohlfest (University of Minnesota). We gratefully acknowledge support for our work received from Philip Jenkins and the Department of Neurosurgery, University of Michigan School of Medicine. We are also grateful to Karin Muraszko for her academic leadership, M. Dahlgren, D. Tomford, and S. Napolitan for their superb administrative support, and Weidong Xiong for the generation of the GL26-Cit cell line. Author contributions: G.J.B, M.G.C., and P.R.L designed the research; G.J.B., V.N.Y, C.K., and Y.M. performed the research; S.M. designed and performed mathematical computations for the agent-based simulation; A.-A.C., S.B., W.S.N., A.C.D, T.M., S.I.C.-P, and D.O. contributed reagents and provided analytical tools; G.J.B., M.G.C., and P.R.L. analyzed the data; G.J.B., M.G.C., and P.R.L. wrote the paper. The authors declare no conflicts of interest.
Footnotes
This article refers to supplementary materials, which are designated by Figures S1 to S7 and Movies S1 and S2 and are available online at www.neoplasia.com.
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke grants 1RO1-NS 054193, 1RO1-NS 061107, and 1RO1-NS082311 to P.R.L. and grants 1UO1-NS052465, 1RO1-NS 057711, and 1RO1-NS074387 to M.G.C.
References
- 1.Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 2012;14(Suppl 5):v1–v49. doi: 10.1093/neuonc/nos218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010;16:2443–2449. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scherer HJ. The forms of growth in gliomas and their practical significance. Brain. 1940;63:1–35. [Google Scholar]
- 4.Scherer HJ. Structural development in gliomas. Am J Cancer. 1938;34:333–351. [Google Scholar]
- 5.Winkler F, Kienast Y, Fuhrmann M, Von Baumgarten L, Burgold S, Mitteregger G, Kretzschmar H, Herms J. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia. 2009;57:1306–1315. doi: 10.1002/glia.20850. [DOI] [PubMed] [Google Scholar]
- 6.Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. 2006;53:799–808. doi: 10.1002/glia.20334. [DOI] [PubMed] [Google Scholar]
- 7.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 8.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, Johnson RS, Bergers G. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 2003;4:133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 10.Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306–314. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, Westphal M, Lamszus K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61:6624–6628. [PubMed] [Google Scholar]
- 12.de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12:233–242. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK. β1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013;73:3145–3154. doi: 10.1158/0008-5472.CAN-13-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark AJ, Lamborn KR, Butowski NA, Chang SM, Prados MD, Clarke JL, McDermott MW, Parsa AT, Berger MS, Aghi MK. Neurosurgical management and prognosis of patients with glioblastoma that progresses during bevacizumab treatment. Neurosurgery. 2012;70:361–370. doi: 10.1227/NEU.0b013e3182314f9d. [DOI] [PubMed] [Google Scholar]
- 15.Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, Long C, Demorest ZL, Zamora EA, Low WC. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009;69:431–439. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 17.Constien R, Forde A, Liliensiek B, Grone HJ, Nawroth P, Hämmerling G, Arnold B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- 18.Motoike T, Loughna S, Perens E, Roman BL, Liao W, Chau TC, Richardson CD, Kawate T, Kuno J, Weinstein BM. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 19.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang ET, Richards HK, Kida S, Weller RO. Directional and compartmentalised drainage of interstitial fluid and cerebrospinal fluid from the rat brain. Acta Neuropathol. 1992;83:233–239. doi: 10.1007/BF00296784. [DOI] [PubMed] [Google Scholar]
- 21.Pollock H, Hutchings M, Weller RO, Zhang ET. Perivascular spaces in the basal ganglia of the human brain: their relationship to lacunes. J Anat. 1997;191(Pt 3):337–346. doi: 10.1046/j.1469-7580.1997.19130337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flanagan SP. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res. 1966;8:295–309. doi: 10.1017/s0016672300010168. [DOI] [PubMed] [Google Scholar]
