

Abstract
The c-jun N-terminal kinase (JNK) signaling pathway is regulated by JNK-interacting protein-1 (JIP1), which is a scaffolding protein assembling the components of the JNK cascade. Overexpression of JIP1 deactivates the JNK pathway selectively by cytoplasmic retention of JNK and thereby inhibits gene expression mediated by JNK, which occurs in the nucleus. Here, we report the crystal structure of human JNK1 complexed with pepJIP1, the peptide fragment of JIP1, revealing its selectivity for JNK1 over other MAPKs and the allosteric inhibition mechanism. The van der Waals contacts by the three residues (Pro157, Leu160, and Leu162) of pepJIP1 and the hydrogen bonding between Glu329 of JNK1 and Arg156 of pepJIP1 are critical for the selective binding. Binding of the peptide also induces a hinge motion between the N- and C-terminal domains of JNK1 and distorts the ATP-binding cleft, reducing the affinity of the kinase for ATP. In addition, we also determined the ternary complex structure of pepJIP1-bound JNK1 complexed with SP600125, an ATP-competitive inhibitor of JNK, providing the basis for the JNK specificity of the compound.
Keywords: docking site, JIP1, JNK, scaffolding protein, SP600125
Introduction
The mitogen-activated protein kinase (MAPK) groups play critical roles in many physiological processes including cell growth, oncogenic transformation, cell differentiation, apoptosis, and the immune response by mediating extracellular stresses to cellular signals. The MAPKs are activated by cascade modules that consist of the upstream kinases of MAPKs, MAPK kinases (MKKs), and the upstream kinase of MKKs, MAPK kinase kinases (MKKKs). Of the MAPK families, Erk, p38 MAPKs, and JNKs have been studied extensively as major stress-activated MAPKs. The diverse combination of MAPKs, MKKs, and MKKKs seems to cause extreme complexity in the cellular responses to a wide range of stimuli. Then, what gives the ability of specific regulation of the MAPK pathways? The specificity is achieved, in part, by the use of scaffolding proteins to coordinate the interaction of the three components of the MAPK modules via direct protein–protein interactions. Scaffolding of multicomponent regulatory systems is now recognized as a major mechanism for controlling signal transduction pathways (Mochly-Rosen, 1995; Pawson, 1995; Faux and Scott, 1996; Pawson and Scott, 1997). Assembly of MAPK modules by scaffolding proteins makes it possible to segregate MAPK signaling components into units that are responsive to independent stimuli, and therefore obtain appropriate subcellular targeting by being insulated from similar modules (Elion, 1998; Garrington and Johnson, 1999).
JNK-interacting protein-1 (JIP1) was identified as a scaffolding protein of the JNK module in yeast two-hybrid analysis, enhancing the correspondent signaling through the pathway (Whitmarsh et al, 1998). However, the overexpression of either the domain within JIP1 that binds to JNK, or the full-length protein, potently inhibits JNK signaling in the cell (Dickens et al, 1997). This is because JIP1 blocks nuclear translocation of JNK with its ability to retain JNK in the cytoplasm and sequesters JNK module components into different JIP1 complexes when it is overexpressed. It has been proposed that JIP1 facilitates mixed-lineage kinase (MLK)-dependent JNK signal transduction by aggregating the three components of the module, MLK, MKK7, and JNK (Tournier et al, 1999; Davis, 2000). JIP1 consists of 707 amino acids and contains an N-terminal JNK-binding domain (residues 1–282) and C-terminal MLK- and MKK7-binding domain (residues 283–660). The C-terminal MLK- and MKK7-binding domain contains a putative phosphotyrosine interaction domain and an Src homology 3 (SH3) domain (Dickens et al, 1997; Whitmarsh et al, 1998). JIP1 is highly concentrated in the adult brain, and particularly enriched in the cerebral cortex and hippocampus (Kim et al, 1999). The minimal region of JIP1 has been identified as retaining the JNK-inhibitory property (Dickens et al, 1997; Bonny et al, 2001; Barr et al, 2002). This peptide, pepJIP1 (a peptide version of JIP1), inhibited JNK activity in vitro toward recombinant c-jun, Elk, and ATF2 up to 90% with significant selectivity of no inhibition of the related Erk and p38 MAPKs.
MAPK docking sites have been identified for substrate transcription factors, MKKs, and scaffolding proteins. It is interesting that the docking sites of substrate transcription factors, MKKs, and scaffolding proteins of MAPKs have a consensus in sequences, (R/K)2–3-X1–6-φA-X-φB, where φA and φB are hydrophobic residues such as Leu, Ile, or Val (Sharrocks et al, 2000). The variability in the number and position of hydrophobic and basic residues within the docking site is known to determine specificity. The number and spacing of basic residues, in particular, make an important contribution but do not appear to be the only specificity determinants (Smith et al, 2000; Tanoue et al, 2001). As a counterpart of the docking sites, MAPKs have surfaces (docking grooves) for docking with the docking sites of MAPK-interacting molecules, which regulate the docking specificity. The docking groove is located on the opposite side of the substrate recognition site. Besides the location of the docking grooves, the docking interaction is different from the transient enzyme–substrate recognition for substrate phosphorylation in the sense that the Ser/Thr-Pro sites, which are phosphorylated in substrates by MAPKs, do not seem to possess a clear consensus motif as found in docking sites. In addition, peptide versions of the Ser/Thr-Pro sites in substrates do not mimic the phosphorylation kinetics of the whole protein, whereas peptides of the minimal regions in docking sites retain properties similar to those of the whole protein (Kemp and Pearson, 1991; Dickens et al, 1997; Bardwell et al, 2001; Barr et al, 2002).
Three distinct genes encoding JNKs, Jnk1, Jnk2, and Jnk3, have been identified and at least 10 different splicing isoforms exist in mammalian cells (Gupta et al, 1996). The JNK isoforms differ in their binding activity with ATF2, Elk-1, and c-jun transcription factors. So, individual members of the JNK group may therefore selectively target specific transcription factors in vivo. JNK1 and JNK2 are widely expressed in a variety of tissues, whereas JNK3 is selectively expressed in the brain and to a lesser extent in the hearts and testis (Mohit et al, 1995; Martin et al, 1996).
Activation of the JNK pathway has been documented in a number of diseases, providing the rationale for targeting this pathway for drug discovery (Manning and Davis, 2003). In rheumatoid arthritis, cartilage and bone erosion is promoted by inducible expression of matrix metalloproteinases (MMPs), which is regulated by activation of AP-1 via the JNK pathway (Gum et al, 1997; Han et al, 2002). The JNK pathway is activated in vulnerable neurons in patients with Alzheimer's disease (AD), suggesting that the JNK pathway is involved in the pathophysiology and pathogenesis of AD (Troy et al, 2001; Bozyczko-Coyne et al, 2002; Zhu et al, 2002). And it has been reported that JNK is a crucial mediator of obesity and insulin resistance and a potential target for type II diabetes (Waeber et al, 2000; Hirosumi et al, 2002). In the hope that therapeutic inhibition of JNK may provide clinical benefit in diverse diseases, JNK inhibitors have been discovered and characterized. As a reversible ATP-competitive JNK inhibitor, SP600125 was reported to inhibit JNK with high potency and selectivity (Bennett et al, 2001).
To investigate the mechanism of selective regulation of JNK by JIP1, we have determined the crystal structure of JNK1 in complex with pepJIP1, the docking site peptide of JIP1 (residues 153–163), at a resolution of 2.35 Å. It is the first structure demonstrating the way by which MAPKs are selectively regulated by their scaffolding proteins. In this structure, it is found that the docking site of the scaffolding protein also binds at the same docking grooves of JNK1 as the upstream kinase and the substrate transcription factor do in p38 MAPK (Chang et al, 2002), but with quite different interaction modes. Interestingly, the dramatic interdomain rearrangement between the N- and C-terminal domains of JNK1 upon pepJIP1 binding distorts the ATP-binding pocket, and thus inhibits the catalytic activity of JNK due to the failure of productive ATP binding. We also determined the structure of the ternary complex JNK1–pepJIP1–SP600125 at a resolution of 2.7 Å. Despite the small size of SP600125 (molecular weight=220), it effectively occupies the hydrophobic pocket of the ATP-binding site in JNK1, and the variations of the crucial hydrophobic residues in other MAP kinases give SP600125 the JNK specificity. These structural studies will facilitate the discovery of more potent and selective JNK inhibitors in the future.
