

Abstract
The Imitation SWItch (ISWI) chromatin remodeling factors have been implicated in nucleosome positioning. In vitro, they can mobilize nucleosomes bi-directionally, making it difficult to envision how they can establish precise translational positioning of nucleosomes in vivo. It has been proposed that they require other cellular factors to do so, but none has been identified thus far. Here, we demonstrate that both ISW2 and TUP1 are required to position nucleosomes across the entire coding sequence of the DNA damage-inducible gene RNR3. The chromatin structure downstream of the URS is indistinguishable in Δisw2 and Δtup1 mutants, and the crosslinking of Tup1 and Isw2 to RNR3 is independent of each other, indicating that both complexes are required to maintain repressive chromatin structure. Furthermore, Tup1 repressed RNR3 and blocked preinitiation complex formation in the Δisw2 mutant, even though nucleosome positioning was completely disrupted over the promoter and ORF. Our study has revealed a novel collaboration between two nucleosome-positioning activities in vivo, and suggests that disruption of nucleosome positioning is insufficient to cause a high level of transcription.
Keywords: chromatin, ISW2, nucleosome positioning, repression, Ssn6–Tup1
Introduction
Chromatin structure plays an essential role in the regulation of eukaryotic gene expression, which is heavily dependent upon the balance between nucleosome positioning and disrupting activities. Our understanding of the latter has increased significantly over the past few years, but the mechanism of nucleosome positioning in vivo is much less clear (for reviews, see Tyler and Kadonaga, 1999; Peterson and Workman, 2000; Becker and Horz, 2002; Narlikar et al, 2002). Changes in nucleosome positioning and structure are carried out by ATP-dependent chromatin remodeling complexes, which include the SWI/SNF family, RSC, the imitation switch (ISWI) group, Chd/Mi-2 and the INO80 complex (for reviews, see Vignali et al, 2000; Langst and Becker, 2001; Narlikar et al, 2002).
Much of what we understand about the mechanism of nucleosome positioning by ATP-dependent remodeling complexes has come from the study of the imitation switch (ISWI) class of chromatin remodeling complexes. The ISWI protein from Drosophila exists in at least three complexes NURF, ACF and CHRAC (Tsukiyama and Wu, 1995; Ito et al, 1997; Varga-Weisz et al, 1997), and three ISWI-containing complexes have been isolated from the yeast Saccharomyces cerevisiae: ISW1a, ISW1b and ISW2 (Tsukiyama et al, 1999; Vary et al, 2003; Iida and Araki, 2004; McConnell et al, 2004). The ISW2 complex, containing Isw2 as the catalytic subunit, displayed only ATP-dependent nucleosome spacing/positioning activity without detectable nucleosome disruption activity in vitro (Tsukiyama et al, 1999), suggesting that it might be involved in transcription repression in vivo. This was supported by later studies showing that deletion of ISW2 weakened repression at many genes (Goldmark et al, 2000; Fazzio et al, 2001; Kent et al, 2001; Sugiyama and Nikawa, 2001; Ruiz et al, 2003). Furthermore, deletion of ISW2 or ISW1 caused a disruption of the chromatin structure over the URS of a variety of genes, and ISW2 may be targeted by Ume6 to some promoters (Goldmark et al, 2000; Kent et al, 2001). However, Drosophila ISWI is abundant and has a role in globally establishing and maintaining chromatin structure of the X chromosome (Deuring et al, 2000), indicating that features other than targeting by gene-specific transcription factors influence its activities. An important unanswered question is how ISWI complexes establish precisely positioned nucleosomes in vivo. In vitro they can slide nucleosomes bi-directionally and independently of DNA sequence composition, although some preferably slide nucleosomes to the ends or center of DNA fragments (Langst and Becker, 2001; Narlikar et al, 2002). Whether recruitment is the sole regulating feature, these complexes are just as likely to slide nucleosomes towards one end of a gene as the other. Thus, other factors must be required for ISWI complexes to position nucleosomes, but these factors or the mechanism have not been identified.
The Ssn6–Tup1 complex is a global corepressor responsible for nucleosome positioning at a number of genes and the recombination enhancer of the silent mating-type loci in budding yeast (Cooper et al, 1994; Weiss and Simpson, 1997; Kastaniotis et al, 2000; Fleming and Pennings, 2001; Li and Reese, 2001). Its nucleosome-positioning ability is proposed to involve interactions of Tup1 with hypoacetylated histone H3 and H4 tails (Edmondson et al, 1996; Ducker and Simpson, 2000; Davie et al, 2002). Mutating histone H3 and H4 tails or deleting genes encoding histone deacetylases (HDACs) weaken Tup1 interaction with promoters, the latter presumably due to the increase in histone tail acetylation, consistent with a model where the binding of Ssn6–Tup1 to histone tails is required to form a domain of repressed chromatin (Roth, 1995; Davie et al, 2002). In support of this idea, Tup1 crosslinking is detected in the coding sequence of the a-cell-specific gene STE6, and two Tup1 molecules per nucleosome were incorporated into the repressive chromatin structure of STE6 on a minichromosome (Ducker and Simpson, 2000), providing the basis for nucleosome positioning over the promoter and the protein-coding region. However, another group did not observe the spreading of Tup1 at STE6 (Wu et al, 2001). Furthermore, the ‘spreading' of Tup1 is not required for it to repress transcription because Tup1 crosslinking is restricted to the upstream regulatory sequence (URS) and promoter regions of RNR3, RNR2 and ENA1 (Wu et al, 2001; Davie et al, 2002), and nucleosome positioning over RNR3 extends into the coding sequence (Li and Reese, 2001). Therefore, Tup1 spreading cannot fully account for its repression and nucleosome-positioning activities.
The genes coding for the enzyme ribonucleotide reductase (RNR) and the similarly regulated gene HUG1 are under the tight control of the Ssn6–Tup1 complex (Zhou and Elledge, 1992; Basrai et al, 1999). Ssn6–Tup1 is recruited to their promoters by the sequence-specific DNA-binding protein Crt1 that recognizes the DNA damage response elements (DREs) in the URS (Huang et al, 1998; Li and Reese, 2001; Davie et al, 2002). Activation of DNA damage response pathways causes the release of Crt1 from the promoter, leading to derepression and chromatin remodeling (Huang et al, 1998; Li and Reese, 2001). Ssn6–Tup1 is required for the establishment of an array of nucleosomes over the promoter of RNR3, with a positioned nucleosome occupying the TATA box and others extending into the coding region (Li and Reese, 2001). Surprisingly, Tup1 recruitment is restricted to the URS of the RNR3 promoter (Davie et al, 2002), again suggesting that the location of Ssn6–Tup1 cannot account for its nucleosome-positioning function, and other factor(s) are required.
In this study, we analyzed the mechanism of nucleosome positioning over RNR3 in an attempt to understand how Ssn6–Tup1 can position nucleosomes from a distance at genes when it is localized over the URS region. We found that, although Ssn6–Tup1 is necessary for nucleosome positioning at RNR3, it is not sufficient. Precise nucleosome positioning is also dependent upon the ISW2 nucleosome-positioning complex. A co-dependence on Ssn6–Tup1 and ISW2 for maintaining nucleosome positioning was also observed at other loci, suggesting that collaboration between different classes of positioning/remodeling activities is a common regulatory mechanism.
