

Abstract
Estrogen antagonists are universally employed in the breast cancer therapy, although antagonist therapy is limited by the inevitable development of cellular resistance. The molecular mechanisms by which these agents inhibit cellular proliferation in breast cancer cells are not fully defined. Recent studies have shown the involvement of the E2F pathway in tamoxifen-induced growth arrest. We show that an E2F repressor, prohibitin, and the chromatin modifiers Brg1/Brm are required for estrogen antagonist-mediated growth suppression through the estrogen receptor, and that their recruitment to native promoter-bound E2F is induced via a JNK1 pathway. In addition, we demonstrate major mechanistic differences among the signaling pathways initiated by estrogen, estrogen deprivation, and estrogen antagonists. Collectively, these findings suggest that the prohibitin/Brg1/Brm node is a major cellular target for estrogen antagonists, and thereby also implicate prohibitin/Brg1/Brm as potentially important targets for breast cancer therapy.
Keywords: E2F, SWI/SNF, tamoxifen, transcription
Introduction
Estrogen antagonists comprise the current endocrine therapy of choice in breast cancer, and lead to highly significant decreases in the rates of both disease recurrence and death. Antagonist therapy is limited, however, by the inevitable development of cellular resistance (Wolczynski et al, 2000). The molecular mechanisms by which estrogen antagonists inhibit cellular proliferation are not fully defined, and this lack of information impedes the rational design of improved anti-breast cancer drugs. Recent studies have shown that the E2F pathway is involved in estrogen antagonist-induced growth arrest in breast cancer cells (Wang et al, 1997; Carroll et al, 2000).
We recently established that a potential tumor suppressor, prohibitin, represses the transcriptional activity of E2F, and this correlates with the ability of prohibitin to induce growth arrest (Wang et al, 1999a, 1999b, 2002a,2002b). The highly evolutionally conserved protein prohibitin was originally identified based on its ability to induce growth arrest at G1/S (Roskams et al, 1993). A role of prohibitin in breast cancer has been suggested by the finding of mutations in prohibitin in certain breast cancers (Sato et al, 1993). We have recently defined the molecular mechanisms of prohibitin-mediated transcription repression and growth suppression, and demonstrated that Brg1 and Brm, members of SWI/SNF family of chromatin-modifying ATPases, are required for these repressive functions of prohibitin (Wang et al, 2002a,2002b). We hypothesized that prohibitin plays a role in the cell cycle regulation of breast cancer cells. We found that estrogen antagonists-induced growth suppression of breast cancer cells requires prohibitin and its co-repressors, Brg1/Brm. JNK1 represses E2F transcriptional activity (Wang et al, 1998, 1999c) and we report here that interfering with the JNK1 pathway reverses estrogen antagonist-induced recruitment of Brg1/Brm to prohibitin/E2F/promoter complexes, suggesting that estrogen antagonists signal to prohibitin via a JNK1 pathway. However, while estrogen induced the recruitment of Brg1 and Brm to an estrogen-responsive promoter, it failed to affect the recruitment of Brg1/Brm to E2F-responsive promoters, suggesting a mechanistic difference between the signaling pathways initiated by estrogen compared to estrogen antagonists.
Results
Treatment of the breast cancer cell line MCF-7 with 4-hydroxytamoxifen (4HT) induced growth arrest in G1 as determined by flow cytometric analysis (FACS) (G0/G1 population of 4HT-treated cells: 81%; vehicle control: 31%), in agreement with previous reports (Coezy et al, 1982). Overexpression of prohibitin similarly induced growth arrest in G1 in certain breast cancer cells, including MCF7 and ZR75-1 (Wang et al, 1999a,1999b, 2002a,2002b). We therefore tested whether these two processes were functionally related, by depleting endogenous prohibitin levels using both antisense and SiRNA strategies. Transfection of a prohibitin antisense vector (data not shown) or prohibitin SiRNA into MCF7 cells ablated expression of prohibitin (Figure 1C) and concomitantly attenuated the growth inhibitory effects of 4HT (as reflected by G0/G1 population detected by FACS: control: 31%; 4HT treated: 81%; 4HT+prohibitin SiRNA: 30%). This result suggests that 4HT-induced growth suppression involves (and requires) prohibitin. Parallel experiments using a structurally distinct pure estrogen antagonist, ICI182780, produced identical results.
Figure 1.
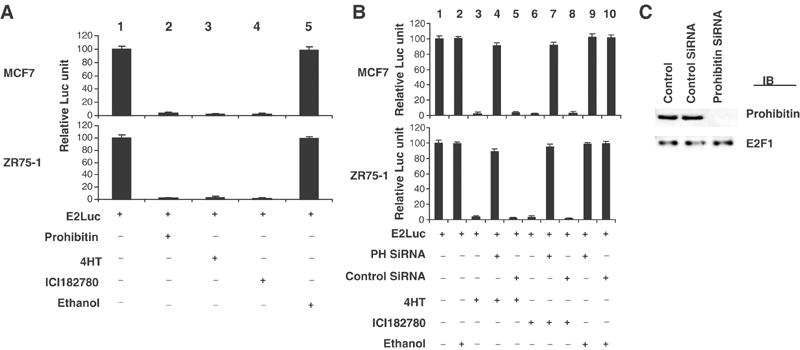
(A) Estrogen antagonists modulate E2F-driven transcription. MCF7 and ZR75-1 cells were transfected with an E2F-responsive reporter (E2Luc, 12 μg). All cells were co-transfected with 1 μg of pSV-βGal followed by βgal assay as an internal control for transcription efficiency, and the activity of a pSVβgal vector was comparable in all samples. The results shown are statistics of four repeated experiments. Cells were treated with 4HT, ICI182780, or ethanol (vehicle control) for 72 h (initiated 24 h prior to transfection). The relative luciferase activities were calculated relative to the ‘E2Luc', which were arbitrarily assigned a value of ‘100'. The basal activity of the E2Luc reporter was repressed by estrogen antagonists (lanes 3 and 4), similar to the effect observed when a prohibitin expression vector was co-transfected (lane 2). Ectopic expression of prohibitin or treatment with estrogen antagonists did not affect the activity of a non-E2F-responsive reporter pSVECG (data not shown). (B) Suppression of prohibitin by prohibitin SiRNA blocks estrogen antagonist-induced E2F transcriptional repression. MCF7 and ZR75-1 cells were transfected with the E2Luc reporter (12 μg), with or without prohibitin SiRNA or control SiRNA. Cells were treated with 4HT (lanes 3–5), ICI182780 (lanes 6–8), or ethanol (vehicle control) (lane 9) for 72 h (initiated 24 h prior to the transfection). The relative luciferase activities were calculated as described in the legend to Figure 2A. Estrogen antagonist-induced repression of activity of the E2F-driven E2Luc reporter was released by co-transfection of prohibitin SiRNA (lanes 4, 7, and 9). (C) Immunoblot analysis of the protein levels of prohibitin and E2F1 (control) in MCF7 cells and the cells transfected with prohibitin SiRNA or nonsilencing control SiRNA.
The growth suppressive effect of prohibitin is mediated through its repression of E2F-specific transcriptional activity (Wang et al, 2002a,2002b). We examined whether estrogen antagonists affect E2F-driven transcription using transient transfection/reporter assays. Treatment of MCF7 or ZR75-1 cells with either 4HT or ICI182780, but not the ethanol vehicle, reduced luciferase (shown) or CAT (not shown) activity driven by an E2F-specific responsive promoter by 98–99% (Figure 1A, lanes 3–5). In contrast, the activity of pSVECG, a non-E2F-driven reporter, was not affected by the estrogen antagonists, indicating that the repressive effect of estrogen antagonists is specific to E2F-driven transcription (data not shown). These results suggested that estrogen antagonists may target E2F to achieve their antiproliferative activity.
