

Abstract
Identification of the signaling pathways that influence the reprogramming of Müller glia into neurogenic retinal progenitors is key to harnessing the potential of these cells to regenerate the retina. Glucocorticoid receptor (GCR) signaling is commonly associated with anti-inflammatory responses and GCR agonists are widely used to treat inflammatory diseases of the eye, even though the cellular targets and mechanisms of action in the retina are not well understood. We find that signaling through GCR has a significant impact upon the ability of Müller glia to become proliferating Müller glia-derived progenitor cells (MGPCs). The primary amino acid sequence and pattern of GCR expression in the retina is highly conserved across vertebrate species, including chickens, mice, guinea pigs, dogs and humans. In all of these species we find GCR expressed by the Müller glia. In the chick retina, we find that GCR is expressed by progenitors in the circumferential marginal zone (CMZ) and is upregulated by Müller glia in acutely damaged retinas. Activation of GCR signaling inhibits the formation of MGPCs and antagonizes FGF2/MAPK signaling in the Müller glia. By contrast, we find that inhibition of GCR signaling stimulates the formation of proliferating MGPCs in damaged retinas, and enhances the neuronal differentiation while diminishing glial differentiation. Given the conserved expression pattern of GCR in different vertebrate retinas, we propose that the functions and mechanisms of GCR signaling are highly conserved and are mediated through the Müller glia. We conclude that GCR signaling directly inhibits the formation of MGPCs, at least in part, by interfering with FGF2/MAPK signaling.
Keywords: Müller glia, Glucocorticoids, Regeneration, Retina, Chick, Mouse
INTRODUCTION
The glucocorticoid receptor (GCR; Nr3c1) is a nuclear hormone receptor with signaling that is associated with suppressing inflammation. Without bound ligand, GCR resides in the cytoplasm as an inactive oligomeric complex with molecular chaperones that maintain the receptor in a high-affinity hormone-binding state. Glucocorticoids bind to GCR to dissociate the receptor from the regulatory complex; GCR becomes phosphorylated at multiple sites and dimerizes to translocate to the nucleus to influence transcription. The GCR dimer interacts with DNA by binding to specific nucleotide palindromic sequences known as Glucocorticoid Response Elements (GRE) or negative GRE (nGRE; reviewed by Schaaf and Cidlowski, 2002). Ligand-bound GCR can activate or repress the expression of genes containing GRE or nGRE sites (reviewed by Schaaf and Cidlowski, 2002).
Previous studies have shown that GCR signaling can suppress neurogenesis (Cameron and Gould, 1994; Gould et al., 1998; Karishma and Herbert, 2002). During development, activation of GCR reduces the proliferation of the neural stem cells in embryonic rat brain (Sundberg et al., 2006). In the central nervous system, GCR is expressed in a context-specific manner with expression in neurons and glial cells in different parts of the brain (reviewed by De Kloet et al., 1998). Furthermore, GCR has been detected in adult mouse hippocampal progenitor cells (Garcia et al., 2004), and activation of GCR signaling decreases the proliferation of these cells (Alonso, 2000; Schroter et al., 2009; Wong and Herbert, 2006). GCR signaling is known to influence late stages of retinal development, i.e. the differentiation of Müller glia. Cortisol, the endogenous ligand of GCR, is known to stimulate the expression of glutamine synthetase (GS) in embryonic chick retina (reviewed by Moscona and Linser, 1983). In the developing chick retina, levels of GS are low until E15 and rapidly increase thereafter (Gorovits et al., 1994, 1996; Grossman et al., 1994), in parallel to adrenal cortex development and elevated systemic levels of glucocorticoid (Marie, 1981). GS expression is a symptom of glial maturation and can be induced in immature Müller glia by exogenous glucocorticoids (Moscona and Linser, 1983); this is probably a direct effect as GCR may be expressed by immature glia or late-stage retinal progenitors (Gorovits et al., 1994). By comparison, FGF2 inhibits the expression of GS in the developing retina (Kruchkova et al., 2001), consistent with the notion that FGF signaling promotes the de-differentiation of Müller glia and maintains neural progenitors (reviewed by Fischer and Bongini, 2010; Gallina et al., 2014). In the retinas of different vertebrate species, proliferating Müller glia-derived retinal progenitor cells (MGPCs) are generated in response to damage or activation of distinct cell-signaling pathways (reviewed by Fischer and Bongini, 2010; Gallina et al., 2014). The purpose of this study was to investigate how GCR signaling influences the formation of MGPCs.
RESULTS
GCR is expressed by Müller glia in the retinas of different vertebrates
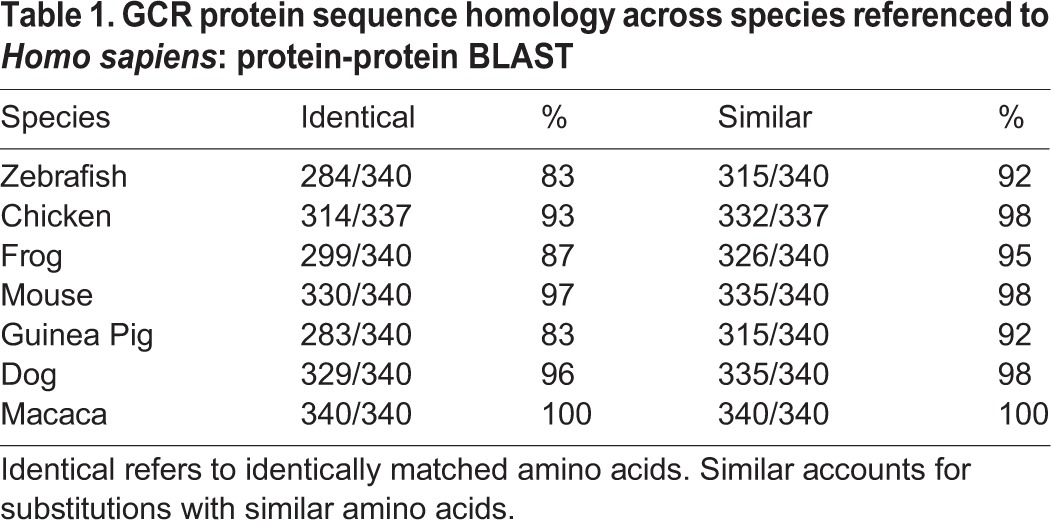
An alignment of primary amino acid sequences indicated that GCR is highly conserved among different vertebrates, including human, monkey, mouse, guinea pig and chick. We found a very high level (83-100%) of sequence identity among vertebrate species when compared with human GCR protein (Table 1). Accounting for conservative amino acid substitutions, we found very high (≥92%) sequence conservation among GCR sequences from different vertebrates (Table 1).
Table 1.
GCR protein sequence homology across species referenced to Homo sapiens: protein-protein BLAST
Given that the primary amino acid sequence of GCR is highly conserved among warm-blooded vertebrates, we expected that pharmacological agents and antibodies designed to act at human GCR would act selectively at GCR in other warm-blooded vertebrates. Accordingly, we used antibodies to GCR to characterize patterns of expression in the retinas of different vertebrates. Retinas were labeled with different antibodies raised to human GCR and Sox2, a well-established marker for glia in normal, healthy mammalian retinas (Fischer et al., 2010). GCR was observed primarily in the Sox2-positive nuclei of Müller glia in mouse, guinea pig, dog and human retina (Fig. 1A-L). The labeling for GCR in the nuclei of Müller glia was uniform across central and peripheral regions of the retina. In addition to labeling the nuclei of Müller glia, we observed GCR immunofluorescence in the nuclei of a few scattered cells in the inner retinal layers (Fig. 1). The identity of these cells remains uncertain.
Fig. 1.
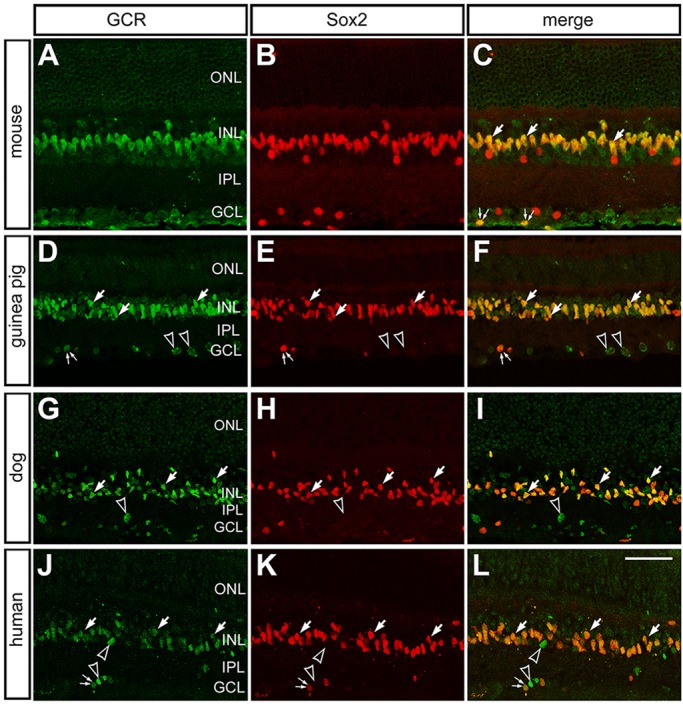
GCR is expressed by Müller glia in the retinas of different vertebrates. (A-L) Retinal sections from adult mouse (A-C), guinea pig (D-F), dog (G-I) and human (J-L) were labeled with antibodies to GCR [green; H-300 polyclonal (A-C) and PA1-511A polyclonal (D-L)] and Sox2 (red). Large arrows indicate Müller glia cells labeled for Sox2 and GCR; pairs of small arrows indicate presumptive cholinergic amacrine cells; hollow arrowheads indicate putative glial cells that are labeled for Sox2 and GCR in the inner layers of the retina; empty arrowheads indicate GCR-positive Sox2-negative cells. ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar: 50 µm.