Results
Ssn6–Tup1 is required for extended nucleosome positioning over RNR3
The Ssn6–Tup1 corepressor complex has been implicated in regulating chromatin structure, but its mechanism is controversial. Our previous studies indicated that nucleosome positioning over the RNR3 promoter is dependent on CRT1, SSN6 and TUP1, and extends at least 750 bp away (∼4–5 nucleosomes) from the upstream regulatory sequence (URS) (Li and Reese, 2001 and Figure 1A). Surprisingly, Tup1 crosslinking is restricted to the URS (Davie et al, 2002; also see below), indicating that the spreading of Tup1 cannot account for its nucleosome-positioning activity and that it requires another factor(s) to do so. As a first step towards understanding how nucleosome positioning is achieved at RNR3, we extended our chromatin-mapping studies to identify the downstream boundary of Ssn6–Tup1-dependent nucleosome positioning. Nuclei from untreated or MMS-treated cells were digested with micrococcal nuclease in situ, and probes were designed to detect the chromatin structure over the entire RNR3 locus. As shown in Figure 1, the digestion pattern generated from chromatin of untreated cells displays the hallmarks of translationally positioned nucleosomes throughout the coding region and beyond the stop codon of RNR3. Specifically, regularly spaced hypersensitive sites with a periodicity of ∼160 base pairs corresponding to internucleosomal regions are observed (lanes 3 and 4, triangles). In addition, the DNA located between the hypersensitive sites is relatively resistant to MNase digestion in the chromatin sample (versus naked DNA), further suggesting that the pattern is the result of nucleosome placement. Treating cells with the DNA-damaging agent methyl methanesulfonate (MMS) caused a loss of the hypersensitive sites and increased digestion of the nucleosomal DNA (lanes 6 and 7), resulting in a digestion pattern strikingly similar to the naked DNA. There is evidence for nucleosome positioning in the repressed state beyond nuc+17, which contains the stop codon, but the effect of derepression (+MMS, Δcrt1 or Δtup1) on the digestion pattern appears to terminate at nuc+17 (Figure 1C and supplementary Figure 1). Furthermore, because the pattern generated after MMS treatment or in the regulatory mutants (see below) is nearly identical to that of digested naked DNA, this indicates that the positions of the nucleosomes are disrupted (or adopt randomized positions) and not simply mobilized to specific alternative positions. From the mapping patterns shown in Figure 1A–C, we conclude that the entire RNR3 gene is packed in an array of at least 20 organized nucleosomes (−3 to +17), and the DNA damage-dependent chromatin disruption extends far beyond the promoter region, up to 2.9 kb away from the URS (Figure 1D).
Figure 1.
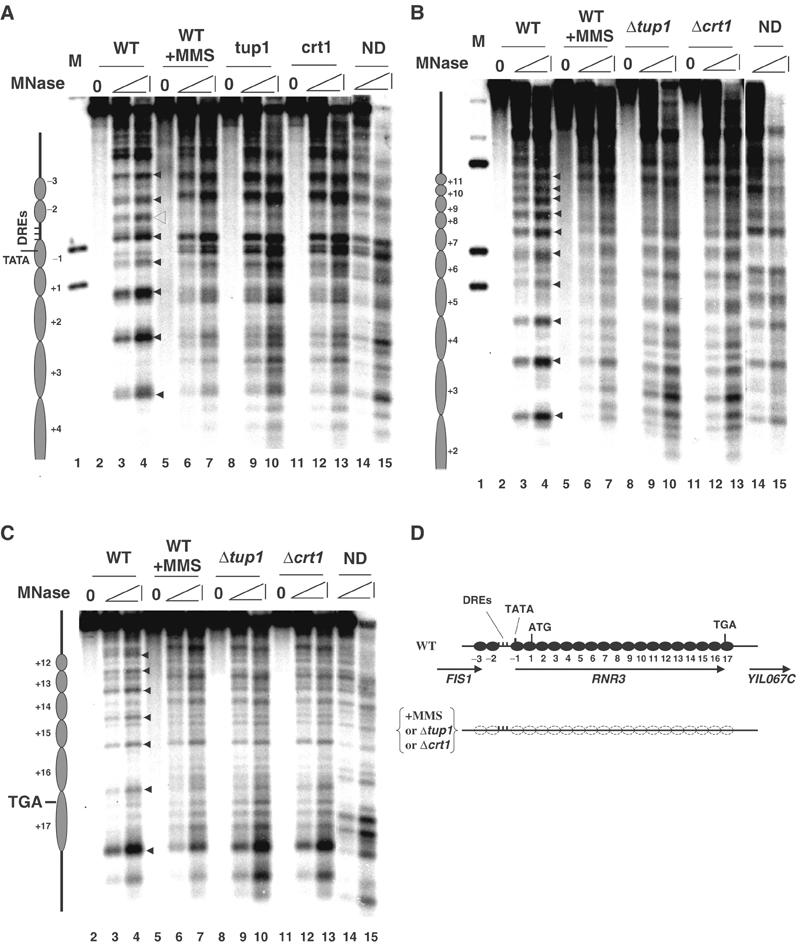
Nucleosome positioning at the RNR3 locus. Nuclei isolated from wild type (WT) with or without MMS (0.03%, 2 h) treatment, Δtup1, and Δcrt1 strains were subjected to micrococcal nuclease (MNase) digestion and detected by indirect end labeling. On the top of each panel, M is a genomic DNA molecular marker digested with the appropriate combinations of restriction enzymes; ND is MNase-digested naked DNA; and 0 represents the undigested sample. Within each panel, the filled triangles represent the internucleosomal hypersensitive sites in the WT chromatin samples; the open triangle indicates the hypersensitive site over the DREs in the WT chromatin; and the open circles indicate the chromatin change in the upstream to the DREs associated with chromatin remodeling. (D) A schematic summary of the chromatin structure shown in (A), (B) and (C), with the position and orientation of RNR3 and the neighboring ORFs indicated by arrows.
Mapping the chromatin from Δtup1 and Δcrt1 strains revealed that the pattern of digestion in these mutants was indistinguishable from wild-type cells treated with MMS and that of naked DNA (Figure 1), indicating a complete loss of the nucleosome positioning. Moreover, as observed in MMS-treated cells, the changes in chromatin structure in both mutants end at nuc+17 (see also supplementary Figure 1). These results clearly show that CRT1- and TUP1-dependent nucleosome positioning extends far beyond the URS, arguing that Tup1 can position nucleosomes from a distance.
The ISW2 complex is required for extended nucleosome positioning
Given that Tup1 spreading cannot account for nucleosome positioning downstream of the URS of RNR3, we hypothesized that some other factor(s) is also required, and it might be an ATP-dependent chromatin remodeling complex. We know that SWI-SNF is not required for the maintenance of nucleosome positioning in the repressed state, and thus is not a candidate (Sharma et al, 2003; data not shown). The RSC, ISW1a/b and ISW2 complexes have been implicated in nucleosome positioning at some yeast genes (Moreira and Holmberg, 1999; Goldmark et al, 2000; Kent et al, 2001). We examined the chromatin structure at RNR3 in mutants with deleted or inactivated catalytic subunits of the RSC (sth1ΔC and sth1-1), ISW1a/b (Δisw1), ISW2 (Δisw2) and CHD1 (Δchd1) complexes. Whereas mutations in STH1 (sth1ΔC or sth1-1; Du et al, 1998) or deleting ISW1 (Vary et al, 2003) or CHD1 (Tran et al, 2000) genes did not cause a disruption of nucleosome positioning at RNR3 (Figure 2A and data not shown), deleting ISW2 completely disrupted nucleosome positioning downstream of the URS to nuc+3, the limit of the resolution of this mapping experiment (Figure 2A). In particular, the nucleosome (nuc−1) embedding the TATA box is disrupted. Thus, ISW2 plays a role in nucleosome positioning over RNR3.
Figure 2.