Results
Overall structure of JNK1 complexed with pepJIP1
As a result of the high homology of sequences with 92% identity, the overall architecture of JNK1 is substantially equal to that of JNK3 (PDB entry 1JNK; Xie et al, 1998). The N-terminal domain (residues 9–112 and 347–363) contains seven β strands (β1, β2, β3, β4, β5, β6, and β7) and two α helices (α1 and α14). The C-terminal domain (residues 113–337) consists of a bundle of seven α helices (α2, α3, α5, α6, α8, α11, and α12) with five short 310-helices (α4, α7, α9, α10, and α13) and three β strands (β8, β9, and β10). The N-terminal domain and the C-terminal domain are linked by two loops, one (residues 108–112) of which is located between β7 and β8 and the other (residues 331–351) is a long connector of α14 to α13 of the C-terminal domain, and some part of the loop is disordered in our model as in the case of the JNK3 structure, implying the high flexibility of the loop (Figure 1A).
Figure 1.
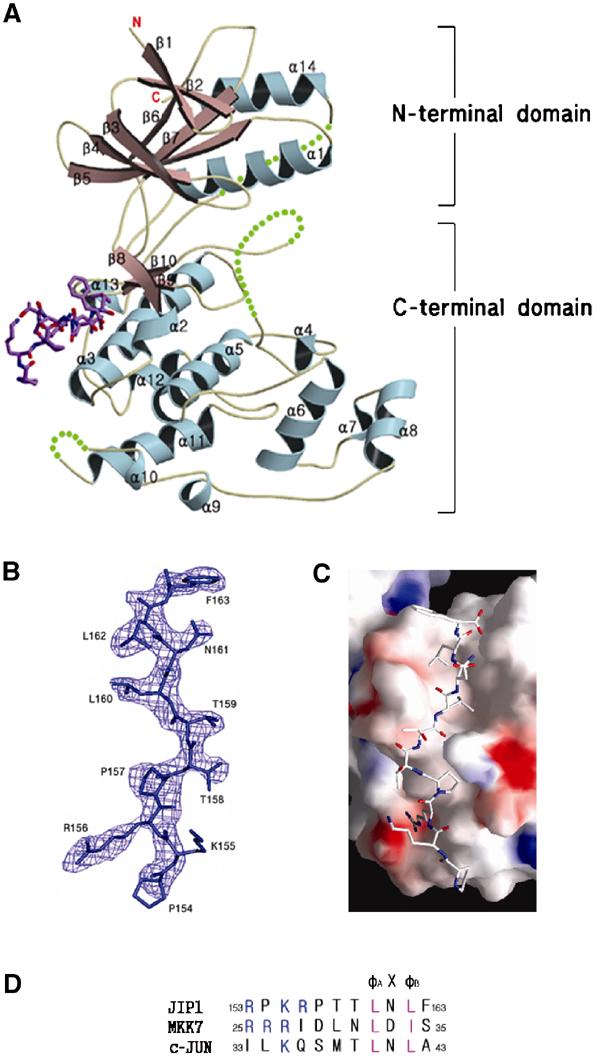
Overall structure of JNK1 complexed with pepJIP1 at a resolution of 2.35 Å. (A) JNK1 is shown in a ribbon model and the disordered regions of JNK1 (residues 173–189, 282–286, and 337–348) are expressed as dotted lines. The bound pepJIP1 is shown in a stick model of atomic color. The regions for the N- and C-terminal domains are indicated. (B) The sigma-A-weighted 2Fo–Fc electron density map calculated with a final refined model without pepJIP1 at a level of 1.2σ. The peptide used in the experiment is RPKRPTTLNLF, but the starting residue, Arg153, is not shown in the density. (C) Surface representation of JNK1 in complex with pepJIP1, colored by electrostatic potential. (D) The docking site sequences of the scaffolding protein (JIP1), the upstream kinase (MKK7), and the substrate transcription factor (c-jun), which are components of the JNK signaling regulated by JIP1 scaffolding protein. The basic residues and φ residues are shown in blue and violet letters, respectively.
PepJIP1 used in this complex study is an 11-mer docking site peptide (residues 153–163 of JIP1 with the sequence of RPKRPTTLNLF) containing the φA-X-φB motif, where φA and φB are Leu160 and Leu162 of JIP1, respectively. The peptide docks to the docking groove of JNK1, a surface of the C-terminal domain covering α2, α3, α13, and β8, which is away from the ATP pocket or substrate recognition site, implying an allosteric inhibition mechanism of pepJIP1. The peptide version of the JIP1 scaffolding protein used in the study has a consensus motif with the upstream kinase (MKK7) and the substrate transcription factor (c-jun) (Figure 1D). Unexpectedly, the side chain of Lys155 (φA−5) of pepJIP1, which is also conserved among the basic residues of the docking sites in both MKK7 and c-jun, is not shown in the electron density map due to the lack of interaction with the docking groove of JNK1 (Figure 1B), implying that the basic residues crucial for binding cannot be predicted only by the sequence comparison at the docking sites.
Interactions of the complex
The complex forms a continuous interface that buries a total surface area of 1034 Å2 (Figure 1C). All of the buried area is contributed by interactions from only the C-terminal domain of JNK1. The interface is mostly nonpolar, relying on hydrophobic interaction. Leu160 and Leu162 of the φA-X-φB motif in pepJIP1 are extensively surrounded by many hydrophobic residues of JNK1 including Met121, Val118, Leu115, Ala113, Leu123, Val59, Leu131, and Cys163 (Figure 2). Asn161 of pepJIP1, the X residue of φA-X-φB motif, does not have any interaction with JNK1 and this is consistent with the fact that the X residues have high diversity among the proteins containing the docking site (Yang et al, 1998; Holland and Cooper, 1999). The side chains of Thr158 (φA−2) and Thr159 (φA−1) are also oriented to the opposite side of the interface, resulting in no interaction. The backbone amide of Leu160 (φA) makes a hydrogen bond with the backbone carbonyl group of Ser161 of JNK1. The oxygen atom of the backbone carbonyl of Thr159 (φA−1) hydrogen bonds to the N ɛ atom of Arg127. Pro157 (φA−3) has van der Waals contact with the side chains of Tyr130, Glu126, and Trp324, and Pro154 (φA−6) makes a weak interaction with Val323. Finally, Arg156 (φA−4) interacts with Glu329 with a bidentate salt bridge of length 2.70 Å. Arg153 (φA−7) and Phe163 (φB+1) are outside of the complex interface and make little or no contribution to the interaction, so it is not surprising that Arg153 (φA−7) is not shown in the electron density map. In the previous study of glycine or alanine replacement and truncation (Dickens et al, 1997; Barr et al, 2002), Arg156 (φA−4), Pro157 (φA−3), Leu160 (φA), and Leu162 (φB) were identified as critical mediators of the JNK inhibition, and a single residue removal from the N-terminus or the C-terminus of pepJIP1 caused no difference in JNK inhibition. The structural features of the JNK1–pepJIP1 complex are completely consistent with the results of alanine scanning replacement and truncation analysis within pepJIP1 to identify the crucial residues for JNK inhibition. To confirm the importance of Arg127 and Glu329 of JNK1 for pepJIP1 binding, we have used isothermal titration calorimetry (ITC) to measure Kd values of pepJIP1 to wild-type JNK1 and mutants of R127A and E329A. The Kd values for pepJIP1 to bind to wild type, R127A, and E329A were found to be 0.42±0.13, 6.4±2.2, and 9.1±3.4 μM, respectively (Figure 2C–E). These Kd values derived from ITC agree well with the above structural description, demonstrating that Arg127 and Glu329 of JNK1 are key residues for pepJIP1 binding.
Figure 2.