To investigate the role of prohibitin in the estrogen antagonist-induced repression of E2F-mediated transcription, the same E2Luc and E2CAT reporter systems were used to test the effect of prohibitin knockdown on cellular responses to estrogen antagonists. Transfection of an antisense prohibitin vector or prohibitin SiRNA released the suppression of E2F activity caused by estrogen antagonists in a dose-dependent fashion (Figure 1B, SiRNA studies are shown). Immunoblot analysis confirmed that prohibitin protein levels were dramatically decreased by the antisense vector and by SiRNA (data not shown).
To demonstrate a requirement for prohibitin–E2F interactions in the effects of estrogen antagonists, we employed a peptide containing the prohibitin-binding domain of E2F to block this interaction. We recently reported that AA304–357 region of E2F is required for the transcriptional repression function of prohibitin and this region alone is sufficient for prohibitin-dependent transcriptional suppression (Wang et al, 2002a,2002b). Region 304–357 represents a highly conserved domain located within the ‘Marked-box' of E2F, and has no other known function. This region of E2F is specifically required for binding to prohibitin and expression of this AA304–357 peptide does not affect repression of E2F by other regulators, such as Rb (Jost et al, 1996). To test whether E2F association is necessary for the prohibitin-dependent response to estrogen antagonists, we established MCF7 cell lines that either constitutively express the prohibitin-binding domain (AA304–357) of E2F1 or a nonrelevant E2F domain (AA263–303) (as a negative control). Both E2F mutants are tagged with Gal4 (Figure 2C; Wang et al, 2002a,2002b). Expression of the prohibitin-binding domain of E2F (AA304–357), but not the nonrelevant domain (AA263–303), effectively blocked the association between prohibitin and E2F1, as demonstrated by the immunoprecipitation–immunoblot analysis (Figure 2A). Furthermore, expression of the prohibitin-binding domain of E2F blocked the repression of E2F-mediated transcription induced by the estrogen antagonist, as demonstrated by an E2F promoter–reporter assay (Figure 2B).
Figure 2.
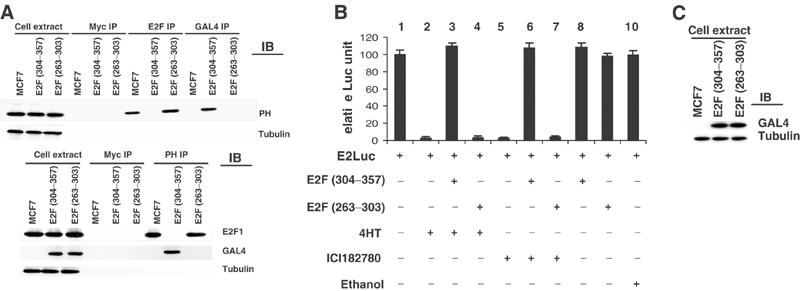
Physical association of prohibitin and E2F1 is required for the repressive function of estrogen antagonists. MCF7 cells were stably transfected with pCR3.1 E2F1 (AA304–357) or pCR3.1 E2F1 (AA263–303), both tagged with Gal4. Expression of the transfected E2F peptides was confirmed by immunoblot analysis (C). (A) Whole-cell extracts, or proteins from cell extracts immunoprecipitated using anti-Gal4 (for the E2F mutants) or control (anti-Myc or -E2F) antibodies, were separated electrophoretically, then identified by immunoblotting using an anti-prohibitin antibody, or anti-tubulin antibody as a loading control (upper panel). The experiments were repeated in a reciprocal fashion (lower panel). (B) The E2F peptide-expressing MCF7 cells were transfected with an E2Luc reporter and treated with ethanol (vehicle control) or estrogen antagonists. Cells were harvested for luciferase activity assay and β-galactosidase assay (transfection control). Luciferase activity calculations are normalized for β-galactosidase activity.
To test whether the growth suppressive actions of estrogen antagonists also require a prohibitin–E2F association, we utilized the same blocking peptides in a colony formation assay. As shown in Table I, transfection of the blocking E2F (AA304–357) peptide, but not the control E2F peptide (263–303), effectively reversed the growth repression induced by estrogen antagonists. These data demonstrate that the physical interaction of prohibitin with E2F is required for the estrogen antagonist-induced transcriptional repression and growth suppression, and that prohibitin is acting through E2F in this pathway.
Table 1.
Reversal of estrogen antagonist-induced repression of colony formation by prohibitin-binding domain of E2F
| Vector transfected | Number of colonies |
|||
|---|---|---|---|---|
| MCF7 |
ZR-75-1 |
|||
| Experiment 1 | Experiment 2 | Experiment 3 | Experiment 4 | |
| pBabePuro+pSVneo | 204 | 215 | 234 | 211 |
| (pBabePuro+pSVneo)+ethanol | 199 | 218 | 194 | 209 |
| (pBabePuro+pSVneo)+4HT | 5 | 11 | 5 | 4 |
| (pBabePuro+pCR3.1 E2F304–357)+4HT | 192 | 209 | 178 | 193 |
| (pBabePuro+pCR3.1 E2F263–303)+4HT | 3 | 8 | 8 | 4 |
| pBabePuro+pSVneo+ICI | 2 | 9 | 2 | 6 |
| (pBabePuro+pCR3.1 E2F304–357)+ICI | 201 | 194 | 189 | 192 |
| (pBabePuro+pCR3.1 E2F263–303)+ICI |
6 |
5 |
10 |
9 |
| Approximately 10 000 MCF7 or ZR-75-1 cells were transfected with 5 μg of the indicated vectors. Colonies with 20 or more cells were scored after 14 days of selection in 5 μg puromycin and 100 μg of neomycin per ml. | ||||
We have recently demonstrated that Brg1/Brm are required for prohibitin-mediated repression of transcription and growth (Wang et al, 2002a,2002b). Brg1 and Brm are ATP-dependent chromatin remodeling enzymes belonging to the SWI–SNF complex, which have been linked to transcriptional regulation. Brg1 is functionally associated with Rb (Strobeck et al, 2000) and BRCA1 (Bochar et al, 2000). Furthermore, Brg1 is mutated in various human tumor cell lines, including breast cancer cells, suggesting a role of Brg1 as a potential tumor suppressor (Wong et al, 2000). We determined whether Brg1/Brm play a role in the transcriptional repression and growth suppression induced by estrogen antagonists by first examining the association between prohibitin and these co-repressors in response to estrogen antagonists (Wang et al, 2002a,2002b). Whole-cell extracts of estrogen antagonist-treated MCF7 (shown) or ZR75-1 (immunoblot not shown) cells were immunoprecipitated using either anti-Brg1 or -Brm antibodies or anti-prohibitin antibody, followed by immunoblot analysis using anti-prohibitin, -Brg1, or -Brm antibodies. An association between prohibitin and Brg1/Brm was rapidly induced by treatment with 4HT or ICI182780, within 20 min (Figure 3A). Anti-cMyc and -p38 antibodies were used for controls in these immunoprecipitation/immunoblot analyses, confirming the specificity of the associations (Figure 3A). Immunoblotting for the target proteins further confirmed the equal loading and the equal immunoprecipitation (Figure 3A).
Figure 3.