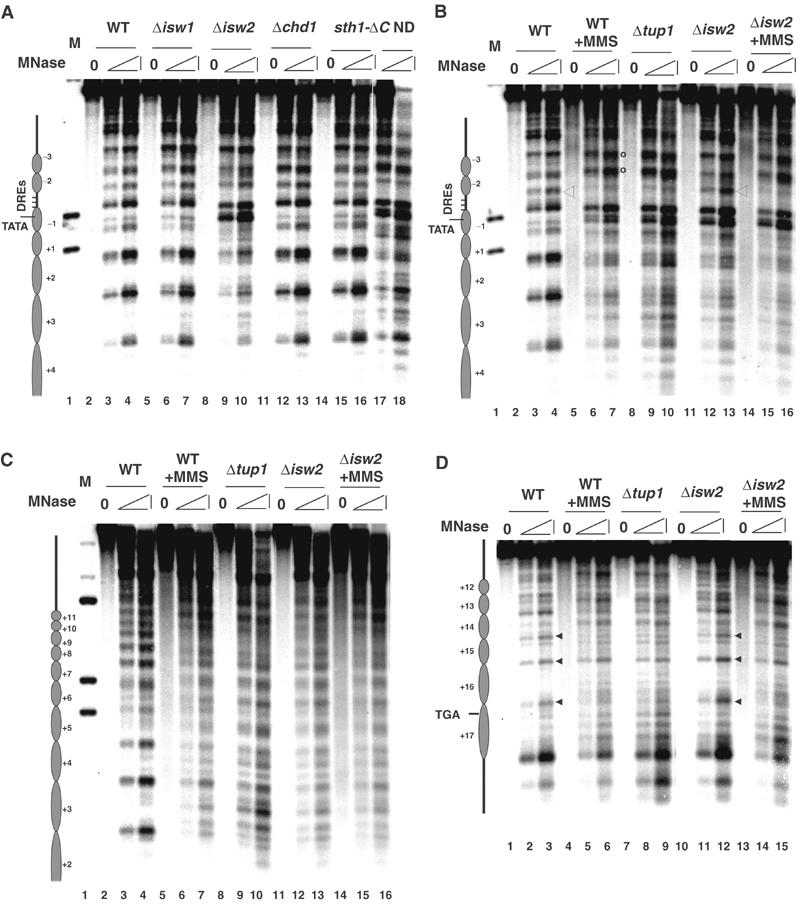
ISW2 is required for nucleosome positioning over RNR3. (A) Chromatin structure around the RNR3 promoter was analyzed in wild type, Δisw1, Δisw2, Δchd1 and sth1-ΔC strains. (B–D) The chromatin structure was analyzed across RNR3 in Δisw2 cells, in parallel with wild-type (−/+ MMS) and Δtup1 strains. See Figure 1 legend for more details.
Next, we examined how far downstream the ISW2-dependent nucleosome positioning extends. Figure 2 shows that deleting ISW2 in fact causes a disruption of nucleosome positioning far into the coding sequence, and the pattern over most of the gene is indistinguishable from MMS-treated cells or in Δcrt1 or Δtup1 mutants. However, despite an overall similarity in the digestion pattern from Δtup1 and Δisw2 mutants, clear differences were observed. First, whereas deleting TUP1 clearly affected chromatin structure up to nuc+17, deleting ISW2 appears to have a less obvious effect between nuc+14 and +17. The hypersensitive sites (HS) observed in wild-type cells between nuc+14 and +15 and between +16 and +17 are largely preserved in the Δisw2 mutant (Figure 2D, lane 12, arrows), and the DNA within nuc+14 and +16 is slightly more resistant to MNase. Note that the HS located between nuc+15 and +16 coincidently overlaps with a HS in naked DNA, so it is difficult to use this as an indicator of positioning. To further underscore the differences, we compared the digestion pattern from Δisw2 cells treated with MMS to those in wild-type cells (−/+ MMS) and a Δtup1 mutant. Significantly, treating Δisw2 mutants with MMS caused the digestion pattern from nuc+14 to +17 to closely match that in a Δtup1 mutant or MMS-treated wild-type cells (compare lanes 2–3, 8–9, 14–15). A second difference in the chromatin structure between Δisw2 cells and the Δcrt1 and Δtup1 mutants is observed upstream of the URS. In the repressed state, an MNase hypersensitive site is located at the downstream edge of the damage response elements, DREs (open triangle, Figures 1A and 2B), and two additional sites are located further upstream. Deleting Δcrt1, Δtup1 or treating cells with MMS causes a loss of the DRE proximal site and broadening of the two further upstream bands (open circles, Figures 1A and 2B), which is identical to the pattern observed in naked DNA digestions. However, these changes are not observed in the Δisw2 mutant in the absence of MMS, but treating the cells with MMS causes the pattern to fully resemble that of a Δcrt1 or Δtup1 mutant (Figure 2B, open circles; also see below). The changes over the DRE elements are likely caused by the binding and release of Crt1–Tup1–Ssn6 at the URS (Li and Reese, 2001); thus, these data suggest that the repressor complex remains bound even though nucleosome positioning is disrupted in Δisw2 cells. Therefore, ISW2 is required to position nucleosomes across most of the RNR3 gene. Most importantly, the pattern of MNase digestion between the URS and nuc+14 in the Δisw2 mutant is essentially identical to that of naked DNA and chromatin from derepressed cells, arguing that the loss of ISW2 results in the disruption of nucleosome positioning or the loss of nucleosomes rather than the sliding of nucleosomes to specific alternate positions.
Next, we examined whether the digestion pattern in the Δisw2 and MMS-treated cells is caused by the removal of nucleosomes from RNR3 or a disruption in positioning, by examining the crosslinking of Myc-tagged histone H4 across its locus. Primers directed to regions of RNR3 were used in PCR amplifications, including primers flanking the DRE region (C) that was proposed to be ‘nucleosome free' (Li and Reese, 2001). The level of crosslinking was largely uniform in repressed cells except for a significant reduction over the DRE (Figure 3), which presumably is due to the lack of a nucleosome within this region. We speculate that a significant portion of the signal amplified using the DRE primers (C) is caused by the limitations of shearing the DNA. The level of histone crosslinking was reduced somewhat over the promoter region in Δisw2 and MMS-treated cells, but was largely unaffected within the ORF. The reduced crosslinking over the promoter may indicate that a nucleosome is ‘lost' over this region of the gene in a small population of cells, or that it is modified in some way to reduce its crosslinking to DNA. Nonetheless, no significant loss of histone H4 was detected within regions of the ORF that MNase mapping indicates have a disrupted chromatin structure (Figures 1and 2), and thus, deleting ISW2 or treating cells with MMS does not cause a widespread loss of nucleosomes from RNR3.
Figure 3.
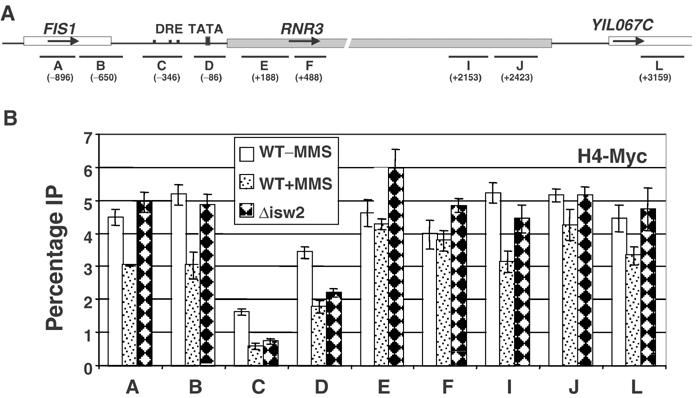
Histone H4 crosslinking to RNR3. Wild-type (WCS484) and Δisw2 (YJR792) strains containing an Myc-tagged version of histone H4 were used in the ChIP assay. (A) Schematic of the PCR probes amplified at the RNR3 locus. The center (in base pairs) of each PCR fragment is indicated in parentheses. (B) Summary of ChIP experiments using anti-Myc monoclonal ascites fluid. For each PCR fragment, the IP signal was normalized to the input signal and expressed as ‘percentage IP'. With the RNR3 translation start site set as +1, the PCR products are: A (−1000–792), B (−788–540), C, URS (−448–236), D, promoter (−179–+8), E (+56–320), F (+333–563), I (+2014–2292), J (+2304–2542) and L (+3055–3263).