- 23.Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell. 2012;22:765–780. doi: 10.1016/j.ccr.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.deCarvalho AC, Nelson K, Lemke N, Lehman NL, Arbab AS, Kalkanis S, Mikkelsen T. Gliosarcoma stem cells undergo glial and mesenchymal differentiation in vivo. Stem Cells. 2010;28:181–190. doi: 10.1002/stem.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS One. 2009;4:e5857. doi: 10.1371/journal.pone.0005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002;3:53–57. doi: 10.1016/s1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 27.Camazine S. Princeton University Press; Princeton, NJ: 2001. Self-Organization in Biological Systems. [Google Scholar]
- 28.Renshaw E, Henderson R. The correlated random-walk. J Appl Probab. 1981;18:403–414. [Google Scholar]
- 29.Kloeden PE, Platen E. Springer-Verlag; Berlin; New York: 1992. Numerical Solution of Stochastic Differential Equations. [Google Scholar]
- 30.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 31.Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 32.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 33.Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 34.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, Böhlen P. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastasis Rev. 1998;17:155–161. doi: 10.1023/a:1006094117427. [DOI] [PubMed] [Google Scholar]
- 36.Jiang F, Zhang X, Kalkanis SN, Zhang Z, Yang H, Katakowski M, Hong X, Zheng X, Zhu Z, Chopp M. Combination therapy with antiangiogenic treatment and photodynamic therapy for the nude mouse bearing U87 glioblastoma. Photochem Photobiol. 2008;84:128–137. doi: 10.1111/j.1751-1097.2007.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martens T, Laabs Y, Günther HS, Kemming D, Zhu Z, Witte L, Hagel C, Westphal M, Lamszus K. Inhibition of glioblastoma growth in a highly invasive nude mouse model can be achieved by targeting epidermal growth factor receptor but not vascular endothelial growth factor receptor-2. Clin Cancer Res. 2008;14:5447–5458. doi: 10.1158/1078-0432.CCR-08-0147. [DOI] [PubMed] [Google Scholar]
- 38.King GD, Muhammad AK, Curtin JF, Barcia C, Puntel M, Liu C, Honig SB, Candolfi M, Mondkar S, Lowenstein PR. Flt3L and TK gene therapy eradicate multifocal glioma in a syngeneic glioblastoma model. Neuro Oncol. 2008;10:19–31. doi: 10.1215/15228517-2007-045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King GD, Muhammad AG, Larocque D, Kelson KR, Xiong W, Liu C, Sanderson NS, Kroeger KM, Castro MG, Lowenstein PR. Combined Flt3L/TK gene therapy induces immunological surveillance which mediates an immune response against a surrogate brain tumor neoantigen. Mol Ther. 2011;19:1793–1801. doi: 10.1038/mt.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L, Wu X, Cheng Z, Lee CV, LeCouter J, Campa C, Fuh G, Lowman H, Ferrara N. Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest Ophthalmol Vis Sci. 2008;49:522–527. doi: 10.1167/iovs.07-1175. [DOI] [PubMed] [Google Scholar]
- 41.Bock F, Onderka J, Dietrich T, Bachmann B, Kruse FE, Paschke M, Zahn G, Cursiefen C. Bevacizumab as a potent inhibitor of inflammatory corneal angiogenesis and lymphangiogenesis. Invest Ophthalmol Vis Sci. 2007;48:2545–2552. doi: 10.1167/iovs.06-0570. [DOI] [PubMed] [Google Scholar]
- 42.Hua J, Spee C, Kase S, Rennel ES, Magnussen AL, Qiu Y, Varey A, Dhayade S, Churchill AJ, Harper SJ. Recombinant human VEGF165b inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010;51:4282–4288. doi: 10.1167/iovs.09-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avisar I, Weinberger D, Kremer I. Effect of subconjunctival and intraocular bevacizumab injections on corneal neovascularization in a mouse model. Curr Eye Res. 2010;35:108–115. doi: 10.3109/02713680903429007. [DOI] [PubMed] [Google Scholar]
- 44.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 45.Folkman J, Hochberg M. Self-regulation of growth in three dimensions. J Exp Med. 1973;138:745–753. doi: 10.1084/jem.138.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60:1388–1393. [PubMed] [Google Scholar]