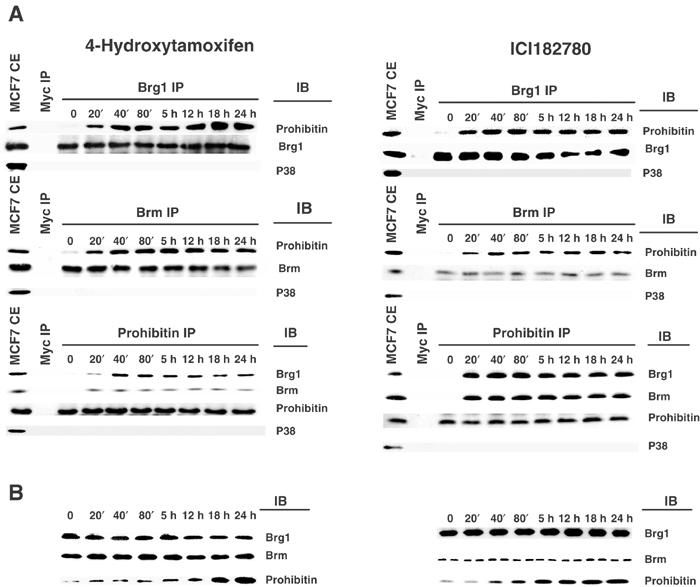
Induction of association of prohibitin with Brg1/Brm by estrogen antagonists. (A) MCF7 cells were treated with 4HT or ICI182780 for the indicated time intervals. Cell extracts were immunoprecipitated (IP) by anti-cMyc (control), -Brg1, or -Brm antibodies, followed by immunoblot analysis (IB), using anti-prohibitin or -p38 (as control) antibodies. The reciprocal IP–immunoblot was performed using anti-cMyc or -prohibitin antibodies for the IP, and anti-Brg1, -Brm, or -p38 antibodies for the immunoblot. Increases in associations between prohibitin and Brg1/Brm were evident as early as 20 min after treatment. Immunoblotting with the anti-p38 antibody failed to detect any protein in the immunoprecipitates, indicating the specificity of the prohibitin–Brm/Brg1 associations. (B) The same cell extracts used in panel A were analyzed by immunoblot using anti-Brg1, -Brm, or -prohibitin antibodies. The results shown are representative of experiments that were repeated four times, which yielded identical results.
One mechanism through which associations between prohibitin and its co-repressors could be enhanced is through increases in the intracellular levels of prohibitin protein. Endogenous prohibitin protein levels, quantitated by immunoblotting, were dramatically elevated (to a maximum of 11-fold (+/−4HT) and 12-fold (+/−ICI182780)) in whole-cell extracts from MCF7 cells (shown) and ZR75-1 cells (immunoblot not shown), within 18 h of treatment with 4HT and within 5 h of treatment with ICI182780 (Figure 3B), confirming a stimulatory effect of estrogen antagonists on endogenous prohibitin protein levels. The levels of Brg1 and Brm proteins remained constant, as demonstrated by four independent immunoblotting studies, which yielded identical data (representative is shown in Figure 3B). To determine the molecular level of this induction, we utilized a transient transfection system (Wang et al, 2002a,2002b) to assess the transcriptional activity of the prohibitin promoter in response to estrogen antagonists. A CAT reporter driven by a rat prohibitin promoter was introduced into MCF7 and ZR75-1 cells. The activity of the prohibitin-CAT reporter was dramatically induced (10-fold) after the cells were treated with 4HT for more than 18 h (Figure 4). A similar magnitude of induction was observed when the cells were treated instead with ICI182780, and the increase in prohibitin promoter activity was observed at even earlier time points (initiating within 5 h) (Figure 4). Thus, although prohibitin gene and protein expression were both markedly enhanced by estrogen antagonists, the enhancement of prohibitin association with Brm–Brg1 occurred much more rapidly, and new transcription of prohibitin could not thus mediate the early increases in the levels of prohibitin–Brm/Brg1 complexes observed.
Figure 4.
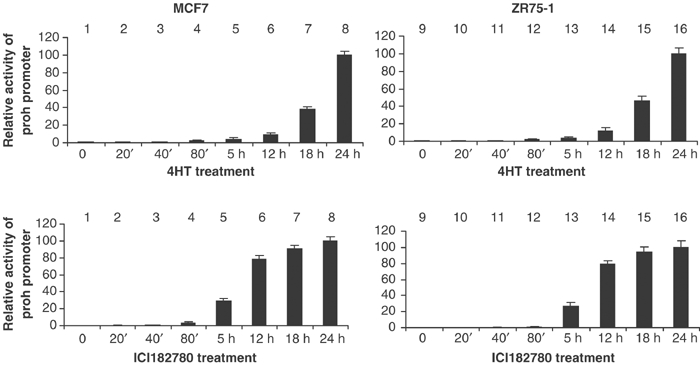
Estrogen antagonists enhance the transcriptional activity of the prohibitin promoter. MCF7 and ZR75-1 cells were transfected with ProhibitinCAT, a CAT reporter gene driven by a rat prohibitin promoter element spanning −485 to −5 bp. Cells were treated with 4HT or ICI182780 for the indicated time intervals, and harvested for CAT activity assay and β-galactosidase assay (transfection control).
As demonstrated above, the repression of E2F-driven transcription induced by estrogen antagonists was released by downregulation of prohibitin via antisense or SiRNA. We next tested whether Brg1 and/or Brm, essential co-repressors of prohibitin, are required for estrogen antagonist-induced repression of E2F-mediated transcription, using the same E2F-responsive E2Luc reporter system described above. Transfection of dominant-negative (ATP-binding-deficient) mutants of Brg1, or Brm (Wang et al, 2002a,2002b), and SiRNA to Brg1 or Brm, both blocked the repression of E2F-driven transcription induced by estrogen antagonists, indicating the necessity of Brg1/Brm in estrogen antagonist-induced E2F transcriptional repression (Figure 5, results using SiRNA are shown).
Figure 5.
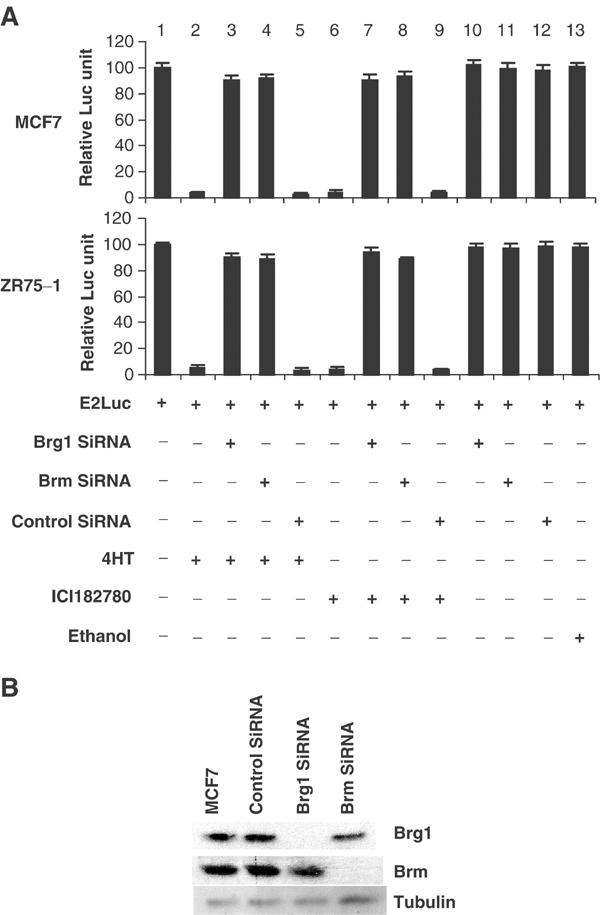
Repression of Brg1 or Brm blocks E2F transcriptional repression induced by estrogen antagonists. (A) MCF7 and ZR75-1 cells were transfected with an E2F-responsive promoter-driven reporter, E2Luc (12 μg/10 cm dish), producing a basal level of luciferase activity (lane 1). Basal luciferase activity was dramatically repressed by treatment of the cells with estrogen antagonists, as indicated (lanes 2 and 5), while treatment with the ethanol vehicle showed no such effect (lane 10). This repression of luciferase activity by estrogen antagonists was reversed by knockdown of Brg1 or Brm using SiRNA (lanes 3, 4, 7 and 8). A 1 μg portion of βGal vector was included in all transfections as a control. βGal values are comparable in all samples. (B) Immunoblot analysis of Brg1, Brm, and tubulin levels in MCF-7 cells or the cells transfected with control SiRNA, or Brg1 or Brm SiRNA.