In the developing chick retina, antibodies raised to human GCR produce nuclear labeling in Sox2-positive Müller glia in late-stage (E14-E16) embryos (not shown), consistent with previous reports (Gorovits et al., 1994). In post-hatch chick retina, immunofluorescence for GCR was elevated in Müller glia in peripheral regions compared with levels seen in central regions of the retina (Fig. 2A). Similar to patterns of expression in mammalian retinas, GCR was detected primarily in the Sox2-positive nuclei of Müller glia and colocalized to TOPAP-positive Müller glia (Fig. 2B,C). TOPAP is a member of sarcolemmal membrane-associated protein family that is expressed by Müller glia and recognized by the 2M6 monoclonal antibody (Ochrietor et al., 2010). In addition, GCR is expressed by the non-astrocytic inner retinal glia (NIRG) cells, as indicated by overlap of labeling for GCR, Sox2 and Nkx2.2 (Fig. 2B). NIRG cells have been identified as a distinct type of glial cell in the retina (Fischer et al., 2010; Zelinka et al., 2012). Levels of GCR expression in Müller glia increase towards the periphery of the retina with the highest levels detected in the progenitors in the circumferential marginal zone (CMZ) and the non-pigmented epithelial cells of the pars plana (Fig. 2A,D,E). We did not observe GCR expression in CD45-positive microglia (not shown). The conserved sequence, as well as the near-identical expression patterns among retinal cell types, implies that the functions of GCR are important and well conserved.
Fig. 2.

In the chick eye, GCR is expressed by Müller glia in peripheral regions of the retina, circumferential marginal zone (CMZ) progenitors and non-pigmented epithelial (NPE) cells of the pars plana of the ciliary body. (A-I) GCR expression is transiently increased in Müller glia in damaged retinas. Retinas were labeled with antibodies to GCR (PA1-511A rabbit polyclonal; green), Sox2 (red; B,D-I), Nkx2.2 (magenta; B) and TopAP (red; C). Sections were obtained from different regions of the retina (A-C), CMZ (D) and pars plana of the ciliary body (E). (F-I) Sox2 (red) is included as a partial field overlay to indicate nuclear colocalization with GCR (green) in Müller glia and NIRG cells. Images were taken from mid-temporal regions of the retina from control (F), or 1 (G), 2 (H) or 3 (I) days after treatment with 2 micromoles of NMDA. Large arrows indicate the nuclei of Müller glia; arrowheads indicate Sox2/Nkx2.2/GCR-positive NIRG cells; pairs of small arrows indicate cholinergic amacrine cells. Scale bar: 50 µm (bar in I applies to F-I). CMZ, ciliary marginal zone; PE, pigmented epithelium of the ciliary body; NPE, non-pigmented epithelium of the ciliary body. (J-L) qRT-PCR was used to measure relative levels mRNA for Gcr (J), Hsd1 (K) and Hsd2 (L) at 4 h, 1 day, 2 days and 3 days after NMDA treatment. Data are mean±s.d. (n≥5) of the relative percentage change in mRNA levels. The significance of differences between control and treated groups was determined by using a two-tailed Mann-Whitney U-test (*P<0.05; **P<0.01; ns, not significant).
GCR expression is dynamic in damaged retinas
We next examined whether expression levels of GCR were influenced by NMDA-induced retinal damage where Müller glia are known to become proliferating progenitor-like cells (reviewed by Fischer and Bongini, 2010; Gallina et al., 2014). Using immunofluorescence, GCR appears to be upregulated by Sox2-positive Müller glia at 1 and 2 days after NMDA treatment (Fig. 2F-H). At 3 days after damage, when the appearance of the MGPCs is observed, and the Sox2-positive nuclei in the INL delaminate, GCR expression is maintained by MGPCs (Fig. 2I). In damaged retinas, we did not observe GCR expression in CD45-positive microglia (data not shown). qPCR analysis showed that GCR mRNA levels after NMDA-induced damage quickly, within 4 h, increase above the pre-damage levels (Fig. 2J). GCR levels remained elevated 1 day after treatment, and then fall to pre-damage levels by day 2, and further decrease below the pre-damage levels by day 3 (Fig. 2J).
To better understand the dynamics of GCR signaling and how cortisol levels are regulated in damaged retinas, we measured the levels of cortisol-processing enzymes 11β-hydroxysteroid dehydrogenase type 1 (HSD1), which converts inactive cortisone to active cortisol, and 11β-hydroxysteroid dehydrogenase type 2 (HSD2), which converts cortisol to cortisone. qPCR analysis demonstrated that, in contrast to GCR, HSD1 levels do not change by 4 h after NMDA treatment, decrease slightly by day 1, increase significantly by day 2 and decrease by day 3 (Fig. 2K). By comparison, HSD2 levels increase 4 h after NMDA treatment and fall to control levels in the following days (Fig. 2L). Collectively, these data indicate that GCR signaling is dynamic following NMDA-induced damage.
Activation of GCR signaling inhibits the formation of MGPCs
To test whether activation of GCR has an impact on the formation of MGPCs, we made intraocular injections of the GCR agonists dexamethasone (Dex) or Compound A (CpdA) following NMDA-induced damage and probed for proliferation (Fig. 3). Cell proliferation is an integral aspect of the transition of Müller glia to MGPCs (reviewed by Fischer and Bongini, 2010; Gallina et al., 2014; Karl and Reh, 2010). Following NMDA-induced damage, the MGPCs enter S-phase of the cell cycle in synchrony at 2 days after treatment, thereby enabling effective BrdU labeling of the MGPCs and their progeny (Fischer and Reh, 2001). In the chick retina, MGPC proliferation is prominent in peripheral regions of the retina, where the Müller glia are believed to be less mature than in central retina (reviewed by Fischer, 2005), and where we find persistent expression of GCR (see Fig. 2A). Application of 100 ng/dose of Dex after NMDA treatment decreased the number of the proliferating MGPCs by about 50% (Fig. 3C), while having no effect on the proliferation of NIRG cells (Fig. 3D). We probed for the proliferation of the NIRG cells because these glia, like the Müller glia, express GCR (see Fig. 2B). Application of 200 ng of Dex after NMDA treatment decreased the number of the proliferating MGPCs by about 90% (Fig. 3E), while having no effect on the proliferation of NIRG cells (Fig. 3F).
Fig. 3.

Activation of GCR signaling inhibits the formation of MGPCs in peripheral regions of damaged retinas. (A,B) Eyes were injected with 2 micromoles of NMDA at P6, and vehicle (DMSO; A), 100 ng Dex (n=13), 200 ng Dex (n=9) (B) or 500 ng CpdA (n=5) at P7 and P8, and BrdU at P8. Retinas were harvested at P9 and processed for immunolabeling. Retinas were labeled for BrdU (green), Nkx2.2 (magenta) and Sox2 (red). Arrows indicate BrdU+/Sox2+/Nkx2.2− MGPCs; hollow arrowheads indicate BrdU+/Sox2+/Nkx2.2+ NIRG cells. Scale bar: 50 µm. (C-H) Data are mean±s.d. The number of BrdU-positive Müller glia (C,E,G) or NIRG cells (D,F,H) in peripheral regions of the retina, where proliferating MGCPs are known to form (Fischer et al., 2002; Fischer and Reh, 2001). The significance of any differences (*P<0.05; **P<0.01; ***P<0.001; ns, not significant) was determined using a two-tailed unpaired t-test.
Dex activates both the GCR dimer and monomer, and does not discriminate between GCR-dependent transcriptional changes and changes in cell signaling pathways. By comparison, CpdA activates only the monomeric form of GCR that interacts with cytoplasmic proteins (De Bosscher et al., 2010, 2005). To better characterize whether activated GCR signaling suppresses the formation of MGPCs because of GCR-dependent transcription or GCR-dependent protein-protein interactions, CpdA was applied after NMDA treatment (Fig. 3). The number of proliferating MGPCs was decreased by about 50% (Fig. 3G), whereas the proliferation of NIRG cells was unaffected (Fig. 3H). These results suggest that GCR-mediated changes in both transcription and cell signaling inhibit the proliferation of MGPCs in damaged retinas.