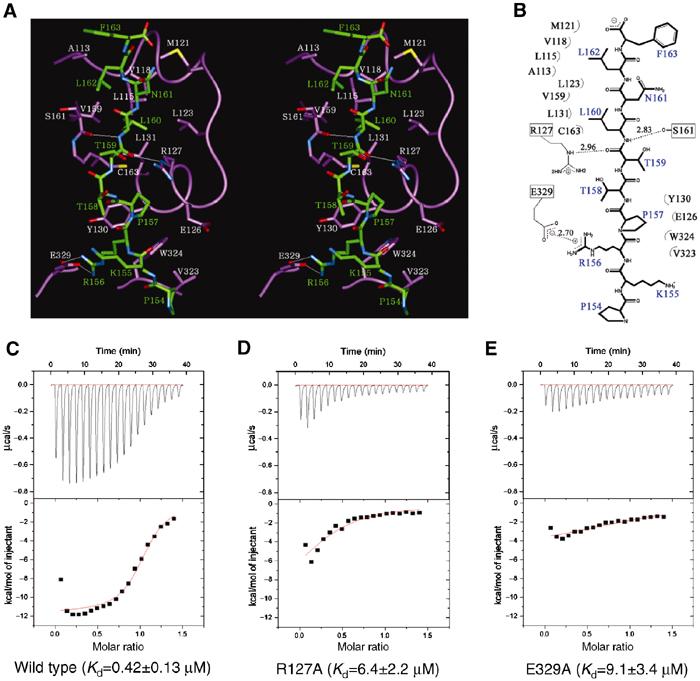
Interactions between JNK1 and pepJIP1. (A) Stereoview of the interactions between JNK1 (violet) and pepJIP1 (green). The residues of JNK1 involved in the interactions are shown in white labels and those of pepJIP1 in green labels. The hydrogen bonds in the interactions are shown as thin white lines. (B) Schematic expression of the interactions. (C) The binding affinity of pepJIP1 to wild-type JNK1 was measured by ITC. The upper panel shows the raw data, a trace of power with time. The lower panel shows the integrated heats from each injection, and the line through the measured points shows the best-fit model for a single binding site. (D, E) The respective Kd values of pepJIP1 to JNK1 mutants, R127A and E329A, were derived from the ITC data.
pepJIP1 as a JNK-specific inhibitor
In the comparison of the complex structure of JNK1–pepJIP1 with that of p38-pepMEF2A or p38-pepMKK3b (PDB entries 1LEW and 1LEZ, respectively; Chang et al, 2002), the overall conformations and binding positions of the bound peptides are quite different from each other when the structures of JNK1 and p38 are superimposed, although the difference between those of pepMEF2A and pepMKK3b is relatively insignificant (Figure 3A). The most notable difference is that the φA-X-φB motif of pepJIP1, especially Leu162 (φB), is shifted by the structural difference between JNK1 and p38 in the region containing the hydrophobic residues critical for the φA-X-φB motif binding. In this region, JNK1 forms a one-turn helix (α2), but a two-turn helix in p38. The structural difference of this region can be predicted from the sequential mismatch due to one insertion in the sequence of p38 (Figure 3D). Another notable difference in the conformation between pepJIP1 and pepMEF2A is that Arg156 (φA−4) interacts with Glu329 of JNK1 through a bidentate salt bridge, which is critical for pepJIP1 binding to JNK1, but the corresponding φA−4 residue in pepMEF2A, which is proline, cannot make such an ionic interaction. Although the residues of p38 and Erk corresponding to Glu329 of JNK1 are also glutamates, they could not interact with Arg156 (φA−4) of pepJIP1 due to structural differences (Figure 3A). And the fact that the structure of Glu329 of JNK1 complexed with pepJIP1 is actually identical to that of Glu367 of free JNK3 indicates that the position of the side chain of Glu329 is not changed to interact with Arg156 (φA−4), suggesting the high specificity of JNK1 to recognize JIP1 (Figure 3A). Although it has been suggested that the CD domain (common docking domain), which is composed of acidic residues in the docking groove of MAPK, is indispensable for binding of the docking site (Tanoue and Nishida, 2002), no possible interaction was observed between the acidic residues of p38 and the basic residues of the MEF2A or MKK3b docking site. However, the distinct interaction between Glu329 of JNK1 and Arg156 of JIP1 shows the importance of the CD domain for the selective binding of a specific docking site. In Figures 1D and 3E, Lys155 (φA−5) is conserved in the docking sites while Arg156 (φA−4) is not. But the key interaction with the CD domain of JNK1 is made only by the unconserved basic residue, Arg156 (φA−4), implying that the number and position of basic residues in the docking sites are important determinants of specificity.
Figure 3.
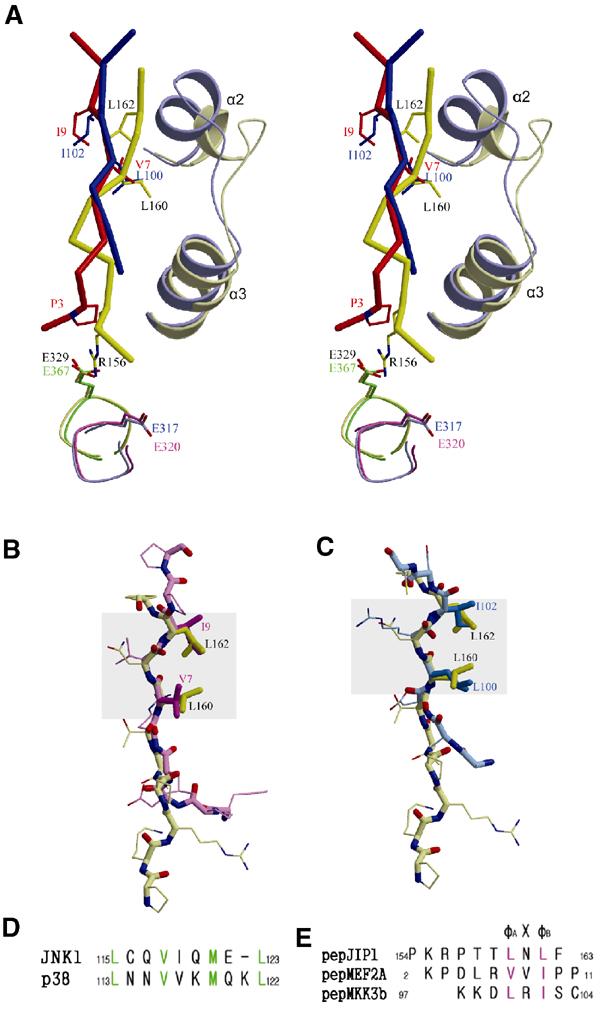
Structural comparison of the docking site peptides bound in the docking grooves of MAPKs. (A) Stereoview of the overlaid structures of pepJIP1 (saturated yellow) bound in JNK1 (whitened yellow) and pepMEF2A (saturated red) or pepMKK3b (saturated blue) bound in p38 MAPK (whitened blue) when the docking grooves of JNK1 and p38 are superimposed. The residues of pepJIP1, pepMEF2A, and pepMKK7 are labeled black, red, and blue, respectively. And the structure of E327 of JNK1 is compared with those of the corresponding glutamates of JNK3 (green), p38 (blue), and Erk2 (pink). (B) The structural deviation between pepJIP1 (yellow) and pepMEF2A (pink) when their φA-X-φB motifs are superimposed. (C) The structural comparison between pepJIP1 (yellow) and pepMKK3b (blue). In (B, C), the conformational conservations of the φA-X-φB motifs are highlighted by gray rectangles. (D) The differences of the sequences in the α2 helices of the docking grooves between JNK1 and p38 MAPK. The residues participating in hydrophobic interactions with the docking site peptides are shown in green letters. (E) The sequences of docking site peptides represented in (A–C). The φ residues are shown in violet letters.
In conclusion, the two major differences, the structural difference in α2 helix and the involvement of the CD domain in docking interaction, provide the rationale for the predominant selectivity of pepJIP1 in inhibition of JNK, but not the related Erk or p38 MAPKs.
Conformational conservation of the φA-X-φB motif
Despite the considerable differences in the overall conformation of the docking site peptides, it is shown that the conformations in the φA-X-φB motifs are conserved (Figure 3B and C). When the φA-X-φB motif of pepJIP1 is superimposed on that of pepMEF2A or pepMKK3b, the major torsional differences are found at the bond outside of the φA-X-φB motifs, and thus the relative orientation of the side chains in the φA-X-φB motifs can be conserved. This result implicates that any conformational change inside of the φA-X-φB motif may decrease the hydrophobic interactions of the two key residues, φA and φB, with the docking grooves of MAPKs, therefore inducing failure in the effective binding of docking sites to MAPKs. In conclusion, the docking site can be characterized by not only the sequential similarity of the φA-X-φB motifs but also the conformational conservation of the motifs in their structures.
