

Abstract
Dosage compensation in flies involves doubling the transcription of genes on the single male X chromosome to match the combined expression level of the two female X chromosomes. Crucial for this activation is the acetylation of histone H4 by the histone acetyltransferase (HAT) MOF. In male cells, MOF resides in a complex (dosage compensation complex, DCC) with MSL proteins and noncoding roX RNA. Previous studies suggested that MOF's localization to the X chromosome was largely RNA-mediated. We now found that contact of the MOF chromo-related domain with roX RNA plays only a minor role in correct targeting to the X chromosome in vivo. Instead, a strong, direct interaction between a conserved MSL1 domain and a zinc finger within MOF's HAT domain is crucial. The functional consequences of this interaction were studied in vitro. Simultaneous contact of MOF with MSL1 and MSL3 led to its recruitment to chromatin, a dramatic stimulation of HAT activity and to improved substrate specificity. Activation of MOF's HAT activity upon integration into the DCC may serve to restrict the critical histone modification to the male X chromosome.
Keywords: chromatin, chromosome, histone acetylation, male-specific lethal, transcription
Introduction
Flies, like humans, have heteromorphic sex chromosomes. Female cells contain two X chromosomes, whereas male cells contain only one in addition to the Y chromosome. Males therefore experience a two-fold reduced ‘dose' of X-linked genes. However, at least some of the gene products encoded on the X chromosome need to be present in comparable concentrations in cells of both sexes. In Drosophila melanogaster, equal levels of such critical factors are achieved by the process of ‘dosage compensation', which increases the specific expression of many X-chromosomal genes in male cells by two-fold. Failure in this adjustment, due to mutation of the regulatory machinery involved, leads to male-specific lethality (Lucchesi, 1996; Stuckenholz et al, 1999). Analysis of these loss-of-function mutations allowed the identification of the major components of the dosage compensation machinery, such as the ‘male-specific lethal' (MSL) proteins. These proteins, called MSL1, MSL2, MSL3, MLE and MOF, form a dosage compensation complex (DCC), which associates with the male X chromosome at hundreds of sites (Kelley et al, 1995). The DCC also contains at least one of the two noncoding roX RNAs (Meller et al, 1997, 2000; Franke and Baker, 1999; Lucchesi, 1999; Kelley and Kuroda, 2000).
The transcription of X-linked genes is regulated according to individual cues. The two-fold increase during dosage compensation involves changes in chromatin structure through epigenetic mechanisms, notably site-specific histone H4 acetylation on lysine 16 (H4K16) (Kelley et al, 1995; Gu et al, 2000). The MYST family member MOF is the histone acetyltransferase (HAT) responsible for this epigenetic mark (Bone et al, 1994; Hilfiker et al, 1997; Akhtar and Becker, 2000; Smith et al, 2000). This mark is present throughout the transcribed regions upregulated by the DCC, suggesting that gene activation does not involve enhanced transcription initiation, but rather facilitates the elongation of the RNA polymerase II (Smith et al, 2001). In a cell-free system, site-specific acetylation of chromatin by MOF is sufficient to derepress transcription from chromatin templates dramatically, documenting a direct, causal effect of this single modification on gene activity (Akhtar and Becker, 2000). While it is clear that MOF is a crucial activator involved in dosage compensation, it is not clear whether the other components of the complex are mainly involved in targeting the HAT to the X chromosome, or whether they have additional functions, for example in the fine-tuning of the transcription. Curiously, even though loss-of-function mutation of MOF leads to male-specific lethality, the enzyme is expressed in female flies as well (Hilfiker et al, 1997), suggesting an alternative function of MOF in females independently of the dosage compensation system. However, the mere presence of MOF does not indicate whether the enzyme is active. It is thus important to determine the principles of integration of MOF into the DCC, and the effects of MSL association for MOF function.
Critical for the selective formation of the DCC in males is the male-specific control of MSL1 and MSL2 expression by post-transcriptional mechanisms (Palmer et al, 1994; Bashaw and Baker, 1995; Kelley et al, 1995, 1997; Chang and Kuroda, 1998). MSL1 and MSL2 interact directly with each other and this interaction is required for the DCC binding to X chromosomal target sites (Lyman et al, 1997; Copps et al, 1998). Three of the MSL proteins, MOF, MSL3 and MLE, are RNA-binding proteins (Gu et al, 1998; Kelley et al, 1999; Akhtar et al, 2000; Gu et al, 2000; Meller et al, 2000; Park et al, 2002, 2003). Our recent observation that MOF interacts with roX RNA via its chromo-related domain combined with the finding that MOF dissociates from the X chromosome after treatment of permeabilized nuclei with RNase (Akhtar et al, 2000) led to the suggestion that MOF's integration into the DCC largely depended on RNA. However, genetic studies indicate that a roX RNA deficiency can be partially overcome by overexpressing MSL1 and MSL2. Under these circumstances, all MSL proteins (with the exception of MLE) can be found on the X chromosome (Oh et al, 2003). We therefore directly investigated the determinants that target MOF to the X chromosome and found that, contrary to our expectations, the RNA-binding function of the chromo-related domain was not important, but rather a strong and direct interaction of MOF with the C-terminus of MSL1. We mapped the binding site to an evolutionary conserved domain of MSL1 and distinguished it from an adjacent MSL3 interaction surface. MOF associates with MSL1 via its CCHC zinc-finger domain. Mutation of this zinc finger prevents localization of MOF to the X chromosome in vivo. Surprisingly, association of MOF with an MSL1–MSL3 heteromeric complex, but not with either single protein alone, leads to 30-fold activation of the enzyme and a refinement of substrate specificity. Thus, integration of MOF into the DCC via protein–protein interactions not only contributes to physical targeting of the HAT enzyme to the X chromosome, but also unleashes the HAT activity of MOF, thereby restricting the critical epigenetic mark to the X chromosome.
Results
MOF's chromodomain is not required for its localization to the X chromosome
We previously documented interaction of MOF with roX RNA via the chromo-related domain (Akhtar et al, 2000) and with histones via the CCHC zinc-finger domain, a hallmark of acetylases of the MYST family (Akhtar and Becker, 2001). During these studies, we generated a number of point mutations in these domains (Figure 1A), which abolish the corresponding interactions.
Figure 1.
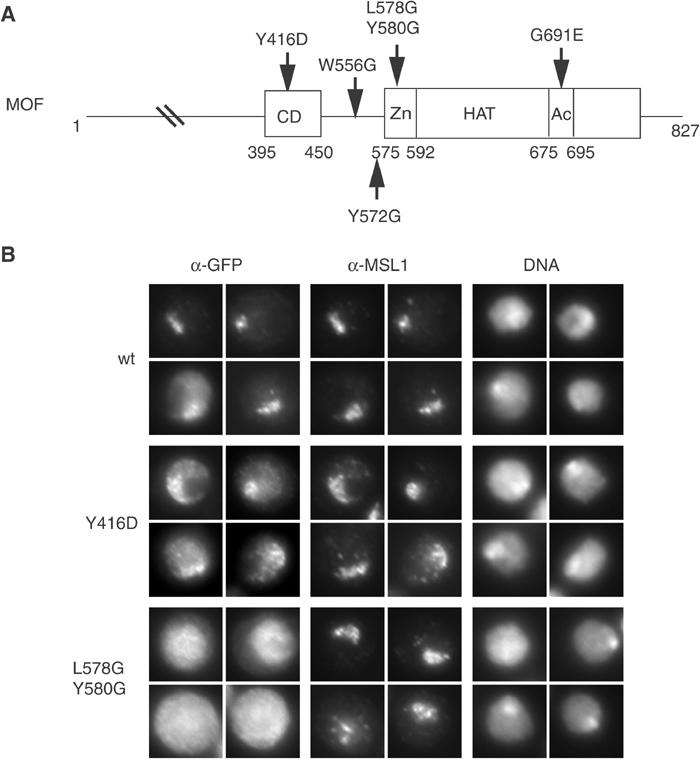
The zinc-finger but not the chromo-related domain of MOF is required for its localization to the X chromosome. (A) Schematic representation of MOF, featuring its domains and the point mutations analysed for MOF localization in living cells and MSL1 binding (see Figure 3). CD: chromo-related domain; Zn: CCHC zinc finger; Ac: acetyl-coA-binding motif (part of HAT domain). Mutated amino acids (aa) are numbered. The letter in front of the number represents the wild-type amino acid, which was changed to the amino acid indicated by the following letter. (B) Drosophila SF4 cells were transiently transfected with either MOF-GFP (wt) or a chromo-related domain mutant (Y416D), or a zinc-finger mutant (L578G/Y580G) derivative. Localization of the fusion proteins was compared to the position of the compensated X chromosome using immunofluorescence staining for GFP and MSL1, respectively. DNA was counterstained with Hoechst 33258. For each staining, four representative nuclei out of three independent experiments are shown.