To better understand how GCR signaling inhibits the formation of MGPCs, we probed for different factors that are known to influence MGCPs. Levels of retinal damage are known to influence the formation of MGPCs (Fischer and Reh, 2001; Fischer et al., 2004b). Therefore, we probed for levels of cell death in retinas treated with Dex or CpdA. Injections of Dex or CpdA into normal, healthy eyes had no effect upon cell survival or the reactivity of Müller glia (data not shown). By contrast, injections of Dex or CpdA following NMDA treatment decreased the number of dying cells by about 50% (Fig. 4A-E).
Fig. 4.

In NMDA-damaged retinas, Dex inhibits microglial reactivity, decreases retinal levels of the complement ligand precursor C3, increases levels of the MAPK inhibitor Spred1 and has little effect on levels of Pax6 in MGPCs. (A-M) Eyes were injected with 2 micromoles of NMDA at P6, and vehicle (DMSO) or 200 ng Dex at P7 and P8. Retinas were harvested at P9 and labeled using TUNEL (A-D) or Draq5 (nuclei; F,G), and antibodies to CD45 (F,G), Sox9 (J,K), Pax6 (J,K) or neurofilament (NF-H, 200 kDa) (L,M). Pairs of small arrows indicate microglia (F,G). (E) Data show the mean±s.d. percentage change in TUNEL-positive cells in the retina. The significance of any differences (*P<0.05) was determined using a two-tailed Mann-Whitney U-test. (H) Data are mean (±s.d.) of total area for pixel intensities, intensity mean and the density sum for CD45 immunofluorescence. The significance of any differences between control and treated groups was determined using a two-tailed t-test (*P<0.05, **P<0.01). (I) qRT-PCR was used to measure the mean (±s.d.; n≥5) percentage change of mRNA levels for Notch1, Hes5, C3, C3aR and Spred1. The significance of any differences between control and treated groups was determined using a two-tailed Mann-Whitney U-test (*P<0.05; ns, not significant). (J,K) Sox9 (red) is included as a partial field overlay to indicate nuclear colocalization with Pax6 (green) in Müller glia. (J-M) Arrows indicate Müller glia. (N) Data are the mean (±s.d.) number of neurofilament-positive MGPCs in the retina. The significance of any differences (**P<0.01) was determined using a two-tailed t-test. Scale bars: 50 µm (bar in G applies to A-G; bar in K applies to J,K; bar in M applies to L,M).
We have found that reactive microglia stimulate the formation of MGPCs in damaged and undamaged retinas (Fischer et al., 2014). As Dex is known to suppress microglial reactivity in the brain (reviewed by Herrera et al., 2005), we probed for changes in the microglia in damaged retinas treated with Dex. We found that Dex inhibited microglia reactivity; levels of CD45 were significantly reduced in Dex/NMDA-treated retinas compared with levels seen in retinas treated with vehicle/NMDA (Fig. 4F-H). Although microglial reactivity was reduced in retinas treated with Dex/NMDA, retinal levels of pro-inflammatory cytokines, including IL1β, IL6, TNFα and the TNFα-processing enzyme Adam17, were unchanged by Dex (data not shown).
We next probed for changes in the expression of different genes known to influence the reprogramming of Müller glia to MGPCs. Activated Notch signaling and upregulation of Ascl1a are known to stimulate the formation of MGPCs in the retina (Fausett et al., 2008; Ghai et al., 2010; Hayes et al., 2007; Pollak et al., 2013). Consistent with previous reports (Hayes et al., 2007), we found increases in retinal levels of chick Delta1, Dll4, Jag (not shown), Notch1, Hes5 and Ascl1a (Fig. 4I) 3 days after NMDA treatment. However, Dex treatment after NMDA treatment did not influence the elevated levels of chick Delta1, Dll4, Jag (not shown), Notch1, Hes5 or Ascl1a (Fig. 4I). Components of the complement system are known to stimulate retinal regeneration in the embryonic chick (Haynes et al., 2013). In NMDA-damaged retinas treated with Dex, we found that retinal levels of C3 were significantly reduced, whereas levels of C3aR were unaffected (Fig. 4I).
Spred1 (sprouty-related, EVH1 domain-containing 1), an intracellular antagonist of MAPK signaling, is expressed by Müller glia in normal and damaged retinas (Roesch et al., 2008, 2012). We found that Spred1 is transiently downregulated at 1 day after NMDA treatment (not shown), whereas levels of Spred1 return to control levels 3 days after NMDA treatment (Fig. 4I). Interestingly, there were significant increases in levels of Spred1 in Dex/NMDA-treated retinas (Fig. 4I). As levels of a the MAPK inhibitor Spred1 were elevated by Dex treatment, and FGF2/MAPK signaling is known to stimulate Pax6 expression in developing ocular tissues (Ashery-Padan and Gruss, 2001; Shaham et al., 2012), we probed for Pax6 in MGPCs in NMDA/Dex-treated retinas. The upregulation of Pax6 is an important step in the reprogramming of glia, including Müller glia, into progenitor-like cells (Heins et al., 2002; Thummel et al., 2010). Levels of Pax6 appeared to be marginally reduced in MGPCs in NMDA/Dex-treated retinas compared with levels seen in MGPCs in NMDA-treated retinas (Fig. 4J,K).
The transient expression of neurofilament is symptomatic of Müller glial reprogramming and the formation of MGPCs in response to damage or treatment with FGF2 (Fischer et al., 2004a; Fischer and Reh, 2001). In addition, there is a near-perfect overlap of neurofilament with Sox2 and phospho-Histone H3, a marker of proliferating cells in M phase (D.G., C.Z. and A.J.F., unpublished). Thus, we examined whether Dex influenced the expression of neurofilament in MGPCs. We found that Dex treatment after NMDA treatment reduced the number of Sox2/neurofilament-positive MGPCs by more than 50% (Fig. 4L-N).
The ligand-binding domain of GCR is similar (58% identities; 76% positives) to that of the mineralocorticoid receptor (MCR or Nr3c2). Therefore, Dex, CpdA and RU486 act at the MCR, albeit with low affinity (Robertson et al., 2010). In addition, the MCR is known to be expressed by Müller glia in the rat retina (Zhao et al., 2010). Thus, we tested whether Mcr expression was affect by NMDA treatment, and whether the MCR-preferring ligand aldosterone influences the formation of MGPCs in NMDA-damaged retinas. We found that levels of mcr where unchanged 4 h after NMDA treatment, and were decreased at 24, 48 and 72 h after NMDA (supplementary material Fig. S1). At doses that provided an initial maximal molar equivalent to that of Dex, we found that aldosterone had no effect upon numbers of proliferating MGPCs or NIRG cells (supplementary material Fig. S1). In addition, aldosterone had no effect upon FGF2-induced accumulations pERK1/2, Fos, Egr1 or pS6 in Müller glia (supplementary material Fig. S1). Collectively, these findings suggest that MCR signaling has little or no effect upon the formation of MGPCs and FGF2/MAPK signaling in the Müller glia.
Activation of GCR signaling inhibits FGF2-induced MAPK and mTor signaling pathways in the Müller glia
Previously, we have shown that FGF2 selectively activates MAPK signaling in Müller glia; intraocular injections of FGF2 stimulate Müller glia to upregulate pERK1/2, Egr1, Fos and pCREB and become primed to form MGPCs (Fischer et al., 2009a). GCR and MAPK signaling can interact antagonistically via transcriptional and cell signaling mechanisms (reviewed by Ayroldi et al., 2012). Accordingly, we tested whether activation of GCR signaling influences the effects of FGF2 on the Müller glia. We found that Dex prevented FGF2-mediated upregulation of pERK1/2, Egr1 and cFos selectively in the Müller glia (Fig. 5A-C,E,F). Although GCR signaling is known to inhibit p38 MAPK (Brewer et al., 2003), we did not find decreased levels of p38 in FGF2/Dex-treated retinas (not shown). These results indicate that FGF2/MAPK signaling in the Müller glia is inhibited by GCR signaling.
Fig. 5.

In uninjured retina, activation of GCR signaling inhibits FGF2-induced MAPK and mTor signaling pathways in the Müller glia. Eyes were injected with FGF2 alone (control) or with 200 ng FGF2+200 ng dexamethasone (treated) at P6 and P7. The retinas were harvested at P8 and labeled for pERK and Sox2 (A), Egr1 and Sox9 (B), Fos and Sox2 (C), and pS6 and Sox2 (D). Arrows indicate the nuclei of Müller glia. (E,F) Data are mean (±s.d.; n=6) area sum and density sum immunofluorescence. The significance of any difference between control and treated groups was determined using a two-tailed t-test (*P<0.05; **P<0.01; ***P<0.001; ns, not significant). Scale bar: 50 µm.