Interdomain rearrangement mediated by pepJIP1 binding
The most interesting result of the pepJIP1 binding to JNK1 is the rotation of the N-terminal domain relative to the C-terminal domain by approximately 15°, thereby distorting the ATP-binding site and inducing disorder of the phosphorylation loop (Figure 4).
Figure 4.
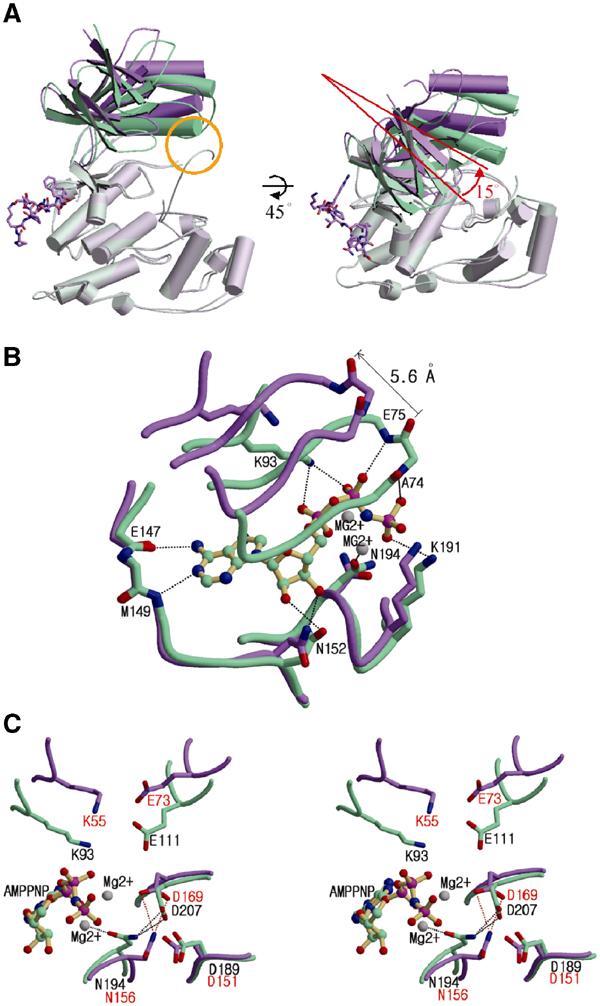
Distortion of the ATP-binding site caused by interdomain rearrangement upon pepJIP1 binding. (A) Structural comparison between JNK3 (green) and pepJIP1-bound JNK1 (violet) when the C-terminal domains of the kinases are superimposed. The conformational differences of the N-terminal domains can be easily distinguished when the conventional view of kinases is rotated by 45° along the horizontal axis. The yellow circle indicates the interaction between the α1 helix and the phosphorylation loop in JNK3, but not existing in JNK1 complexed with pepJIP1. (B) Comparison of ATP-binding sites between the JNK1–pepJIP1 (violet) and JNK3–AMPPNP (green) complexes. The AMPPNP bound in JNK3 is shown in a ball-and-stick model. The residues of JNK3 involved in the hydrogen bonding with AMPPNP are labeled. The side chains of the residues in the glycine-rich loop including E75 and A74 of JNK3 are omitted for clarity because the backbone amide groups only are involved in the hydrogen bonds with the phosphate groups of AMPPNP. (C) The structural comparison of the residues crucial for the catalytic activity between the JNK1–pepJIP1 (violet) and JNK3–AMPPNP (green) complexes. The residues in JNK1 and JNK3 are labeled red and black, respectively. In (B, C), hydrogen bonds are indicated by dashed lines.
The root-mean-square (r.m.s.) deviations for Cα atoms of the N- and C-terminal domain between the structures of JNK1–pepJIP1 and JNK3 are 1.27 and 0.79 Å, respectively. These values show that there is no significant structural difference between the corresponding domains. However, when the full structures of combined N- and C-terminal domains are superimposed, the r.m.s. deviation for Cα atoms runs to 3.84 Å, indicating an extensive hinge motion between the two domains. The hinge motion is triggered in β8, which interacts with Leu162 (φB) by van der Waals contacts, and is propagated to the N-terminal domain via the single loop, which connects β7 and β8. The fact that the N- and C-terminal domains are linked by only two loops, the loop connecting β7 and β8 and the disordered 331–351 loop, implies that the intrinsic flexibility may exist in the kinase, and the inherent structural flexibility may play a central role in its regulation upon JIP1 binding to JNK1.
Inhibition of JNK1 by pepJIP1 is mainly due to the competition of pepJIP1 with other docking sites of the substrates or the upstream kinases (Dickens et al, 1997; Ho et al, 2003). However, this is not the only mechanism through which pepJIP1 inhibits the kinase. A close-up look at the catalytic cleft shows that the ATP-binding site is distorted on binding of pepJIP1 to JNK1 (Figure 4B). In the JNK3–AMPPNP complex structure, AMPPNP is bound to the ATP pocket by residues in the N-terminal domain (Ala74, Glu75, and Lys93), by residues in the linker loop (Glu147 and Met149), and by residues in the C-terminal domain (Asn152, Asn194, and Lys191). The residues in the C-terminal domain and the linker loop are in similar positions in both structures of JNK1–pepJIP1 and JNK3, whereas the residues in the N-terminal domain are in different positions. The glycine-rich loop in the N-terminal domain is involved in several hydrogen bonds with the phosphate groups of AMPPNP. The orientation of ATP in JNK3 may be determined by the hydrogen bonds between the phosphate groups of ATP and the backbone nitrogen atoms of Ala74 and Glu75 in JNK3. Lys93 of JNK3 also makes good hydrogen bonds with the phosphate groups, thereby contributing to the proper positioning of ATP in the kinase. The above-mentioned residues, which belong to the N-terminal domain, are moved by roughly 4–6 Å as a result of the interdomain rearrangement upon pepJIP1 binding, and this movement makes the ATP-binding site distorted. This distortion would not eliminate the ATP binding, but it would significantly reduce ATP affinity and may well disorient any bound ATP with respect to the kinase catalysis, and thus inhibit the JNK activity to phosphorylate the substrates. The reduced binding affinity of ATP to JNK1 was observed by ITC. The Kd value was increased by three times when pepJIP1 is bound to JNK1 (Kd=55±16 μM) compared to free JNK1 (Kd=19±4.2 μM) (Figure 5A and B). We have also observed that pepJIP1 inhibits JNK1 to phosphorylate myelin basic protein (MBP), which is a substrate not containing a docking site, and that this inhibition is dose-dependent. But, a mutated pepJIP1, where the four key residues for interaction (R156, P157, L160, and L162) are replaced with alanine, does not inhibit the JNK1 activity due to the failure of binding to JNK1 (Figure 5C).
Figure 5.