- 47.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 48.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373–1382. doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolfenson H, Lavelin I, Geiger B. Dynamic regulation of the structure and functions of integrin adhesions. Dev Cell. 2013;24:447–458. doi: 10.1016/j.devcel.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- 51.Wolburg H, Noell S, Fallier-Becker P, Mack AF, Wolburg-Buchholz K. The disturbed blood-brain barrier in human glioblastoma. Mol Aspects Med. 2012;33:579–589. doi: 10.1016/j.mam.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Huveldt D, Lewis-Tuffin LJ, Carlson BL, Schroeder MA, Rodriguez F, Giannini C, Galanis E, Sarkaria JN, Anastasiadis PZ. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS One. 2013;8:e56505. doi: 10.1371/journal.pone.0056505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 55.Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15:4589–4599. doi: 10.1158/1078-0432.CCR-09-0575. [DOI] [PubMed] [Google Scholar]
- 56.Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, McNiel EA, Ohlfest JR, Freese AB, Moore PF. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85:133–148. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alternative methods for labeling brain microvasculature. (A) Fluorescence confocal microscopy of GL26-Cit glioma (left image) in the RAG1−/− mouse striatum (72 hpi). Blood vessels (center image) were labeled by transcardial delivery of lysine-fixable tetramethylrhodamine-conjugated dextrans immediately before animal perfusion with heparinized Tyrode’s solution and 4% PFA. Merged image (right image) reveals tumor cells surrounding dextran-perfused microvessels. (B) Confocal micrographs of GL26-Cit tumors. Blood vessels were labeled with anti-CD31 (endothelium; magenta), anti-laminin (vascular basement membrane; cyan), and anti–α-SMA (SMA/pericytes; red) primary antibodies. White arrows show vascular contacts and perivascular elongations made by glioma cells.
Labeling of glioma cells with USPIO nanoparticles permits the identification of tumor cells in the mouse brain at the ultrastructural level. (A) Cultured GL26-Cit glioma cells after overnight incubation with rhodamine B–conjugated USPIO. Phase contrast (top left) and fluorescence (top right) images are shown. Lower images show individual Gl26-Cit cells in suspension after labeling: glioma cells (left), rhodamine B–conjugated USPIOs (center), and merged image (right). Note the presence of USPIOs in all tumor cells. (B–G) TEM micrographs demonstrating the presence of USPIOs in GL26-Cit cells in the mouse brain. Note also the direct vascular apposition between all tumor cells (pseudocolored green; tumor nuclei pseudocolored blue) and capillary-sized microvessels (pseudocolored red). White arrows identify electron-dense USPIO nanoparticles within tumor cell cytoplasm. Np, neuropil, L, blood vessel lumen. Panels F and G are areas outlined by the white boxes in E shown at higher magnification to clearly illustrate parenchymal displacement due to invading tumor cells.
Exponential growth fitting. The number of tumor cells in the model grows exponentially. We plot the number of glioma cells as a function of time (in hours) since the start of the simulation (blue curve) and compare this result with an exponential fitting (green curve) to show that the simulation exhibits an exact fit with exponential growth, thus demonstrating that the simulated tumor grows exponentially with time, as would be biologically expected. (A) Glioma cell number on a linear scale. (B) Glioma cell number on a log-transformed scale.
Tumor cell density distribution. The density set of tumor cells greater than the threshold h = 0.5 cell per μm2 is shown as a color map (A) or as a contour outline (B). After an initially rapid spread (0 ≤ t ≤ 60 hours), glioma cells adhere to nearby blood vessels, which reduces their migration speed. Both the x and y axes are given in units of μm. The proportion of tumor cells on blood vessels therefore increases dramatically over the first 8 hours to reach a constant value of approximately 96% by t = 60 hours. (C) The proportion of glioma cells on a blood vessel over time on a linear scale. (D) The proportion of glioma cells on a blood vessel over time on a log-transformed scale to demonstrate the rapid initial accumulation of glioma cells on blood vessels over the first 8 hours.