Akt/mTor signaling is known to stimulate cell growth and proliferation in many different cell types, including neural stem cells (Magri and Galli, 2013; van Wijngaarden and Franklin, 2013). There is also evidence that the Akt/mTor-pathway can be activated by FGF2/MAPK signaling via crosstalk (reviewed by Ciuffreda et al., 2014; De Luca et al., 2012). A read-out of Akt-mTor signaling is pS6, a component of the 40S ribosomal subunit that is phosphorylated at multiple sites by S6 kinase (S6K), downstream of mTor activation (Dufner and Thomas, 1999). In untreated retinas, levels of pS6 in Müller glia are below levels of detection (not shown), whereas levels of pS6 are dramatically upregulated in Müller glia by FGF2 (Fig. 5D). We found that FGF2-induced accumulation of pS6 in the Müller glia is inhibited by Dex (Fig. 5D-F).
We next tested whether activation of GCR signaling inhibits FGF2-induced proliferation of MGPCs. A low dose of NMDA followed by injections of FGF2 stimulates the formation of proliferating MGPCs (Ghai et al., 2010). As activation of GCR inhibited FGF2/MAPK signaling in undamaged retinas, we hypothesized that Dex would inhibit the formation of MGPCs formed by FGF2 treatment in damaged retinas. Indeed, the number of proliferating MGPCs was significantly decreased, by nearly 90%, by Dex in FGF2/NMDA-treated retinas, whereas numbers of proliferating NIRG cells were unaffected (Fig. 6A-D). These findings indicate that activation of GCR signaling overrides FGF2-mediated formation of MGPCs in damaged retinas. We next tested whether GCR signaling overrides FGF2-induced formation of MGPCs in the absence of retinal damage. We have found that four consecutive daily injections of FGF2 stimulates the formation of proliferating MGPCs, this proliferation requires the presence of reactive microglia and occurs in the absence of damage (Fischer et al., 2014). We found that co-application of Dex with FGF2 reduced the number of proliferating MGPCs by nearly 75%, whereas numbers of proliferating NIRG cells were unaffected (Fig. 6E-H). To examine how Dex might influence FGF2-mediated reprogramming of Müller glia we probed for changes in gene expression by using real-time RT-PCR. FGF2 is known to upregulate levels of Notch1, Hes5 and Ascl1a in Müller glia in the absence of retinal damaged (Ghai et al., 2010). We found that co-application of Dex with FGF2 significantly reduced levels of Notch1, Hes5, Ascl1a and components of the complement system, C3 and C3aR (Fig. 6I). Unlike Dex treatment after NMDA treatment, co-application of Dex with FGF2 decreased levels of the MAPK antagonist Spred1 (Fig. 6I).
Fig. 6.

Activation of GCR signaling inhibits FGF2-induced formation of proliferating MGPCs. (A,B) Eyes were injected with 200 nanomoles of NMDA at P6, 200 ng FGF2±200 ng Dex at P7 and P8, and BrdU at P8. Retinas were harvested at P9. (E,F) Eyes were injected FGF2±Dex at P6, P7, P8 and P9, with BrdU included with the injection at P9. Retinas were harvested at P10. Retinas were labeled for BrdU (green), Sox9 (red) and Nkx2.2 (magenta). (A,B,E,F) Arrows indicate BrdU+/Sox2+/Nkx2.2− MGPCs; hollow arrowheads indicate BrdU+/Sox2+/Nkx2.2+ NIRG cells. Scale bar: 50 µm. (C,D,G,H) Data are mean [±s.d.; n=6 (C,D); n=8 (G,H)] BrdU-positive Müller glia or NIRG cells in peripheral regions of the retina. The significance of any difference (*P<0.05; **P<0.01; ***P<0.001; ns, not significant) was determined using a two-tailed t-test. To assess how Dex influenced FGF2-induced reprogramming of Müller glia, we probed for changes in mRNA levels of Notch1, Hes5, Ascl1a, C3, C3aR and Spred1. (I) Data are mean (±s.d.; n≥4) percentage change of mRNA in retinas treated with FGF2 versus FGF2+Dex. (J) Data are mean (±s.d.; n=4) percentage change of mRNA from retinas treated with saline versus FGF2. The significance of differences was determined using a two-tailed Mann-Whitney U-test (*P<0.05, ns, not significant).
Given that GCR signaling inhibits FGF2/MAPK signaling in Müller glia (see Fig. 5) and FGF2 primes Müller glia to become more progenitor like (Fischer et al., 2009a,b), we tested whether there is a reciprocal influence of FGF2 upon GCR signaling in the retina. We found that three consecutive daily injections of FGF2 had no influence on retinal levels of GCR, whereas levels of the cortisol-activation enzyme HSD1 are downregulated and of the cortisol-inactivating enzyme HSD2 are upregulated (Fig. 6J). This finding suggests that FGF2/MAPK signaling may, at least in part, prime Müller glia to become proliferating MGPCs by decreasing retinal levels of cortisol and decreasing GCR signaling.
Inhibition of GCR signaling induces the proliferation of MGPCs
As activation of GCR inhibited the formation of MGPCs, we tested whether inhibition of GCR stimulated the formation of MGPCs. We applied four consecutive daily injections of RU486, an antagonist at both progesterone and GCR receptors, and found that proliferating MGPCs were not stimulated (not shown). This finding suggests that blockade of GCR is not sufficient to induce the formation of MGPCs in undamaged retinas.
Previously, we have shown that a relatively low dose of NMDA primes Müller glia to become MGPCs, without leading to proliferation (Fischer and Reh, 2001; Ghai et al., 2010). Therefore, we sought to test whether inhibition of GCR signaling induces the formation of MGPCs following a moderate insult. The number of the proliferating MGPCs was increased nearly sixfold when RU486 was applied following a moderate NMDA insult (Fig. 7A-C). By comparison, treatment with RU486 following NMDA had no effect on the number of proliferating NIRG cells (Fig. 7D). Injections with RU486 following NMDA treatment had no significant effect upon cell death (Fig. 7E-G) and no effect upon microglial reactivity (not shown), suggesting the RU486-mediated increases in MGPC proliferation are not secondary to increases in retinal damage or microglial reactivity.
Fig. 7.

Inhibition of GCR signaling induces the proliferation of MGPCs in peripheral regions of damaged retinas and influences the differentiation of Müller glia-derived cells. (A-G) Eyes were injected with a low dose (83 nanomoles) of NMDA at P6, vehicle (30% DMSO in saline) or 1 µg RU486 (n=8) at P7/P8, and BrdU at P8. (H,I) Eyes were injected with 83 nanomoles of NMDA at P6, RU486 or RU486+200 ng Dex (n=5) at P7/P8, and BrdU at P8. Retinas were harvested at P9. (J-S) Eyes were injected with 83 nanomoles of NMDA at P6, and vehicle or 1 µg RU486 at P6-P9. BrdU was included with each injection of vehicle/RU486. Retinas were harvested at P10. Retinas were labeled for BrdU (green), Sox9 and Nkx2.2 (red and magenta, respectively; A,B), TUNEL (E,F), HuD/C (J-M) or GS (N-Q). (A,B) Arrows indicate the nuclei of BrdU+/Sox2+/Nkx2.2− MGPCs; hollow arrowheads indicate the nuclei of BrdU+/Sox2+/Nkx2.2+ NIRG cells. (C,D,H,I) Data show the mean (±s.d.) number of BrdU-positive Müller glia (C,H) or NIRG cells (D,I) in peripheral regions of the retina. (G) Data are mean (±s.d.) percentage change in TUNEL-positive cells in retinas treated with NMDA±RU486. (J-Q) Arrows indicate cells double-labeled for BrdU and HuD/C; hollow arrowheads indicate cells double-labeled for BrdU and Sox9/GS; hollow arrows indicate cells labeled for BrdU alone; pairs of small arrows indicate cells labelled for BrdU and Sox9, but not for GS. The areas boxed in yellow in L and P are enlarged twofold in M and Q, respectively. Scale bars: 50 µm (bar in B applies to A,B; bar in L applies to J-L; bar in P applies to N-P). (R,S) Data are mean (±s.d.) percentage change BrdU+HuD/C+ and BrdU+GS+ cell numbers for treated minus control treatments in retinas treated with NMDA±RU486. A total of 5680 cells were counted for BrdU/HuD/C-labeled cells and 3529 cells for BrdU/GS-labeled cells. (C,D,H,I) The significances of differences for treated versus control datasets were determined using a two-tailed t-test. (**P<0.01; ***P<0.001; ns, not significant). (G,R,S) The significance of differences for the percent change was determined using a two-tailed Mann-Whitney U-test (*P<0.05; ns, not significant).
We next investigated the specificity of RU486 given that this molecule acts at both GCR and the progesterone receptor. Accordingly, we applied Dex, which has no demonstrated specificity for the progesterone receptor, with RU486 following a relatively low dose of NMDA. We found that Dex prevented the proliferation of MGPCs that otherwise resulted from RU486 treatment in NMDA-damaged retinas (Fig. 7H). The proliferation of NIRG cells was not influenced by treatments with Dex and RU486 in damaged retinas (Fig. 7I). These results suggest that RU486-mediated formation of MGPCs results from the inhibition of GCR signaling.