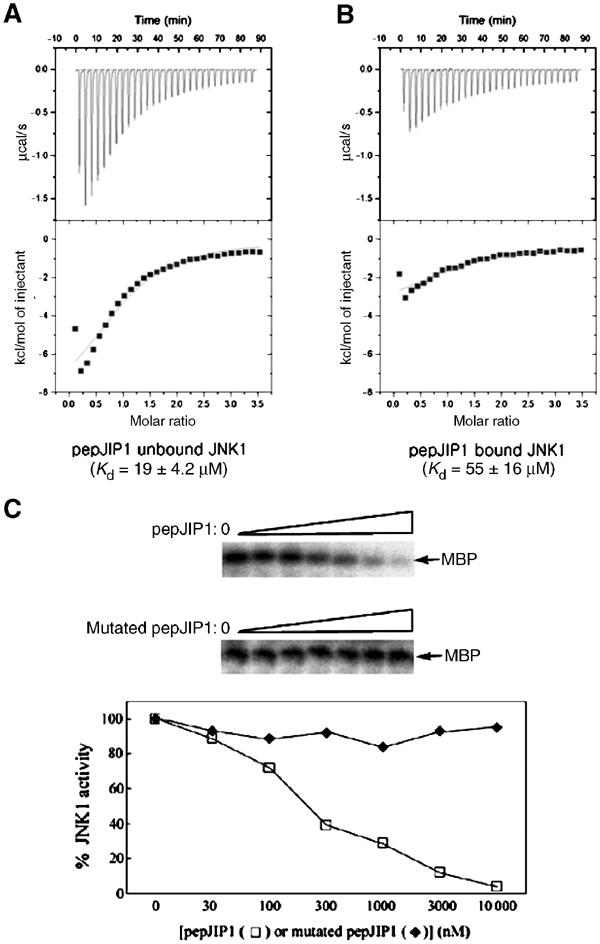
The inhibited phosphorylation of MBP, the docking site-independent substrate, due to the reduced ATP binding affinity to JNK1 by pepJIP1 binding. (A, B) The binding affinities of ATP to JNK1 were measured by ITC when pepJIP1 was unbound (A) and bound (B) to JNK1. (C) Dose-dependent inhibition of the kinase activity of JNK1 by pepJIP1 using MBP as substrates. The mutated pepJIP1 used for control experiment has the sequence of RPKAATTANAF.
In the structure of JNK3 complexed with AMPPNP, Asp189, Asp207, Glu111, and Lys93 are known to be catalytically important residues and Asp207 is connected to two magnesium ions by a water molecule and by an interaction with Asn194 (Xie et al, 1998). But, in the structure of the JNK1–pepJIP1 complex, the structure of the interaction between Asp169 and Asn156 is different from that of JNK3 due to the lack of the fixed magnesium ions. In addition, Lys55 and Glu73 are located 4 Å further away from the position of the corresponding residues of JNK3 due to the interdomain rearrangement by pepJIP1 binding (Figure 4C). These differences also may play a role in the reduction of the catalytic activity of JNK1 by pepJIP1 binding.
These structural changes by pepJIP1 binding in the inactive form of JNK1 might be quite different when pepJIP1 binds to the active form of JNK1, in which Thr or Tyr is phosphorylated. But, from the result of the BIAcore-based assay that the binding between JNK and pepJIP1 is not dependent on the kinase activity of JNK (Barr et al, 2002), the similar mode of interaction is expected when pepJIP1 would bind to the active JNK1.
Disorder of the phosphorylation loop
Much more residues of the phosphorylation loop of the JNK1 complex are disordered than the JNK3 structure (missing residues 175–187 in JNK1–pepJIP1 and 212–216 in JNK3) although their ordered regions adopt a similar conformation (Figure 4A). Even if it is not clear whether the increased disorder is a crystallographic artifact or not, the similar increased disorder of the phosphorylation loop of p38 MAPK was also found when MEF2A or MKK3b peptide binds (Chang et al, 2002). This implies that JNK and p38 may act as chemomechanical energy transducers, using peptide binding energy to generate allosteric conformational changes (Goldsmith, 1996).
In the structure of unphosphorylated JNK3, the relatively more ordered conformation of the phosphorylation loop could be achieved by interactions between this loop and the α1 helix of the N-terminal domain including two hydrogen-bonding interactions of Thr103–Ser217 and Asn101 (Nδ2 atom)–Phe218 (carbonyl oxygen atom) and one van der Waals contact of His104–Met219. However, in the structure of the JNK1–pepJIP1 complex, the α1 helix moved too far away for such interactions to occur as a result of the interdomain rearrangement upon pepJIP1 binding, and this loss of interactions released the phosphorylation loop of JNK1–pepJIP1 from the N-terminal domain, resulting in the increased disorder of the loop. The ordered phosphorylation loop in the structure of JNK3 partially blocks the substrate-binding site. This structural feature of JNK3 suggests that refolding of this loop is required to relieve steric constraints to the substrate binding and the activation of the substrate by JNK3 (Xie et al, 1998). As shown in the structure of the JNK1–pepJIP1 complex, the release of the phosphorylation loop, when JNK binds to the JIP1 scaffolding protein, may give a chance for the loop to refold and take an appropriate conformation to be recognized and phosphorylated by MKKs, which are also scaffolded in JIP1, and be ready to recognize the substrates.
The ternary complex JNK1–pepJIP1–SP600125
A small molecule, which acts as a selective inhibitor of JNK catalytic activity, was identified and characterized. SP600125, an anthrapyrazole, was identified in a high-throughput biochemical screen by using purified recombinant JNK2 and c-jun (Bennett et al, 2001). SP600125 is a reversible ATP-competitive inhibitor with more than 20-fold selectivity over other kinases including Erk, p38 MAPKs, MKKs, and PKCs. In cells, SP600125 dose-dependently inhibited the phosphorylation of c-jun, the expression of inflammatory genes COX-2, IL-2, IHN-γ, and TNF-α, and prevented the activation and differentiation of human CD4 cell. In in vivo studies in mice, SP600125 inhibited lipopolysaccharide-induced expression of TNF-α and prevented anti-CD3-mediated thymocyte apoptosis (Bennett et al, 2001). These previous studies imply that therapeutic inhibition of JNK has significant potential in inflammatory disease, acute organ failure due to apoptotic cell death, and cancer.
Recently, the structure of JNK3 in complex with SP600125 was reported (PDB entry 1PMV; Scapin et al, 2003). So, we have tried the structure of the ternary complex JNK1–pepJIP1–SP600125, by soaking the crystal of JNK1–pepJIP1 in the solution of SP600125. Surprisingly, the difference electron density corresponding to SP600125 clearly appeared on the adenine-binding pocket of the ATP-binding site (Figure 6). As an ATP-competitive inhibitor, SP600125 was not expected to bind to the complex form of JNK1–pepJIP1 with such a high occupancy in the crystal because the distortion of the ATP-binding site by pepJIP1 was supposed to block productive binding of ATP or ATP-like molecules, including ATP-competitive inhibitors. Fortunately, the small molecule, SP600125, occupied the deepest adenine-binding site only, not expanding to the phosphate-binding site, whereby the binding of SP600125 could not be influenced much by the distortion of the ATP-binding site, which moved the residues of JNK1 interacting with the phosphate groups of ATP far away from the active site (Figure 4B). In this structure, a little shift of SP600125 from the position of the inhibitor bound to JNK3 was found as a result of the movement of the residues in the hydrophobic pocket (Figure 6D). The structure of this ternary complex can be considered as a structural model for the state where SP600125 blocks JNK1 when it is bound to JIP1 scaffolding protein in vivo. Table I summarizes the statistics for data collection and refinement.
Figure 6.
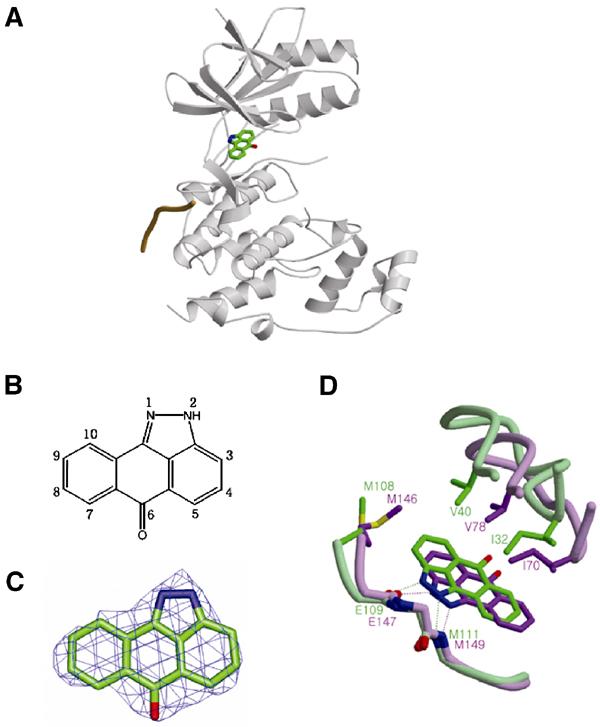
The ternary complex JNK1–pepJIP1–SP600125. (A) SP600125 is bound at the hydrophobic pocket of the ATP-binding site. pepJIP1 is depicted as a single coil of orange color. (B) The numbering scheme of the compound SP600125. (C) The Fo–Fc map calculated with a final refined model without SP600125 at the 3σ contour level. The refined atomic model of SP600125 is superimposed on the map. (D) Comparison of the positions of SP600125 when it is bound to JNK1 complexed with pepJIP1 (green) and free JNK3 (violet). The residues related with this shift are labeled and hydrogen bonds are represented by dashed lines.
