

Abstract
Mitogen-activated protein kinases (MAPKs) are integral to the mechanisms by which cells respond to physiological stimuli and to a wide variety of environmental stresses. MAPK cascades can be inactivated at the MAPK activation step by members of the MAPK phosphatase (MKP) family. However, the components that act in MKP-regulated pathways have not been well characterized in the context of whole organisms. Here we characterize the Caenorhabditis elegans vhp-1 gene, encoding an MKP that acts preferentially on the c-Jun N-terminal kinase (JNK) and p38 MAPKs. We found that animals defective in vhp-1 are arrested during larval development. This vhp-1 defect is suppressed by loss-of-function mutations in the kgb-1, mek-1, and mlk-1 genes encoding a JNK-like MAPK, an MKK7-type MAPKK, and an MLK-type MAPKKK, respectively. The genetic and biochemical data presented here demonstrate a critical role for VHP-1 in the KGB-1 pathway. Loss-of-function mutations in each component in the KGB-1 pathway result in hypersensitivity to heavy metals. These results suggest that VHP-1 plays a pivotal role in the integration and fine-tuning of the stress response regulated by the KGB-1 MAPK pathway.
Keywords: C. elegans, heavy metal stress, MAP kinase, MAPK phosphatase
Introduction
Mitogen-activated protein kinases (MAPKs) belong to a family of evolutionarily conserved protein kinases that regulate cellular fates and responses to a variety of extracellular signals. Three subgroups of MAPKs have been identified: the extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 kinases (Chang and Karin, 2001). Extracellular signals activate MAPKs through a specific dual-specificity MAPK kinase (MAPKK), which phosphorylates MAPKs on critical threonine and tyrosine residues within a Thr-X-Tyr motif. Several subgroups of the MAPKK superfamily have been identified, including MEK1, MEK2, MKK3, MKK4, MKK6, and MKK7 (Chang and Karin, 2001). MEK1 and MEK2 activate the ERK group. MKK4 can activate both the JNK and p38 subgroups and MKK7 is specific for the JNK subgroup, while MKK3 and MKK6 act solely as activators of the p38 subgroup. The specific MAPKKs are themselves phosphorylated and activated by specific MAPKK kinases (MAPKKKs) such as Raf, TAK1, Tpl2, ASK1, and the MEKK and MLK group of proteins.
The JNK pathway has been implicated in a variety of biological functions in mammalian cells, including apoptosis and the responses to stress. Recent studies using model genetic organisms have revealed some of the physiological roles of the JNK signaling pathway. In the insect Drosophila melanogaster, the JNK pathway is required for mid-embryonic development (Martin and Wood, 2002). Mutants for two components of the Drosophila JNK (D-JNK) pathway, hemipterous (hep) and basket (bsk), have been identified and shown to encode Drosophila homologs of MKK7 and JNK, respectively (Noselli and Agnes, 1999; Stronach and Perrimon, 1999). In the absence of Hep or Bsk function, lateral epithelial cells fail to stretch, and the embryo develops a hole in its dorsal cuticle. Therefore, the D-JNK pathway plays a critical role in the dorsal closure process. D-JNK is also required for some forms of developmental apoptosis in Drosophila (Adachi-Yamada et al, 1999; Igaki et al, 2002; Moreno et al, 2002).
In Caenorhabditis elegans, genetic studies demonstrate that the JNK pathway, composed of the MKK7-type MAPKK, JKK-1, and the JNK-type MAPK, JNK-1, regulates coordinated movement via type D GABAergic (GABA: γ-aminobutyric acid) motor neurons, and, in contrast to Drosophila, does not appear to be essential for embryonic morphogenesis (Kawasaki et al, 1999). Furthermore, the C. elegans JNK signaling pathway also has a role in synaptic vesicle transport. A loss-of-function mutation in jnk-1 results in the mislocalization of a synaptic vesicle marker (Byrd et al, 2001). C. elegans also possesses another MKK7-type MAPKK called MEK-1, disruption of which results in hypersensitivity to heavy metals, suggesting that MEK-1 plays a pivotal role in the stress response (Koga et al, 2000).
The duration and extent of MAPK activation are governed by a balance between MAPKK-mediated phosphorylation and protein phosphatase-mediated dephosphorylation on the threonine and tyrosine residues in the Thr-X-Tyr motif. Thus, protein phosphatases are critical regulators of MAPK-dependent signaling events. These phosphatases are specific for phosphorylated tyrosine, serine/threonine, or both, as in the case of the dual-specificity MAPK phosphatases (MKPs), which are highly specific for MAPKs (Camps et al, 2000; Keyse, 2000). MKPs, which can remove phosphate groups from both the critical threonine and tyrosine residues of activated MAPKs, contain a specific MAPK-binding domain located in the N-terminal half and a catalytic phosphatase domain located in the C-terminal portion. The binding of an MKP to an activated MAPK induces a conformational change in the MKP that stimulates its catalytic activity (Camps et al, 2000; Keyse, 2000). Overexpression studies have suggested that different MKPs display different activities toward the MAPKs ERK, JNK, and p38 (Camps et al, 2000). These experiments show that MKP-5 and MKP-7 have higher activity against JNK and p38 than against ERK, whereas MKP-3, MKP-4, and PAC1 appear more specific for ERK. MKP-1 appears to have equal activity against ERK, JNK, and p38. Thus, the mammalian members of the MKP family appear to be selective in their inactivation of distinct MAPK isoforms. In addition, each MKP exhibits distinguishing features in terms of subcellular localization, tissue distribution, and transcriptional induction pattern (Camps et al, 2000).
Recent genetic studies in multicellular eukaryotes have reinforced biochemical studies, demonstrating the important role of MKPs in the precise regulation of MAPK activities (Keyse, 2000). A null mutant of the Drosophila MKP puckered (puc) exhibits severe developmental defects, resulting in embryonic lethality due to hyperactivation of D-JNK and failure of dorsal closure (Martin-Blanco et al, 1998). The C. elegans MKP, LIP-1, regulates the G2/M meiotic arrest of developing oocytes by dephosphorylating MPK-1/SUR-1, an ERK-type MAPK (Berset et al, 2001; Hajnal and Berset, 2002). The Arabidopsis mkp1 mutation results in hypersensitivity to genotoxic stress (Ulm et al, 2001).
To further understand the biological function of MKPs in the context of a whole organism, we isolated and characterized the vhp-1 gene in the genetically amenable organism C. elegans. The vhp-1 gene encodes an MKP that is most similar to mammalian MKP-7, which acts preferentially on JNK and p38 MAPKs. Here we show that VHP-1 negatively regulates a novel JNK-like MAPK pathway composed of MLK-1 (MAPKKK), MEK-1 (MAPKK), and KGB-1 (JNK-like MAPK) that is involved in a stress response to heavy metals. Moreover, worms harboring the null allele for vhp-1 undergo larval arrest, most likely due to hyperactivation of the KGB-1 pathway. Our analysis provides the first demonstration of an in vivo function for a dual-specificity phosphatase in regulating a stress response, and establishes a specific genetic link between MKPs and the JNK-like MAPK pathway.