Disruption of nucleosome positioning is insufficient for derepression
The loss of nucleosome positioning at RNR3 correlates with a high level of derepression in Δcrt1, Δssn6, Δtup1, or MMS-induced cells (Huang et al, 1998; Li and Reese, 2001). In contrast, the level of RNR3 mRNA was only slightly (∼2-fold) increased in Δisw2 cells (Figure 4A), only 5% of that caused by MMS treatment. Furthermore, Δisw2 cells can be fully derepressed by MMS. Thus, disruption of nucleosome positioning is insufficient to cause significant derepression of transcription. However, as ISWI factors may play a role in transcription elongation and RNA processing (Alen et al, 2002; Morillon et al, 2003; Santos-Rosa et al, 2003; Simic et al, 2003), the failure to observe large increases in RNR3 mRNA in Δisw2 cells could result from elongation or RNA-processing defects, rather than a block in preinitiation complex assembly per se. So, we analyzed the crosslinking of TBP and RNA polymerase II to RNR3 promoter in Δisw2 cells. MMS-induced derepression of RNR3 correlated with more than a three- and eight-fold increase in the level of TBP and RNA polymerase II crosslinking, respectively, in wild-type cells. In contrast, deleting ISW2 did not increase crosslinking above the level observed in untreated wild-type cells (Figure 4B). Therefore, disrupting chromatin over the TATA box and coding sequence (Δisw2) is not sufficient to promote the recruitment of TBP or RNA polymerase II to the promoter, and mechanisms other than nucleosome positioning inhibit their recruitment in Δisw2 cells. The data presented in Figure 4 argue that the disrupted chromatin structure in Δisw2 cells is not the result of transcriptional elongation or factors associated with elongating polymerase, as we detect no PIC formation. Furthermore, deleting CHD1 or ISW1 in a Δisw2 background, two factors proposed to disrupt chromatin during elongation (Morillon et al, 2003; Santos-Rosa et al, 2003; Simic et al, 2003), failed to suppress the disruption in chromatin structure caused by the Δisw2 mutation (not shown).
Figure 4.

Transcription and PIC formation in repressed Δisw2 cells. (A) Northern blot analysis of RNR3 expression in wild-type and Δisw2 strain. The small cellular RNA (scR1) was used as a loading control. (B) Chromatin IP analysis of TBP and RNA polymerase recruitment to the RNR3 promoter. Primers amplifying RNR3 promoter (−179 to +8) were used for the PCR analysis. The signals from the untreated wild-type strain were arbitrarily set as 1.
Transcription and PIC formation are repressed in Δisw2 cells and the digestion pattern over the URS is consistent with the presence of Ssn6–Tup1 at the promoter. To verify the level of Tup1 at RNR3 locus in the Δisw2 mutant, the ChIP assay was carried out using a Tup1 polyclonal antibody. The results in Figure 5B show that Tup1 crosslinking is localized over the URS region in wild-type cells, as previously reported (Davie et al, 2002). Moreover, Tup1 crosslinking was reduced about four-fold upon MMS treatment (Figure 5B), consistent with the dissociation of Crt1 (Huang et al, 1998; supplementary Figure 2). Unexpectedly, we found about a 4-fold increase in Tup1 crosslinking to RNR3 in the Δisw2 cells compared to wild-type cells (Figure 5B). This is unlikely to result from the cross-reactivity of the antibody to other proteins or changes in epitope accessibility, because similar results were obtained using Myc antibody in strains containing Tup1Myc-tagged at the C-terminus (data not shown). Furthermore, whereas the level of Tup1 crosslinking is increased in the Δisw2 strain, its relative distribution across RNR3 was not significantly different from that observed in wild-type cells, that is, the peak of crosslinking is still over the URS. Thus, the increased signal is unlikely to result from the ‘spreading' of Tup1. As a more than two-fold increase in Crt1 crosslinking was also observed in this mutant (supplementary Figure 2), it is possible that the randomized chromatin structure may expose additional repressor binding sites or improve crosslinking efficiencies. Nonetheless, the disrupted nucleosome structure of RNR3 in the Δisw2 mutant is not due to reduced Tup1 binding, and, importantly, Tup1 is insufficient to position nucleosomes at RNR3 in the absence of ISW2, but it continues to repress through other mechanisms. Furthermore, as Tup1 and Crt1 remain associated with RNR3 in Δisw2 cells, it is unlikely that the chromatin changes are due to indirect effects caused by the generation of a DNA damage or replication block signal, as this would result in the release of Crt1 and Tup1 (Huang et al, 1998 and Figure 5B).
Figure 5.
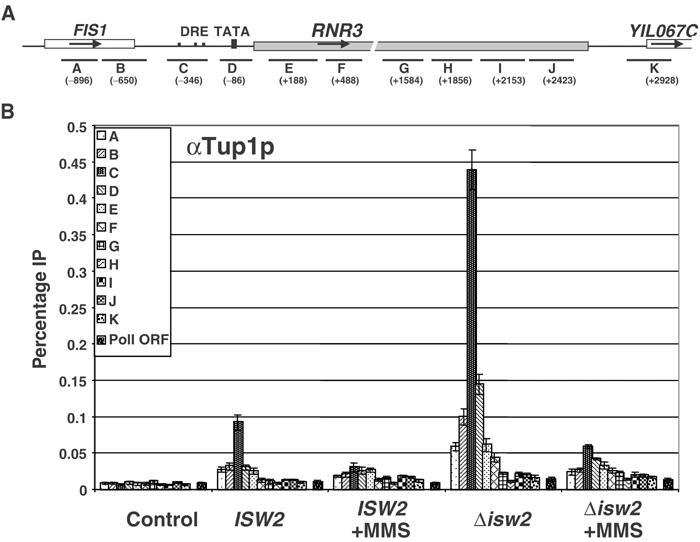
Tup1 is recruited to RNR3 independent of ISW2. (A) Schematic of the PCR probes amplified at the RNR3 locus. (B) Summary of results using anti-serum raised to full-length Tup1. The preimmune serum was used as an IP background control. A pair of primers directed to the coding region of POL1 was used as a control for the specificity of Tup1 localization. The location of PCR products over RNR3 is indicated in the legend of Figure 3, with the addition of regions G (+1465–1703), H (+1742–1968) and K (+2811–3045).
ISW2 is associated with RNR3 and independently of Ssn6–Tup1
Deletion of ISW2 primarily affected chromatin structure adjacent to the promoters of its target genes, and it was proposed that the complex is recruited to the promoter by the sequence-specific repressor Ume6 based on their interaction in vitro and an ISW2-induced supershift of a Ume6–DNA complex in gel mobility shift assays (Goldmark et al, 2000). Isw2 crosslinks to promoter regions in vivo, but its distribution across each locus was not examined (Kent et al, 2001; Fazzio and Tsukiyama, 2003). As chromatin structure was disrupted far into the coding region of RNR3 in Δisw2 cells, we examined Isw2 crosslinking across the entire RNR3 locus and its dependence on Ssn6–Tup1. In contrast to the localized recruitment of Tup1 to the URS, Isw2–Myc crosslinking was detected across the entire RNR3 locus and extended into the flanking ORFs (Figure 6A, about a 15-fold enrichment over background levels). Isw2's localization is fully consistent with its ability to position nucleosomes far downstream of the RNR3 URS. However, Isw2 is also crosslinked to regions where deleting ISW2 had no detectable effect on chromatin structure (Figure 6A, primer sets A, B, I and J). Others likewise reported crosslinking of Isw1, Chd1 and Isw2 to regions where deleting these genes had no effect on chromatin structure (Alen et al, 2002). IPs from extracts prepared from uncrosslinked Isw2–Myc cells pulled down less DNA than the crosslinked untagged controls (data not shown). As a positive control, we examined the crosslinking of Isw2 to the promoters of two genes previously reported to be regulated by ISW2, SUC2 and INO1 (Fazzio et al, 2001; Kent et al, 2001), and found comparable levels of crosslinking. We found a significant level of Isw2 crosslinking at many loci, including telomeres and centromeres, suggesting that it is an abundant complex of widespread distribution (Zhang and Reese, unpublished data, and supplementary Figure 3). It is important to note that published Isw2 crosslinking experiments used untagged controls to determine the background and only examined crosslinking over the URS region of target genes (Kent et al, 2001; Fazzio and Tsukiyama, 2003). Ours is the first to report Isw2 crosslinking across an entire gene. The crosslinking results in Figure 6A, and that of others, indicate that recruitment or physical location is not the only mechanism regulating the function of the ISW2 complex and related factors, and further underscores the importance of identifying factors that allow ISWI complexes to regulate chromatin structure at specific locations within the genome.