Table 1.
Statistics on data collection and structure refinement
| Binary complex (JNK1–pepJIP1) | Ternary complex (JNK1–pepJIP1–SP600125) | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.0000 | 1.1271 |
| Space group | P212121 | P212121 |
| Unit cell (a, b, c) (Å) | 62.40, 80.84, 83.74 | 61.69, 79.46, 82.86 |
| Resolution (Å) | 2.35 | 2.70 |
| Observations | 45 475 | 33 160 |
| Unique reflections | 16 980 | 10 605 |
| Completeness (%) | 91.9 (77.7) | 90.4 (73.5) |
| Average I/σ(I) | 25.43 (2.53) | 15.31 (2.30) |
| Rsym (%)a | 6.4 (29.7) | 7.8 (33.4) |
| Structure refinement | ||
| Resolution (Å) | 20.0–2.35 | 20.0–2.70 |
| Rcryst b (%) | 22.6 | 21.6 |
| Rfree c (%) | 24.5 | 24.1 |
| R.m.s.d.d | ||
| Bonds (Å) | 0.008 | 0.009 |
| Angles (deg) | 1.2 | 1.3 |
| Reflections (∣F∣>0σ) | 16 720 | 10 497 |
| Average B-factor (Å2) | ||
| JNK1 | 61.4 | 48.8 |
| PepJIP1 | 60.1 | 49.2 |
| SP600125 | — | 41.7 |
| Water |
57.8 |
28.8 |
| Values in parentheses are for the outer resolution shell. | ||
| a Rsym=∑h∑i∣Ih,i−〈Ih,i〉∣/∑h∑i∑h,i for the intensity (I) of i observations of reflection h. | ||
| b Rcryst=∑∣Fobs−Fcalc∣/∑∣Fobs∣, where Fobs and Fcalc are the observed and calculated structure factors, respectively. | ||
| c Rfree=R-factor calculated using 5% of the reflections data chosen randomly and omitted from the start of refinement. | ||
| d Root-mean-square deviations from ideal geometry. | ||
Selectivity of SP600125 over other kinases
In the ternary complex structure, SP600125 makes two hydrogen bonds with JNK1 between the N atom at the 1-position and the backbone amide group of Met111, and between the NH group at the 2-position and the backbone carbonyl oxygen atom of Glu109 (Figure 7A). And these hydrogen bonds are common in many structures of inhibitor-bound kinases. This result is very consistent with the previous structure–activity relationship studies, which revealed that all of the derivatives having N-alkyl substituents instead of the free NH group at the 2-position showed significant loss in inhibitory activity (Bennett et al, 2001). In addition, SP600125 is surrounded by hydrophobic residues in the adenine-binding pocket (Ile32, Val40, Ala53, Ile86, Met108, Leu110, Val158, and Leu168) with effective van der Waals contacts, and the JNK specificity of SP600125 seems to come from the interactions with these hydrophobic residues. SP600125 inhibited JNK1, 2, and 3 isoforms with similar potency, and all of the hydrophobic residues are conserved in JNK isoforms. The latest result demonstrates that the JNK specificity of SP600125 is not so excellent and this compound inhibits some protein kinases with similar or greater potency than JNK (Bain et al, 2003). SP600125 exhibited selectivity against protein kinases including p38, ERK2, MKK1, GSK3β, CSK, CK2, LCK, PDK1, PKBα, and PKA. However, other protein kinases including PHK, CK1, CDK2, CHK1, AMP-activated protein kinase, and p70 ribosomal protein S6 kinase 1 were inhibited to similar or greater extent than JNK. To investigate the importance of the above hydrophobic residues for the selectivity of SP600125, the amino acids are compared by superimposing the structures of the hydrophobic pockets of the protein kinases (Table II). The distinct variations of amino acids are found at the amino acids corresponding to Met108 and Leu168 of JNK1. The variation in the position of Leu168 does not provide any rationale for the selectivity. But, the variation in the amino acids corresponding to Met108 may lead us to the conclusion that the hydrophilic residues such as threonine or glutamine could repel the aromatic ring of SP600125 and thus reduce the inhibitory potency even though there are some kinases showing low inhibition by SP600125 despite hydrophobic residues at this position. Mutation of M108T or M108Q caused the complete removal of inhibition by SP600125 (Figure 7B and C). The aromatic side chain of phenylalanine at Met108 position could interact with the aromatic ring of SP600125 by a T-shaped interaction.
Figure 7.
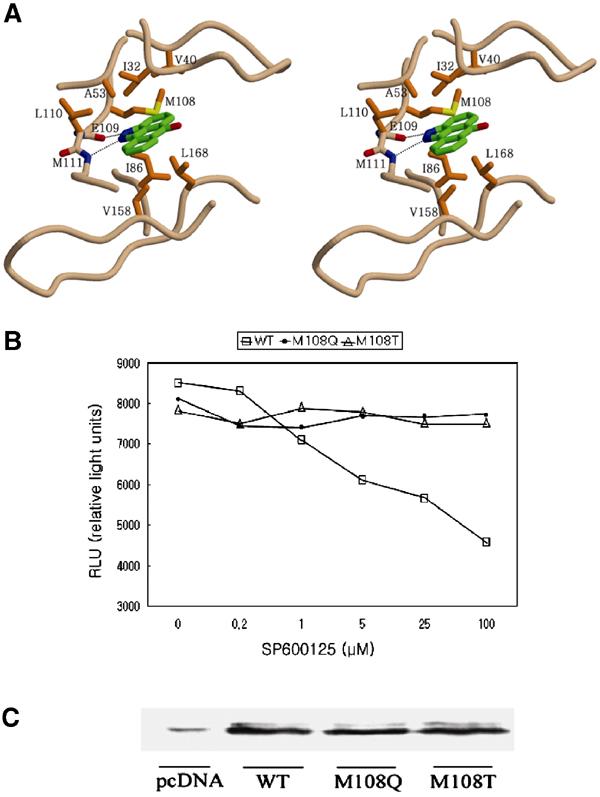
The importance of the hydrophobic residues of JNK1 for binding of SP600125. (A) Stereoview of the binding mode of SP600125 in the JNK1–pepJIP1 complex. The residues important for inhibitor binding are represented and labeled. The side chains of E109 and M111 are omitted for clarity. (B) Cell-based assay of the JNK1 wild type and mutants using SP600125 as an inhibitor. The kinase activity was measured in HeLa cells using the PathDetect® c-jun trans-reporting systems (Stratagene). RLU is proportional to the kinase activity. (C) The physical amounts of the JNK1 wild type and mutants used in cell-based assay were measured by Western blot analysis. The weak band labeled as pcDNA means the amount of JNK1 isoforms existing in HeLa cells when the pcDNA3.1A expression vector only was transfected, where the cDNA of JNK1 was not inserted.
Table 2.