In order to test whether MOF is tethered to the X chromosome via RNA interaction with its chromo-related domain, we analysed the localization of different MOF–GFP fusion proteins in a male Drosophila cell line (SF4) (Figure 1B). Transient transfection of wild-type MOF (wt) revealed a distribution pattern of exogenous protein that resembles that of endogenous MOF: a preferential enrichment on the X chromosome, which was marked by counterstaining MSL1. Y416D is a mutation in the chromo-related domain of MOF that abolishes MOF–RNA interaction in vitro and MOF–roX2 interaction in vivo (Akhtar et al, 2000). Surprisingly, the Y416D–GFP protein still localizes preferentially to the X chromosome, although the definition of the territory was not as distinct and a fraction of MOF appeared delocalized in other areas of the nucleus. This result suggests that chromo-related domain–RNA interactions are not solely responsible for targeting to the X chromosome and pointed to the importance of an additional, hitherto unappreciated determinant of MOF localization. This prompted us to investigate the protein interactions that might contribute to specific MOF localization.
MOF interacts directly with MSL1
We limited our analysis to MSL1, MSL2, MSL3 and MOF, since it has been observed that MLE associates less tightly with the MSLs and was mainly important for guiding roX RNA into the DCC (Copps et al, 1998; Smith et al, 2000). We coexpressed various combinations of the four MSL proteins in SF9 cells by coinfection with the respective baculoviruses and verified correct expression (data not shown). In order to facilitate the interaction studies, we added an HA tag to MOF and a flag tag to MSL3. We then used anti-flag and anti-HA affinity beads to purify the tagged proteins and visualized associated proteins by Coomassie blue staining (Figure 2). Notably, after complex formation, we subjected the beads to stringent washes with concentrations of up to 1 M monovalent cations and 1% detergent in order to focus on tight interactions.
Figure 2.
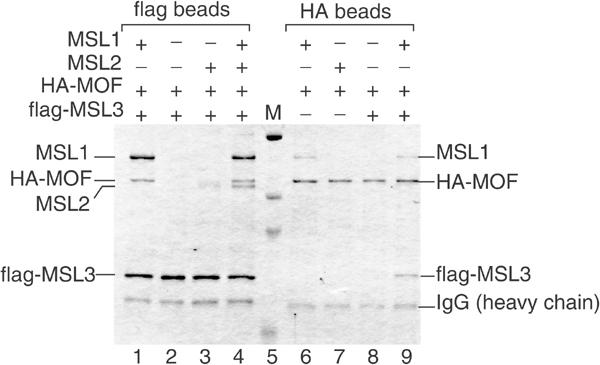
MOF associates with the DCC through a stable interaction with MSL1. SF9 cells were coinfected with various baculoviruses expressing HA-tagged MOF (HA-MOF), MSL1, MSL2 and flag-tagged MSL3 in the combinations indicated by ‘+' above each lane. Protein complexes were purified from total cell extracts by chromatography on beads coated with flag- or HA-specific antibodies (flag beads, HA beads, respectively). After binding, the beads were washed stringently (1 M NaCl, 1% NP40), and the adsorbed proteins were analysed by SDS–PAGE and Coomassie blue staining. The lane marked ‘M' displays protein size standards in kDa (206, 119, 91 and 51.4 kDa, respectively).
Coinfection with all four expression viruses allowed purification of a four-subunit DCC via the flagged MSL3 (Figure 2, lane 4). Omission of MSL2 did not affect the binding of MOF to the MSL1–MSL3 complex (Figure 2, compare lanes 1 and 4). By contrast, if MSL1 was missing, the association of MOF with the remaining MSL proteins was not detectable (Figure 2, lanes 2 and 3). We also confirmed the known interactions between MSL1 and MSL2 as well as MSL3 in this system (data not shown).
These results were corroborated using the HA epitope of MOF for the ‘pull-down' experiments. Coexpression of MOF with individual MSL proteins showed a selective association of MOF with MSL1 only (Figure 2, lanes 6–9). MSL3 could associate with MOF only in the presence of MSL1 (Figure 2, compare lanes 8 and 9). We concluded that, under stringent conditions, MOF interacts with MSL1 but not with MSL2 or MSL3. This confirms prior observations of Scott et al (2000) and extends them by documenting a direct interaction between MSL1 and MOF.
MOF interacts with MSL1 and localizes to the X chromosome via its CCHC zinc-finger domain
We next tested the point-mutated MOF derivatives (Figure 1A) for their ability to interact with MSL1. Wild-type MOF and MOF mutants were incubated with affinity beads coated with MSL3–MSL1 complex (immobilized via the flag MSL3) and bound MOF was visualized by SDS–PAGE and Western blotting (Figure 3). The controls for protein input (In) and the amount of interacting wild-type MOF (lanes 1 and 2) provide references for quantification. Point mutations that either abolish the RNA-binding capacity of the chromo-related domain (Y416D), or prevent acetyl-CoA binding (G691E), or a substitution of tryptophan 556 (W556G) did not affect the interaction of MOF with MSL1–MSL3. Strikingly, however, mutation of tyrosine 572 just in front of the CCHC zinc finger, or a double mutation (L578G/Y580G) in two loop positions of the zinc-finger structure drastically reduced this interaction (Figure 3, lanes 4 and 6). In order to test whether the surface involving the zinc-finger motif that contacts MSL1 contributes to localization of MOF in vivo, we expressed a MOF L578G/Y580G–GFP fusion protein in SF4 cells as before (Figure 1B). In contrast to wild-type MOF and the Y416D chromo-related domain mutant, this mutant was never found enriched on the male X chromosome. In a substantial proportion of transfected cells, mutant protein could be detected in high amounts in the cytoplasm, which could point to a general intracellular localization defect. In any case, the data suggest that the MSL1 contact is crucial for correct targeting of MOF.
Figure 3.
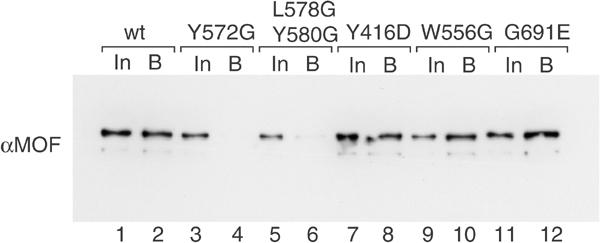
MOF interacts with MSL1 through its zinc-finger region. Wild-type MOF or derivatives (100 ng) bearing the point mutations indicated in Figure 1A were incubated with affinity beads, on which the flag-MSL3/MSL1 complex had been immobilized via the flag tag. After washing, bound protein was eluted with denaturing loading buffer, and half of the eluted proteins were resolved by SDS–PAGE. Bound MOF (B) was detected by Western blotting and compared to 1/10 of the corresponding input (In).