Figure 6.
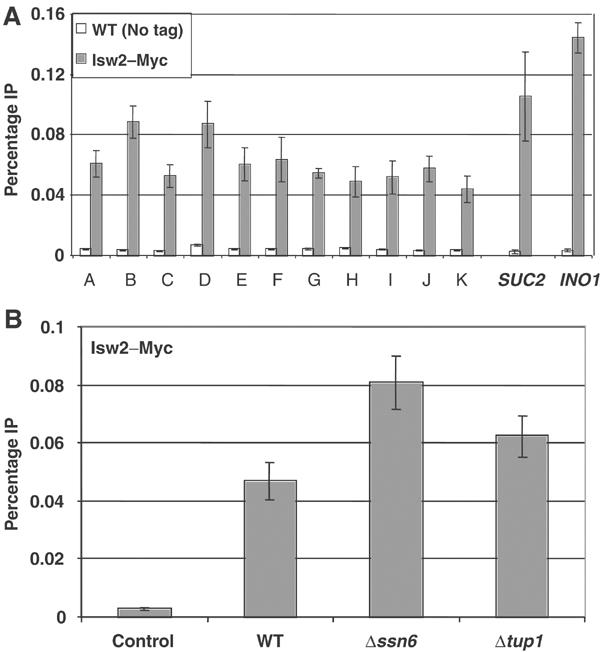
Isw2 crosslinking to the RNR3 locus is widespread and independent of Ssn6–Tup1. The crosslinking of Isw2 containing nine Myc epitopes at the C-terminus (ISW2-MYC9) was examined by the ChIP assay. IPs from an untagged strain were used as the negative control. (A) Isw2–Myc crosslinking was detected across the RNR3 locus. Primers for SUC2 (−298 to +19) and INO1 (−112 to +102) promoters were used as positive controls. The RNR3 primers are described in the legends of Figures 3 and 5. (B) Crosslinking of Isw2–Myc in Δssn6 and Δtup1 strains using the URS PCR primer pair C.
Our ChIP data argued against a recruitment model in the regulation of the ISW2 complex function at the RNR3 locus; however, the essential role of ISW2 in nucleosome positioning and the inability of Tup1 alone to do so in a Δisw2 mutant still suggested one possibility: the disruption of chromatin in Δtup1 cells was due to a loss of ISW2 complex at RNR3. To clarify this issue, we determined whether the binding of Isw2 requires Ssn6–Tup1. The results in Figure 6B show that deleting SSN6 or TUP1 did not reduce the crosslinking of Isw2 to the URS of RNR3, but rather increased it somewhat. Likewise, Isw2 was crosslinked to RNR3 independent of Crt1 (data not shown). Therefore, Ssn6–Tup1 is not required for the association of ISW2 with RNR3, and it is present under both repressive and derepressive conditions. Taken together, the nuclease mapping and ChIP results suggest that nucleosome positioning requires the actions of both the Ssn6–Tup1 and ISW2 complexes, and the presence of one complex in the absence of the other is insufficient to maintain chromatin structure at RNR3.
Regulation of nucleosome positioning by ISW2 is a feature of other Ssn6–Tup1-regulated genes
Ssn6–Tup1 regulates many genes controlled by different cellular pathways. To address whether it functions with ISW2 at other loci, we examined the chromatin structure over the osmotic stress response gene ENA1. Similar to RNR3, Tup1 has been shown to be localized over the URS region of the ENA1 promoter, where the sequence-specific repressor Sko1 and Mig1/2 bind (Wu et al, 2001). It is shown that Tup1 is capable of recruiting HDACs and deacetylating histones at ENA1, but it was not known as to whether it is required for nucleosome positioning at this locus or whether positioning extends into the coding sequence. MNase mapping of ENA1 reveals a pattern consistent with the presence of an array of at least ∼6 nucleosomes extending well into the coding region up to 1 kb away from the URS, the resolution limit of this Southern Blot (Figure 7A). Deletion of either ISW2 or TUP1 caused dramatic chromatin changes over the promoter and loss of nucleosome positioning downstream. The changes in the digestion pattern downstream of the promoter at ENA1 are not as dramatic as those at RNR3, but, as the digestion pattern in these mutants is very similar to that of digested naked DNA (compares lanes 6, 7, 9, 10 versus 11 and 12), we believe that the pattern is consistent with a disrupted chromatin structure. As observed for RNR3, ENA1 is not derepressed in Δisw2 mutants (not shown), suggesting that events in addition to chromatin disruption are required for its expression. We next examined the interdependence of the crosslinking of Isw2 and Tup1 to the ENA1 promoter. As we observed at RNR3, deletion of either factor individually did not significantly affect the crosslinking of the other (Figure 7B), indicating that both are required for nucleosome positioning.
Figure 7.
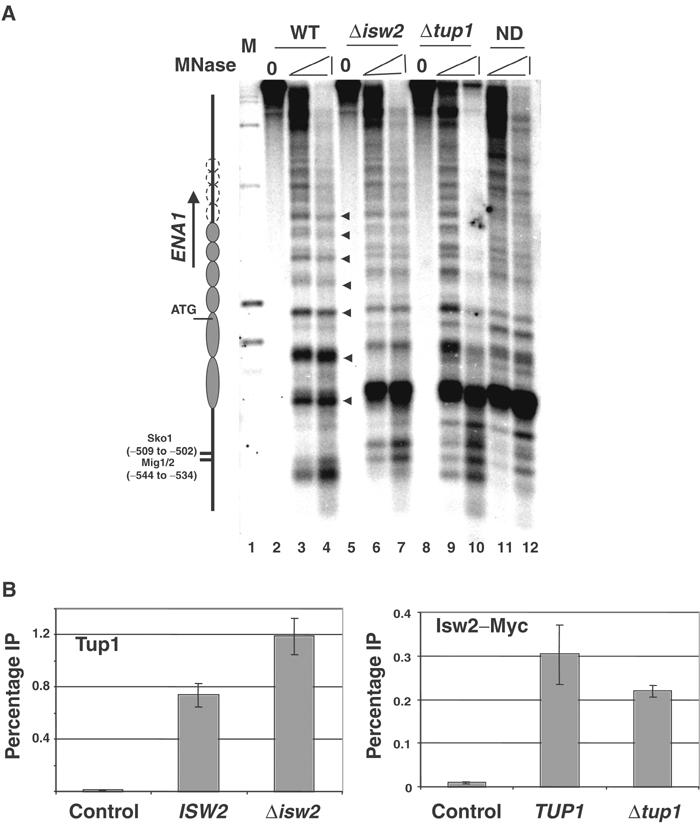
Collaboration of Ssn6–Tup1 and ISW2 at the ENA1 locus. (A) MNase mapping of the ENA1 promoter detected by indirect end labeling. Filled triangles represent the regularly spaced hypersensitive sites in the wild-type chromatin, which are interpreted as internucleosomal sites. The open circles extending beyond nucleosome 6 (left of panel) indicate the possibility that nucleosome positioning continues, but could not be resolved by this gel. (B) ChIP assay for Tup1 and Isw2-Myc crosslinking to the ENA1 URS. Tup1 and Myc antibody were used, with the preimmune or IPs from an untagged strain used as negative controls, respectively. The PCR fragment corresponding to −632 to −316 relative to the translation start site flanks the Sko1 and Mig1/2 binding sites.