Results
Isolation of the vhp-1 gene
A search of the C. elegans genome revealed seven MKP-like genes. Among them, we identified a candidate, termed vhp-1 (VH1-like phosphatase1; corresponding to F08B1.1a), that is most similar to human MKP-7 (53% identity and 72% similarity in the catalytic domain), which acts preferentially on JNK and p38 MAPKs (Masuda et al, 2001; Tanoue et al, 2001). The predicted VHP-1 protein contains an N-terminal rhodanase homology domain, which contains an MAPK-interacting motif, and an extended active-site sequence motif Ile/Val-His-Cys-X-X-Gly-X-Ser-Arg-Ser (where X is any amino acid) that is conserved in all MKPs (Figure 1A). Within this latter motif, the two amino acids Cys-262 and Ser-269, together with Asp-231, are likely to participate in the catalytic mechanism of dual-specificity phosphatase activity (Camps et al, 2000).
Figure 1.
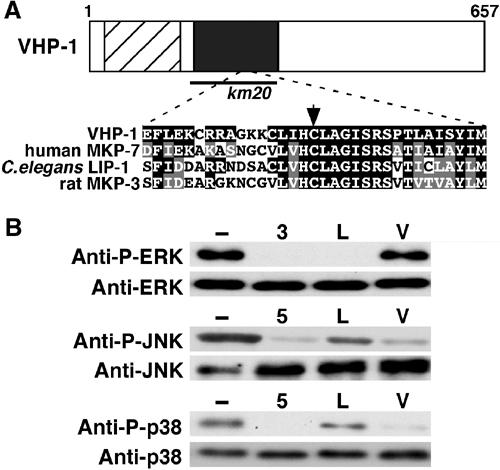
Structure and enzymatic properties of VHP-1. (A) Schematic representation of VHP-1. Hatched and dark boxes represent the rhodanase homology and dual-specificity phosphatase catalytic domains, respectively. The bold line underneath shows the extent of the km20 deletion. Sequence alignment of the catalytic domains of VHP-1, human MKP-7, C. elegans LIP-1, and rat MKP-3 is shown below. Identical and similar residues are highlighted with black and gray shading, respectively. The arrow indicates the essential Cys-262 residue. The DDBJ/EMBL/GenBank number for the VHP-1 sequence is AY585194. (B) In vitro phosphatase assay. Myc-tagged MKPs, rat MKP-3 (3), human MKP-5 (5), LIP-1 (L), and VHP-1 (V), were expressed in COS7 cells and immunoprecipitated with anti-Myc antibody. They were then used for in vitro phosphatase assays utilizing GFP-phospho-ERK, GST-phospho-JNK and GST-phospho-p38 as exogenous substrates. Reaction mixtures were immunoblotted with anti-phospho-MAPKs and anti-MAPKs antibodies. In all experiments, the amounts of immunoprecipitated Myc-MKPs were verified by immunoblotting, confirming that similar amounts of MKPs were used for each assay (data not shown).
Comparison of the database genomic DNA and the cloned cDNA sequences revealed that the vhp-1 gene has seven exons (Figure 2A). To determine the expression pattern of vhp-1, we constructed a translational fusion between vhp-1 (including both promoter and coding sequences) and green fluorescent protein (GFP) to generate vhp-1∷gfp (Figure 2A). This fusion protein has functional activity in C. elegans (see below). Transgenic C. elegans bearing the vhp-1∷gfp fusion showed that VHP-1∷GFP is expressed throughout development in the pharynx, intestine, neurons, and vulval hypodermal cells (data not shown).
Figure 2.

The vhp-1 deletion. (A) Structure of the vhp-1 gene. Exons are indicated by boxes. The shaded and open boxes are the translated and untranslated regions, respectively. The black boxes indicate the phosphatase domains. vhp-1(km20) is an 876 bp deletion mutation. The GFP fusion construct is also indicated. (B) Phenotype of vhp-1 mutants. N2 (wild type; WT), vhp-1(km20), vhp-1(km20); Ex[vhp-1∷gfp], and vhp-1(km20); mlk-1(km701) worms were grown for 4 days after hatching. Bar: 100 μm.
VHP-1 is a phosphatase specific for JNK and p38 MAPKs
We examined the substrate specificities of VHP-1 and LIP-1 for representatives of the three major classes of MAPKs: ERK2, p38α, and JNK2. Each MAPK was expressed in Escherichia coli, isolated, and phosphorylated in vitro on the Thr-X-Tyr motif by MAPKKs (see Materials and methods). It is known that mammalian MKP-3 specifically acts on ERK, whereas MKP-5 has higher activity against JNK and p38 than against ERK (Camps et al, 2000). The phosphorylated MAPKs were then incubated with immunoprecipitated Myc-VHP-1, Myc-LIP-1, Myc-MKP-3, or Myc-MKP-5, and the phosphorylation status of the MAPKs was followed by immunoblotting with anti-phospho-MAPKs antibodies that specifically recognize the phosphorylated form of each MAPK. As shown in Figure 1B, Myc-VHP-1 dephosphorylated JNK2 and p38α, but not ERK2. In contrast, Myc-LIP-1 dephosphorylated ERK2, but did not efficiently dephosphorylate JNK2 or p38α. Thus, VHP-1 acts preferentially on JNK and p38 MAPKs, whereas LIP-1 is specific for ERK. These results are consistent with the observation that VHP-1 is homologous to MKP-7 and that LIP-1 regulates the G2/M meiotic arrest of developing oocytes by dephosphorylating ERK-type MPK-1 (Berset et al, 2001; Hajnal and Berset, 2002).
Isolation of a vhp-1 deletion mutant
To gain an insight into the physiological role of vhp-1 in the context of an intact organism, we undertook a reverse genetic approach to isolate loss-of-function deletion mutations in the vhp-1 gene. Using a PCR-based assay, we screened a library of worms mutagenized by a TMP/UV method, and isolated a presumptive null mutation in vhp-1 (km20: Figure 2A) that removes 876 base pairs encoding a section of the catalytic domain of VHP-1 (Figure 1A). Progeny homozygous for vhp-1(km20) appeared normal during embryogenesis and were indistinguishable from the wild type in morphology and movement at the time of hatching (data not shown). However, vhp-1(km20) mutants did not become larger than the L2 or L3 larvae in body size even after 8 days following hatching, whereas wild-type animals became adults in 4 days, passing through four larval stages (L1–L4) (Figure 2B). This vhp-1(km20) phenotype was rescued by the vhp-1∷gfp transgene, supporting the idea that the larval arrest phenotype of the vhp-1(km20) mutant was indeed caused by loss of vhp-1 function. RNAi of vhp-1 has been reported to result in the following phenotypes: poor health, exploded vulva, and sluggish behavior (Maeda et al, 2001; Kamath et al, 2003). Thus, vhp-1 may exert different effects on worms when the function of vhp-1 is partially defective.