Inhibition by SP600125 and amino-acid comparison in the hydrophobic cleft
| Protein kinasea | % Activityb | ||||||||
| JNK1 | 38±5 | I32 | V40 | A53 | I86 | M108 | L110 | V158 | L168 |
| P38 | 86±3 | V | V | A | I | T | L | A | L |
| ERK2 | 55±3 | I | V | A | I | Q | L | L | C |
| GSK3β | 60±5 | I | V | A | V | L | Y | L | C |
| CSK | 71±6 | L | V | A | V | T | Y | L | S |
| CK2 | 63±1 | V | V | I | V | F | Y | M | I |
| LCK | 53±1 | L | V | A | V | T | Y | L | A |
| PDK1 | 62±5 | L | V | A | V | L | Y | L | T |
| PKBα | 95±2 | L | V | A | V | M | Y | M | T |
| PKA | 82±1 | L | V | A | V | M | Y | L | T |
| PHK | 34±1 | L | V | A | I | F | L | L | T |
| CDK2/cyclin A | 20±1 | I | V | A | V | F | F | L | A |
| CK1 | 10±1 | I | I | A | P | I | L | L | V |
| CHK1 |
39±0 |
L |
V |
A |
V |
L |
Y |
L |
S |
| a Abbreviations are not defined in the text: GSK3β, glycogen synthase kinase 3β; CSK, C-terminal Src kinase; CK, casein kinase; LCK, lymphocyte kinase; PDK1, 3-phosphoinositide-dependent protein kinase 1; PKBα, protein kinase Bα; PKA, cAMP-dependent protein kinase; PHK, phosphorylase kinase; CDK2, cyclin-dependent protein kinase 2; CHK1, checkpoint kinase 1. | |||||||||
| b Kinase activity as a percentage of that in control after incubation with SP600125 of 10 μM. The data are from the previously published result (Bain et al, 2003). | |||||||||
It is thought that the selectivity of SP600125 may be determined by the size of the hydrophobic pocket, which varies in diverse protein kinases, and not merely by the amino acids themselves of the hydrophobic pocket. And the size of the pocket may also be connected with the intrinsic flexibility of the loops around the catalytic cleft in protein kinases, and the diverse modes of the flexibility dependent on each protein kinases could be important determinants of the selectivity of SP600125.
Discussion
The structural analysis in this study demonstrates that the peptide derived from the docking site of the scaffolding protein also binds at the same docking grooves of JNK1 as those of the upstream kinase and the substrate transcription factor do in p38 MAPK. However, similar but nonidentical docking grooves in each MAPKs appear to be the important determinant of pathway specificity in addition to the variations in sequences of docking sites. The docking groove of JNK1 is expected to accommodate the docking sites of MKK7 or c-jun as that of p38 does, but there may exist other important determinants than the interaction by Arg156 (φA−4) of JIP1 for specific recognition of the docking sites of MKK7 or c-jun because this residue is not conserved in the sequential comparison (Figure 1D). But, we cannot exclude the possibility that the basic residues at the position of φA−5 of MKK7 or c-jun may be involved in such an interaction as Arg156 (φA−4) of JIP1 as a result of any conformational change of the backbones outside of the φA-X-φB motifs.
The most striking feature of the present structure is the distortion of the ATP-binding site induced by the interdomain rearrangement upon pepJIP1 binding, providing another mechanism of allosteric inhibition of the kinase activity by interfering with productive binding of ATP. Although we could not get other crystals of unrelated lattices, this hinge motion is not likely to be a packing artifact because this structural feature is consistent with the experimental results including ITC measurement of ATP binding affinity and the inhibition of phosphorylation of a docking site-independent substrate by pepJIP1. A similar structural distortion of the ATP-binding site has been reported in the crystal structure of CDK6 bound to the tumor suppressor p16INK4a or p19INK4d (Brotherton et al, 1998; Russo et al, 1998). In the structure, the INK4 inhibitors bind next to the ATP-binding site, opposite where the activating cyclin binds, and thereby not only prevent cyclin binding indirectly but also reduce the affinity of the kinase for ATP by causing extensive allosteric changes in CDK6.
Overexpression of the cytoplasmic protein JIP1 inhibits JNK activity by blocking nuclear translocation of JNK and sequestering JNK module components into different JIP1 complexes. In other words, the inhibition of the full-length JIP1 in vivo is more complicated than that of pepJIP1, and the structural feature of the inhibition by full-length JIP1 might have significant differences from that of pepJIP1.
The ability of JNK to bind different proteins including upstream kinases, substrate transcription factors, and scaffolding proteins with high specificity may be mediated by the conformational flexibility of the loop connecting β7 and β8 and the disordered 331–351 loop. Phosphorylation of JNK by MKKs and activation of c-jun by JNK also might undergo such an interdomain rearrangement as the JNK1–pepJIP1 complex due to the intrinsic flexibility of the loops around the catalytic cleft. This inherent structural flexibility may play a central role in allowing the JNK pathway to be regulated by specific interactions with many docking site proteins. This flexibility may have been evolution's answer to the need that JNK should play its diverse roles in many physiological processes including cell growth, oncogenic transformation, cell differentiation, apoptosis, and the immune response by mediating extracellular stresses to cellular signals.
Although further work will be required to verify whether the docking site interactions could be used as targets for non-ATP-competitive drugs against protein kinases, the information from this structural study can contribute to the optimization of JNK inhibitors of high affinity and specificity, which can be derived from the docking site peptide of JIP1.
Inspection of the active site occupied by SP600125 provides some suggestions for improvement of inhibitor binding affinity. Although most of the surface of SP600125 is surrounded by the hydrophobic surface of the adenine-binding site of JNK1, expansion of SP600125 may be advantageous by adding some functional groups at the 5-, 6-, and 7-positions, which look toward the phosphate group-binding site through the ATP-binding cleft, and the conquest of the phosphate group-binding site by expanding through the crevice could improve the binding affinity. Specifically, adding a long polar group at the 5-position may provide additional hydrogen bonds with the polar surface of the phosphate group-binding site and higher water solubility of the derivative than that of SP600125 itself, which is poorly soluble in aqueous solvents (0.0012 mg/ml in water). We believe that these structural studies can provide clues for development of more potent and selective JNK inhibitors with better pharmacological profiles than SP600125.
Materials and methods
HeLa cell culture, Western blot analysis, protein expression and purification, crystallization, and data collection
See Supplementary material available at The EMBO Journal Online.
In vitro JNK1 inhibition studies
The inhibition of JNK1 to phosphorylate MBP by pepJIP1 was measured in duplicate using the active JNK1α1 (Upstate) with MBP (Upstate) as a substrate and pepJIP1 (RPKRPTTLNLF) as an inhibitor and the mutated pepJIP1 (RPKAATTANAF) as a control inhibitor. All reactions were performed for 60 min at 30°C in the solution containing 100 nM JNK1α1, 18 μM MBP, 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 0.4 mM DTT, 1 mM EGTA, and 1 mM EDTA and assay volume of 50 μl. The reaction was initiated by the addition of 1 μCi of [32P]ATP and unlabeled ATP to a final concentration of 1 μM ATP with a range of the peptide inhibitor concentrations (0, 30, 100, 300, 1000, 3000, and 10 000 nM). Each reaction was terminated by the addition of 10 μl of 5% phosphoric acid. 32P-incorporated MBP was resolved by SDS–PAGE and the gels were dried and exposed to a phosphorimager plate, and 32P incorporation was quantified by integration of the bands with background correction.
Cell-based assay for JNK1 activity
We carried out transfections using lipofectamine Plus™ reagent (Invitrogen) with a luciferase vector as a normalization control. The transfection mixes included 1 μg pFR-Luc plasmid, 50 ng pFA2 c-jun plasmid, 50 ng pFC2-dbd plasmid, and 50 ng JNK1 wild-type or mutants (M108Q and M108T) plasmid. Cell-based assay for the inhibition of JNK1 wild type and mutants by SP600125 was performed in HeLa cells using the PathDetect® c-jun trans-reporting systems (Stratagene). The physical amount of JNK1 was measured by Western blot analysis.
Structure determination and refinement
The structure of the binary complex JNK1–pepJIP1 was solved by molecular replacement with the unphosphorylated JNK3 structure (PDB entry 1JNK) using CNS (Brünger et al, 1998). Due to the interdomain conformational change of JNK1 induced by pepJIP1 binding, MR search with the full domain structure of JNK3 as a search model gave no solution. After deleting many parts of the N-terminal domain of the JNK3 model, which were supposed to be flexible, we could get the MR solution. The model was refined with the program CNS and manual model building was performed using the program QUANTA (Accelrys Inc., San Diego). The Fo–Fc electron density corresponding to pepJIP1 was shown clearly after a full domain model of JNK1 was built. Subsequent rounds of model adjustment, simulated annealing, and thermal parameter refinement were performed, and during the final stage of refinement water molecules were inserted in the protein model. The final model contained one JNK1 molecule of residues 9–172, 190–281, 287–336, and 349–363, one pepJIP1 molecule of residues 154–163, and 97 water molecules.