We tested whether activation of MAPK signaling with FGF2 and inhibition of GCR signaling with RU486 acts synergistically to stimulate the formation of MGPCs. Three consecutively daily injections of FGF2 and RU486 failed to stimulate the formation of MGPCs (not shown). As FGF2 modulates retinal levels of HSD1 and HSD2 (see Fig. 6), which should decrease levels of cortisol, we speculate that there was little GCR signaling for the RU486 to antagonize and enable FGF2/MAPK-induced formation of MGPCs.
We next examined whether inhibition of GCR with RU486 influenced the types of cells derived from MGPCs. Activation of GCR with cortisol during retinal development is known to stimulate the maturation of Müller glia (Gorovits et al., 1994, 1996). Therefore, we hypothesized that RU486-mediated inhibition of GCR and the enhanced formation of proliferating MGPCs would also result in diminished formation of new Müller glia and increased formation of new neurons. Indeed, we found that RU486 increased the percentage of MGPC-derived cells that differentiated into HuD/C-positive neurons by about 50% (Fig. 7J-M,R). By comparison, the RU486 treatment decreased the percentage of MGPC-derived cells that differentiated into GS-positive glia by more than 20% (Fig. 7N-Q,S).
DISCUSSION
We find that GCR signaling has significant impacts upon retinal Müller glia. The activation of GCR in Müller glia inhibits the proliferation of MGPCs by inhibiting signaling through MAPK. It is known that glucocorticoids can negatively regulate MAPK signaling (Bruna et al., 2003). For example, activation of MAPK signaling through ERK1/2 and p38 MAPK can be inhibited by GCR signaling (Brewer et al., 2003), and p38 MAPK signaling enhances the expression of pro-inflammatory cytokines such as TNFα, interleukin 6 (IL6) and IL8 in immune cells (Inoue et al., 1996). Although we failed to detect decreases in p38 MAPK when Dex was applied with FGF2, it remains possible that targets downstream of p38 were influenced by Dex. Consistent with our findings, Dex inhibits the accumulation of pERK in Müller glia in retinas with endotoxin-induced uveitis to suppress glial reactivity (Takeda et al., 2002). Glucocorticoids can enhance the production of anti-inflammatory cytokines including IL10 (Schaaf and Cidlowski, 2002), while inhibiting the production of pro-inflammatory cytokines, including IL1β, IL6, IL8 and TNFα (Bladh et al., 2005; Brewer et al., 2003; Stellato, 2004). However, in NMDA-damaged retinas, we did not find decreases in IL1β, IL6 or TNFα in response to Dex, even though the microglial reactivity was suppressed. In the rodent retina, Müller glia are known to produce pro-inflammatory cytokines in response to NMDA treatment, and elevated production of TNFα can render neurons more susceptible to excitotoxic damage (Lebrun-Julien et al., 2009). The mechanisms by which Dex suppresses the reactivity of microglia in NMDA-damaged retinas remain uncertain, but are likely to be secondary to signaling in Müller glia given that GCR was not detected in microglia.
Patterns of GCR expression in the chick retina coincide with the level of Müller glia maturation: nuclear GCR levels in the central retina are lower where the Müller glia are more mature and nuclear GCR levels increase towards the periphery where the Müller glia are less mature (Anezary et al., 2001; Fischer et al., 2002). The nuclear localization of GCR suggests ligand binding and translocation (reviewed by Schaaf and Cidlowski, 2002). The nuclear localization of GCR in Müller glia in peripheral retinal regions may represent increased expression and/or increased ligand binding and nuclear translocation. In central regions of the chick retina, GCR may be expressed at low levels or may be expressed at high levels but remains diffusely distributed in the peripheral processes of Müller glia, below levels of detection. Alternatively, in the cytoplasm GCR is part of a complex with chaperones that may mask the epitopes that are recognized by antibodies. Following acute damage, GCR immunolabeling was increased in the nuclei of Müller glia across all regions of the retina (not shown); this may represent a combination of increased expression, dissociation from the chaperone complex and ligand-binding/nuclear translocation. Glucocorticoid signaling in the retina is dynamically modulated after acute retinal injury. The cortisone-to-cortisol converting enzyme HSD1 is expressed at diminished levels 1 day after NMDA treatment, and then at elevated levels at 2 days after NMDA treatment. By comparison, the cortisol-to-cortisone converting enzyme HSD2 is elevated within 4 h of NMDA treatment, and is downregulated thereafter. These findings suggest that soon after NMDA treatment, retinal levels of cortisol and GCR signaling are reduced, whereas cortisol levels are elevated 2 days after treatment when de-differentiation of Müller glia leads to re-entry into the cell cycle (Fischer and Reh, 2001). The observation that increased levels of HSD2 coincide with increased levels of GCR, whereas increased HSD1 levels follow the spikes in HSD2 and GCR, suggests local modulation of cortisol levels and GCR signaling in the retina. Collectively, these findings suggest that after acute retinal injury there is a transient drop, followed by a transient spike, in GCR signaling in Müller glia. Furthermore, we find that levels of HSD1 are decreased, while levels of HSD2 are increased, by FGF2 treatment, after which MGPCs are stimulated to form. This finding is consistent with the hypothesis that reduced GCR signaling in Müller glia permits the formation of proliferating MGPCs.
We found that application of Dex/CpdA after NMDA reduced the reactivity of microglia and reduced numbers of dying cells. Reactive microglia (Fischer et al., 2014) and levels of retinal damage (Fischer et al., 2004b) stimulate the formation of MGPCs. Therefore, it is possible that suppressed formation of MGPCs in CpdA/NMDA- or Dex/NMDA-treated retinas resulted, at least in part, secondarily through diminished microglial reactivity and/or diminished levels of neuronal damage. Nevertheless, we propose that GCR signaling primarily inhibits the formation of MGPCs by directly influencing the Müller glia for the following reasons: (1) GCR is expressed by Müller glia, not by retinal microglia or neurons; (2) FGF/MAPK signaling is known to directly stimulate Müller glia (Fischer et al., 2009b) and activation of GCR signaling blocks FGF2-mediated formation of MGPCs in damaged retinas (current study); and (3) in the absence of retinal damage, activation of GCR signaling inhibits the formation of MGPCs in response to FGF2. In addition to the inhibition of MAPK signaling, activation of GCR inhibited FGF2-mediated accumulation of pS6, a readout of mTor signaling. mTor signaling has well-known roles in cell growth and proliferation of neural stem cells and tumors (reviewed by Ito et al., 2009). Collectively, these findings are consistent with the hypothesis that GCR signaling directly inhibits the dedifferentiation of Müller glia and formation of MGPCs.
We propose that Dex-mediated inhibition of MGPCs is primarily manifested through the inhibition of MAPK signaling in the Müller glia. Previous studies have indicated that GCR signaling can negatively impact MAPK signaling in different types of cells (reviewed by Ayroldi et al., 2012). Dex suppresses FGF2-mediated activation of MAPK signaling in the Müller glia and suppresses the formation of MGPCs. In addition, injections of Dex following NMDA treatment increased retinal levels of the intracellular MAPK antagonist Spred1. By comparison, co-application of Dex with FGF2, in the absence of retina damage, resulted in reduced expression of Spred1. Spred1 and Spred2 are known to be expressed by Müller glia, and levels of Spred1 are influenced by retinal degeneration (Roesch et al., 2008, 2012). Interestingly, Spred1 is enriched in neural stem cells in the rodent ventricular zone and acts to suppress proliferation (Phoenix and Temple, 2010). Collectively, our data suggest that activation of GCR may upregulate transcription of Spred1 to dampen MAPK signaling in Müller glia in damaged retinas, but not in FGF2-treated retinas in the absence of damage. GCR-mediated transcriptional repression is due in part to direct interactions between monomeric GCR and transcription factors such as Jun-Fos and NF-κB, which synergistically coordinate the transcriptional activation of many genes involved in inflammation (Bladh et al., 2005). However, we find that FGF2-induced expression of cFos in Müller glia was inhibited by Dex. Collectively, these findings suggest that GCR signaling influences MAPK signaling by modulating protein-protein interactions and by influencing transcriptional activation of cFos. However, we cannot exclude the possibility that MCR signaling influences the formation of MGPCs. Although the MCR-preferring ligand aldosterone failed to influence FGF2/MAPK signaling in Müller glia and did not influence the proliferation of MGPCs in damaged retinas, the GCR-preferring agents that we used are known to have some affinity for MCR (Robertson et al., 2010) and MCR is likely to be expressed by Müller glia (Zhao et al., 2010). Consistent with our findings, a recent report by Anacker and colleagues (Anacker et al., 2013) indicates that low levels of cortisol, acting through MCR, increased proliferation of hippocampal progenitors and decreased neuronal differentiation whereas high levels of cortisol, acting through GCR, decreased proliferation and decreased neural differentiation.