Isolation of mlk-1 as a vhp-1 suppressor
The above results suggested that hyperactivation of an MAPK pathway results in early larval arrest. If this is true, a mutation that causes inactivation of the relevant MAPK pathway should suppress the growth arrest caused by the loss-of-function vhp-1 mutation and may identify the components involved in VHP-1-mediated signaling. In total, we isolated seven independent extragenic suppressor mutations (Figure 2B). Complementation analysis revealed that they were classified into three groups. The first and second groups were mapped to Linkage Group IV and X, respectively (data not shown). The remaining one, km701, was mapped to Linkage Group V between −4.4 and −2.7, a region that contains the mlk-1 gene encoding an MAPKKK homologous to mammalian MLK, a mixed-lineage kinase subfamily of the MAPKKK superfamily (Gallo and Johnson, 2002). We found that, compared to wild-type animals, km701 animals harbor a single-base pair change within the mlk-1-coding region that results in an amino-acid substitution in kinase domain V, thereby changing a conserved glycine to arginine (Figure 3A). To confirm the suppression of vhp-1 by the mlk-1 mutation, we constructed the deletion mutant mlk-1(km19) (Figure 3B). We found that the growth arrest phenotype of vhp-1(km20) was suppressed by the mlk-1(km19) deletion mutant (Figure 4).
Figure 3.
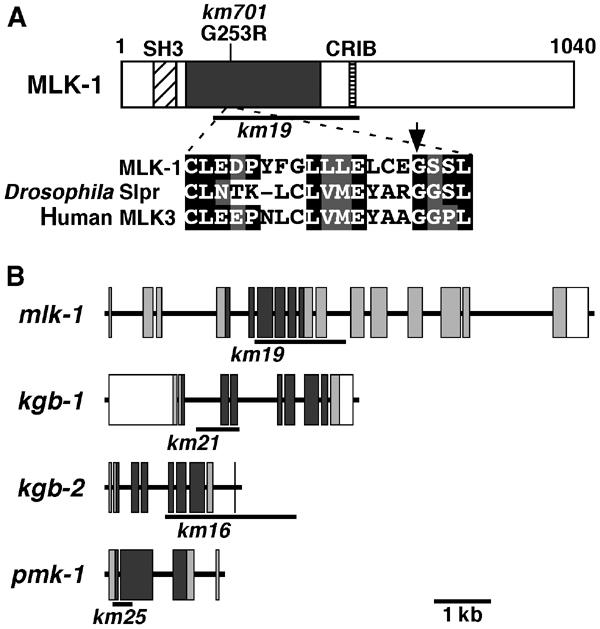
Structures of MLK-1 and genes encoding MAPKs. (A) Schematic representation of MLK-1. Composite structure of MLK-1 shows an N-terminal SH3 domain, a catalytic kinase domain (black box), and a Cdc42/Rac-interacting binding motif (CRIB). The km701 allele has a mutation within the kinase domain V as indicated. The bold line underneath shows the extent of the km19 deletion. A sequence alignment of the kinase domain V of MLK-1, Drosophila Slpr, and human MLK3 is shown below. Identical and similar residues are highlighted with black and gray shading, respectively. The arrow indicates the mutation site in the km701 allele. (B) Structures of the mlk-1, kgb-1, kgb-2, and pmk-1 genes. Exons are indicated by boxes. The shaded and open boxes are the translated and untranslated regions, respectively. The black boxes indicate kinase domains. The bold lines underneath show the extent of each deletion allele.
Figure 4.
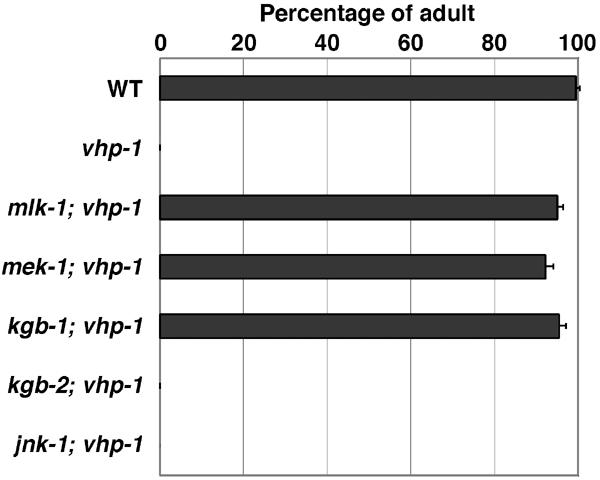
Suppression of the vhp-1 larval arrest phenotype. The percentage of each worm that reached adulthood 4 days after egg laying is shown with standard errors.
kgb-1 and mek-1 deletion mutations suppress the vhp-1 defect
Mammalian members of the MLK family have been implicated in the regulation of JNK signal transduction at the MAPKKK level (Gallo and Johnson, 2002). Consistent with this observation, we found that the growth arrest phenotype caused by the vhp-1(km20) mutation was suppressed by deletion of the C. elegans JNK homolog KGB-1, kgb-1(km21) (Figures 3B and 4). Interestingly, however, deletion of two other JNK homologs JNK-1, jnk-1(gk7) (Villanueva et al, 2001), or KGB-2, kgb-2(km16) (Figure 3B), did not suppress the vhp-1(km20) mutation (Figure 4). Thus, vhp-1 suppression is specific to the KGB-1 JNK-like MAPK. Mammals have two MAPKKs that act on JNKs: MKK4 and MKK7 (Weston and Davis, 2002). C. elegans MEK-1 is homologous to MKK7 (Koga et al, 2000). Indeed, we found that the vhp-1 growth defect was suppressed by a mek-1(ks54) deletion mutation (Figure 4). Taken together, these results suggest that VHP-1 negatively regulates the JNK-like MAPK pathway composed of KGB-1 (MAPK), MEK-1 (MAPKK), and MLK-1 (MAPKKK), and that hyperactivation of this pathway causes growth arrest in the early larval stage.
KGB-1 activity is regulated by MEK-1 and VHP-1
To determine whether KGB-1 is activated by MEK-1, 293 cells were co-transfected with mammalian expression vectors encoding Flag epitope-tagged MEK-1 (Flag-MEK-1) and HA epitope-tagged KGB-1 (HA-KGB-1). HA-KGB-1 was then immunoprecipitated from cell lysates and used in a protein kinase assay with GST-c-Jun protein as a substrate. The c-Jun transcription factor is known to be phosphorylated by JNK in mammalian cells (Chang and Karin, 2001; Weston and Davis, 2002). Transfection with MEK-1 resulted in strong activation of KGB-1 (Figure 5A). Transfection with a kinase-inactive form of MEK-1, in which Lys-99 in the ATP-binding domain had been mutated to arginine, abrogated KGB-1 activation. This indicates that the kinase activity of MEK-1 is required for activation of KGB-1. Western blot analysis showed that the kinase-inactive mutant was expressed at levels comparable to that of the wild-type MEK-1.
Figure 5.