In situ detection of bevacizumab within the mouse brain. Immunohistochemistry on brain tissue from RAG1−/− mice treated with control IgG (left) or bevacizumab (right) at 120 hpi using Alexa Fluor 546–conjugated goat anti-human IgG (H + L) secondary antibodies. Bevacizumab was detected due to the presence of its humanized Fc region, an epitope absent from control IgG, which allows for the specific recognition of bevacizumab in mouse brain tissue. Bevacizumab (cyan) was found throughout the tumor and the surrounding normal brain tissue of mice treated with the antibody (left), while no staining was seen in the brains of control IgG–treated mice (right). White boxes outline the areas shown below at high magnification at a single point in the Z axis. These micrographs reveal large bevacizumab aggregates between adjacent tumor cells within the tumor center in mice treated with the drug.
Bevacizumab increases the invasion of HF2303 primary human GBM stem cells; 5 × mosaic epifluorescence micrographs of bevacizumab- (left) or control IgG– (right) treated HF2303 brain tumors immunolabeled with human-specific nestin (hNestin) antibodies in the RAG1−/− mouse brain at the moribund state. Corresponding high-magnification scanning fluorescence confocal micrographs show hNestin and CD31 to reveal vasculature-associated tissue invasion. Bevacizumab treatment was associated with highly diffuse tissue invasion crossing into the contralateral striatum, while control IgG–treated tumors failed to diffusely invade, grew in a more nodular fashion, and appeared to compress the contralateral striatum. Control IgG–treated tumors also contain fragmented microvessels, while bevacizumab-treated tumors contain well-preserved microvessels both inside and outside of the tumor mass.
α-SMA morphology and vessel diameter distinguish vessel-type within the CNS. (A) Tumor-naïve C57BL/6J brain tissue sections were immunolabeled with vessel-specific markers: CD31 (endothelium), α-SMA (pericytes/smooth muscle cells), and laminin (LAM) (vessel basement membrane). 4',6-diamidino-2-phenylindole (DAPI) was used as a nuclear counterstain. Representative fluorescence scanning confocal micrographs are shown demonstrating the distinctive morphologic difference between arterioles, capillaries, and venules (top to bottom) within the mouse brain. Each fluorescence channel has been shown on its own to highlight the morphologic characteristics of these three vessel classifications. Merged images are shown with (right) and without (left) DAPI counterstain. SMA morphology is indicative of vessel class. Closely wound spiraling of SMA characterizes arterioles, while the presence of single SMA+ cells (i.e., pericytes) characterizes capillaries. Less structured and sparser SMA arrangement is associated with venules (white arrowheads). Average vessel diameters have also been included in this classification. (B) Flow chart further classifying microvessel types based on approximate vessel diameter. (D) High-magnification fluorescence scanning confocal micrograph of a C57BL/6J mouse brain arteriole showing the microanatomic arrangement of the vascular markers LAM, SMA, and CD31. L, vessel lumen.
Real-time intravital multiphoton laser scanning microscopy of perivascular GL26-Cit glioma cell migration. GL26-Cit glioma cells were imaged at the infiltrative tumor margin for ~ 2.5 hours beginning 24 hpi. Note the exclusive migration of glioma cells along the capillary plexus and the interactions between glioma cells and brain microvessels.
Agent-based model simulating perivascular growth and invasion. The model begins by simulating a tight group of 1 × 103 tumor cells implanted into the center of the microvessel domain at t = 0. The model is then allowed to evolve over time for 240 hours. Note the gradual transition of the simulated brain tumor through the three distinctive phases of early perivascular growth and invasion empirically described in Figure 1.
Supplemental Materials and Methods