We recently reported that Brg1 and Brm are recruited by prohibitin to native E2F-responsive promoters (Wang et al, 2002a,2002b). We therefore asked whether the enhanced association between prohibitin and Brg1/Brm in response to estrogen antagonists demonstrated in Figure 3 reflects the recruitment of Brg1 and Brm to native E2F-responsive promoters, using an in vivo CHIP assay. Cell extracts from MCF7 cells treated with estrogen antagonists, estradiol, or both were immunoprecipitated using anti-Brg1, -Brm, -prohibitin, -E2F1, or -p38 (control) antibodies (Figure 6A), followed by PCR, using primers covering a region of the E2F1 promoter, TK promoter, or cFos promoter (as a control) (Wang et al, 2002a,2002b). A six- to eight-fold increase in the level of amplified PCR product was detected in the anti-Brg1 and -Brm precipitates for the E2F-responsive promoters from the cells that had received more than 40 min of tamoxifen treatment, indicating an elevated recruitment of these factors to the endogenous E2F1 promoter. This recruitment was not affected by estradiol (Figure 6A). Results of RT–PCR analyses of levels of the corresponding gene transcripts, and a CHIP assay using an anti-PolII antibody as a marker for active transcription (Li et al, 2001), were consistent with a decrease in transcription of these E2F-responsive promoters in the tamoxifen-treated cells, which was not affected by co-treatment with estradiol. Interestingly, estradiol alone induced the level of transcript of the E2F-responsive promoters, although the recruitment of Brg1 and Brm were not affected, as shown in Figure 6A. However, estradiol did induce Brg1 recruitment to an estrogen-responsive promoter, pS2 (Figure 6B), as previously reported by others (DiRenzo et al, 2000) and estradiol had a similar effect on recruitment of Brm (Figure 6B).
Figure 6.
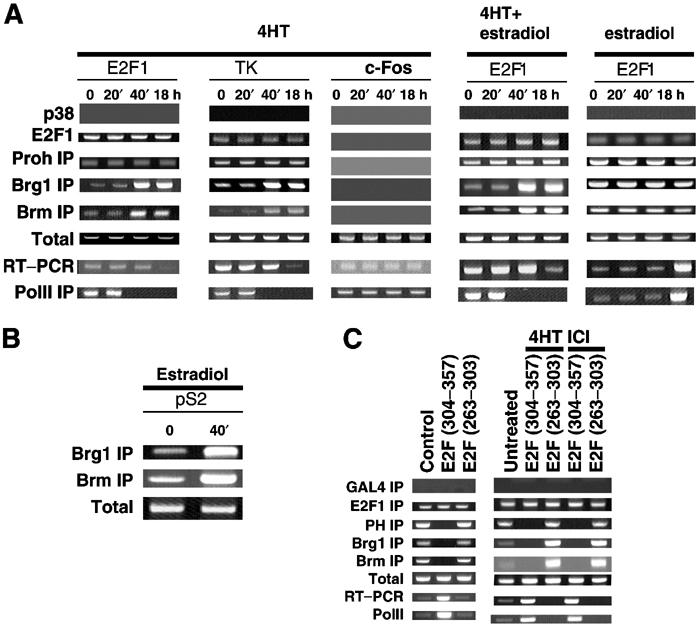
Estrogen antagonists induce the recruitment of Brg1 and Brm to native E2F-responsive promoters for transcriptional repression. (A) MCF7 cells were cultured in an estrogen-free medium for 3 days. The cells were then treated with tamoxifen alone, tamoxifen plus estradiol, or Estradiol alone, in separate experiments. Cell extracts were collected at the different time points as indicated and analyzed using an in vivo CHIP assay. The DNA recovered from the immunoprecipitates by the indicated antibodies was PCR-amplified, using primers against a region on each of the two E2F-responsive promoters (E2F1 and TK), and one non-E2F-responsive promoter (c-Fos), as a control. Higher levels of amplified products from the Brg1 or Brm antibodies were detected in the CHIP assays of the E2F-responsive promoters in the cells treated with 4HT for more than 40 min. This enhanced recruitment of Brm and Brg1 was not affected by co-treatment with estradiol. Amplified products from CHIP using E2F1 or prohibitin antibodies did not show variations in levels regardless of treatment. Control CHIP assay using p38 antibody, and CHIP assay on the c-Fos promoter using prohibitin, Brg1, Brm, and E2F1 antibodies, failed to generate any product, confirming the specificity of this assay. PCR using DNA directly isolated from the cell extracts produced products in all the lanes tested, serving as a positive control for the PCR reaction (Total). RT–PCR assays demonstrated a relative decrease in the levels of transcripts from the E2F-responsive genes in the tamoxifen-treated cells, but not the estradiol-treated cells, which instead produced increased levels of transcripts from E2F-responsive promoters. CHIP assay using PolII antibody demonstrated the transcriptional repression of E2F-responsive genes by tamoxifen and transcriptional induction by estradiol. (B) Positive control CHIP assay using anti-Brg1 and -Brm antibodies was performed on an estrogen-responsive promoter (pS2). Recruitment of Brm and Brg1 to the pS2 promoter was induced by estradiol. (C) MCF7 cells stably transfected with a vector encoding the prohibitin-binding domain of E2F (304–357) or control peptide (263–303). The expression of the transfected genes was confirmed by immunoblot shown in Figure 2C. A CHIP assay was performed on endogenous E2F1 promoter using Brg1, Brm, E2F1 prohibitin antibodies, and control antibody (Gal4). A significantly lower amount of PCR product associated with Brg1 or Brm was found when E2F (304–357) was present in the cells (left). The induction of Brg1 or Brm recruitment to the promoter by estrogen antagonists was blocked when E2F (304–357) was expressed.
As demonstrated in Table I, the growth suppressive functions of prohibitin in response to estrogen antagonists require interaction with E2F and repression of E2F-mediated transcription. To further analyze the basis for this specificity for E2F, we tested the recruitment of prohibitin, Brg1, and Brm to natural, native E2F1 promoters in the presence of prohibitin-binding domain of E2F (AA304–357), which disrupts the prohibitin–E2F association and blocks the E2F repression induced by estrogen antagonists, as shown in Figure 2. An in vivo CHIP assay was performed using MCF7 cells stably expressing the E2F (AA304–357) peptide or the nonrelevant (AA263–303) E2F peptide (as a negative control). The associations of prohibitin, Brg1, and Brm with the E2F1 promoter were blocked when the blocking E2F peptide (AA304–357), but not the control E2F peptide (AA263–303), was expressed (Figure 6C, left). Furthermore, estrogen antagonists were unable to induce the recruitment of Brg1/Brm to the E2F1 promoter in the presence of prohibitin-binding domain of E2F1 (Figure 6C, right).