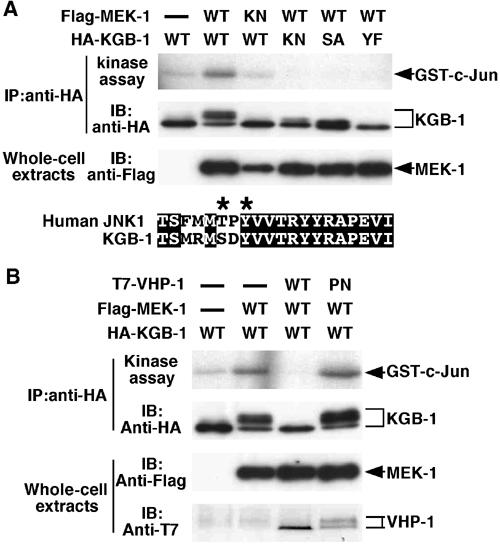
Regulation of KGB-1 activity by MEK-1 and VHP-1. (A) Activation of KGB-1 by MEK-1. 293 cells were transfected with Flag-MEK-1 (WT), Flag-MEK-1(K99R) (KN), HA-KGB-1 (WT), HA-KGB-1(K67R) (KN), HA-KGB-1(S198A) (SA), and HA-KGB-1(Y200F) (YF) as indicated. Immunoprecipitated complexes obtained with anti-HA antibodies were used for in vitro kinase reactions with GST-c-Jun as a substrate (top panel). The amounts of immunoprecipitated HA-KGB-1 were determined with anti-HA antibodies (middle panel). Whole-cell extracts were also immunoblotted with anti-Flag antibodies (bottom panel). Comparison of the phosphorylation lip regions of KGB-1 and human JNK1 is shown below. The Thr-X-Tyr motif of JNK1 is indicated by asterisks. (B) Inactivation of KGB-1 by VHP-1. 293 cells were transfected with Flag-MEK-1 (WT), HA-KGB-1 (WT), T7-VHP-1 (WT), and T7-VHP-1(C262S) (PN) as indicated. Immunocomplex kinase assays were carried out as described above.
All known MAPKs have a conserved Thr-X-Tyr motif in kinase domain VIII that is phosphorylated by an MAPKK (Camps et al, 2000; Keyse, 2000; Chang and Karin, 2001). The JNK subgroup of the MAPK superfamily has the sequence Thr-Pro-Tyr (Camps et al, 2000; Keyse, 2000; Chang and Karin, 2001), whereas KGB-1 possesses the sequence Ser-Asp-Tyr at the analogous site (aa 198–200: Figure 5A). To investigate whether the Ser-198 and Tyr-200 residues are required for activation of KGB-1, we transiently co-transfected 293 cells with plasmids encoding Flag-MEK-1 and two mutants of KGB-1: HA-KGB-1(S198A) and HA-KGB-1(Y200F), in which Ser-198 is changed to alanine, and Tyr-200 is changed to phenylalanine, respectively. Immune complex kinase assays showed that neither mutant form of KGB-1 was activated by MEK-1 (Figure 5A). Thus, the Ser-Asp-Tyr motif in KGB-1 is essential for its kinase activity.
Next, we examined the catalytic activity of VHP-1 toward KGB-1. Co-expression of VHP-1 efficiently suppressed the activation of KGB-1 by MEK-1 (Figure 5B). It is known that dual-specificity phosphatases can be rendered catalytically inactive by replacing a cysteine residue in the catalytic active site with serine (Masuda et al, 2001; Tanoue et al, 2001). We therefore introduced this mutation into VHP-1, generating VHP-1(C262S), and found that this mutant could not inactivate KGB-1 (Figure 5B). These results demonstrate that VHP-1 efficiently inactivates KGB-1. When co-expressed with active KGB-1, the catalytically inactive VHP-1(C262S) protein migrated as two bands, one of which had a slightly higher mass than the wild-type protein (Figure 5B). This suggests that VHP-1 is phosphorylated by its target KGB-1 as the activity of KGB-1 is unaffected by the expression of VHP-1(C262S) but completely suppressed by the wild-type VHP-1.
KGB-1 activity is dependent on MLK-1 and MEK-1 in C. elegans
To determine whether KGB-1 is regulated by MLK-1 and MEK-1 in C. elegans, KGB-1 kinase activity was tested in wild-type N2, mlk-1(km19), and mek-1(ks54) animals. If MLK-1 and MEK-1 function as an MAPKKK and an MAPKK, respectively, in activating the KGB-1 pathway in C. elegans, KGB-1 should show reduced kinase activity in loss-of-function mlk-1(km19) or mek-1(ks54) mutant animals. KGB-1 proteins were immunoprecipitated with anti-KGB-1 antibodies and tested in a protein kinase assay using GST-c-Jun as a substrate. KGB-1 immunocomplexes prepared from wild-type animals were found to have clearly detectable kinase activity (Figure 6A). However, when KGB-1 was immunoprecipitated from cell extracts of mlk-1(km19) or mek-1(ks54) mutant animals, its kinase activity was significantly decreased (Figure 6A). This indicates that MLK-1 and MEK-1 are required for the full activation of KGB-1 in C. elegans. Taken together, these results indicate that MLK-1 MAPKKK and MEK-1 MAPKK act in the same pathway upstream of KGB-1 and function to activate KGB-1 kinase activity.
Figure 6.
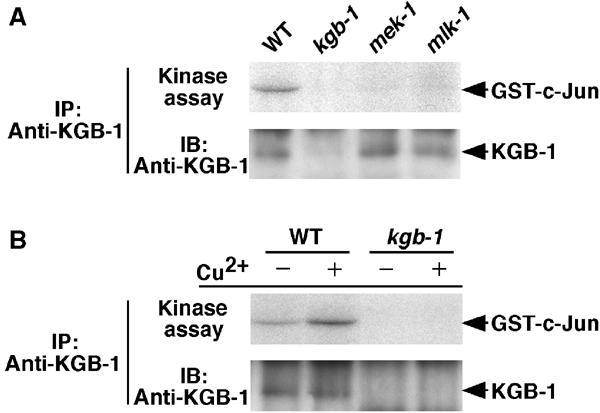
Regulation of KGB-1 activity in C. elegans. (A) Effects of mek-1 and mlk-1 mutations on KGB-1 activity in C. elegans. Cell lysates from N2 (WT), kgb-1(km21), mek-1(ks54), and mlk-1(km19) animals were immunoprecipitated with anti-KGB-1 antibodies. The immunoprecipitates were used for in vitro kinase reactions with GST-c-Jun as a substrate (upper panel). The immunoprecipitates were immunoblotted with anti-KGB-1 antibodies (lower panel). (B) Activation of KGB-1 by copper ion. N2 (WT) and kgb-1(km21) animals were treated with copper sulfate (500 μM) for 12 h. Extracts prepared from each animal were immunoprecipitated with anti-KGB-1 antibodies. The immunoprecipitates were used for in vitro kinase reactions as described above.
The MLK-1-MEK-1-KGB-1 pathway confers tolerance to heavy metal stress
What kind of signal is transduced by the MLK-1-MEK-1-KGB-1 MAPK cascade? Recently, it has been shown that MEK-1 is involved in the response to different types of stress, including heavy metals (Koga et al, 2000). We therefore tested whether the KGB-1 pathway also regulates a stress response to heavy metals using kgb-1(km21) and mlk-1(km19) mutant animals. These strains were placed on agar plates containing heavy metal ions and their development was monitored for any signs of an altered response to these toxic compounds. We found that kgb-1(km21) and mlk-1(km19) mutants were hypersensitive to copper (Cu2+) and cadmium (Cd2+) ions, similar to the mek-1(ks54) null allele (Figure 7A). In contrast to N2 wild-type animals, which grew well, becoming adults by 4 days, on plates containing Cu2+ (100 μM) or Cd2+ (100 μM), kgb-1(km21) and mlk-1(km19) mutants grew poorly. In fact, most of the mutant animals failed to reach the adult stage within 4 days. These results suggest that the MLK-1-MEK-1-KGB-1 MAPK pathway regulates the response to heavy metal stress.