MOF and MSL3 interact with adjacent MSL1 domains in vitro and in cells
We mapped the surfaces on MSL1 that contact MOF and MSL3 using in vitro-translated MSL1 derivatives and affinity beads, on which full-length MOF or MSL3 were immobilized through their respective HA and flag tags. Various portions of MSL1 were produced by in vitro translation and labelled by inclusion of [35S]methionine in the reaction. These proteins were allowed to bind to immobilized MOF and MSL3 and complexes were washed stringently (500 mM salt, 1% NP40). Bound and unbound protein was subsequently visualized by electrophoresis and autoradiography (Figure 4). Plain anti-HA and anti-flag beads served as controls for nonspecific adsorption (Ctr1 and Ctr2). An MSL1 derivative containing sequences between amino acids (aa) 70 and 1039 interacted well with both MOF and MSL3, but not with the control beads (Figure 4A). Fragments derived from the N-terminus of MSL1 interacted neither with MOF nor with MSL3 (Figure 4B and C), as expected (Scott et al, 2000). Also, an internal fragment between aa 634 and 750 did not interact with MOF or MSL3 (Figure 4D). By contrast, fragment E covering aa 766–939 and containing the conserved PEHE domain (Marin, 2003) bound tightly to MOF but not to MSL3 (Figure 4E). The site of MSL3 interaction could be localized to fragment F (aa 973–1039; Figure 4F). In conclusion, MOF contacts the conserved PEHE domain of MSL1 and MSL3 binds directly adjacent to the very C-terminus of MSL1.
Figure 4.
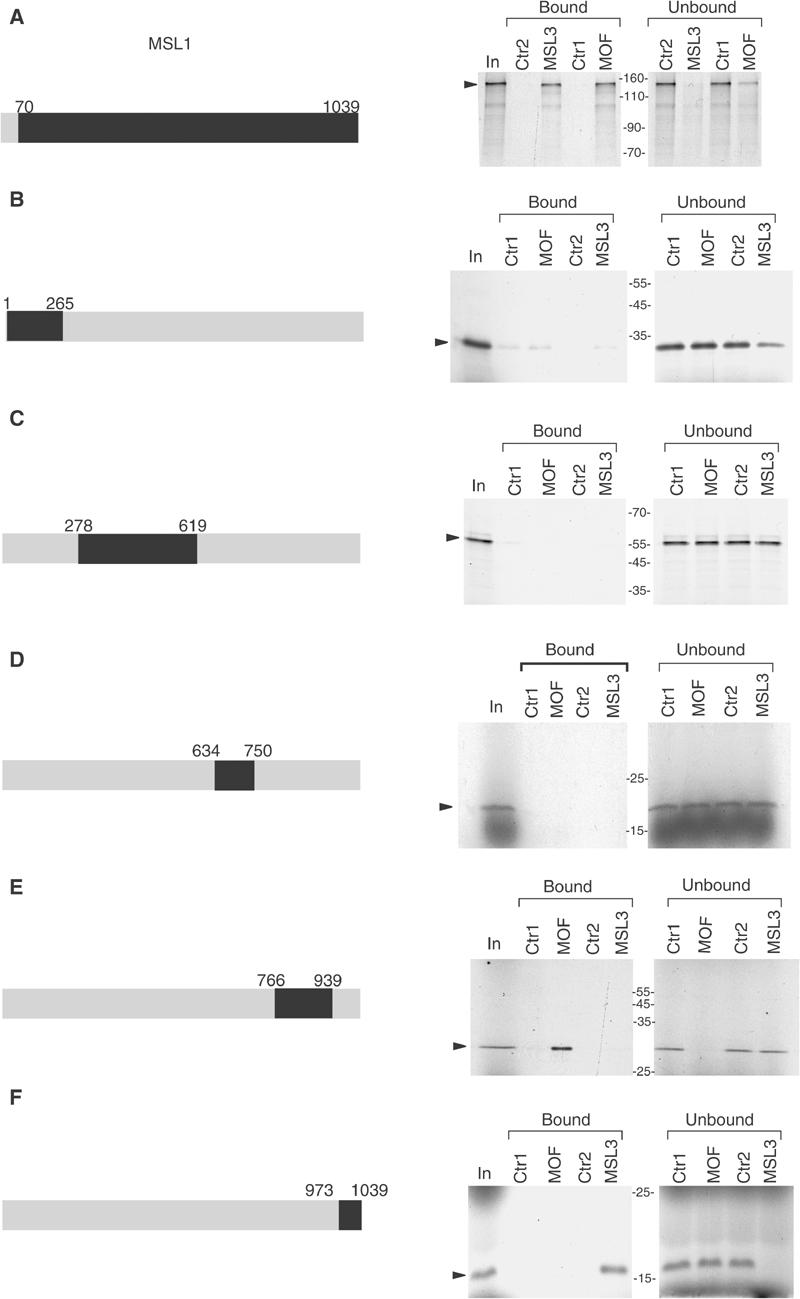
MOF and MSL3 interact with different domains of MSL1. Different fragments (A–F) of MSL1 (highlighted in black on the grey bar, which represents full-length MSL1) were in vitro translated in the presence of [35S]methionine and incubated with affinity beads, on which either HA-MOF (MOF) or flag-MSL3 (MSL3) had been immobilized. Uncharged anti-HA and anti-flag beads alone were used as specificity controls (Ctr1 and Ctr2, respectively). MSL1 derivatives were allowed to bind to the beads, and bound or unbound fractions were separated by SDS–PAGE (8% polyacrylamide gel for (A), 12% for (B, C) and 15% for (D–F)) and visualized by autoradiography of the dried gel. Input: In. The MSL1 translation products are indicated by arrows.
In order to verify the binding specificity of MOF and MSL3 to these distinct MSL1 domains in vivo, we expressed HA-tagged MSL1 fragments covering either aa 618–938 (containing fragment E) or aa 940–1039 (containing fragment F) in Drosophila SF4 cells. As shown in Figure 5A, the two MSL1 fragments were barely detectable in whole-cell extracts of transfected cells (Figure 5A, lanes 2 and 3) but were enriched to different degrees after anti-HA immunoprecipitation (Figure 5A, lanes 5 and 6). To study the interactions of MOF and MSL3 with these MSL1 fragments, we then probed for associated endogenous MSL1, MOF and MSL3 in the precipitate with specific antibodies (Figure 5B). We observed a specific binding of endogenous MSL3 with HA-MSL1(940–1039) (Figure 5B, lane 2) and binding of endogenous MOF with HA-MSL1(618–938) (lane 3) with amounts roughly coinciding with the expression levels of the domains. We did not detect any endogenous MSL1 in the immunoprecipitate.
Figure 5.
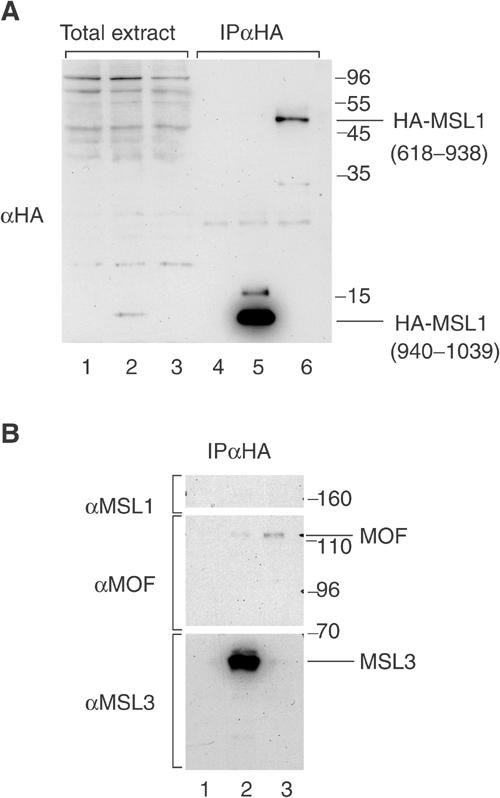
Specific interaction of MOF and MSL3 with different domains of MSL1 in living cells. (A) SF4 cells were transfected with a plasmid expressing the HA-tagged MSL1 domains containing either MSL3-binding domain (HA-MSL1(940–1039)) (lanes 2 and 5) or MOF-binding region (HA-MSL1(618–938)) (lanes 3 and 6). Total extracts from transfected cells (lanes 2, 3, 5 and 6) and untransfected cells (lanes 1 and 4) were immunoprecipitated with anti-HA beads (lanes 4–6). The MSL1 fragments were detected in the pellet by Western blotting using an anti-HA antibody. (B) The immunoprecipitate was resolved by 8% SDS–PAGE. The Western blot membrane was cut in three segments and separately probed with antibodies directed against MSL1 (upper part), MOF (middle part) and MSL3 (lower part).