Notch signaling is largely unaffected by GCR signaling and changes in Notch signaling probably do not underlie the diminished formation of MGPC in Dex/NMDA-treated retinas. We find that Dex-mediated inhibition of MGPCs in damaged retinas is not correlated with decreased Notch signaling. Mature Müller glia express Notch and related genes, and maintain relatively low Notch signaling in normal healthy retinas (Ghai et al., 2010; Hayes et al., 2007; Nelson et al., 2011; Roesch et al., 2008). In the chick retina, Notch signaling stimulates the formation of MGPCs (Ghai et al., 2010; Hayes et al., 2007) and is required for FGF2/MAPK-mediated formation of MGPCs (Ghai et al., 2010). However, we found fewer proliferating MGPCs with NMDA/Dex treatment. As we cannot determine the number of transcripts in individual MGPCs, it is possible that levels of Notch-related genes may have been elevated in many cells that failed to progress through the cell cycle because of GCR-mediated inhibition. Although Dex may not have affected Notch signaling in damaged retinas, Notch signaling was suppressed by Dex in FGF2-treated retinas. These findings suggest contextual differences in the effects of Dex/GCR signaling on Notch and on the formation of MGPCs. Activation of GCR appears to directly inhibit FGF2/MAPK signaling and the initiation of Müller glial de-differentiation in undamaged retinas, whereas Müller glia in NMDA-damaged retinas appear to be influenced by many signaling pathways in addition to GCR and MAPK. These pathways probably include Wnt/β-catenin (Osakada et al., 2007; Ramachandran et al., 2011), Jak/Stat3 (Nelson et al., 2012), TGFβ/Smad (Close et al., 2005; Lenkowski et al., 2013) and TNFα (Nelson et al., 2013). Any one of these pathways in the damaged retina could influence Notch signaling independently of MAPK and GCR signaling. In damaged retinas, activation of GCR signaling with Dex inhibited the formation of MGPCs, similar to the inhibition of Notch (Ghai et al., 2010; Hayes et al., 2007), without influencing levels of Notch. Thus, Notch and GCR signaling pathways appear to influence MGPCs via independent, parallel pathways in damaged retinas, whereas Notch signaling in FGF2-induced MGPCs, in the absence of retinal damage, is inhibited by activation of GCR.
Conclusions
The expression pattern of GCR in the retina is highly conserved across warm-blooded vertebrates, with expression residing almost exclusively within the Müller glia. The conserved nature of GCR suggests that GCR signaling in chick Müller glia acts similarly in mammalian glia. In the chick retina, activation of GCR inhibits the proliferation of MGPCs in damaged and undamaged retinas; this occurs by overriding FGF2/MAPK signaling in the Müller glia. In a complementary manner, inhibition of GCR enhances the formation of neuro-competent MGPCs in damaged retinas. GCR signaling has a significant impact upon the de-differentiation of Müller glia and transition into proliferating MGPCs. We propose that the effects of GCR signaling on suppressed microglial reactivity and enhanced neuronal survival are mediated indirectly through the Müller glia.
MATERIALS AND METHODS
Animals
The use of animals in these experiments was in accordance with the guidelines established by the National Institutes of Health and the Ohio State University. Newly hatched wild-type leghorn chickens (Gallus gallus domesticus) were obtained from the Department of Animal Sciences at the Ohio State University or Meyer Hatchery (Polk, OH, USA). Postnatal chicks were kept on a cycle of 12 h light, 12 h dark (lights on at 8:00 AM). Chicks were housed in a stainless steel brooder at about 25°C and received water and Purina chick starter ad libitum.
Eyes were obtained post-mortem from C57BL/6 mice (Mus musculata; Dr Karl Obrietan, Department of Neuroscience, The Ohio State University, Columbus, USA), guinea pigs (Cavia porcellus; Dr Jackie Wood, Department of Physiology and Cell Biology, Ohio State University), dogs (Canis familiaris; Simon Petersen-Jones, Veterinary Sciences, Michigan State University, East Lansing, USA) and human tissues (Drs Cynthia Roberts and Colleen Cebulla, Department of Ophthalmology, The Ohio State University).
Intraocular injections
Chickens were anesthetized and intraocular injections were performed as described previously (Fischer et al., 2008, 2009a). Injected compounds included NMDA (6.4 or 154 μg/dose; Sigma-Aldrich), dexamethasone (100-200 ng/dose; Sigma-Aldrich), CpdA (500 ng/dose; Millipore), aldosterone (200 ng/dose; Sigma-Aldrich), RU486 (1 μg/dose; Sigma-Aldrich), FGF2 (200 ng/dose; Sigma-Aldrich) and BrdU (1 μg/dose; Sigma-Aldrich). Hydrophobic compounds were diluted and injected in 30% DMSO in saline. Injection paradigms are included in the figures and legends.
PCR
Tissue dissections, RNA isolation, reverse transcriptase reactions and PCR reactions were performed as described previously (Fischer et al., 2004a, 2010; Ghai et al., 2010). PCR primers were designed by using the Primer-BLAST primer design tool at NCBI (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primer sequences and predicted product sizes are listed in supplementary material Table S1.
For qPCR, reactions were performed using SYBR Green Master Mix and StepOnePlus Real-Time system (Applied BioSystems). Samples were run in triplicate on at least four different samples. Ct values obtained from real-time PCR were normalized to GAPDH for each sample and the fold change between control and treated samples was determined using the 2–ΔΔCt method [=fold change 2(−ΔΔCt)] and represented as a percentage change from the control, which was assigned a value of 100. The significance of any differences in percentage change was determined using a non-parametric Mann-Whitney U-test.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)
To identify dying cells that contained fragmented DNA, the TUNEL method was used. We used an In Situ Cell Death Kit (TMR Red; Roche Applied Science), as per the manufacturer's instructions.
Fixation, sectioning and immunocytochemistry
Tissues were fixed, sectioned and immunolabeled as described previously (Fischer et al., 2008, 2009b). Working dilutions and sources of primary and secondary antibodies used in this study are listed in supplementary material Table S2. In the chick retina, the specificity of the GCR immunolabeling was based on comparisons of expression patterns seen in a previous report (Gorovits et al., 1994) and the identical patterns of labeling observed with two different polyclonal antibodies raised to different GCR epitopes.
Photography, cell counts and statistics
Digital photomicroscopy was performed as described in previous studies (Fischer et al., 2008, 2010; Ghai et al., 2009, 2010). Similar to previous studies (Fischer et al., 2009a,b, 2010; Ghai et al., 2009), immunofluorescence was quantified using ImagePro 6.2 (Media Cybernetics). The mean area, intensity and density sum was calculated for the pixels within threshold regions from at least four different retinas for each experimental condition. GraphPad Prism 6 was used for statistical analyses. The significance of any differences was determined between two treatment groups using a two-tailed, unpaired t-test.
Supplementary Material
Acknowledgements
The antibodies to transitin, Nkx2.2, Pax6, neurofilament and BrdU developed by Drs G. J. Cole, T. M. Jessell, A. Kawakami, J. Wood and S. J. Kaufman, respectively, were obtained from the Developmental Studies Hybridoma Bank, which was developed under the auspices of the NICHD and is maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242, USA.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
D.G. designed and executed experiments, gathered data, made figures and contributed to writing the manuscript. C.Z. designed and executed experiments, gathered data and made figures. A.J.F. designed and executed experiments, gathered data, made figures and contributed to writing the manuscript.