Figure 7.
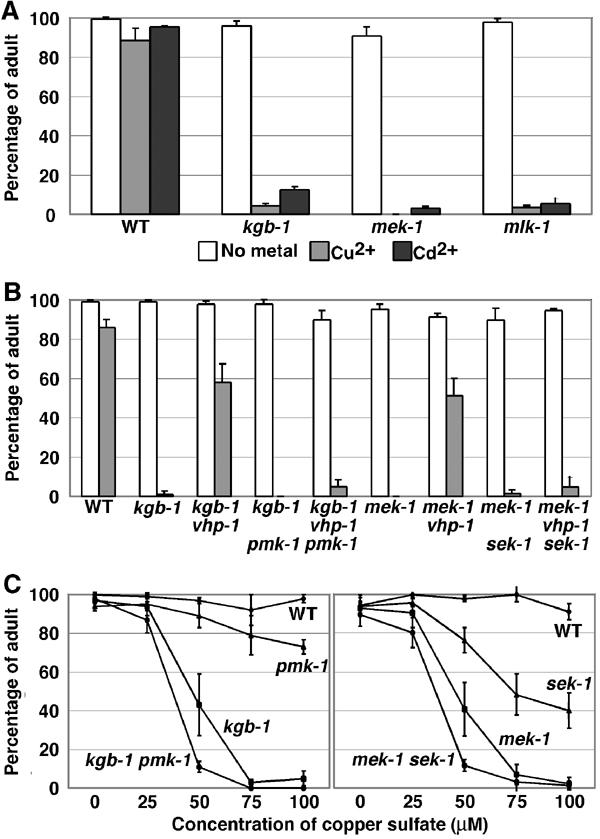
Effects of heavy metals on growth. Each worm was cultured from embryogenesis on normal plates containing copper sulfate (100 μM) and cadmium chloride (100 μM) (A, B) or the indicated concentrations of copper sulfate (C). The percentage of worms reaching adulthood 4 days after egg laying is shown with standard errors.
To confirm that KGB-1 is involved in the heavy metal stress response, we examined the effect of heavy metal exposure on KGB-1 protein kinase activity. N2 wild-type animals were treated with Cu2+; the lysates were subjected to immunoprecipitation with anti-KGB-1 antibodies, and in vitro protein kinase assays were performed using GST-c-Jun as a substrate. We found that KGB-1 activity was indeed upregulated by treatment of animals with Cu2+ (Figure 6B).
VHP-1 negatively regulates the KGB-1 and PMK-1 pathways in response to heavy metal stress
To determine whether VHP-1 is involved in the response to heavy metal stress, we examined the effect of the vhp-1(km20) mutation on Cu2+ sensitivity in a kgb-1(km21) or mek-1(ks54) background. The vhp-1 mutation partially suppressed the Cu2+ sensitive phenotype of kgb-1 and mek-1 mutants (Figure 7B), indicating that VHP-1 functions as a negative regulator of stress tolerance. If KGB-1 is the only MAPK involved in heavy metal stress response, we would not have expected vhp-1(km20) to suppress kgb-1(km21). Therefore, this result raised the possibility that another MAPK pathway that is negatively regulated by VHP-1 may play a redundant role with the KGB-1 pathway in mediating the heavy metal stress response. We looked for another MAPK that might participate in the stress response. As the mammalian MKP-7 acts preferentially on p38 MAPK as well as JNK (Masuda et al, 2001; Tanoue et al, 2001), we investigated the C. elegans p38 homolog pmk-1 (Berman et al, 2001) by examining the effect of the loss-of-function mutation pmk-1(km25) (Figure 3B) in animals having a wild-type, kgb-1(km21), or kgb-1(km21); vhp-1(km20) background. The pmk-1(km25) mutation caused a modest sensitivity to Cu2+ ion, enhanced the heavy metal sensitivity of kgb-1(km21) mutants (Figure 7C), and suppressed stress resistance in the kgb-1(km21); vhp-1(km20) double mutant (Figure 7B).
It is known that SEK-1 MAPKK functions upstream of PMK-1 in an innate immune response (Kim et al, 2002), suggesting that SEK-1 may also regulate heavy metal stress by activating PMK-1. Supporting this possibility, the loss-of-function mutation sek-1(km4) (Tanaka-Hino et al, 2002) exhibited phenotypes similar to those of the pmk-1(km25) mutation with regard to the heavy metal response: sek-1(km4) mutants were slightly sensitive to heavy metals; the sek-1(km4) mek-1(ks54) double mutant was more sensitive than the mek-1(ks54) single mutant; and a sek-1(km4) mek-1(ks54); vhp-1(km20) triple mutant was markedly sensitive to heavy metals (Figure 7B and C). Taken together, these data suggest that the SEK-1-PMK-1 p38-type MAPK pathway serves a role that is redundant with the MEK-1-KGB-1 JNK-like MAPK pathway in the response to heavy metals, and that VHP-1 negatively regulates both the KGB-1 and PMK-1 pathways.
Discussion
An important determinant of the biological response to stress is the magnitude and duration of MAPK activation, which is governed by upstream activating MAPKKs and deactivating MKPs. However, there have been relatively few genetic studies of this response in the context of a whole multicellular eukaryote organism. Some members of the MKP family display specificity with regard to the MAPKs they inactivate. The C. elegans VHP-1 protein is an MKP that is specific for JNK and p38 MAPKs in vitro. This substrate selectivity of VHP-1 is similar to that of the mammalian MKP-7, which is the mammalian homolog most similar to C. elegans VHP-1. In this study, we show that VHP-1 plays an important role in the heavy metal stress response in C. elegans by negatively regulating the KGB-1 (JNK-like) and the PMK-1 (p38-type) MAPK signaling pathways (Figure 8). This is the first report demonstrating an in vivo function for a dual-specificity phosphatase in regulating a stress response, and establishes a specific genetic link between MKPs and the JNK-like MAPK pathway.
Figure 8.
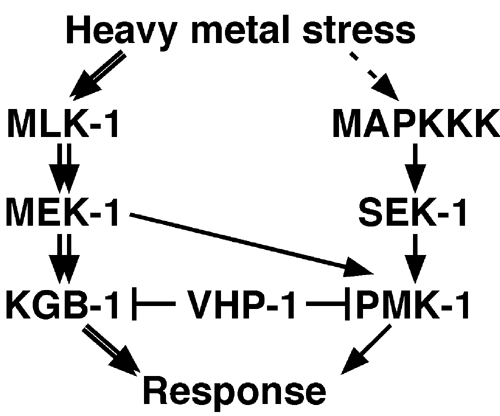
Relationship between VHP-1 and MAPK pathways. See text for details.