To establish whether Brg1 and/or Brm are required for the growth suppression induced by estrogen antagonists in susceptible cells, MCF7 and ZR75-1 cells were transfected with vectors expressing Brg1 SiRNA or Brm SiRNA, or with control SiRNA. The transfected cells were continuously exposed to either 4HT or ICI182780, and to G418 for selection, and colonies were enumerated to quantitate the effects on growth (Wang et al, 2002a,2002b). Estrogen antagonists repressed the growth of the breast cancer cells, as assessed by substantial relative decreases in the number of colonies compared to control cultures (Table II). Expression of Brg1 SiRNA or Brm SiRNA, however, dramatically reversed the growth suppression induced by the estrogen antagonists, demonstrating a requirement for Brg1 and Brm in estrogen antagonist-induced growth suppression of breast cancer cells. Repeat experiments using dominant-negative Brg1 or dominant-negative Brm expression in the place of SiRNA confirmed the effect (data not shown).
Table 2.
Reversal of estrogen antagonist-induced repression of colony formation by Brg1 SiRNA and Brm SiRNA
| Vector-based SiRNA transfection | Number of colonies |
|||
|---|---|---|---|---|
| MCF7 |
ZR-75-1 |
|||
| Experiment 1 | Experiment 2 | Experiment 3 | Experiment 4 | |
| Control SiRNA+ethanol | 245 | 243 | 269 | 265 |
| Brg1 SiRNA+ethanol | 255 | 241 | 272 | 271 |
| Brm SiRNA+ethanol | 242 | 246 | 261 | 261 |
| Control SiRNA+4HT | 5 | 3 | 11 | 12 |
| Brg1 SiRNA+4HT | 263 | 258 | 275 | 261 |
| Brm SiRNA+4HT | 239 | 244 | 256 | 251 |
| Control SiRNA+ICI | 7 | 5 | 8 | 6 |
| Brg1 SiRNA+ICI | 238 | 244 | 254 | 255 |
| Brm SiRNA+ICI |
249 |
240 |
261 |
258 |
| Approximately 10 000 MCF7 or ZR-75-1 cells were transfected with 10 μg of the indicated vector-based SiRNA (in pRNAT-U6.1/Neo). Colonies with 20 or more cells were scored after 14 days of selection in 100 μg of neomycin per ml. | ||||
The early signal transduction mechanism utilized by estrogen antagonists is not yet understood, but recent studies indicate the involvement of the stress-activated protein kinase JNK1 in mediating the growth-regulating effect of tamoxifen (Duh et al, 1997; Mandlekar et al, 2000). Tamoxifen stimulates JNK1 activity, and interference with the JNK pathway by overexpression of a dominant-negative JNK1 (DNJNK1) mutant attenuated tamoxifen-induced apoptosis (Mandlekar et al, 2000). We recently reported that JNK1 activity represses the transcriptional activity of E2F (Wang et al, 1999c). Furthermore, prohibitin interacts with MLK2, a binding partner of, activator of, and substrate for JNK1 (Rasmussen et al, 1997; Nagata et al, 1998; Phelan et al, 2001). We therefore hypothesized that the growth regulatory effect of estrogen antagonists via prohibitin may require JNK1. We first tested whether JNK1 could affect the association between prohibitin and Brg1/Brm in a fashion similar to estrogen antagonists. MCF7 cells were transfected with wild-type JNK1 or kinase-deficient, mutant JNK1 genes (Minden et al, 1994). As shown in Figure 7A, overexpression of wild-type JNK1 (confirmed by immunoblot shown in Figure 7B), but not the mutant JNK1, dramatically increased the association between prohibitin and Brg1/Brm. This effect is strikingly similar to that induced by treatment with estrogen antagonists (see Figure 3). We then tested the necessity of the JNK1 pathway in the induction of prohibitin–Brg1/Brm association by estrogen antagonists, using either expression of a DNJNK1 or vector-based JNK1 SiRNA (not shown) or treatment with a specific inhibitor of JNK1 (Bennett et al, 2001), to interfere with JNK1 signaling. The prohibitin/Brg1 and prohibitin/Brm associations induced by 4HT were reversed by the JNK1 inhibitor and also by expression of JNK1 SiRNA (not shown) or DNJNK1 (Figure 7C). An identical effect was observed when using ICI182780 as the antagonist (data not shown), suggesting that JNK1 is required for the regulation of prohibitin by estrogen antagonists. We also repeated the experiment using JNK1 SiRNA to knock down JNK1 protein levels, and confirmed the requirement for JNK1 (data not shown).
Figure 7.
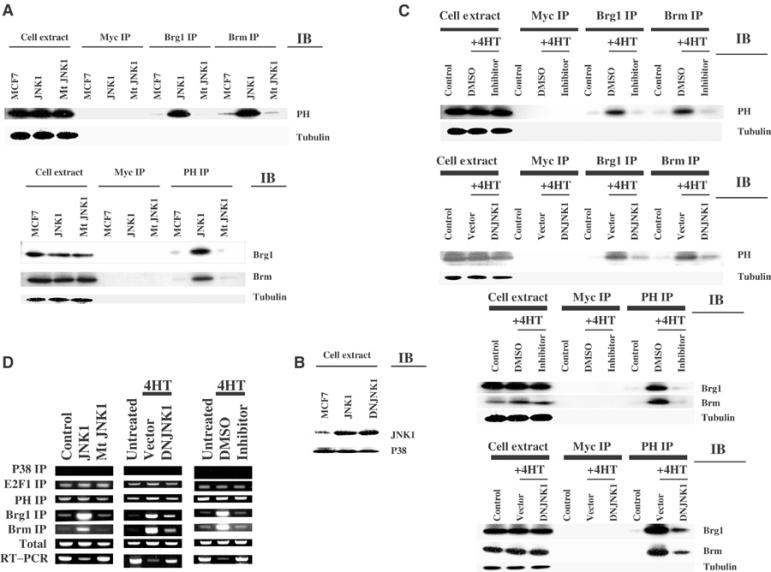
JNK1 mediates estrogen antagonist signaling to the prohibitin/E2F axis. (A) MCF7 cells were transfected with JNK1 or mutant (kinase-deficient) JNK1. Immunoprecipitation/immunoblot analysis (IP/IB), and the reciprocal IP–IB, was performed using the indicated antibodies. Anti-Myc and -tubulin antibodies were used as controls for IP and IB. Equal loading and precipitation are further confirmed by immunoblotting using antibodies for the target proteins (data not shown). JNK1 overexpression induced association between prohibitin and Brg1/Brm, just as tamoxifen does (see Figure 1). (B) The same cell extracts used in panel A were analyzed by immunoblotting using anti-JNK1 antibody or -p38 antibody, confirming the overexpression of the products of the transfected vectors. (C) JNK1 activity was suppressed either by transfection of DNJNK1 or treatment with a JNK1 inhibitor. The cells were co-treated with tamoxifen (‘4HT') or vehicle controls as indicated for 3 h. DMSO was vehicle control for JNK1 inhibitor. Empty vector (‘vector') was transfected as a control for the DNJNK1 vector. Immunoprecipitation/immunoblot analysis (IP/IB), and the reciprocal IP/IB, was performed using the indicated antibodies. Equal loading and precipitation are further confirmed by immunoblotting using antibodies for the target proteins (data not shown). DNJNK1 and JNK1 inhibitor each blocked the enhanced association between prohibitin and Brg1/Brm induced by tamoxifen. (D) In vivo CHIP assay. Left column: MCF7 cells were transfected with empty vector control, JNK1, or mutant (kinase-deficient) JNK1 vectors. Cell extracts were analyzed using an in vivo CHIP assay on the endogenous, native E2F1 promoter, described in Figure 3 (above). Higher levels of amplified products precipitated by the Brg1 or Brm antibodies were detected when JNK1 (but not mutant JNK1) was transfected. This enhanced recruitment of Brm and Brg1 by JNK1 was similar to that induced by tamoxifen treatment, as shown in Figure 3. RT–PCR assays demonstrated a relative decrease in the levels of transcripts from the E2F1 genes when JNK1, but not the mutant JNK1, was transfected. Middle column: MCF7 cells were transfected with control vector or DNJNK1. Cells were treated with ethanol vehicle or tamoxifen as indicated. CHIP assay was performed on the native E2F1 promoter. Transfection of the DNJNK1 blocked the tamoxifen-induced recruitment of BRG1 and BRM to the E2F1 promoter, and reversed the repression of E2F1 transcript levels. Right column: MCF7 cells were treated with ethanol (for tamoxifen vehicle control), DMSO (for JNK1 inhibitor vehicle control), or JNK1 inhibitor. Cells were collected after 3 h of treatment. Extracts were analyzed by CHIP assay. Similar to the effects of DNJNK1 shown in the middle panel, the JNK1 inhibitor also blocked the tamoxifen-induced Brg1/Brm recruitment to the native E2F1 promoter, and the tamoxifen-induced repression of E2F1 transcript levels was reversed by the inhibitor.