Interaction of MOF with an MSL1–MSL3 subcomplex increases efficiency and substrate specificity of the acetyltransferase
In order to determine the functional consequences of MOF association with the MSL complex, we monitored the HAT activity of MOF on nucleosomes or free histones in the absence or the presence of purified MSL3 or MSL1 proteins (Figure 6). Half of each reaction was used to determine the incorporation of [3H]acetate as a general measure of HAT activity (left panels), and the other half was analysed by SDS–PAGE and autoradiography to visualize specifically acetylated proteins (right panels). We first monitored the ability of Escherichia coli-expressed MOF to acetylate histones in nucleosomes (Figure 6A). The effect of addition of three different MSL1 proteins was scored: an MSL1 with N-terminal GST tag or with a C-terminal TAP tag, both expressed in SF9 cells and purified independently, or an untagged MSL1 coexpressed and co-purified with flag-MSL3. The MSL proteins themselves, in the absence of MOF, had negligible HAT activity (Figure 6A, lanes 2–6). Under the conditions of these experiments, MOF alone had barely detectable acetylase activity (lanes 7 and 13), which could be stimulated by addition of either GST-MSL1 or TAP-MSL1 (Figure 6A, lanes 8 and 9). In this reaction, autoacetylation of MOF and acetylation of MSL3, MSL1 as well as of histone H4 were observed, but overall MOF activity remained low. Interestingly, most of the acetyl groups were incorporated into MSL1 (lanes 14 and 15). Addition of purified MSL3 to MOF had only a minor effect on activity (Figure 6A, lanes 10 and 16). However, if MSL3 was added together with GST-MSL1, a significant shift of substrate specificity away from MSL1 and towards histone H4 was observed (Figure 6A, compare lanes 14 and 17). Strikingly, addition of a coexpressed MSL1–flag-MSL3 subcomplex to MOF led to a 30-fold stimulation of its acetylase activity (lanes 12 and 18) with exclusive specificity for histone H4 (lane 18).
Figure 6.
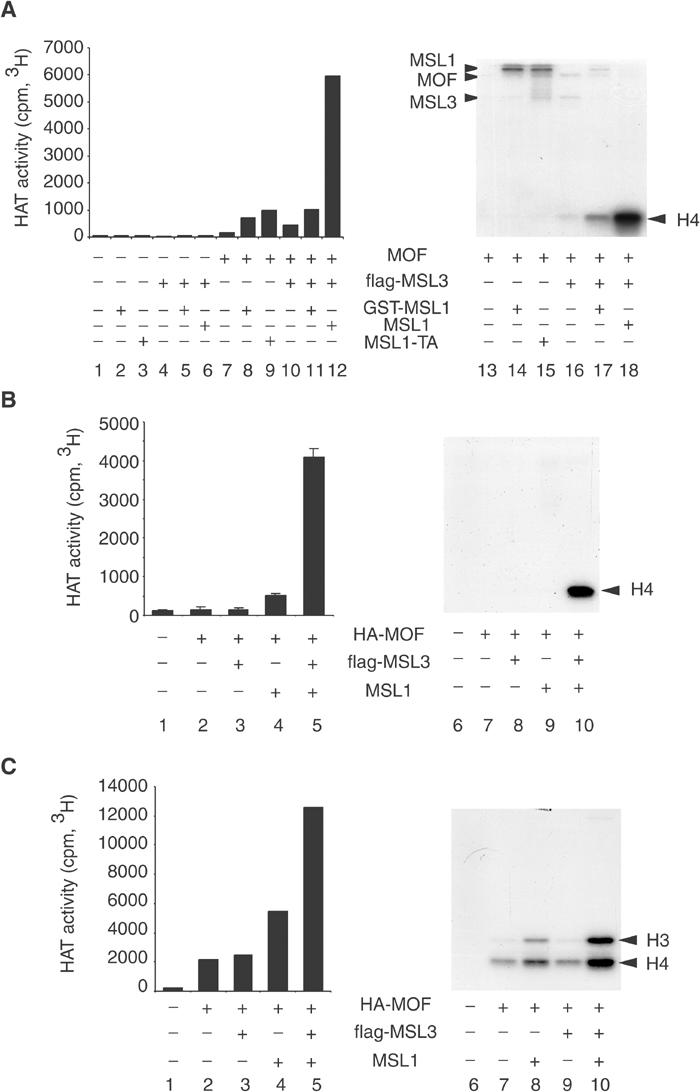
MSL1 and MSL3 stimulate the HAT activity of MOF on nucleosome templates. (A) HAT assays were performed by measuring transfer of [3H]acetyl from [3H]acetyl-coenzyme A to nucleosomal histones by recombinant (E. coli) MOF in the absence or presence of MSL3 or MSL1 expressed in SF9 cells as indicated (+). In this experiment, three different MSL1 proteins were tested: an N-terminal GST-tagged MSL1, a C-terminal TAP-tagged MSL1, both expressed and purified independently, or an untagged MSL1 coexpressed and co-purified with flag-MSL3. Half of the HAT reaction was used to determine the incorporation of isotope as a measure of general HAT activity (left panel), and the other half was analysed by 15% SDS–PAGE in order to determine the substrate specificity (right panel). The migration of GST-MSL1, TAP-MSL1 (MSL1), MOF or flag-MSL3 is indicated on the side. (B) HAT assays were performed as in (A) but with different combinations of MSL proteins coexpressed in SF9 cells and co-purified via the HA tag of MOF. (C) HAT assays using the same MSL proteins as in (B) but with free histone complexes as substrates.
We considered two possible explanations for this latter effect. Either activation of MOF required that MSL1 was coexpressed and co-purified with MSL3, or only untagged MSL1 was active whereas the activity of GST- or TAP-tagged MSL1 was compromised. To test these different hypotheses, we repeated the assay but with MSL1, MSL3 and MOF coexpressed from baculoviruses and co-purified (Figure 6B). Coexpression of either MSL3 or (untagged) MSL1 with MOF led to relatively minor activation (Figure 6B, lanes 2–4). However, coexpression of MSL1 and MSL3 with MOF stimulated its activity about 30-fold (lane 5). Under these conditions, only acetylation of histone H4, and no acetylation of either MSL1 or MSL3 were observed (lane 10). Identical results were obtained if a tagged version of MSL1 was coexpressed with MSL3 (data not shown). Therefore, these experiments strongly suggest that coexpression of the MSLs is necessary for a higher activation of MOF. Addition of MSL2 to MSL1 and MSL3 did not lead to further changes in MOF activity (data not shown).
So far we had used nucleosomal histones as substrates. When assayed with free histone substrates (Figure 6C), MOF was more active, but at the expense of specificity, in agreement with earlier observations (Akhtar and Becker, 2000). In addition to histone H4, also histone H3 was modified. Under these circumstances, the MSL1–MSL3 subcomplex stimulated the HAT activity of MOF to a lesser degree (Figure 6C, compare lanes 3–5), and it did not improve the specificity of acetylation (lanes 8–10).