Relationship between VHP-1 and MAPK pathways in C. elegans
Here, we examined the relationship between VHP-1 and the C. elegans MAPKs, and found that VHP-1 can genetically interact with the KGB-1 JNK-like MAPK. Disruption of the vhp-1 gene results in an early larval arrest phenotype. The vhp-1 larval arrest phenotype is rescued by a loss-of-function mutation of kgb-1. Furthermore, the vhp-1 phenotype is suppressed by loss-of-function mutations in the mlk-1 and mek-1 genes, which encode an MLK-type MAPKKK and an MKK7-type MAPKK, respectively. Endogenous KGB-1 kinase activity is greatly decreased in mlk-1 and mek-1 null mutant animals compared with wild-type animals. These results suggest that MLK-1, MEK-1, and KGB-1 function in the same signal transduction pathway, and that disruption of vhp-1 causes hyperactivation of the MLK-1-MEK-1-KGB-1 signaling pathway, resulting in early larval arrest.
Several lines of evidence indicate that the MLK-1-MEK-1-KGB-1 signaling cascade and its negative regulator VHP-1 are involved in the regulation of the response to heavy metal stress. First, Koga et al (2000) have demonstrated that mek-1 null mutants exhibit hypersensitivity to several heavy metals. We showed that animals harboring a null allele for kgb-1 or mlk-1 are hypersensitive to heavy metals, similar to mek-1 null mutants. Second, treatment of animals with copper ion stimulates the kinase activity of endogenous KGB-1, implicating KGB-1 as a heavy metal-responsive MAPK in C. elegans. Third, deletion of vhp-1 partially suppresses the hypersensitivity of kgb-1 mutant animals to heavy metals. This suggests that VHP-1 functions as a negative regulator of heavy metal resistance. Thus, the MLK-1-MEK-1-KGB-1 pathway is required for tolerance to heavy metal stress, and VHP-1 negatively regulates the KGB-1 pathway. One model of VHP-1 action is that it dephosphorylates KGB-1 at a constant rate, but that heavy metal stress stimulates KGB-1 phosphorylation at a rate that overrides the effect of VHP-1. Whereas heavy metal-induced activation of the KGB-1 pathway contributes to tolerance to stress in C. elegans, prolonged and high-level activation triggers an early larval arrest. It is not clear at present as to how the larval arrest caused by the hyperactivation of the KGB-1 pathway is related to the heavy metal stress response. Previously, Villanueva et al (2001) have reported that JNK-1 regulates the heavy metal stress response by functioning downstream of MEK-1. However, we observed no difference in the sensitivity of jnk-1(gk7) null mutants to heavy metals compared with wild-type animals (data not shown). The reason for this discrepancy is not clear. Furthermore, a jnk-1 null mutation failed to suppress the vhp-1 larval arrest phenotype. These results indicate that JNK-1 is not involved in the heavy metal response.
Suppression of the kgb-1 hypersensitivity to heavy metals by vhp-1 suggested the possibility that VHP-1 negatively regulates another MAPK that functions in a redundant manner to mediate tolerance to heavy metals. We found that a pmk-1 null mutation diminishes the ability of vhp-1 to suppress the kgb-1 heavy metal-sensitive phenotype. Furthermore, the pmk-1 null mutation enhances the heavy metal sensitivity of kgb-1 mutant animals. These results suggest that PMK-1 is another target of VHP-1. Supporting this possibility, Kim et al (submitted) have demonstrated that VHP-1 acts on PMK-1 in the innate immune response. Thus, VHP-1 negatively regulates both the KGB-1 JNK-like MAPK and the p38-type PMK-1. The results presented here link MAPK signaling pathways to the C. elegans stress response and demonstrate a genetically defined role for an MKP in stress signaling in the context of a multicellular eukaryote.
The MLK-1-MEK-1-KGB-1 signaling cascade in C. elegans
The C. elegans mlk-1 gene encodes a mixed-lineage kinase related to the mammalian MLKs and Drosophila Slipper (Slpr), which have been shown to stimulate JNK activity (Gallo and Johnson, 2002). One interesting observation is that the homology between MLK-1 and the MLKs is found not only in their kinase domains, but also in their non-catalytic domains. C. elegans MLK-1 contains an N-terminal SH3 domain, followed sequentially by a kinase domain and a Cdc42/Rac-interactive binding (CRIB) motif, common to the MLK subfamily. This similarity may indicate that these MLKs utilize a common regulatory mechanism. Components that are known or have been suggested to operate upstream of MLKs include MAPKKK kinases (MAPKKKKs) and low-molecular-weight GTP-binding proteins, including Rac and Cdc42 (Gallo and Johnson, 2002). Indeed, Drosophila Slpr is regulated by D-Rac1 and an MAPKKKK called Misshapen (Msn) (Stronach and Perrimon, 2002). In addition, mutations in the C. elegans homolog of Msn, mig-15(rh148), have no effect on the sensitivity to heavy metals, whereas mutations in the C. elegans Rac proteins ced-10(n3246) and mig-2(mu28) cause sensitivity to heavy metals (data not shown). This supports the physiological relevance of Rac in regulating the activity of MLK-1. The identification of other components upstream of MLK-1 in C. elegans will undoubtedly provide valuable insights into the signaling pathway regulating the response to heavy metal stress.
The vhp-1 deletion suppressed the hypersensitivity to heavy metals of mek-1 and kgb-1 mutants, but not that of mek-1 sek-1 and kgb-1 pmk-1 double mutants. Furthermore, sek-1 and pmk-1 mutations enhanced the heavy metal sensitivity in mek-1 and kgb-1 mutants, respectively. These results suggest that the MEK-1-KGB-1 and SEK-1-PMK-1 pathways regulate heavy metal stress response redundantly. Do these pathways crosstalk? For example, do MEK-1 and SEK-1 activate both KGB-1 and PMK-1? Biochemical analysis revealed that the sek-1(km4) mutation had no effect on the kinase activity of KGB-1 in C. elegans (data not shown). Thus, SEK-1 does not seem to be required for KGB-1 activation. On the other hand, Kim et al (submitted) have demonstrated that MEK-1 acts on PMK-1 in the innate immune response. Thus, KGB-1 is specifically activated by MEK-1, whereas PMK-1 is activated by two different MAPKKs, SEK-1 and MEK-1 (Figure 8). This suggests that crosstalk occurs at the MAPKK step in the signaling pathway.
MAPKs are activated by dual phosphorylation on a conserved Thr-X-Tyr motif in their activation loop by an upstream MAPKK (Camps et al, 2000; Keyse, 2000). KGB-1 is categorized as a member of the JNK subfamily, based on characteristic features of its sequence. However, KGB-1 has a Ser-Asp-Tyr motif, which differs from the normal JNK sequence (Thr-Pro-Tyr). We observed that both the Ser-198 and Tyr-200 residues are required for KGB-1 activation by MEK-1. This suggests either that MEK-1 can catalyze phosphorylation of both serine and tyrosine residues, resulting in KGB-1 activation, or that autophosphorylation contributes to activation. In the latter case, MEK-1 would catalyze the phosphorylation of KGB-1 on either Tyr-200 or Ser-198 and activate KGB-1 autophosphorylation of the other residue.