Funding
This work was supported by a grant from the National Institutes of Health, National Eye Institute [EY022030-02]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.109835/-/DC1
References
- Alonso G. (2000). Prolonged corticosterone treatment of adult rats inhibits the proliferation of oligodendrocyte progenitors present throughout white and gray matter regions of the brain. Glia 31, 219-231 [DOI] [PubMed] [Google Scholar]
- Anacker C., Cattaneo A., Luoni A., Musaelyan K., Zunszain P. A., Milanesi E., Rybka J., Berry A., Cirulli F., Thuret S., et al. (2013). Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology 38, 872-883 10.1038/npp.2012.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anezary L., Medina J. I., Sánchez-Nogueiro J., López-Gallardo M., Prada C. (2001). Shape diversity among chick retina Müller cells and their postnatal differentiation. J. Comp. Neurol. 438, 32-49 10.1002/cne.1300 [DOI] [PubMed] [Google Scholar]
- Ashery-Padan R., Gruss P. (2001). Pax6 lights-up the way for eye development. Curr. Opin. Cell Biol. 13, 706-714 10.1016/S0955-0674(00)00274-X [DOI] [PubMed] [Google Scholar]
- Ayroldi E., Cannarile L., Migliorati G., Nocentini G., Delfino D. V., Riccardi C. (2012). Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 26, 4805-4820 10.1096/fj.12-216382 [DOI] [PubMed] [Google Scholar]
- Bladh L.-G., Lidén J., Pazirandeh A., Rafter I., Dahlman-Wright K., Nilsson S., Okret S. (2005). Identification of target genes involved in the antiproliferative effect of glucocorticoids reveals a role for nuclear factor-(kappa)B repression. Mol. Endocrinol. 19, 632-643 10.1210/me.2004-0294 [DOI] [PubMed] [Google Scholar]
- Brewer J. A., Khor B., Vogt S. K., Muglia L. M., Fujiwara H., Haegele K. E., Sleckman B. P., Muglia L. J. (2003). T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat. Med. 9, 1318-1322 10.1038/nm895 [DOI] [PubMed] [Google Scholar]
- Bruna A., Nicolàs M., Muñoz A., Kyriakis J. M., Caelles C. (2003). Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 22, 6035-6044 10.1093/emboj/cdg590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron H. A., Gould E. (1994). Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 61, 203-209 10.1016/0306-4522(94)90224-0 [DOI] [PubMed] [Google Scholar]
- Ciuffreda L., Incani U. C., Steelman L. S., Abrams S. L., Falcone I., Curatolo A. D., Chappell W. H., Franklin R. A., Vari S., Cognetti F., et al. (2014). Signaling intermediates (MAPK and PI3K) as therapeutic targets in NSCLC. Curr. Pharm. Des. 20, 3944-3957 10.2174/13816128113196660763 [DOI] [PubMed] [Google Scholar]
- Close J. L., Gumuscu B., Reh T. A. (2005). Retinal neurons regulate proliferation of postnatal progenitors and Muller glia in the rat retina via TGF beta signaling. Development 132, 3015-3026 10.1242/dev.01882 [DOI] [PubMed] [Google Scholar]
- De Bosscher K., Vanden Berghe W., Beck I. M. E., Van Molle W., Hennuyer N., Hapgood J., Libert C., Staels B., Louw A., Haegeman G. (2005). A fully dissociated compound of plant origin for inflammatory gene repression. Proc. Natl. Acad. Sci. USA 102, 15827-15832 10.1073/pnas.0505554102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K., Beck I. M., Haegeman G. (2010). Classic glucocorticoids versus non-steroidal glucocorticoid receptor modulators: survival of the fittest regulator of the immune system? Brain Behav. Immun. 24, 1035-1042 10.1016/j.bbi.2010.06.010 [DOI] [PubMed] [Google Scholar]
- De Kloet E. R., Vreugdenhil E., Oitzl M. S., Joels M. (1998). Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 19, 269-301. [DOI] [PubMed] [Google Scholar]
- De Luca A., Maiello M. R., D'Alessio A., Pergameno M., Normanno N. (2012). The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 16 Suppl. 2, S17-S27 10.1517/14728222.2011.639361 [DOI] [PubMed] [Google Scholar]
- Dufner A., Thomas G. (1999). Ribosomal S6 kinase signaling and the control of translation. Exp. Cell Res. 253, 100-109 10.1006/excr.1999.4683 [DOI] [PubMed] [Google Scholar]
- Fausett B. V., Gumerson J. D., Goldman D. (2008). The proneural basic helix-loop-helix gene ascl1a is required for retina regeneration. J. Neurosci. 28, 1109-1117 10.1523/JNEUROSCI.4853-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J. (2005). Neural regeneration in the chick retina. Prog. Retin. Eye Res. 24, 161-182 10.1016/j.preteyeres.2004.07.003 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Bongini R. (2010). Turning Müller glia into neural progenitors in the retina. Mol. Neurobiol. 42, 199-209 10.1007/s12035-010-8152-2 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Reh T. A. (2001). Muller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat. Neurosci. 4, 247-252 10.1038/85090 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., McGuire C. R., Dierks B. D., Reh T. A. (2002). Insulin and fibroblast growth factor 2 activate a neurogenic program in Muller glia of the chicken retina. J. Neurosci. 22, 9387-9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Omar G., Eubanks J., McGuire C. R., Dierks B. D., Reh T. A. (2004a). Different aspects of gliosis in retinal Muller glia can be induced by CNTF, insulin and FGF2 in the absence of damage. Mol. Vis. 10, 973-986. [PubMed] [Google Scholar]
- Fischer A. J., Schmidt M., Omar G., Reh T. A. (2004b). BMP4 and CNTF are neuroprotective and suppress damage-induced proliferation of Müller glia in the retina. Mol. Cell. Neurosci. 27, 531-542 10.1016/j.mcn.2004.08.007 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Ritchey E. R., Scott M. A., Wynne A. (2008). Bullwhip neurons in the retina regulate the size and shape of the eye. Dev. Biol. 317, 196-212 10.1016/j.ydbio.2008.02.023 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Scott M. A., Ritchey E. R., Sherwood P. (2009a). Mitogen-activated protein kinase-signaling regulates the ability of Müller glia to proliferate and protect retinal neurons against excitotoxicity. Glia 57, 1538-1552 10.1002/glia.20868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Scott M. A., Tuten W. (2009b). Mitogen-activated protein kinase-signaling stimulates Müller glia to proliferate in acutely damaged chicken retina. Glia 57, 166-181 10.1002/glia.20743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Scott M. A., Zelinka C., Sherwood P. (2010). A novel type of glial cell in the retina is stimulated by insulin-like growth factor 1 and may exacerbate damage to neurons and Müller glia. Glia 58, 633-649 10.1002/glia.20950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Zelinka C., Gallina D., Scott M. A., Todd L. (2014). Reactive microglia and macrophage facilitate the formation of Muller glia-derived retinal progenitors. Glia (in press). 10.1002/Glia.22703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina D., Todd L., Fischer A. J. (2014). A comparative analysis of Müller glia-mediated regeneration in the vertebrate retina. Exp. Eye Res. 123, 121-130 10.1016/j.exer.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A., Steiner B., Kronenberg G., Bick-Sander A., Kempermann G. (2004). Age-dependent expression of glucocorticoid- and mineralocorticoid receptors on neural precursor cell populations in the adult murine hippocampus. Aging Cell 3, 363-371 10.1111/j.1474-9728.2004.00130.x [DOI] [PubMed] [Google Scholar]
- Ghai K., Zelinka C., Fischer A. J. (2009). Serotonin released from amacrine neurons is scavenged and degraded in bipolar neurons in the retina. J. Neurochem. 111, 1-14 10.1111/j.1471-4159.2009.06270.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K., Zelinka C., Fischer A. J. (2010). Notch signaling influences neuroprotective and proliferative properties of mature Muller glia. J. Neurosci. 30, 3101-3112 10.1523/JNEUROSCI.4919-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovits R., Ben-Dror I., Fox L. E., Westphal H. M., Vardimon L. (1994). Developmental changes in the expression and compartmentalization of the glucocorticoid receptor in embryonic retina. Proc. Natl. Acad. Sci. USA 91, 4786-4790 10.1073/pnas.91.11.4786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovits R., Yakir A., Fox L. E., Vardimon L. (1996). Hormonal and non-hormonal regulation of glutamine synthetase in the developing neural retina. Brain Res. Mol. Brain Res. 43, 321-329 10.1016/S0169-328X(96)00213-6 [DOI] [PubMed] [Google Scholar]
- Gould E., Tanapat P., McEwen B. S., Flugge G., Fuchs E. (1998). Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc. Natl. Acad. Sci. USA 95, 3168-3171 10.1073/pnas.95.6.3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman R., Fox L. E., Gorovits R., Ben-Dror I., Reisfeld S., Vardimon L. (1994). Molecular basis for differential expression of glutamine synthetase in retina glia and neurons. Brain Res. Mol. Brain Res. 21, 312-320 10.1016/0169-328X(94)90262-3 [DOI] [PubMed] [Google Scholar]