MOF activation requires direct interaction between MSL1 and MSL3
The above-mentioned results indicated that MSL3 was important for MOF activity. Although we did not observe a strong interaction between MOF and MSL3 under our stringent conditions, others have described a direct binding of MOF and MSL3 (Buscaino et al, 2003), raising the issue as to whether MSL3 needed to be in a complex with MSL1 or simply present in the reaction for direct and perhaps transient contact with MOF. To clarify this point, we coexpressed MSL1 and three derivatives lacking either the N- or the C-terminal parts or a stretch of internal acidic residues (Figure 7A) with TAP-tagged MSL2 in order to facilitate their purification (see Figure 7B for a comparison of protein amounts). We then compared the influence of these MSL1–MSL2 complexes on MOF activity in the presence or absence of MSL3, which had been expressed separately (Figure 7C). Wild-type MSL1 and MSL3 stimulated the HAT activity of MOF as before (compare lanes 4 and 5, 15 and 16). The deletion of the first 84 aa or the acidic domain of MSL1 did not affect this stimulation (Δnt and Δac in Figure 7C; note the reduced MSL1 input in the Δac reaction; Figure 7B). In contrast, deletion of the MSL3-binding domain (Δct; Figure 7C) completely abolished the activation of MOF, indicating that MSL3 and MSL1 have to interact physically in order to cooperate for MOF activation.
Figure 7.
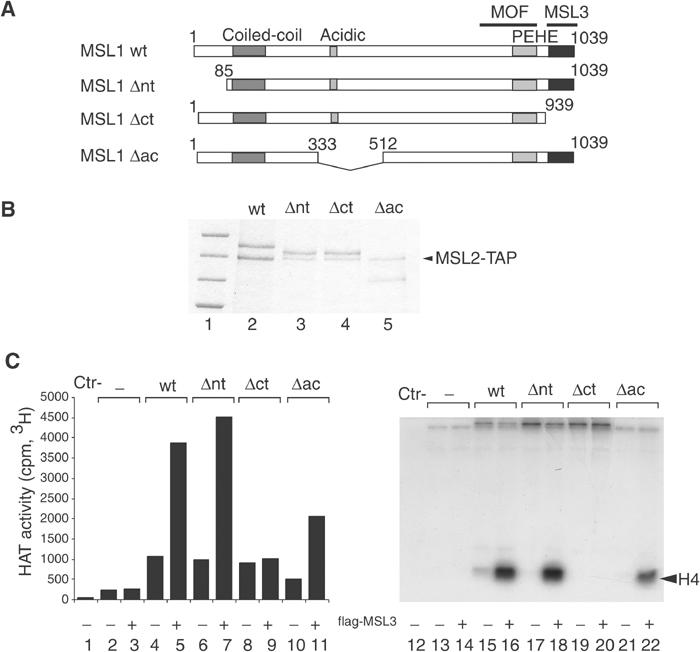
MSL3 binding to MSL1 is required for MOF activation on nucleosomes. (A) Schematic representation of MSL1: wild type (wt) and deletion mutants (Δnt, Δct and Δac). Deleted amino acids are numbered. Characteristic domains of MSL1, like the coiled-coil, the acidic and PEHE domains are indicated. (B) SF9 cells were coinfected with baculoviruses expressing TAP-MSL2 and either wild-type (wt) MSL1 or the MSL1 mutants described in (A). The MSL1 derivatives were purified via the TAP tag of MSL2, and analysed by SDS–PAGE and Coomassie blue staining (lanes 2–5). Lane 1 displays protein size standards in kDa (200, 150, 120, 100, respectively). (C) HAT assays on nucleosome templates were performed in the presence of MOF with or without MSL1 derivatives purified in (B) and in the absence or the presence of purified flag-MSL3. As in Figure 6, the left panel represents the general HAT activity and the right panel the substrate specificity.
In summary, tight substrate specificity of MOF HAT activity is only observed with the native, nucleosomal substrate. Faithful and efficient acetylation of nucleosomal histone H4 is only observed upon integration of MOF into an MSL1–MSL3 subcomplex, where the presence, correct folding and interactions of both subunits are crucial.
Recruitment of MOF to the chromatin substrate
The stable interaction of MOF with an MSL complex in solution suggests a way by which MOF is targeted to the X chromosome. We previously showed that MOF is able to interact with nucleosomes reconstituted with native Drosophila histones, but not free DNA (Akhtar and Becker, 2001). We now repeated these experiments in the presence of MSL1 and MSL3. In order to control the modification status of the substrate, we reconstituted chromatin on linear DNA using recombinant Drosophila histones. Free or nucleosomal DNA was attached with one end to paramagnetic beads. These beads were incubated with MOF in the presence or absence of MSL1, MSL3 or both proteins (Figure 8, lanes 5–12), washed and the interaction of MOF was measured by Western blotting. Consistent with earlier observations (Akhtar and Becker, 2000), we detected the interaction of MOF alone with nucleosomal DNA (lane 6), but this interaction was relatively weak. Both MSL1 and MSL3 were able to bind to free DNA and chromatin, but MSL3 profited from interaction with MSL1 (Figure 8, lanes 7, 8, 11 and 12). The interaction of MSL1 with DNA and chromatin was particularly striking (Figure 8, lanes 9 to 12) and this interaction also occurred in the absence of other proteins (data not shown). Remarkably, MOF was recruited to free and nucleosomal DNA in the presence of the MSL proteins (Figure 8, lanes 11 and 12) and both MSL1 and MSL3 were required for this recruitment (Figure 8, lanes 7–12).
Figure 8.
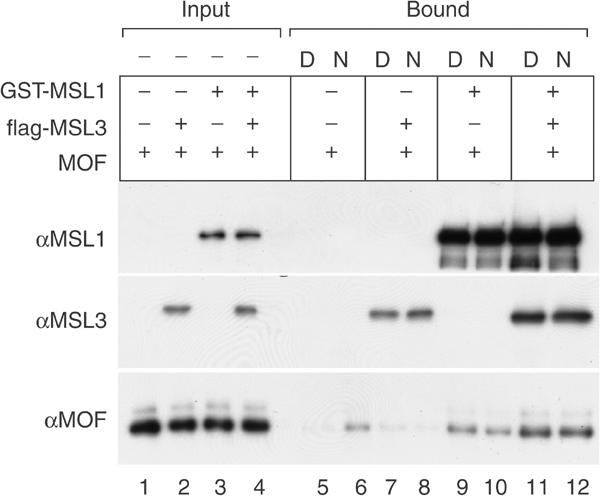
MSL1 and MSL3 together promote the binding of MOF to DNA and chromatin. Free (D) or nucleosomal (N) linear DNA was immobilized on paramagnetic beads and incubated either with MOF alone (lanes 5 and 6) or MOF and flag-MSL3 (lanes 7 and 8), MOF and GST-MSL1 (lanes 9 and 10), or all three proteins (lanes 11 and 12). After washes, the bound proteins were detected by Western blots using specific antibodies. Input: 5% of input (lanes 1–4); bound: 50% of bound proteins (lanes 5–12).
The synergistic activation of HAT activity by a complex of MSL1 and MSL3 is thus paralleled by a requirement of both proteins for enhanced association with the chromatin substrate.
Discussion
Activation of the male X chromosome in Drosophila requires acetylation of H4K16 by MOF. In vitro, untargeted acetylation of H4K16 is sufficient to activate any chromatin template. Targeting MOF to a promoter in yeast via fusion to a heterologous DNA-binding domain also leads to derepression of transcription (Akhtar and Becker, 2000). Given the potential of MOF to activate transcription, fine-tuning the expression of X-linked genes crucially relies on restricting MOF activity to the X chromosome.
Our current study highlights the protein–protein interactions that dictate the incorporation of MOF into the DCC. More importantly, it illustrates a novel principle of conditional activation of the enzyme. Because MSL proteins are limiting in male cells (Oh et al, 2003) and their association with the X chromosome is stable (T Straub, MF Neumann and PB Becker, unpublished results), essentially all MSL complexes are chromosomal. Activation of MOF requires interaction with MSL1, which initiates the DCC assembly, and MSL3, which is thought to associate with the complex after MSL1 and MSL2, suggesting that MOF may sense completed complex assembly. Integration into the complex unleashes MOF activity thereby restricting acetylation of H4K16 to the X chromosome. Rendering a regulatory acetylase activity dependent on the appropriate molecular context may be a more widespread principle. Recombinant Tip60 acetylase, another MYST family member, is unable to acetylate its physiological nucleosome substrate unless incorporated into a native complex (Ikura et al, 2000). The HAT activity of the MYST member Sas2 absolutely requires Sas4 and is stimulated by Sas5 (Sutton et al, 2003).