We have shown that nucleosome positioning at RNR3 and ENA1 requires both Ssn6–Tup1 and ISW2, and deleting ISW2 affects the chromatin structure at SUC2 (Fazzio et al, 2001; Li and Reese, unpublished data), suggesting that ISW2 regulates chromatin structure at multiple Tup1-dependent genes. We extended our analysis to STE6, ANB1 and HUG1, and found that ISW2 is required for nucleosome positioning at these genes also (supplementary Figure 4), indicating that the ISW2 complex regulates chromatin structure at multiple Ssn6–Tup1-dependent genes and that our observations are not unique to RNR3 or its DNA sequence.
Discussion
Identification of a factor required for ISW2-dependent nucleosome positioning in vivo
It was recently reported that the ISW2 complex can slide nucleosomes towards the URS of the meiotic genes POT1and REC104 in vivo, and it was proposed that ‘other cellular factors' are working with it to position nucleosomes (Fazzio and Tsukiyama, 2003). Our study has identified Ssn6–Tup1 as a factor that influences ISW2 nucleosome-positioning activity in vivo at five different genes. Even though ISW2 maintains the repressive chromatin structure at meiotic genes and RNR3, our data suggest that it is utilized differently at these loci. A strikingly distinguishing feature of the regulation of nucleosome positioning at RNR3 is that ISW2-dependent nucleosome positioning extends well into the coding region and the nucleosomes to adopt either random positions or a disrupted/remodeled configuration in an Δisw2 mutant. In contrast, the nucleosomes adjacent to the promoters of meiotic genes adopt new and stable translational positions in Δisw2 cells (Goldmark et al, 2000; Fazzio and Tsukiyama, 2003). Furthermore, we find that Isw2 crosslinks across the entire RNR3 locus, without an obvious peak over the URS, which argues against a directed recruitment mechanism. It was proposed that Ume6 recruits ISW2 to specific genes (Goldmark et al, 2000; Kent et al, 2001; Fazzio and Tsukiyama, 2003), which is consistent with its ability to position only 1–2 nucleosomes adjacent to the URS of these genes. Thus, the ISW2 complex appears to act more ‘globally' at RNR3 than at meiotic genes, indicating that it affects chromatin structure by more than one mechanism in vivo. This also suggests that gene-specific factors have a profound effect on how ISW2 regulates chromatin structure in vivo. The repressors or corepressors that are recruited to the promoter or the underlying DNA sequence may confer the information for gene-specific regulation of ISW2 function, although we favor the former.
Is Ssn6–Tup1 a barrier to ISW2-mediated nucleosome sliding in vivo?
The next interesting question is how cellular factors, such as Ssn6–Tup1, regulate ISW2 function. In the case of RNR3, ISW2-dependent nucleosome positioning is detected up to 14 nucleosomes downstream from the site of Tup1 crosslinking, arguing against any model that requires the colocalization of these two complexes, such as Tup1 acting in a structural capacity to immobilize nucleosomes positioned by ISW2. Likewise, even though metazoan ISWI complexes require intact histone H4 tails to slide nucleosomes in vitro (Georgel et al, 1997; Clapier et al, 2001; Hamiche et al, 2001; Loyola et al, 2001), a model where Tup1 facilitates the interaction of ISW2 with the tails also cannot account for the collaboration as it is difficult to imagine how Tup1 might do so without binding to each nucleosome across RNR3 via a spreading mechanism. Therefore, Tup1 affects the ability of ISW2 to position nucleosomes from a distance. Given that Ssn6–Tup1 recruits HDACs (Watson et al, 2000; Wu et al, 2001; Davie et al, 2003), part of the mechanism may involve creating a region of histone hypoacetylation. However, broadly increasing histone acetylation cannot cause a disruption of nucleosome positioning by Tup1 at RNR3 because nucleosomes remain tightly positioned in Δhda1 and Δrpd3/Δhos2 (Sharma, Zhang and Reese, unpublished data) mutants, which display equal or even higher levels of histone acetylation than the fully derepressed cells (MMS). We cannot rule out that specific patterns of acetylation or other Tup1-dependent modifications play a role, however.
Based on our data and that of others, we speculate that Ssn6–Tup1 acts as a barrier to ISW2-dependent nucleosome mobilization beyond the URS, causing nucleosomes to adopt precise translational positions downstream. There is a clear correlation between the presence of Tup1 at the promoter and the ability of ISW2 to position nucleosomes. The binding of Crt1 alone is not sufficient to act as a barrier because it remains associated with the DREs in Δssn6 or Δtup1 cells, and the chromatin structure upstream of the URS is fully disrupted in these mutants (Li and Reese, 2001). Thus, it is the release of Ssn6–Tup1 that renders ISW2 incapable of maintaining nucleosome positioning. The association of Tup1 with nucleosomes located near the URS may position the nucleosome immediately upstream of the URS, preventing the ISW2 complex from sliding nucleosomes beyond this barrier, and therefore causing nucleosomes to be positioned downstream. This model would also explain why the nucleosomes upstream of the promoter remain positioned in the Δisw2 mutant until Tup1 is released by MMS treatment (Figures 2 and 5). DNA damage signals cause the release of the ‘barrier' imparted by Crt1–Ssn6–Tup1, allowing greater degrees of freedom for the downstream nucleosomes to adopt randomized spacing even though ISW2 is present, resulting in an MNase digestion pattern similar to naked DNA. Implicit in this model is that ISW2 is sliding nucleosomes in a unidirectional manner toward the URS, or that another ‘barrier' exists downstream of the coding sequence. Although these possibilities remain to be examined, directional sliding of nucleosomes towards the URS of POT1 and REC104 was also observed (Fazzio and Tsukiyama, 2003).
A Crt1–Ssn6–Tup1-nucleosome barrier model is also consistent with the activities of ISWI-containing complexes in vitro. An adjacent nucleosome can act as a barrier to hSnf2-dependent nucleosome sliding on a trinucleosomal template in vitro (Fan et al, 2003), and the DNA-binding proteins lac repressor and GAL4 restrict nucleosome mobilization by ACF and NURF, respectively (Pazin et al, 1997; Kang et al, 2002). Perhaps the most relevant experiments are those conducted with ACF. The binding of lac repressor to templates in vitro affected the ability of ACF to maintain nucleosome positioning, and its removal by the addition of IPTG caused the randomization of nucleosome positions even when ACF was present, suggesting that a boundary imposed by the lac repressor was required to maintain nucleosome positioning (Pazin et al, 1997). The release of the lacI repressor by IPTG in this in vitro system is analogous to the DNA damage-dependent release of Crt1–Ssn6–Tup1 from RNR3 in vivo (Huang et al, 1998; Li and Reese, 2001; this study). However, a recent paper showed that yeast ISW2 can slide nucleosomes through a single GAL4 site bound by GAL4–VP16 (Kassabov et al, 2002). The differences may be attributable to the inherent properties of the complexes themselves or the strength of the binding of the DNA binding factor. The studies on NURF were performed on a template that contained five Gal4 sites, whereas those with yeast ISW2 contained a single Gal4 site. This is also consistent with our finding that stable Crt1–Ssn6–Tup1 complex(es) at the RNR3 URS is required for the nucleosome positioning by ISW2, while the binding of Crt1 alone is not sufficient (Li and Reese, 2001).