KGB-1 has been previously identified as a protein that interacts with the GLH proteins, C. elegans P granule components (Smith et al, 2002). The kgb-1(um3) null allele was reported to exhibit temperature-sensitive sterility at 25–26°C, characterized by meiotic defects, including endoreplicating oocytes and nonfunctional sperm. We observed similar phenotypes in kgb-1(km21), mek-1(ks54), and mlk-1(km19) mutants (data not shown). Smith et al (2002) have observed that kgb-1(um3) worms raised at high temperatures are less healthy than their wild-type counterparts, suggesting that KGB-1 may respond to the stress of temperature extremes. These results suggest that the MLK-1-MEK-1-KGB-1 pathway appears to affect other cellular processes in addition to the heavy metal stress response.
Materials and methods
Isolation of deletion mutants of vhp-1, mlk-1, kgb-1, kgb-2, and pmk-1
Deletion mutants vhp-1(km20), mlk-1(km19), kgb-1(km21), kgb-2(km16), and pmk-1(km25) were generated by a TMP/UV method (Gengyo-Ando and Mitani, 2000) and isolated using a sib-selection protocol. Direct sequencing of the PCR products verified the deleted region. These deletion mutants were backcrossed more than twice to animals of a wild-type N2 background. The km20 mutation was balanced by a balancer chromosome (mln1), because vhp-1(km20) homozygotes arrested in the early larval stage. All strains were maintained on nematode growth medium (NGM) plates and fed with bacteria of the OP50 strain, as described by Brenner (1974).
Construction of vhp-1∷gfp
A SmaI–BalI 1.7 kbp genomic fragment, which contains the fifth to seventh exons of vhp-1, was obtained by PCR using the cosmid F08B1 as a template. The 5′ end of this fragment (SmaI site) is within the fourth intron of vhp-1, and the 3′ end (BalI site) is just upstream of the stop codon. This fragment was subcloned into the SmaI–BalI site of the GFP reporter vector pPD95.75. Then, a 16.7 kbp SmaI fragment from the cosmid K10C6, whose 5′ end is 9.6 kbp upstream of the first exon and the 3′ end SmaI site is within the fourth intron (described above), was subcloned, yielding vhp-1∷gfp.
Screen for vhp-1 suppressor
km701 was isolated in a screen for vhp-1 suppressors performed as follows. Hermaphrodites of vhp-1(km20)/mln1 genotype were mutagenized with ethylmethane sulfonate (EMS). Worms bearing the mln1 chromosome express GFP in the pharynx. Therefore, adults not expressing GFP were isolated from the F2 progeny as candidates of a vhp-1 suppressor. The km701 mutation was mapped using single-nucleotide polymorphisms (SNP). To identify the km701 mutation, genomic DNA was prepared from km701 and subjected to PCR amplification with mlk-1 gene-derived primers. Direct sequencing of the PCR products verified the km701 mutation.
Strain construction
We generated vhp-1(km20)/mln1 hermaphrodites carrying a mutation in individual components of the MAPK cascades such as mlk-1(km19); vhp-1(km20)/mln1, kgb-1(km21); vhp-1(km20)/mln1, jnk-1(gk7); vhp-1(km20)/mln1, kgb-2(km16); vhp-1(km20)/mln1 and mek-1(ks54); vhp-1(km20)/mln1. These hermaphrodites were allowed to self-fertilize to investigate whether the growth arrest induced by vhp-1(km20) is suppressed by each mutation.
Growth arrest
vhp-1(km20)/mln1 hermaphrodites were grown and allowed to lay eggs on seeded NGM plates. About 200 embryos were transferred to fresh NGM plates. After incubation for 1 day, the numbers of hatched embryos were determined by counting unhatched embryos. After additional incubation for 1 day, the numbers of worms homozygous for vhp-1(km20) were determined by counting worms expressing GFP, which are vhp-1(km20)/mln1 or mln1. Worms expressing GFP were removed after counting. At 4 days after egg laying, worms that developed into adulthood were counted. The percentage of adults was calculated by multiplying the number of adults by 100 and dividing by the number of worms homozygous for vhp-1(km20).
Heavy metal sensitivity
Worms were grown and allowed to lay eggs on normal NGM plates seeded with Escherichia coli OP50 bacteria. About 50 embryos were transferred to NGM plates with or without heavy metal. After incubation for 1 day, the numbers of hatched embryos were determined by counting unhatched embryos. The worms that developed into adulthood were counted 4 days after egg laying. The percentage of adults was calculated by multiplying the number of adults by 100 and dividing by the number of hatched worms.
Construction of cDNAs
Full-length kgb-1 and vhp-1 cDNAs were amplified by PCR from a C. elegans cDNA library. The amplified cDNAs were subcloned into pBluescript and completely sequenced. Full-length mek-1 cDNA was provided by Y Ohshima. The mammalian expression vectors for HA epitope-tagged KGB-1, Flag epitope-tagged MEK-1, and T7 epitope-tagged VHP-1 were constructed by inserting each coding sequence into a vector expressing epitope-tagged protein under the control of the cytomegalovirus (CMV) promoter. Each coding sequence was amplified by PCR using primer sets to create restriction sites immediately before the first codon and after the stop codon. Mutated forms of KGB-1, MEK-1, and VHP-1 were made by oligonucleotide-directed PCR and the mutations were verified by DNA sequencing.
In vitro phosphatase assay
Recombinant proteins, GFP-phospho-ERK, GST-phospho-JNK, and GST-phospho-p38 were prepared as follows. His-tagged GFP-phospho-ERK (Xenopus ERK2) was prepared as described (Matsubayashi et al, 2001). Bacterially expressed GST-JNK (rat JNK2) and GST-p38 (human p38α) were purified and phosphorylated in vitro by MKK7 and MKK6, respectively. For the in vitro phosphatase assay, COS7 cells were transfected with Myc-tagged MKP constructs. After 24 h, cells were collected and lysed. Proteins tagged by the Myc epitope were immunoprecipitated with anti-Myc monoclonal antibody 9E10 (Santa Cruz Biotechnology, Inc.). Aliquots of immunoprecipitates were incubated with substrates in reaction buffer (20 mM Tris–HCl (pH 7.5), 1 mM DTT) at 37°C for 1 h. Samples were analyzed by 10% SDS–PAGE and immunoblotting. The phosphorylation state of substrates was examined using anti-phospho-ERK antibody E10 (New England Biolabs Inc.), anti-phospho-JNK antibody (New England Biolabs Inc.), and anti-phospho-p38 antibody (Sigma).
Assays for KGB-1 kinase activity in 293 mammalian cells
Immunoprecipitation from human embryonic kidney 293 cells was carried out as described previously (Kawasaki et al, 1999). Aliquots of immunoprecipitates were incubated with 2 mg of bacterially expressed GST-c-Jun in 10 μl of kinase buffer containing 20 mM Tris–HCl (pH 7.5), 20 mM MgCl2, 20 mM β-glycerophosphate, 20 mM ρ-nitrophenylphosphate, 1 mM EDTA, 1 mM sodium orthovanadate, 0.4 mM PMSF, 20 mM creatine phosphate, and 5 μCi of [γ-32P] ATP (3000 Ci/mmol) at 30°C for 30 min. Samples were analyzed by 10% SDS–PAGE and autoradiography. For immunoblotting, anti-HA rabbit polyclonal antibody Y-11 (Santa Cruz Biotechnology, Inc.), anti-Flag mouse monoclonal antibody M2 (Sigma), and anti-T7 mouse monoclonal antibody (Novagen) were used.