We discovered earlier that faithful association of MOF with the X chromosomal territory was lost upon RNase treatment of nuclei in permeabilized cells and that the chromo-related domain of MOF was important for interaction with roX RNA in vivo, suggesting that targeting of MOF relied heavily on RNA interactions (Akhtar et al, 2000). Our current results suggest that chromo-related domain–RNA interactions contribute to targeting but are not the primary targeting determinants for MOF. The RNase treatment not only results in displacement of MOF but also of MLE and MSL3 (Richter et al, 1996; Akhtar et al, 2000; Buscaino et al, 2003) and might also affect other, yet unknown factors in the complex. RNA degradation thus leads to the simultaneous disruption of many protein–RNA interactions, which collectively may be required for complex integrity. Although roX RNA improves the assembly of the DCC and its distribution over the X chromosome under conditions of limiting MSL proteins in wild-type flies (Meller et al, 2000; Meller and Rattner, 2002; Park et al, 2002), the deficiency due to the absence of both roX RNAs can be partially overcome by overexpressing MSL1 and MSL2 in flies (Oh et al, 2003). This finding is consistent with our observation that protein–protein interactions are essential for targeting MOF to the X chromosome. How the incorporation of roX RNA into the DCC modulates the protein interactions studied here and the dynamics of chromatin association remains to be explored.
Extending previous observations (Lyman et al, 1997; Copps et al, 1998; Scott et al, 2000), our study emphasizes the central role of MSL1 in the DCC complex formation and chromatin recruitment. In addition to its well-documented association with MSL2, MSL1 directly interacts with MOF and MSL3 via two distinct surfaces. Interestingly, two phylogenetically conserved regions of MSL1 have recently been identified (Marin, 2003). The first one corresponds to an N-terminal coiled-coil domain involved in the interaction of MSL1 with the ring-finger domain of MSL2. The second one, called PEHE domain, overlaps with our fragment E. We therefore consider it likely that a MOF interaction surface is present within the N-terminal part of this sequence conservation. The conservation of the PEHE region and the existence of MOF and MSL3 homologues in yeast and humans (Pannuti and Lucchesi, 2000; Eisen et al, 2001) underline the functional importance of the observed interactions.
MOF interacts with MSL1 via the zinc-finger domain, a hallmark of MYST-type HAT domains. While this interaction is necessary for targeting MOF to the X chromosome, we do not know whether it is sufficient. Molecular modelling suggests that the zinc finger is an integral part of the HAT domain and hence additional surfaces may be involved in the contact (T Straub, unpublished observation). Interestingly, the very same mutations that abolish this interaction with MSL1 also led to reduced acetylation of histones (Akhtar and Becker, 2001). Since the zinc finger is not close to the substrate-binding pocket, we consider that modulation of the zinc-finger structure, either through mutation or MSL1–MSL3 interaction, may have an allosteric negative or positive effect, respectively, on the ability of the catalytic site to interact with the histone tail substrate productively. Although the interactions of MSL3 and MOF are weak by comparison, MSL3 regulates MOF activity quantitatively and qualitatively. MSL3 and MOF interact with adjacent regions in the C-terminus of MSL1, which may promote their direct interaction. Alternatively, MSL3 may modulate MOF activity indirectly, through changes in MSL1 conformation.
In order to explore whether the activation of MOF's HAT activity by association with MSL1–MSL3 was due to enhanced binding to the chromatin substrate or an allosteric activation of catalysis, we carried out chromatin binding experiments. MSL1 interacted with chromatin and free DNA particularly well, and it helped both MSL3 and MOF to associate with chromatin. This observation lends additional support to the earlier notion of a central ‘platform' function of MSL1. MSL1 interacts with MSL2, MSL3, MOF and chromatin and thus is ideally suited to function as a nucleation factor for the DCC complex assembly on chromatin. Interestingly, MSL3 also assisted MOF's chromatin association, in keeping with the functional interactions between the two proteins observed in vitro and in vivo (Buscaino et al, 2003). Since the magnitude of the stimulation of chromatin binding was still an order of magnitude less than the observed stimulation of HAT activity, it is quite possible that allosteric effects of MSL protein association on the catalytic center of MOF contribute to activation of the HAT upon incorporation into the complex.
Interestingly, interaction of MOF with MSL1 and MSL3 also led to a change in substrate specificity. In the absence of MSL3, MOF activity was mainly directed towards MSL1, even though the nucleosomal substrate was present. In reactions containing only MSL3 and MOF, acetylation of MSL3 can also be detected (Buscaino et al, 2003). In the presence of both MSL1 and MSL3, MOF did not acetylate either protein significantly, but histone H4 was the exclusive substrate. Whether acetylation of MSL1 occurs in the context of DCC assembly or its distribution over the X chromosome in vivo remains to be seen. The sensitivity of our metabolic labelling strategy did not suffice to detect acetylation of endogenous MSL1 in cells. We envision the interaction of the DCC subunits to be dynamic during the initial assembly of the complex, its propagation over the X chromosome and its perpetuation through replication and mitosis. Conceivably, MSL1 acetylation may occur transiently at one stage, may signal a particular functional status or be involved in feedback loops fine-tuning the two-fold enhancement of transcription from the male X chromosome.
Materials and methods
Localization of wild-type and mutant MOF–GFP fusion proteins
Wild-type MOF was fused to GFP by cloning its coding sequence (aa 1–827) into pEGFP-1 (Clontech). Expression of MOF–GFP was driven by an HSP70 promoter placed upstream of the construct. MOF mutants Y416D and L578G/Y580G were fused to GFP by exchanging a BamHI/MroI fragment (aa 210–712) in the wild-type construct with fragments derived from the published plasmids for expression of MOF derivatives (Akhtar et al, 2000; Akhtar and Becker, 2001).
Drosophila SF4 cells were obtained from D Arndt-Jovin (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany). Cells were kept at 26°C in Schneider's Drosophila medium (Invitrogen) supplemented with penicillin, streptomycin, glutamine and 10% fetal calf serum. SF4 cells (1 × 106) were transiently transfected with 0.4 μg plasmid DNA in six-well plates using Effectene reagent (Qiagen). After 48 h of incubation, cells were seeded onto coverslips and allowed to settle for 2 h. Subsequently, cells were fixed in 2% paraformaldehyde in PBS for 7 min on ice. Following permeabilization with 0.25% Triton-X and 1% PFA in PBS for 7 min on ice, cells were washed twice in PBS. All subsequent steps were at room temperature. Coverslips were blocked with 2% BSA and 5% goat serum in PBS for 1 h. Cells were incubated with mouse anti GFP antibody (Molecular Probes) and rabbit anti-MSL1 antibody (M Kuroda) for 1 h at room temperature. After washing in PBS, slides were stained for 1 h with Cy3 and Cy2 labelled secondary antibodies (Jackson) diluted in blocking buffer. Cells were washed four times in PBS. DNA was counterstained with 1 μg/ml bisbenzimide (Hoechst 33258). Slides were mounted using 1.5% n-propyl gallate and 60% glycerol in PBS. Cells were examined and pictures were taken at × 1200 magnification using a Zeiss Axiophot microscope coupled to a Retiga Exi CCD Camera (Qimaging, Burnaby, Canada). Images were cropped and level-adjusted in Adobe Photoshop.
Expression of proteins in SF9 cell via baculovirus vectors
Recombinant baculoviruses expressing the MSL proteins were produced using the ‘Bac-to-Bac' expression system (Invitrogen) after cloning the cDNAs into pFastBac (for MSL1, MSL2, MSL3) or PVL1392.2 (for MOF) vectors. MSL2 was expressed untagged or with a C-terminal TAP tag, MSL1 was expressed untagged or with either an N-terminal GST tag or a C-terminal TAP tag, MSL3 was expressed with an N-terminal flag tag and MOF with an N-terminal HA tag.