Implications for Tup1 repression
Our study has addressed some outstanding questions regarding Tup1 repression and nucleosome positioning, especially at genes where it is localized over the URS. The first is, does Tup1 position nucleosomes across an extended region of a gene when it is localized over the promoter and how can it do so? Here we show that Ssn6–Tup1 is required to position nucleosomes, but it is not sufficient; it requires the ISW2 complex to do so. Therefore, the localization of Tup1 is not necessarily an indicator of whether or not it is required for nucleosome positioning, but rather only how positioning is achieved. Further, our results by no means refute the nucleosome-positioning activity of the Ssn6–Tup1 complex or the possibility that Tup1 can spread through the interaction with histone tails. At some genes, but apparently not at RNR3, ISW2 may be required to position the nucleosomes initially, and Tup1 affixes them into a stable configuration by binding to histone tails and spreading. Second, is nucleosome positioning absolutely required for Tup1 to repress transcription in vivo? Attempts to address this question using HDAC or histone tail mutants is complicated by the fact that mutations causing derepression could, and in some cases did, result in a release of Tup1 from the promoters of target genes (Huang et al, 1997; Kastaniotis et al, 2000; Davie et al, 2002). In addition, deleting histone tails affects gene activation as well (Durrin et al, 1991). Using the Δisw2 mutant has provided an elegant strategy to show that Tup1 can repress transcription even when chromatin structure is disrupted over the promoter (TATA) and coding sequence. This suggests that Tup1 can block PIC formation, even though the positioning of the TATA-containing nucleosome is disrupted. It has been proposed that Tup1 represses haploid-specific genes in the absence of nucleosome positioning (Huang et al, 1997). However, unlike RNR3, the authors observed no derepression-dependent changes in nucleosome positioning at these genes, which is quite unusual for Ssn6–Tup1 regulated genes. While deleting Δisw2 causes only a modest level of derepression of RNR3, we believe that the establishment of a nucleosomal array plays a role in repression nonetheless. Logically, nucleosome positioning contributes to repression because the transcriptional apparatus must access the underlying DNA. Thus, in the absence of nucleosome positioning (Δisw2), at least one other mechanism is required to repress transcription at Tup1 target genes (Smith and Johnson, 2000).
Materials and methods
Strains and media
The strains used in this study are listed in supplementary Table 1. Gene deletions were carried out by a one-step replacement using PCR-generated cassettes (Brachmann et al, 1998). Detailed information on their construction will be provided upon request. In all cases, cells were grown in 2% peptone, 1% yeast extract, 20 μg/ml adenine sulfate and 2% dextrose (YPAD) at 30°C. The induced cells were treated with MMS at a concentration of 0.03% for 2 h. RNA was isolated, fractionated on formaldehyde-containing agarose gels and detected by Northern blotting using standard techniques.
Nuclease mapping
Nuclei preparation and MNase mapping were carried out essentially as described (Li and Reese, 2001). In brief, 1 L of cells were grown in YPAD to an OD600 of around 1.0, harvested and digested with Zymolyase T100 (Seikagaku). The nuclei were isolated by differential centrifugation and resuspended in digestion buffer according to the size of the nuclei pellet, and digested by 0, 2, 4 and 8 unit/ml of micrococcal nuclease (MNase, Worthington) for 10 min at 37°C. For the naked DNA, the treatment was the same, except that the MNase digestion was conducted after the purification of the DNA from the nuclei and less enzyme was used. The DNA was digested with restriction enzyme and subjected to Southern blotting and detection by indirect end labeling. Three probes were used to map the chromatin across RNR3 (with the translation start site as +1): (1) PstI site at +731 (probe +486 to +725); (2) EagI site at +21 and probe (+25 to +257); and (3) PstI site at +3054 and probe (+2811 to +3045). ENA1 was mapped using the BglII site at −751 and a probe corresponding to (−755 to −491).
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described in previous publications with minor changes (Hecht and Grunstein, 1999; Sharma et al, 2003). In all, 50 ml of cells were grown in YPAD media to an OD600 of 0.5–1.0 and treated with formaldehyde (1% v/v) for 15 min at 23°C, followed by a 15 min treatment with glycine (125 mM final). The induced cells were treated at an OD600 of 0.7 with 0.03% MMS and incubated for 2 h before crosslinking. Lysates were prepared by glass bead disruption and the chromatin was sheared by sonication into fragments ranging in size from 200 to 1000 bp. The lysates were then clarified by centrifugation, and 200 μl was incubated with anti-TBP polyclonal antiserum, anti-Myc (9E10, Covance), 8WG16 monoclonal antibody (Covance) or anti-Tup1 polyclonal antiserum. Myc-tagged Histone H4 was precipitated from 100 μl of extract using 3 μl of 9E10. The immune complexes were recovered by incubation with 25 μl of protein A sepharose CL-4B beads (Amersham), washed and the DNA eluted. After reversing the crosslinks at 65°C overnight, the IPed and input DNA were analyzed by semiquantitative PCR. The PCR products were loaded into 2% agarose gel, stained with ethidium bromide, scanned with Typhoon system (Molecualr Dynamics) and quantified by ImageQuant software (Molecular Dynamics). The amplified IP DNA was normalized to DNA amplified from input samples. The results are averages and standard errors from at least three independent experiments.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Table 1
Acknowledgments
We acknowledge Bing Li for the initial mapping in the ISW2 mutant. We thank Drs Robert Simpson, Jerry Workman, Song Tan, Alan Hinnebusch and members of the Reese lab and the Penn State gene regulation group for advice and comments on this work, and Toshio Tsukiyama, Brehon Laurent, Winston Shen and Richard Zitomer for yeast strains. This research was supported by funds provided by the National Institutes of Health (GM58672) and by an Established Investigator Grant from the American Heart Association to JCR.
References
- Alen C, Kent NA, Jones HS, O'Sullivan J, Aranda A, Proudfoot NJ (2002) A role for chromatin remodeling in transcriptional termination by RNA polymerase II. Mol Cell 10: 1441–1452 [DOI] [PubMed] [Google Scholar]
- Basrai MA, Velculescu VE, Kinzler KW, Hieter P (1999) NORF5/HUG1 is a component of the MEC1-mediated checkpoint response to DNA damage and replication arrest in Saccharomyces cerevisiae. Mol Cell Biol 19: 7041–7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker PB, Horz W (2002) ATP-dependent nucleosome remodeling. Annu Rev Biochem 71: 247–273 [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132 [DOI] [PubMed] [Google Scholar]
- Clapier CR, Langst G, Corona DF, Becker PB, Nightingale KP (2001) Critical role for the histone H4 N terminus in nucleosome remodeling by ISWI. Mol Cell Biol 21: 875–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JP, Roth SY, Simpson RT (1994) The global transcriptional regulators, SSN6 and TUP1, play distinct roles in the establishment of a repressive chromatin structure. Genes Dev 8: 1400–1410 [DOI] [PubMed] [Google Scholar]
- Davie JK, Edmondson DG, Coco CB, Dent SY (2003) Tup1–Ssn6 interacts with multiple class I histone deacetylases in vivo. J Biol Chem 278: 50158–50162 [DOI] [PubMed] [Google Scholar]
- Davie JK, Trumbly RJ, Dent SYR (2002) Histone-dependent association of Tup1–Ssn6 with repressed genes in vivo. Mol Cell Biol 22: 693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuring R, Fanti L, Armstrong JA, Sarte M, Papoulas O, Prestel M, Daubresse G, Verard M, Moseley SL, Berloco M, Tsukiyama T, Wu C, Pimpinelli S, Tamkun JW (2000) The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol Cell 5: 355–365 [DOI] [PubMed] [Google Scholar]
- Du J, Nasir I, Benton BK, Kladde MP, Laurent BC (1998) Sth1p, a Saccharomyces cerevisiae Snf2p/Swi2p homolog, is an essential ATPase in RSC and differs from Snf/Swi in its interactions with histones and chromatin-associated proteins. Genetics 150: 987–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker CE, Simpson RT (2000) The organized chromatin domain of the repressed yeast a-specific gene STE6 contains two molecules of the corepressor Tup1p per nucleosome. EMBO J 19: 400–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrin LK, Mann RK, Kayne PS, Grunstein M (1991) Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell 65: 1023–1031 [DOI] [PubMed] [Google Scholar]