Assays for KGB-1 kinase activity in C. elegans
Anti-KGB-1 rabbit polyclonal antisera (KGB-1 C1-1 and KGB-1 C2-2) were raised against the synthetic polypeptides, SENRYDQEIDFADKTL (KGB-1 C1-1) and NHPYVKLWFKDDEV (KGB-1 C2-2), corresponding to the C-terminal portions of KGB-1. Anti-KGB-1 antibody KGB-1 C1-1 was affinity purified from its antiserum. For in vitro kinase assays, age-asynchronous worms were collected and washed twice with PBS, and sonicated in 0.5% Triton X-100 lysis buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 2 mM DTT, 1 mM sodium orthovanadate, 1 mM PMSF, and 20 μM aprotinin. Debris was removed by centrifugation. KGB-1 protein was immunoprecipitated with anti-KGB-1 antibody KGB-1 C1-1. Kinase assay was performed as above. For immunoblotting, we used a 1:500 dilution of anti-KGB-1 antiserum KGB-1 C2-2. To test the activation of KGB-1 in response to heavy metal, animals were incubated in NGM buffer (0.3% NaCl, 0.25% peptone, 5 μg/ml cholesterol, 1 mM CaCl2, 1 mM MgSO4, and 25 mM potassium phosphatase (pH 6.0)) with or without 500 μM CuSO4 for 12 h and washed twice with NGM buffer.
Acknowledgments
We thank I Mori and T Inada for scientific advice, M Noguchi for technical support, and A Coulson, A Fire, Y Kohara, Y Ohshima, and the Caenorhabditis Genetics Center for materials. DHK is supported by a postdoctoral fellowship from the Howard Hughes Medical Institute, by an NIH K08 Career Development Award, and by a Burroughs Wellcome Fund Career Award in the biomedical sciences. This work was supported by special grants for CREST and Advanced Research on Cancer from the Ministry of Education, Culture and Science of Japan (KM), and by NIH grant GM48707 (FMA).
References
- Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K (1999) Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400: 166–169 [DOI] [PubMed] [Google Scholar]
- Berman K, McKay J, Avery L, Cobb M (2001) Isolation and characterization of pmk-(1–3): three p38 homologs in Caenorhabditis elegans. Mol Cell Biol Res Commun 4: 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berset T, Hoier EF, Battu G, Canevascini S, Hajnal A (2001) Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science 291: 1055–1058 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y (2001) UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron 32: 787–800 [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Arkinstall S (2000) Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J 14: 6–16 [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37–40 [DOI] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL (2002) Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol 3: 663–672 [DOI] [PubMed] [Google Scholar]
- Gengyo-Ando K, Mitani S (2000) Characterization of mutations induced by ethyl methanesulfonate, UV, and trimethylpsoralen in the nematode Caenorhabditis elegans. Biochem Biophys Res Commun 269: 64–69 [DOI] [PubMed] [Google Scholar]
- Hajnal A, Berset T (2002) The C. elegans MAPK phosphatase LIP-1 is required for the G2/M meiotic arrest of developing oocytes. EMBO J 21: 4317–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M (2002) Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 21: 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Bot NL, Moreno S, Sohrmann M, Welchman DP, Zipperien P, Ahringer J (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237 [DOI] [PubMed] [Google Scholar]
- Kawasaki M, Hisamoto N, Iino Y, Yamamoto M, Ninomiya-Tsuji J, Matsumoto K (1999) A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J 18: 3604–3615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse SM (2000) Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol 12: 186–192 [DOI] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, Ausubel FM (2002) A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297: 623–626 [DOI] [PubMed] [Google Scholar]
- Koga M, Zwaal R, Guan KL, Avery L, Ohshima Y (2000) A Caenorhabditis elegans MAP kinase kinase, MEK-1, is involved in stress responses. EMBO J 19: 5148–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda I, Kohara Y, Yamamoto M, Sugimoto A (2001) Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr Biol 11: 171–176 [DOI] [PubMed] [Google Scholar]
- Martin P, Wood W (2002) Epithelial fusions in the embryo. Curr Opin Cell Biol 14: 569–574 [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A (1998) puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 12: 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda K, Shima H, Watanabe M, Kikuchi K (2001) MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J Biol Chem 276: 39002–39011 [DOI] [PubMed] [Google Scholar]
- Matsubayashi Y, Fukuda M, Nishida E (2001) Evidence for existence of a nuclear pore complex-mediated, cytosol-independent pathway of nuclear translocation of ERK MAP kinase in permeabilized cells. J Biol Chem 276: 41755–41760 [DOI] [PubMed] [Google Scholar]
- Moreno E, Yan M, Basler K (2002) Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12: 1263–1268 [DOI] [PubMed] [Google Scholar]
- Noselli S, Agnes F (1999) Roles of the JNK signaling pathway in Drosophila morphogenesis. Curr Opin Genet Dev 9: 466–472 [DOI] [PubMed] [Google Scholar]
- Smith P, Leung-Chiu WM, Montgomery R, Orsborn A, Kuznicki K, Gressman-Coberly E, Mutapcic L, Bennett K (2002) The GLH proteins, Caenorhabditis elegans P granule components, associate with CSN-5 and KGB-1, proteins necessary for fertility, and with ZYX-1, a predicted cytoskeletal protein. Dev Biol 251: 333–347 [DOI] [PubMed] [Google Scholar]
- Stronach BE, Perrimon N (1999) Stress signaling in Drosophila. Oncogene 18: 6172–6182 [DOI] [PubMed] [Google Scholar]
- Stronach BE, Perrimon N (2002) Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev 16: 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Hino M, Sagasti A, Hisamoto N, Kawasaki M, Nakano S, Ninomiya-Tsuji J, Bargamnn CI, Matsumoto K (2002) SEK-1 MAPKK mediates Ca2+ signaling to determine neuronal asymmetric development in C. elegans. EMBO Rep 3: 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Yamamoto T, Maeda R, Nishida E (2001) A Novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38α and β MAPKs. J Biol Chem 276: 26629–26639 [DOI] [PubMed] [Google Scholar]
- Ulm R, Revenkova E, di Sansebastiano GP, Bechtold N, Paszkowski J (2001) Mitogen-activated protein kinase phosphatase is required for genotoxic stress relief in Arabidopsis. Genes Dev 15: 699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A, Lozano J, Morales A, Lin X, Deng X, Hengartner MO, Kolesnick RN (2001) jkk-1 and mek-1 regulate body movement coordination and response to heavy metals through jnk-1 in Caenorhabditis elegans. EMBO J 20: 5114–5128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston CR, Davis RJ (2002) The JNK signal transduction pathway. Curr Opin Genet Dev 12: 14–21 [DOI] [PubMed] [Google Scholar]