SF9 infections were carried out under standard conditions in 15 cm diameter dishes and the optimal amount of each virus was determined empirically. The cells were collected after 2 days incubation at 26°C. In general, the cell pellets were frozen in liquid nitrogen and stored at −80°C prior to protein purification.
Total cell extract preparation and MSL protein purification
Total cell extract was prepared by suspending SF9 cell pellets in 1 ml lysis buffer (50 mM Hepes (pH 7.6), 300 mM KCl, 0.5 mM EDTA, 0.1% NP40, 1 mM DTT, protease inhibitor mix (PMSF, leupeptine, aprotinin, pepstatin)) per dish and incubated for 15 min on ice. The cell suspension was sonicated by pulses of 15 s at 20% amplitude (Branson digital sonifier model 250-D), and centrifuged for 30 min at 14 krpm at 4°C. The supernatant was used directly for protein purification or stored at −80°C.
For purification, soluble cell extracts (0.5 ml) were incubated for 2 h on a rotating wheel with affinity beads (25 μl) according to the tag of the protein: anti-flag M2 beads (Sigma), anti-HA (Roche) beads or glutathione beads (Amersham). The beads were washed five times with 0.5 ml of each of the three following buffers: buffer I: 0.5 M KCl, 50 mM Hepes (pH 7.6), 0.5 mM EDTA, 1% NP40, 0.5 mM DTT, protease inhibitor mix; buffer II: buffer I with 1 M KCl; buffer III: 0.1 M KCl, 10 mM Hepes (pH 7.6), 0.5 mM EDTA, 0.1% NP40, 10% glycerol, 0.5 mM DTT, protease inhibitor mix. Bound proteins were boiled for 5 min in denaturing loading buffer and analysed by SDS–PAGE and Coomassie staining.
For the HAT assay (Figures 6 and 7), the MSL proteins were eluted (three times for 4–8 h on a rotating wheel at 4°C) in 100 μl buffer III supplemented with flag peptides (0.25 mg/ml) and HA peptides (0.3 mg/ml) for flag-MSL3 or HA-MOF, respectively. For GST-MSL1, elution was achieved with 20 mM glutathione at pH 8.0. TAP-tagged MSL1 and TAP-tagged MSL2 were purified only via the calmodulin-binding domain of the tag following the protocol of Rigaut et al (1999).
Wild-type and mutants MOF were expressed in E. coli and purified as described (Akhtar and Becker, 2000).
In vitro transcription and translation
MSL1 cDNA fragments from MSL1-pFastBac were subcloned into pGEM-7Zf (Promega). The HindIII fragment (base pairs (bp) 213–3237; with reference to the transcription start site at +1; Figure 4A) and the BamHI fragment (bp 2813–3258; Figure 4F) were transcribed from the SP6 promoter. The EcoRI fragment (bp −9 to 791), the EcoRI–BamHI fragment (bp 791–1853), the BamHI–HincII fragment (bp 1853–2248) and the HincII–BamHI fragment (bp 2248–2813) were transcribed from the T7 promoter (Figure 4B–E, respectively). In vitro transcription and translation (IVT) reactions were performed for 3 h at 30°C with the TNT System (Promega) using 1 μg of plasmid and 10 μCi of [35S]methionine in 25 μl reaction volume.
Pull-down experiments
A 16 μl measure of each IVT reaction was mixed with 344 μl of buffer III. A 1/20 portion of the mix was removed (input), and the remainder was split into four 85 μl aliquots, incubated for 2–4 h at 4°C (rotating wheel) with either anti-HA or anti-flag beads incubated with mock extracts as negative controls or HA-MOF or flag-MSL3 beads (5 μl) corresponding to anti-HA and anti-flag beads coated with HA-MOF and flag-MSL3, respectively (see MSL purification). The beads (bound fraction) were washed five times with 0.5 ml of buffer I and III. Bound and unbound materials were analysed by SDS–PAGE.
To test for interaction of MSL1 with MOF mutants, flag beads coated with flag-MSL3–MSL1 (see MSL purification) were incubated (2–4 h, 4°C) with 100 ng of either wild-type MOF or MOF derivatives in 200 μl of 25 mM Hepes (pH 7.6), 150 mM KCl, 2 mM MgCl2, 0.25 mM EDTA, 0.01% NP40, 0.5 mM DTT and protease inhibitors. After five 0.5 ml washes with the same buffer, bound MOF was detected by Western blotting.
Stable transfection of SF4 cells and anti-HA immunoprecipitation
Two MSL1 BamHI fragments (bp 1853–2813, 2813–3258) were filled in with Klenow enzyme and cloned into the filled-in AvaI site of pRMHN downstream of a copper-inducible Drosophila metallothionein promoter. pRMHN is a derivative of pRMHA (Bunch et al, 1988), which fuses the expressed protein to an HA tag. The expression led to two HA-tagged MSL1 fragments (HA-MSL1(618–938) and HA-MSL1(940–1039)). Details are available upon request.
SF4 cells (4 × 106/10 cm diameter dish) were cotransfected with each plasmid (1 μg) and 0.2 μg pUCHSNEO (coding for neomycin resistance) using the Effectene Kit (Qiagen). After 48 h at 26°C, 1 mg/ml neomycin was added and selective growth was maintained for 4 weeks. To induce the expression of HA-MSL1 domains, 0.5 mM copper sulphate was added. Cells were collected after 6 days incubation at 26°C. As a control nontransfected SF4 cells were incubated under the same induction conditions.
Anti-HA beads were incubated with cell extracts containing HA-MSL1(618–938) and HA-MSL1(940–1039), washed five times with 0.5 ml of 25 mM Hepes (pH 7.6), 150 mM KCl, 2 mM MgCl2, 0.25 mM EDTA, 0.01% NP40, 0.5 mM DTT and protease inhibitors. The beads were resuspended in SDS loading buffer, boiled for 5 min and eluted proteins were resolved by 12 or 8% SDS–PAGE.
Chromatin assembly and interaction studies
cDNAs for Drosophila histones (H2A, H2B, H3 and H4) were cloned from genomic DNA by PCR into pET3a and pET3c, expressed in E. coli BL21-CodonPlus-RIL and purified according to Luger et al (1997). Nucleosomal arrays were assembled onto plasmid using these recombinant histones (histone octamer:DNA ratio of 1.3:1) dialysing through a salt gradient from 2 M to 50 mM NaCl in 10 mM Tris (pH 7.6), 1 mM EDTA, 1 mM β-mercaptoethanol and 0.01% NP40. The quality of the nucleosome assembly was checked by microccocal nuclease digestion and the histone stoichiometry by SDS–PAGE and Coomassie staining.
HAT assays
Reconstituted nucleosomal arrays (corresponding to 1.5 μg of histone octamer) were incubated for 1 h at 26°C, in 20 μl final volume with 0.25 μCi of [3H]acetyl-CoA (4.1 Ci/mmol), 100 ng of each MSL protein (HA-MOF±MSL1±flag-MSL3) in 50 mM KCl, 50 mM Tris (pH 7.8) and 0.1 mM EDTA. A 10 μl portion of each reaction was spotted onto p81 filters, washed three times with 50 mM NaCarbonate (pH 9.2) and the filters were counted in a scintillation counter. The remainder of the reactions was analysed by 15% SDS–PAGE. The gel was treated with Amplify (Amersham Biosciences) for 30 min, dried and autoradiographed.
MSLs binding assay to nucleosomal array on paramagnetic beads
Linear DNA was immobilized on paramagnetic beads (Dynal) according to Sandaltzopoulos and Becker (1999) and assembled into nucleosomes as described above. Chromatin beads containing 200 ng of free DNA or nucleosomal DNA were incubated with either bacterially expressed MOF alone or in the presence of 50–100 ng of flag-MSL3 and GST-MSL1 (both from baculovirus expression) as indicated in the legend to Figure 8 for 2 h at 4°C on a rotating wheel in 200 μl of a buffer composed of 20 mM Tris–HCl (pH 7.6), 50 mM NaCl, 0.1 mM EDTA, 1 mM MgCl2, 0.1 mg/ml BSA, 0.05% NP40, 1 mM DTT and protease inhibitors. The beads were then washed (4 × 0.3 ml) with the same buffer and treated as previously for further Western blot analysis.