The structure of the ternary complex JNK1–pepJIP1–SP600125 was solved after one cycle of rigid-body refinement using the structure of the binary complex as a probe. A strong and planar density was found in the deep adenine pocket of JNK1, and SP600125 was easily positioned on the planar density due to its obvious orientation and structural rigidity. Additional residues can be built from the disordered residues of the model of the binary complex and the thermal parameters were lowered roughly by 13 Å2. The long-time soaking is supposed to make the crystal shrink a little and make it more ordered through the dehydration effect (Heras et al, 2003). The final model contained one JNK1 molecule (residues 9–174, 188–281, 287–337, and 347–363), one pepJIP1, one SP600125 molecule, and 15 water molecules.
Isothermal titration calorimetry
The binding affinities of pepJIP1 to wild-type JNK1 and to mutants R127A and E329A were determined by titration of pepJIP1 solution to the protein solutions, and the binding affinities of ATP to the JNK1–pepJIP1 complex and to free JNK1 were measured by titration of ATP solution at 20°C using a MicroCal VP instrument (MicroCal). Protein solutions were dialyzed extensively in 25 mM HEPES buffer, pH 7.0, 100 mM NaCl, 10% glycerol, and 5 mM β-mercaptoethanol, filtered and degassed before measurement. The data were analyzed by MicroCal Origin software.
Accession numbers
Coordinates have been deposited in the Protein Data Bank under accession codes 1UKH (the binary complex JNK1–JIP1) and 1UKI (the ternary complex JNK1–JIP1–SP600125).
Supplementary Material
Supplementary Material
Acknowledgments
We are grateful to Dr Seong-Eon Ryu in KRIBB for a critical reading of the manuscript and Ho Min Kim in KAIST for assistance with phosphorimager. We also thank the staffs of the beamline 6B1 during data collection at Pohang Light Source (PLS). Experiment at PLS was supported, in part, by MOST and POSCO. This work was supported by a grant from the National Research Laboratory Program and the 21C Frontier R&D Program, subsidized by Korea Ministry of Science and Technology. This work was also supported by a grant from Korea Ministry of Commerce, Industry and Energy, and the Brain Korea 21 program in part.
References
- Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem J 371: 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell AJ, Flatauer LJ, Matsukuma K, Thorner J, Bardwell L (2001) A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J Biol Chem 276: 10374–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr RK, Kendrick TS, Bogoyevitch MA (2002) Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem 277: 10987–10997 [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 98: 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50: 77–82 [DOI] [PubMed] [Google Scholar]
- Bozyczko-Coyne D, Saporito MS, Hudkins RL (2002) Targeting the JNK pathway for therapeutic benefit in CNS disease. Curr Drug Target CNS Neurol Disord 1: 31–49 [DOI] [PubMed] [Google Scholar]
- Brotherton DH, Dhanaraj V, Wick S, Brizuela L, Domaille PJ, Volyanik E, Xu X, Parisini E, Smith BO, Archer SJ, Serrano M, Brenner SL, Blundell TL, Laue ED (1998) Crystal structure of the complex of the cyclin D-dependent kinase Cdk6 bound to the cell-cycle inhibitor p19INK4d. Nature 395: 244–250 [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell 9: 1241–1249 [DOI] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- Dickens M, Roger JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyer CL, Davis RJ (1997) A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277: 693–696 [DOI] [PubMed] [Google Scholar]
- Elion EA (1998) Routing MAP kinase cascade. Science 281: 1625–1626 [DOI] [PubMed] [Google Scholar]
- Faux MC, Scott JD (1996) Molecular glue: kinase anchoring and scaffold proteins. Cell 85: 9–12 [DOI] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL (1999) Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol 11: 211–218 [DOI] [PubMed] [Google Scholar]
- Goldsmith EJ (1996) Allosteric enzymes as models for chemomechanical energy transducing assemblies. FASEB J 10: 702–708 [DOI] [PubMed] [Google Scholar]
- Gum R, Wang H, Lengyel E, Juarez J, Boyd D (1997) Regulation of 92 kDa type IV collagenase expression by the jun aminoterminal kinase- and the extracellular signal-regulated kinase-dependent signaling cascades. Oncogene 14: 1481–1493 [DOI] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 15: 2760–2770 [PMC free article] [PubMed] [Google Scholar]
- Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS (2002) Joint damage and inflammation in c-jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum 46: 818–823 [DOI] [PubMed] [Google Scholar]
- Heras B, Edeling MA, Byriel KA, Jones A, Raina S, Martin JL (2003) Dehydration converts DsbG crystal diffraction from low to high resolution. Structure 11: 139–145 [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420: 333–336 [DOI] [PubMed] [Google Scholar]
- Ho DT, Bardwell AJ, Abdollahi M, Bardwell L (2003) A docking site in MKK4 mediates high-affinity binding to JNK MAP kinases and competes with similar docking sites in JNK substrates. J Biol Chem 278: 32662–32672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PM, Cooper JA (1999) Protein modification: docking sites for kinases. Curr Biol 9: R329–R331 [DOI] [PubMed] [Google Scholar]
- Kemp BE, Pearson RB (1991) Design and use of peptide substrates for protein kinases. Methods Enzymol 200: 121–134 [DOI] [PubMed] [Google Scholar]
- Kim IJ, Lee KW, Park BY, Lee JK, Park J, Choi IY, Eom SJ, Chang TS, Kim MJ, Yeom YI, Chang SK, Lee YD, Choi EJ, Han PL (1999) Molecular cloning of multiple splicing variants of JIP-1 preferentially expressed in brain. J Neurochem 72: 1335–1343 [DOI] [PubMed] [Google Scholar]
- Manning AM, Davis RJ (2003) Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov 2: 554–565 [DOI] [PubMed] [Google Scholar]
- Martin JH, Mohit AA, Miller CA (1996) Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Brain Res Mol Brain Res 35: 47–57 [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D (1995) Localization of protein kinases by anchoring protein: a theme in signal transduction. Science 268: 247–251 [DOI] [PubMed] [Google Scholar]
- Mohit AA, Martin JH, Miller CA (1995) p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 14: 67–78 [DOI] [PubMed] [Google Scholar]
- Pawson T (1995) Protein modules and signaling networks. Nature 373: 573–580 [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD (1997) Signalling through scaffold, anchoring, and adaptor proteins. Science 278: 2075–2080 [DOI] [PubMed] [Google Scholar]
- Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP (1998) Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395: 237–243 [DOI] [PubMed] [Google Scholar]
- Scapin G, Patel SB, Lisnock J, Becker JW, LoGrasso PV (2003) The structure of JNK3 in complex with small molecule inhibitors: structural basis for potency and selectivity. Chem Biol 10: 705–712 [DOI] [PubMed] [Google Scholar]
- Sharrocks AD, Yang SH, Galanis A (2000) Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci 25: 448–453 [DOI] [PubMed] [Google Scholar]
- Smith JA, Poteet-Smith CE, Lannigan DA, Freed TA, Zoltoski AJ, Sturgill TW (2000) Creation of a stress-activated p90 ribosomal S6 kinase. The carboxyl-terminal tail of the MAPK-activated protein kinases dictates the signal transduction pathway in which they function. J Biol Chem 275: 31588–31593 [DOI] [PubMed] [Google Scholar]
- Tanoue T, Maeda R, Adachi M, Nishida E (2001) Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J 20: 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Nishida E (2002) Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol Ther 93: 193–202 [DOI] [PubMed] [Google Scholar]
- Tournier C, Whitmarsh AJ, Cavanagh J, Barrett T, Davis RJ (1999) The MKK7 gene encodes a group of c-jun NH2-terminal kinase kinases. Mol Cell Biol 19: 1569–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA (2001) beta-Amyloid-induced neuronal apoptosis requires c-jun N-terminal kinase activation. J Neurochem 77: 157–164 [DOI] [PubMed] [Google Scholar]
- Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, Hani EH, Vionnet N, Nicod P, Boutin P, Froguel P (2000) The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet 24: 291–295 [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ (1998) A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281: 1671–1674 [DOI] [PubMed] [Google Scholar]
- Xie X, Gu Y, Fox T, Coll JT, Fleming MA, Markland W, Caron PR, Wilson KP, Su MS (1998) Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure 6: 983–991 [DOI] [PubMed] [Google Scholar]
- Yang S-H, Whitmarsh AJ, Davis RJ, Sharrocks AD (1998) Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J 17: 1740–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Raina AK, Perry G, Smith MA (2002) The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals 11: 270–281 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material