We next studied the effect of JNK1 on the recruitment of Brg1 and Brm to the endogenous, natural E2F1 promoter. Similar to the effects of estrogen antagonist treatment, expression of activated JNK1 also induced the recruitment of Brg1 and Brm to the endogenous E2F1 promoter and decreased E2F1 transcript levels (Figure 7D, left). Furthermore, the enhanced recruitment of Brg1 and Brm, as well as the repression of E2F1 transcription by tamoxifen, was reversed by the JNK1 inhibitor and also by DNJNK1 (Figure 7D, middle and right). Similar results were observed when using ICI182780 as the antagonist (data not shown). Collectively, these data demonstrate the necessity of JNK1 in estrogen antagonist-mediated prohibitin/E2F regulation. To test whether JNK2, a closely related member of the c-Jun N-terminal kinase family, has a similar effect, we repeated these experiments using constitutively active JNK2 and DNJNK2 constructs (Gupta et al, 1996; Lei et al, 2002). Unlike JNK1, JNK2 did not promote the association of prohibitin with Brm or Brg1, and DNJNK2 did not interfere with the association of prohibitin/Brm–Brg1/E2F complexes on E2F-responsive promoters induced by estrogen antagonists (data not shown).
The major effects of estrogen antagonists on the growth of breast cancer cells are mediated through the estrogen receptor. To determine whether the regulation of prohibitin by estrogen antagonists is estrogen receptor (ER)-dependent, the ER alpha-negative cell line MDA-MB-231 was transfected with either a control vector or an ER alpha expression construct (Fujita et al, 2003). The cell lines were then treated with vehicle control or estrogen antagonists. Proteins from cell extracts were immunoprecipitated by control antibody or anti-Brg1 or -Brm antibodies, followed by immunoblotting using an anti-prohibitin antibody. The experiments were also performed in a reciprocal fashion. Estrogen antagonists failed to induce an association between prohibitin and Brg1/Brm in the ER-negative cells (Figure 8, ‘vector' for control). However, when the cells were forced to express the ER (expression confirmed by immunoblot; Figure 8C), the association of prohibitin/Brg1/Brm was dramatically increased by estrogen antagonist treatment (Figure 8A and B, ‘+ER').
Figure 8.
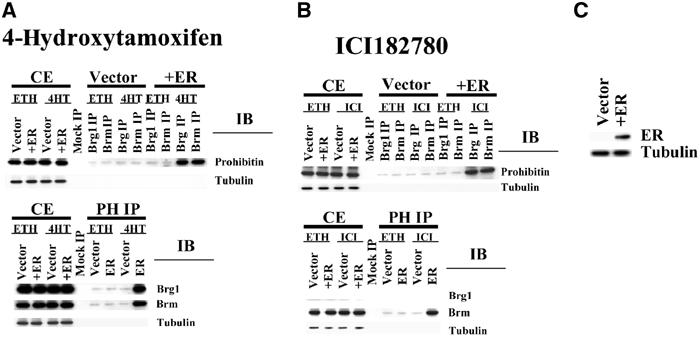
Regulation of prohibitin by estrogen antagonist is ER-dependent. (A, B) MDA-MB-231, an ER-negative cell line, was stably transfected with an ER expression vector, or an empty vector as a control. The empty vector-transfected cells and the ER-expressing cells (‘+ER') were treated with vehicle control (‘ETH') or estrogen antagonists (‘4HT' or ‘ICI'), as indicated. Whole-cell extracts (CE), or proteins immunoprecipitated from cell extracts with control antibody (myc) (‘mock IP') or anti-Brg1 or -Brm antibodies, were electrophoretically separated, followed by immunoblotting with an anti-prohibitin antibody (upper panels). The IP:IB analysis was repeated in a reciprocal fashion (lower panels). Equal loading and precipitation were further confirmed by immunoblotting using antibodies for the target proteins (data not shown). (C) Expression of the transfected ER was detected by immunoblot.
Discussion
Estrogen antagonists are widely used in the treatment of hormone-responsive breast cancers, but are limited by the inevitable development of estrogen independence and cellular resistance to antihormonal agents (Carroll et al, 2000; Wolczynski et al, 2000). Full elucidation of the mechanism of estrogen antagonist-induced growth repression will likely provide pivotal information as to the etiology of resistance, as well as providing new molecules and pathways for therapeutic targeting. Data presented in this report demonstrate that prohibitin and its co-repressors, Brg1/Brm, are required for growth suppressive signaling by estrogen antagonists. Prohibitin has been found to be mutated in sporadic breast cancers, suggesting a role of prohibitin as a tumor suppressor in the development of breast cancer (Sato et al, 1993).
The fundamental importance of the E2F–prohibitin axis in signaling by estrogen antagonists is further suggested by the dual level of regulation imposed by these agents—estrogen antagonists both increase the expression of the prohibitin protein at the level of transcription and enhance prohibitin–Brg1/Brm associations. The higher levels of prohibitin protein induced by estrogen antagonists would likely reinforce E2F repression and growth suppression. However, induction of the prohibitin promoter became apparent only after extended treatment (between 5 and 18 h), which is far later than the elevated prohibitin/Brg1/Brm associations and transcriptional repression (demonstrated by the PolII CHIP assay in Figure 6) induced by estrogen antagonists, which occur within 20 min of estrogen antagonist exposure (Figure 3). Furthermore, the prohibitin association with E2F-responsive promoters remains constant (Figure 6A), even when the total amount of cellular prohibitin protein was elevated at 18 h time point (Figure 3). The temporal differences found in these two assays suggest that elevation of prohibitin levels by estrogen antagonists may not be necessary for E2F transcriptional repression/growth suppression (at least initially). The increases in cellular levels of prohibitin may be a ‘secondary' effect of estrogen antagonist treatment, unrelated to transcription repression/growth suppression. An alternate possibility is that higher amounts of prohibitin would serve to sustain the effect of estrogen antagonists (to maintain constant high-level associations of prohibitin/Brg1/Brm with promoter).
A recent report demonstrates that Brg1 physically and functionally associates with BRCA1 (Bochar et al, 2000). Furthermore, Brg1 was recently found to associate with the estrogen receptor and is required for the activation of estrogen agonist-responsive promoters (DiRenzo et al, 2000). Data presented in this study indicate that estrogen antagonists may utilize Brg1/Brm by modulating their recruitment to prohibitin complexes for the repression of breast cancer cell growth via the E2F pathway. Brg1 and Brm have recently attracted significant attention in the study of E2F regulation. Brg1 and Brm are ATP-dependant chromatin remodeling molecules originally related to transcriptional activation. Recent reports, however, indicate that Brg1 and Brm are also involved in transcriptional repression (Strobeck et al, 2000). For example, Brg1 and Brm both interact with Rb and participate in the E2F repression (Strober et al, 1996; Zhang et al, 2000; Wang et al, 2002a,2002b). How can the same molecules function as both activator and repressor of transcription? One possibility is that Brg1/Brm actually function as neutral co-regulators capable of synergizing with other co-regulator molecules. The activating or repressing mode would be determined by the transcription factor or repressor they are associated with at the promoter. Further study is under way to elucidate the mechanism underlying this hypothesized context dependency of Brg1 and Brm function.