- Edmondson DG, Smith MM, Roth SY (1996) Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4. Genes Dev 10: 1247–1259 [DOI] [PubMed] [Google Scholar]
- Fan HY, He X, Kingston RE, Narlikar GJ (2003) Distinct strategies to make nucleosomal DNA accessible. Mol Cell 11: 1311–1322 [DOI] [PubMed] [Google Scholar]
- Fazzio TG, Kooperberg C, Goldmark JP, Neal C, Basom R, Delrow J, Tsukiyama T (2001) Widespread collaboration of Isw2 and Sin3-Rpd3 chromatin remodeling complexes in transcriptional repression. Mol Cell Biol 21: 6450–6460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, Tsukiyama T (2003) Chromatin remodeling in vivo: evidence for a nucleosome sliding mechanism. Mol Cell 12: 1333–1340 [DOI] [PubMed] [Google Scholar]
- Fleming AB, Pennings S (2001) Antagonistic remodeling by Swi–Snf and Tup1–Ssn6 of an extensive chromatin region forms the background for FLO1 gene regulation. EMBO J 20: 5219–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgel PT, Tsukiyama T, Wu C (1997) Role of histone tails in nucleosome remodeling by Drosophila NURF. EMBO J 16: 4717–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmark JP, Fasio TG, Estep PW, Church GM, Tsukiyama T (2000) The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 103: 423–433 [DOI] [PubMed] [Google Scholar]
- Hamiche A, Kang JG, Dennis C, Xiao H, Wu C (2001) Histone tails modulate nucleosome mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc Natl Acad Sci USA 98: 14316–14321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A, Grunstein M (1999) Mapping DNA interaction sites of chromosomal proteins using immunoprecipitation and polymerase chain reaction. Methods Enzymol 304: 399–414 [DOI] [PubMed] [Google Scholar]
- Huang L, Zhang W, Roth SY (1997) Amino termini of histones H3 and H4 are required for a1-alpha2 repression in yeast. Mol Cell Biol 17: 6555–6562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Zhou Z, Elledge SJ (1998) The DNA replication and damage check point pathways induce transcription by inhibiting the Crt1 repressor. Cell 94: 595–605 [DOI] [PubMed] [Google Scholar]
- Iida T, Araki H (2004) Noncompetitive counteractions of DNA polymerase epsilon and ISW2/yCHRAC for epigenetic inheritance of telomere position effect in Saccharomyces cerevisiae. Mol Cell Biol 24: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin MJ, Ryuji K, Kadonag JT (1997) ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 90: 145–155 [DOI] [PubMed] [Google Scholar]
- Kang JG, Hamiche A, Wu C (2002) GAL4 directs nucleosome sliding induced by NURF. EMBO J 21: 1406–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassabov SR, Henry NM, Zofall M, Tsukiyama T, Bartholomew B (2002) High-resolution mapping of changes in histone–DNA contacts of nucleosomes remodeled by ISW2. Mol Cell Biol 22: 7524–7534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastaniotis AJ, Mennelia TA, Konrad C, Torres AM, Zitomer RS (2000) Roles of transcription factor Mot3 and chromatin in repression of the hypoxic gene ANB1 in yeast. Mol Cell Biol 20: 7088–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent NA, Karabetsou N, Politis PK, Mellor J (2001) In vivo chromatin remodeling by yeast ISW1 homologs Isw1p and Isw2p. Genes Dev 15: 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langst G, Becker PB (2001) Nucleosome mobilization and positioning by ISWI-containing chromatin remodeling factors. J Cell Sci 114: 2561–2568 [DOI] [PubMed] [Google Scholar]
- Li B, Reese JC (2001) Ssn6–Tup1 regulates RNR3 by positioning nucleosomes and affecting the chromatin structure at the upstream repression sequence. J Biol Chem 276: 33788–33797 [DOI] [PubMed] [Google Scholar]
- Loyola A, LeRoy G, Wang YH, Reinberg D (2001) Reconstitution of recombinant chromatin establishes a requirement for histone-tail modifications during chromatin assembly and transcription. Genes Dev 15: 2837–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell AD, Gelbart ME, Tsukiyama T (2004) Histone fold protein Dls1p is required for Isw2-dependent chromatin remodeling in vivo. Mol Cell Biol 24: 2605–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira JM, Holmberg S (1999) Transcriptional repression of the yeast CHA1 gene requires the chromatin-remodeling complex RSC. EMBO J 18: 2836–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, Karabetsou N, O'Sullivan J, Kent N, Proudfoot N, Mellor J (2003) Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell 115: 425–435 [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE (2002) Cooperation between complexes that regulate chromatin structure and transcription. Cell 108: 475–487 [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Bhargava P, Geiduschek EP, Kadonaga JT (1997) Nucleosome mobility and the maintenance of nucleosome positioning. Science 276: 809–812 [DOI] [PubMed] [Google Scholar]
- Peterson CL, Workman JL (2000) Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr Opin Genet Dev 10: 187–192 [DOI] [PubMed] [Google Scholar]
- Roth SY (1995) Chromatin-mediated transcriptional repression in yeast. Curr Opin Genet Dev 5: 168–173 [DOI] [PubMed] [Google Scholar]
- Ruiz C, Escribano V, Morgado E, Molina M, Mazon MJ (2003) Cell-type-dependent repression of yeast a-specific genes requires Itc1p, a subunit of the Isw2p–Itc1p chromatin remodeling complex. Microbiology 149: 341–351 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bernstein BE, Karabetsou N, Morillon A, Weise C, Schreiber SL, Mellor J, Kouzarides T (2003) Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell 12: 1325–1332 [DOI] [PubMed] [Google Scholar]
- Sharma VM, Li B, Reese JC (2003) SWI/SNF-dependent chromatin remodeling of RNR3 requires TAF(II)s and the general transcription machinery. Genes Dev 17: 502–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, Arndt KM (2003) Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J 22: 1846–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RL, Johnson AD (2000) Turning genes off by Ssn6–Tup1: a conserved system of transcriptional repression in eukaryotes. Trens Biochem Sci 25: 325–330 [DOI] [PubMed] [Google Scholar]
- Sugiyama M, Nikawa JI (2001) The Saccharomyces cerevisiae Isw2p–Itc1p complex represses INO1 expression and maintaines cell morphology. J Bacteriol 183: 4985–4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HG, Steger DJ, Iyer VR, Johnson AD (2000) The chromo domain protein Chd1p from budding yeast is an ATP-dependent chromatin-modifying factor. EMBO J 19: 2323–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama T, Palmer J, Landel CC, Shiloach J, Wu C (1999) Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev 13: 686–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama T, Wu C (1995) Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 83: 1011–1020 [DOI] [PubMed] [Google Scholar]
- Tyler JK, Kadonaga JT (1999) The ‘dark side' of chromatin remodeling: repressive effects on transcription. Cell 99: 443–446 [DOI] [PubMed] [Google Scholar]
- Varga-Weisz PD, Wilm M, Bonte E, Dumas K, Mann M, Becker PB (1997) Chromatin-remodeling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature 388: 598–602 [DOI] [PubMed] [Google Scholar]
- Vary JC Jr, Gangaraju VK, Qin J, Landel CC, Kooperberg C, Bartholomew B, Tsukiyama T (2003) Yeast Isw1p forms two separable complexes in vivo. Mol Cell Biol 23: 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali M, Hassan AH, Neely KE, Workman JL (2000) ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 20: 1899–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AD, Edmondson DG, Bone JR, Mukai Y, Yu Y, Du W, Stillman DJ, Roth SY (2000) Ssn6–Tup1 interacts with class I histone deacetylases required for repression. Genes Dev 14: 2737–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss K, Simpson RT (1997) Cell type-specific chromatin organization of the region that governs directionality of yeast mating type switching. EMBO J 16: 4352–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Suka N, Carlson M, Grunstein M (2001) TUP1 utilizes histone H3/H2B-specific HDA1 deacetylase to repress gene activity in yeast. Mol Cell 7: 117–126 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Elledge SJ (1992) Isolation of crt mutants constitutive for transcription of the DNA damage inducible gene RNR3 in Saccharomyces cerevisiae. Genetics 131: 851–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Table 1