- Hayes S., Nelson B. R., Buckingham B., Reh T. A. (2007). Notch signaling regulates regeneration in the avian retina. Dev. Biol. 312, 300-311 10.1016/j.ydbio.2007.09.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes T., Luz-Madrigal A., Reis E. S., Echeverri Ruiz N. P., Grajales-Esquivel E., Tzekou A., Tsonis P. A., Lambris J. D., Del Rio-Tsonis K. (2013). Complement anaphylatoxin C3a is a potent inducer of embryonic chick retina regeneration. Nat. Commun. 4, 2312 10.1038/ncomms3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heins N., Malatesta P., Cecconi F., Nakafuku M., Tucker K. L., Hack M. A., Chapouton P., Barde Y.-A., Götz M. (2002). Glial cells generate neurons: the role of the transcription factor Pax6. Nat. Neurosci. 5, 308-315 10.1038/nn828 [DOI] [PubMed] [Google Scholar]
- Herrera A. J., Tomás-Camardiel M., Venero J. L., Cano J., Machado A. (2005). Inflammatory process as a determinant factor for the degeneration of substantia nigra dopaminergic neurons. J. Neural Transm. 112, 111-119 10.1007/s00702-004-0121-3 [DOI] [PubMed] [Google Scholar]
- Inoue M., Nakayama C., Noguchi H. (1996). Activating mechanism of CNTF and related cytokines. Mol. Neurobiol. 12, 195-209 10.1007/BF02755588 [DOI] [PubMed] [Google Scholar]
- Ito K., Bernardi R., Pandolfi P. P. (2009). A novel signaling network as a critical rheostat for the biology and maintenance of the normal stem cell and the cancer-initiating cell. Curr. Opin. Genet. Dev. 19, 51-59 10.1016/j.gde.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karishma K. K., Herbert J. (2002). Dehydroepiandrosterone (DHEA) stimulates neurogenesis in the hippocampus of the rat, promotes survival of newly formed neurons and prevents corticosterone-induced suppression. Eur. J. Neurosci. 16, 445-453 10.1046/j.1460-9568.2002.02099.x [DOI] [PubMed] [Google Scholar]
- Karl M. O., Reh T. A. (2010). Regenerative medicine for retinal diseases: activating endogenous repair mechanisms. Trends Mol. Med. 16, 193-202 10.1016/j.molmed.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruchkova Y., Ben-Dror I., Herschkovitz A., David M., Yayon A., Vardimon L. (2001). Basic fibroblast growth factor: a potential inhibitor of glutamine synthetase expression in injured neural tissue. J. Neurochem. 77, 1641-1649 10.1046/j.1471-4159.2001.00390.x [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F., Duplan L., Pernet V., Osswald I., Sapieha P., Bourgeois P., Dickson K., Bowie D., Barker P. A., Di Polo A. (2009). Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J. Neurosci. 29, 5536-5545 10.1523/JNEUROSCI.0831-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski J. R., Qin Z., Sifuentes C. J., Thummel R., Soto C. M., Moens C. B., Raymond P. A. (2013). Retinal regeneration in adult zebrafish requires regulation of TGFbeta signaling. Glia 61, 1687-1697 10.1002/glia.22549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri L., Galli R. (2013). mTOR signaling in neural stem cells: from basic biology to disease. Cell. Mol. Life Sci. 70, 2887-2898 10.1007/s00018-012-1196-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie C. (1981). Ontogenesis of the adrenal glucocorticoids and of the target function of the enzymatic tyrosine transaminase activity in the chick embryo. J. Endocrinol. 90, 193-200 10.1677/joe.0.0900193 [DOI] [PubMed] [Google Scholar]
- Moscona A. A., Linser P. (1983). Developmental and experimental changes in retinal glia cells: cell interactions and control of phenotype expression and stability. Curr. Top. Dev. Biol. 18, 155-188 10.1016/S0070-2153(08)60582-7 [DOI] [PubMed] [Google Scholar]
- Nelson B. R., Ueki Y., Reardon S., Karl M. O., Georgi S., Hartman B. H., Lamba D. A., Reh T. A. (2011). Genome-wide analysis of Müller glial differentiation reveals a requirement for Notch signaling in postmitotic cells to maintain the glial fate. PLoS ONE 6, e22817 10.1371/journal.pone.0022817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. M., Gorsuch R. A., Bailey T. J., Ackerman K. M., Kassen S. C., Hyde D. R. (2012). Stat3 defines three populations of Müller glia and is required for initiating maximal müller glia proliferation in the regenerating zebrafish retina. J. Comp. Neurol. 520, 4294-4311 10.1002/cne.23213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. M., Ackerman K. M., O'Hayer P., Bailey T. J., Gorsuch R. A., Hyde D. R. (2013). Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. J. Neurosci. 33, 6524-6539 10.1523/JNEUROSCI.3838-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochrietor J. D., Moroz T. P., Linser P. J. (2010). The 2M6 antigen is a Muller cell-specific intracellular membrane-associated protein of the sarcolemmal-membrane-associated protein family and is also TopAP. Mol. Vis. 16, 961-969. [PMC free article] [PubMed] [Google Scholar]
- Osakada F., Ooto S., Akagi T., Mandai M., Akaike A., Takahashi M. (2007). Wnt signaling promotes regeneration in the retina of adult mammals. J. Neurosci. 27, 4210-4219 10.1523/JNEUROSCI.4193-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoenix T. N., Temple S. (2010). Spred1, a negative regulator of Ras-MAPK-ERK, is enriched in CNS germinal zones, dampens NSC proliferation, and maintains ventricular zone structure. Genes Dev. 24, 45-56 10.1101/gad.1839510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak J., Wilken M. S., Ueki Y., Cox K. E., Sullivan J. M., Taylor R. J., Levine E. M., Reh T. A. (2013). Ascl1 reprograms mouse Müller glia into neurogenic retinal progenitors. Development 140, 2619-2631 10.1242/dev.091355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Zhao X.-F., Goldman D. (2011). Ascl1a/Dkk/beta-catenin signaling pathway is necessary and glycogen synthase kinase-3beta inhibition is sufficient for zebrafish retina regeneration. Proc. Natl. Acad. Sci. USA 108, 15858-15863 10.1073/pnas.1107220108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson S., Allie-Reid F., Vanden Berghe W., Visser K., Binder A., Africander D., Vismer M., De Bosscher K., Hapgood J., Haegeman G., et al. (2010). Abrogation of glucocorticoid receptor dimerization correlates with dissociated glucocorticoid behavior of compound A. J. Biol. Chem. 285, 8061-8075 10.1074/jbc.M109.087866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch K., Jadhav A. P., Trimarchi J. M., Stadler M. B., Roska B., Sun B. B., Cepko C. L. (2008). The transcriptome of retinal Müller glial cells. J. Comp. Neurol. 509, 225-238 10.1002/cne.21730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch K., Stadler M. B., Cepko C. L. (2012). Gene expression changes within Muller glial cells in retinitis pigmentosa. Mol. Vis. 18, 1197-1214. [PMC free article] [PubMed] [Google Scholar]
- Schaaf M. J. M., Cidlowski J. A. (2002). Molecular mechanisms of glucocorticoid action and resistance. J. Steroid Biochem. Mol. Biol. 83, 37-48 10.1016/S0960-0760(02)00263-7 [DOI] [PubMed] [Google Scholar]
- Schröter A., Lustenberger R. M., Obermair F. J., Thallmair M. (2009). High-dose corticosteroids after spinal cord injury reduce neural progenitor cell proliferation. Neuroscience 161, 753-763 10.1016/j.neuroscience.2009.04.016 [DOI] [PubMed] [Google Scholar]
- Shaham O., Menuchin Y., Farhy C., Ashery-Padan R. (2012). Pax6: a multi-level regulator of ocular development. Prog. Retin. Eye Res. 31, 351-376 10.1016/j.preteyeres.2012.04.002 [DOI] [PubMed] [Google Scholar]
- Stellato C. (2004). Post-transcriptional and nongenomic effects of glucocorticoids. Proc. Am. Thorac. Soc. 1, 255-263 10.1513/pats.200402-015MS [DOI] [PubMed] [Google Scholar]
- Sundberg M., Savola S., Hienola A., Korhonen L., Lindholm D. (2006). Glucocorticoid hormones decrease proliferation of embryonic neural stem cells through ubiquitin-mediated degradation of cyclin D1. J. Neurosci. 26, 5402-5410 10.1523/JNEUROSCI.4906-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M., Takamiya A., Yoshida A., Kiyama H. (2002). Extracellular signal-regulated kinase activation predominantly in Muller cells of retina with endotoxin-induced uveitis. Invest. Ophthalmol. Vis. Sci. 43, 907-911. [PubMed] [Google Scholar]
- Thummel R., Enright J. M., Kassen S. C., Montgomery J. E., Bailey T. J., Hyde D. R. (2010). Pax6a and Pax6b are required at different points in neuronal progenitor cell proliferation during zebrafish photoreceptor regeneration. Exp. Eye Res. 90, 572-582 10.1016/j.exer.2010.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijngaarden P., Franklin R. J. M. (2013). Ageing stem and progenitor cells: implications for rejuvenation of the central nervous system. Development 140, 2562-2575 10.1242/dev.092262 [DOI] [PubMed] [Google Scholar]
- Wong E. Y. H., Herbert J. (2006). Raised circulating corticosterone inhibits neuronal differentiation of progenitor cells in the adult hippocampus. Neuroscience 137, 83-92 10.1016/j.neuroscience.2005.08.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelinka C. P., Scott M. A., Volkov L., Fischer A. J. (2012). The reactivity, distribution and abundance of Non-Astrocytic Inner Retinal Glial (NIRG) cells are regulated by microglia, acute damage, and IGF1. PLoS ONE 7, e44477 10.1371/journal.pone.0044477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Valamanesh F., Celerier I., Savoldelli M., Jonet L., Jeanny J.-C., Jaisser F., Farman N., Behar-Cohen F. (2010). The neuroretina is a novel mineralocorticoid target: aldosterone up-regulates ion and water channels in Muller glial cells. FASEB J. 24, 3405-3415 10.1096/fj.09-154344 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.