A traditional mechanism postulated for the repressive function of estrogen antagonists is that of competitive binding to the estrogen receptor, thereby reversing estrogen signaling events or mimicking an estrogen-free state. We demonstrate that the effect of estrogen antagonists on prohibitin/Brm–Brg1 is indeed ER-dependent. A recent report indicates that estrogen induces the recruitment of Brg1 to estrogen-responsive promoters (DiRenzo et al, 2000). The data presented in Figure 3B demonstrate that estrogen induces recruitment of not only Brg1 but also Brm to the estrogen-responsive pS2 promoter. The recruitment of Brg1/Brm to native E2F-responsive promoters was not affected by estrogen however, and co-treatment of cells with estradiol did not release the repressive effects of estrogen antagonists, nor affect the recruitment of Brg1/Brm to the native E2F-responsive promoter. These differential effects between estrogen and estrogen antagonists suggest that estrogen antagonists, even ‘pure' antagonists like ICI182780, may not be functionally equivalent to an estrogen-free state. Rather, their occupancy of the ER, enhancement of dimerization, and binding to estrogen-response elements (EREs) may differentially regulate estrogen-responsive genes and target specific signaling pathway(s), which utilize prohibitin/Brg1/Brm as intermediates, to achieve their growth suppression function.
The central role of the E2F transcription factors in cell cycle regulation has been well documented (Harbour and Dean, 2000). E2F complexes are altered in response to a wide array of signals and we recently demonstrated that JNK1 modulates E2F activity (Wang et al, 1999c). The molecular mechanism of this regulation is not yet elucidated. In unpublished studies, we have demonstrated that purified JNK1 can efficiently phosphorylate prohibitin in vitro. The finding that JNK1 both phosphorylates prohibitin (in vitro) and regulates prohibitin/Brg1/Brm associations in vivo now adds a testable potential mechanistic link to this pathway. Unlike JNK1, JNK2 is not involved in this regulation, suggesting that the signal transduction pathway used by the estrogen antagonist is highly specific. The data presented here now link the activation of JNK1 by estrogen antagonists with the recruitment of the co-repressors Brg1/Brm to the growth inhibitory prohibitin/E2F axis in the regulation of breast cancer cell growth.
Independent reports recently established that tamoxifen induces JNK1 activity and that interfering with the JNK1 pathway reverses the growth inhibitory activity of tamoxifen (Duh et al, 1997; Mandlekar et al, 2000). The mechanism of JNK activation by estrogen antagonist is as yet undefined, but recent findings suggest that the generation of reactive oxygen species mediates this process (Mandlekar et al, 2000). Despite the finding that both estrogen and estrogen antagonists work through the estrogen receptor, it is noteworthy that the regulation of prohibitin/Brg1/Brm by estrogen antagonists cannot be reversed by estrogen, suggesting possible differences in the function of estrogen receptor when associated with estrogen or estrogen antagonists. It is plausible to hypothesize that estrogen antagonists may differentially alter ER function, leading to activation of an alternate signaling pathway involving components of JNK pathway and impinging on the prohibitin/Brm–Brg1/E2F axis. Future studies will focus on the very early signaling events initiated after ligand binding, to discern where ER-generated signals bifurcate into proliferative or antiproliferative pathways.
Materials and methods
Cell lines, vectors, and transfections
Cells were maintained in Dulbecco's modified Eagle's medium (MCF7), RPMI1640 medium (ZR75-1), or DME (MDA-MB-231) containing 10% fetal bovine serum (FBS). A total of 2 μg of plasmid vectors was used in all transfections for reporter analyses, unless otherwise noted. A 1 μg portion of a pSV-βGal vector was included as internal control in all transfections, and the β-galactosidase activity varied only slightly (<5%) within each experiment. In all cases, representative chloramphenicol acetyltransferase (CAT) assay results from multiple experiments are shown. The total amount of DNA used in each transfection was normalized with salmon sperm DNA. The ProhibitinCAT reporter was generated by inserting the rat prohibitin promoter (Altus et al, 1995) (spanning −485 to −5 bp, generated by PCR) between the SphI/XbaI sites of the pCAT-Basic vector (Promega). A 12 μg portion of DNJNK1 or DNJNK2 was used for transfection (Minden et al, 1994; Gupta et al, 1996). JNK1 vector was described before (Wang et al, 1999c). Constitutively activated form of JNK2 and DNJNK2 constructs were gifts from Dr Roger J Davis (Gupta et al, 1996; Lei et al, 2002). The estrogen receptor expression vector pCDNA3-ER was a gift from Dr Junn Yanagisawa at University Tsukuba (Fujita et al, 2003). Stable transfections were described before (Wang et al, 2002a,2002b).
Estrogen and estrogen antagonists
The cells were treated with 4HT (100 nM) (Sigma), ICI182780 (100 nM) (TOCRIS, Cookson Inc.), or β-estradiol (100 nM) (Sigma) in phenol red-free DMEM or RPMI (both from Gibco) with 10% charcoal-treated FBS (Hyclone).
JNK1 inhibitor
Cells were treated with 30 μM SP600125 (from Calbiochem) in DMSO, or 0.3% DMSO as a vehicle control.
Flow cytometric analysis
Cells were harvested with 0.5 ml trypsin/EDTA after washing twice with ice-cold PBS, followed by one more wash with 5 volumes of ice-cold PBS supplemented with 2% FBS, and gently resuspended in PBS–EtOH (50%) and fixed overnight at 4°C. The fixed cells were then washed once in ice-cold PBS and resuspended in PBS with 550 μg RNase A/ml and incubated at 4°C for 1 h. Cells were washed again in PBS and re-suspended in 50 μg propidium iodide/ml and 50 μg RNase A/ml, and incubated at 4°C for 1 h. DNA profiles of the cells were analyzed by FACScan (Becton-Dickinson, San Jose, CA), using the Cell Quest program.
Immunoprecipitation, immunoblotting, and CHIP assay techniques
These were described previously (Wang et al, 2002a,2002b). Brg1 (H-88), Brm (N-19), tubulin, JNK1, PolII, and cMyc antibodies were purchased from Santa Cruz. Specificity of Brg1 and Brm was confirmed by immunoblotting and transfection of Brg1 or Brm in a Brg1/Brm-negative SW13 cell. Prohibitin antibody was purchased from NeoMarkers (via Lab Vision Corporation).
SiRNA
SiRNA were designed using the ‘SiRNA design tool' at www.qiagen.com/SiRNA. Prohibitin SiRNA and nonsilencing sequences were purchased from Qiagen. Transfections were carried out by following the protocol provided by the company. Vector (pRNAT-U6.1/Neo)-based SiRNA of Brg1, Brm, and JNK1 and control SiRNA (pRNA-U6.1/Neo/siLuc) were purchased from GenScript Corporation (www.genscript.com). Transcription of the vector-based SiRNA was performed according to the suggestion of the company.
cDNA target sequences of SiRNA:
Prohibitin: AATGTGGATGCTGGGCACAGA
Brg1: AACATGCACCAGATGCACAAG
Brm: AAGTCCTGGACCTCCAAGTGT
JNK1: AACGTGGATTTATGGTCTGTG
Luciferase assay
This was performed by using luciferase assay system from Promega according to the standard protocol of the company using a TD-20/20 Luminometer (Turner Designs).