Acknowledgments
This work was supported by a Training and Mobility Network grant HPRN-CT-2000-00078 from the European Union and by DFG through Transregio5. GM was supported by the Human Frontier Science Program. We thank Irene Vetter for help with cloning. We thank G Gilfillan, I Dahlsveen, A Eberharter, C Regnard and R Ferreira for critical reading of the manuscript and helpful discussions. We thank D Arndt-Jovin for providing the SF4 cell line and M Kuroda for antibodies against MSL proteins. We thank the Friedrich-Baur Stiftung for the donation of a Qimaging Retiga Exi CCD Camera.
References
- Akhtar A, Becker PB (2000) Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol Cell 5: 367–375 [DOI] [PubMed] [Google Scholar]
- Akhtar A, Becker PB (2001) The histone H4 acetyltransferase MOF uses a C2HC zinc finger for substrate recognition. EMBO Rep 2: 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar A, Zink D, Becker PB (2000) Chromodomains are protein–RNA interaction modules. Nature 407: 405–409 [DOI] [PubMed] [Google Scholar]
- Bashaw GJ, Baker BS (1995) The msl-2 dosage compensation gene of Drosophila encodes a putative DNA-binding protein whose expression is sex specifically regulated by Sex- lethal. Development 121: 3245–3258 [DOI] [PubMed] [Google Scholar]
- Bone JR, Lavender J, Richman R, Palmer MJ, Turner BM, Kuroda MI (1994) Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev 8: 96–104 [DOI] [PubMed] [Google Scholar]
- Bunch TA, Grinblat Y, Goldstein LS (1988) Characterization and use of the Drosophila metallothionein promoter in cultured Drosophila melanogaster cells. Nucleic Acids Res 16: 1043–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscaino A, Kocher T, Kind JH, Holz H, Taipale M, Wagner K, Wilm M, Akhtar A (2003) MOF-regulated acetylation of MSL-3 in the Drosophila dosage compensation complex. Mol Cell 11: 1265–1277 [DOI] [PubMed] [Google Scholar]
- Chang KA, Kuroda MI (1998) Modulation of MSL1 abundance in female Drosophila contributes to the sex specificity of dosage compensation. Genetics 150: 699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps K, Richman R, Lyman LM, Chang KA, Rampersad-Ammons J, Kuroda MI (1998) Complex formation by the Drosophila MSL proteins: role of the MSL2 RING finger in protein complex assembly. EMBO J 17: 5409–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A, Utley RT, Nourani A, Allard S, Schmidt P, Lane WS, Lucchesi JC, Cote J (2001) The yeast NuA4 and Drosophila MSL complexes contain homologous subunits important for transcription regulation. J Biol Chem 276: 3484–3491 [DOI] [PubMed] [Google Scholar]
- Franke A, Baker BS (1999) The rox1 and rox2 RNAs are essential components of the compensasome, which mediates dosage compensation in Drosophila. Mol Cell 4: 117–122 [DOI] [PubMed] [Google Scholar]
- Gu W, Szauter P, Lucchesi JC (1998) Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Dev Genet 22: 56–64 [DOI] [PubMed] [Google Scholar]
- Gu W, Wei X, Pannuti A, Lucchesi JC (2000) Targeting the chromatin remodeling MSL complex of Drosophila to its sites of action on the X chromosome requires both acetyl transferase and ATPase activities. EMBO J 19: 5202–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker A, Hilfiker KD, Pannuti A, Lucchesi JC (1997) mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J 16: 2054–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y (2000) Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102: 463–473 [DOI] [PubMed] [Google Scholar]
- Kelley RL, Kuroda MI (2000) Noncoding RNA genes in dosage compensation and imprinting. Cell 103: 9–12 [DOI] [PubMed] [Google Scholar]
- Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI (1999) Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98: 513–522 [DOI] [PubMed] [Google Scholar]
- Kelley RL, Solovyeva I, Lyman LM, Richman R, Solovyev V, Kuroda MI (1995) Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell 81: 867–877 [DOI] [PubMed] [Google Scholar]
- Kelley RL, Wang J, Bell L, Kuroda MI (1997) Sex lethal controls dosage compensation in Drosophila by a non-splicing mechanism. Nature 387: 195–199 [DOI] [PubMed] [Google Scholar]
- Lucchesi JC (1996) Dosage compensation in Drosophila and the ‘complex' world of transcriptional regulation. BioEssays 18: 541–547 [DOI] [PubMed] [Google Scholar]
- Lucchesi JC (1999) Dosage compensation: roX marks the spot. Curr Biol 9: R807–R808 [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Flans AJ, Waye MMY, Richmond TJ (1997) Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol 272: 301–311 [DOI] [PubMed] [Google Scholar]
- Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI (1997) Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL-1 for chromosome association. Genetics 147: 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin I (2003) Evolution of chromatin-remodeling complexes: comparative genomics reveals the ancient origin of ‘Novel' compensasome genes. J Mol Evol 56: 527–539 [DOI] [PubMed] [Google Scholar]
- Meller VH, Gordadze PR, Park Y, Chu X, Stuckenholz C, Kelley RL, Kuroda MI (2000) Ordered assembly of roX RNAs into MSL complexes on the dosage-compensated X chromosome in Drosophila. Curr Biol 10: 136–143 [DOI] [PubMed] [Google Scholar]
- Meller VH, Rattner BP (2002) The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J 21: 1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, Wu KH, Roman G, Kuroda MI, Davis RL (1997) roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell 88: 445–457 [DOI] [PubMed] [Google Scholar]
- Oh H, Park Y, Kuroda MI (2003) Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev 17: 1334–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Richman R, Richter L, Kuroda MI (1994) Sex-specific regulation of the male-specific lethal-1 dosage compensation gene in Drosophila. Genes Dev 8: 698–706 [DOI] [PubMed] [Google Scholar]
- Pannuti A, Lucchesi JC (2000) Recycling to remodel: evolution of dosage-compensation complexes. Curr Opin Genet Dev 10: 644–650 [DOI] [PubMed] [Google Scholar]
- Park Y, Kelley RL, Oh H, Kuroda MI, Meller VH (2002) Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins. Science 298: 1620–1623 [DOI] [PubMed] [Google Scholar]
- Park Y, Mengus G, Bai X, Kageyama Y, Meller VH, Becker PB, Kuroda MI (2003) Sequence-specific targeting of Drosophila roX genes by the MSL dosage compensation complex. Mol Cell 11: 977–986 [DOI] [PubMed] [Google Scholar]
- Richter L, Bone JR, Kuroda MI (1996) RNA-dependent association of the Drosophila maleless protein with the male X chromosome. Genes Cells 1: 325–336 [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Sandaltzopoulos R, Becker PB (1999) A solid phase approach for the analysis of reconstituted chromatin. In Chromatin Protocols, Becker PB (ed) pp 195–206. Totowa, USA: Humana Press [DOI] [PubMed] [Google Scholar]
- Scott MJ, Pan LL, Cleland SB, Knox AL, Heinrich J (2000) MSL1 plays a central role in assembly of the MSL complex, essential for dosage compensation in Drosophila. EMBO J 19: 144–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Allis CD, Lucchesi JC (2001) Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem 276: 31483–31486 [DOI] [PubMed] [Google Scholar]
- Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, Lucchesi JC (2000) The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol 20: 312–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckenholz C, Kageyama Y, Kuroda MI (1999) Guilt by association: non-coding RNAs, chromosome-specific proteins and dosage compensation in Drosophila. Trends Genet 15: 454–458 [DOI] [PubMed] [Google Scholar]
- Sutton A, Shia WJ, Band D, Kaufman PD, Osada S, Workman JL, Sternglanz R (2003) Sas4 and Sas5 are required for the histone acetyltransferase activity of Sas2 in the SAS complex. J Biol Chem 278: 16887–16892 [DOI] [PubMed] [Google Scholar]