Acknowledgments
We thank Dr Srikumar P Chellappan for his continuous support. We thank Dr Roger J Davis (Howard Hughes Medical Institute, University of Massachusetts), Dr Ulla Hansen (Boston University), and Dr Junn Yanagisawa (University of Tsukuba) for reagents. This work was partially supported by grants from the National Cancer Institute (1 R03 CA102940-01) (SW), the American Cancer Society (IRG-72-001-27-IRG) (SW), the National Cancer Institute (CA 84193) (DVF), and the US Army Breast Cancer Research Program (Contract DAMD17-00-1-0160) (DVF). SW is the recipient of a Susan G Komen Breast Cancer Foundation Breast Cancer Research Grant, BUSM Department of Medicine Pilot Project Grant Award, and an Aid for Cancer Research grant award.
References
- Altus MS, Wood CM, Stewart DA, Roskams AJ, Friedman V, Henderson T, Owens GA, Danner DB, Jupe ER, Dell'Orco RT, McClung JK (1995) Regions of evolutionary conservation between the rat and human prohibitin-encoding genes. Gene 158: 291–294 [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 98: 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R (2000) BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer [In Process Citation]. Cell 102: 257–265 [DOI] [PubMed] [Google Scholar]
- Carroll JS, Prall OW, Musgrove EA, Sutherland RL (2000) A pure estrogen antagonist inhibits cyclin E-Cdk2 activity in MCF-7 breast cancer cells and induces accumulation of p130–E2F4 complexes characteristic of quiescence. J Biol Chem 275: 38221–38229 [DOI] [PubMed] [Google Scholar]
- Coezy E, Borgna JL, Rochefort H (1982) Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res 42: 317–323 [PubMed] [Google Scholar]
- DiRenzo J, Shang Y, Phelan M, Sif S, Myers M, Kingston R, Brown M (2000) BRG-1 is recruited to estrogen-responsive promoters and cooperates with factors involved in histone acetylation. Mol Cell Biol 20: 7541–7549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duh JL, Yu R, Jiao JJ, Matwyshyn GA, Li W, Tan TH, Kong AN (1997) Activation of signal transduction kinases by tamoxifen. Pharm Res 14: 186–189 [DOI] [PubMed] [Google Scholar]
- Fujita T, Kobayashi Y, Wada O, Tateishi Y, Kitada L, Yamamoto Y, Takashima H, Murayama A, Yano T, Baba T, Kato S, Kawabe YI, Yanagisawa J (2003) Full activation of estrogen receptor alpha (ER alpha) activation function-1 (AF-1) induces proliferation of breast cancer cells. J Biol Chem 278 (29): 26704–26714 [DOI] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 15: 2760–2770 [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Dean DC (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 14: 2393–2409 [DOI] [PubMed] [Google Scholar]
- Jost CA, Ginsberg D, Kaelin WG Jr (1996) A conserved region of unknown function participates in the recognition of E2F family members by the adenovirus E4 ORF 6/7 protein. Virology 220: 78–90 [DOI] [PubMed] [Google Scholar]
- Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ (2002) The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol 22: 4929–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Gorospe M, Hutter D, Barnes J, Keyse SM, Liu Y (2001) Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation–acetylation. Mol Cell Biol 21: 8213–8224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandlekar S, Yu R, Tan TH, Kong AN (2000) Activation of caspase-3 and c-Jun NH2-terminal kinase-1 signaling pathways in tamoxifen-induced apoptosis of human breast cancer cells. Cancer Res 60: 5995–6000 [PubMed] [Google Scholar]
- Minden A, Lin A, McMahon M, Lange-Carter C, Derijard B, Davis RJ, Johnson GL, Karin M (1994) Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science 266: 1719–1723 [DOI] [PubMed] [Google Scholar]
- Nagata K, Puls A, Futter C, Aspenstrom P, Schaefer E, Nakata T, Hirokawa N, Hall A (1998) The MAP kinase kinase kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO J 17: 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan DR, Price G, Liu YF, Dorow DS (2001) Activated JNK phosphorylates the c-terminal domain of MLK2 that is required for MLK2-induced apoptosis. J Biol Chem 276: 10801–10810 [DOI] [PubMed] [Google Scholar]
- Rasmussen RK, Ji H, Eddes JS, Moritz RL, Reid GE, Simpson RJ, Dorow DS (1997) Two-dimensional electrophoretic analysis of human breast carcinoma proteins: mapping of proteins that bind to the SH3 domain of mixed lineage kinase MLK2. Electrophoresis 18: 588–598 [DOI] [PubMed] [Google Scholar]
- Roskams AJ, Friedman V, Wood CM, Walker L, Owens GA, Stewart DA, Altus MS, Danner DB, Liu XT, McClung JK (1993) Cell cycle activity and expression of prohibitin mRNA. J Cell Physiol 157: 289–295 [DOI] [PubMed] [Google Scholar]
- Sato T, Sakamoto T, Takita K, Saito H, Okui K, Nakamura Y (1993) The human prohibitin (PHB) gene family and its somatic mutations in human tumors. Genomics 17: 762–764 [DOI] [PubMed] [Google Scholar]
- Strobeck MW, Knudsen KE, Fribourg AF, DeCristofaro MF, Weissman BE, Imbalzano AN, Knudsen ES (2000) BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci USA 97: 7748–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober BE, Dunaief JL, Guha S, Goff SP (1996) Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Mol Cell Biol 16: 1576–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Fusaro G, Padmanabhan J, Chellappan SP (2002a) Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene 21: 8388–8396 [DOI] [PubMed] [Google Scholar]
- Wang S, Ghosh RN, Chellappan SP (1998) Raf-1 physically interacts with Rb and regulates its function: a link between mitogenic signaling and cell cycle regulation. Mol Cell Biol 18: 7487–7498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Nath N, Adlam M, Chellappan S (1999a) Prohibitin, a potential tumor suppressor, interacts with RB and regulates E2F function. Oncogene 18: 3501–3510 [DOI] [PubMed] [Google Scholar]
- Wang S, Nath N, Fusaro G, Chellappan S (1999b) Rb and prohibitin target distinct regions of E2F1 for repression and respond to different upstream signals. Mol Cell Biol 19: 7447–7460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Nath N, Minden A, Chellappan S (1999c) Regulation of Rb and E2F by signal transduction cascades: divergent effects of JNK1 and p38 kinases. EMBO J 18: 1559–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang B, Faller DV (2002b) Prohibitin requires Brg-1/Brm for the repression of E2F and cell growth. EMBO J 21: 3019–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Smith R III, Burghardt R, Safe SH (1997) 17 beta-Estradiol-mediated growth inhibition of MDA-MB-468 cells stably transfected with the estrogen receptor: cell cycle effects. Mol Cell Endocrinol 133: 49–62 [DOI] [PubMed] [Google Scholar]
- Wolczynski S, Swiatecka J, Anchim T, Dabrowska M, Dzieciol J (2000) [Biochemical mechanism of raloxifen and tamoxifen action for the prevention of breast cancer. Studies in vitro]. Ginekol Pol 71: 1147–1152 [PubMed] [Google Scholar]
- Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, Ghaffari S, Iliev D, Penn B, Woodland AM, Smith R, Salada G, Carillo A, Laity K, Gupte J, Swedlund B, Tavtigian SV, Teng DH, Lees E (2000) BRG1, a component of the SWI–SNF complex, is mutated in multiple human tumor cell lines. Cancer Res 60: 6171–6177 [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 101: 79–89 [DOI] [PubMed] [Google Scholar]