

Abstract
Targeting of cellular histone acetyltransferases (HATs) by viral proteins is important in the development of virus-associated diseases. The immediate-early 2 protein (IE2) of human cytomegalovirus (HCMV) binds to the tumor suppressor, p53, and inactivates its functions by unknown mechanisms. Here, we show that IE2 binds to the HAT domain of the p53 coactivators, p300 and CREB-binding protein (CBP), and blocks their acetyltransferase activity on both histones and p53. The minimal HAT inactivation region on IE2 involves the N-terminal 98 amino acids. The in vivo DNA binding of p53 and local histone acetylation on p53-dependent promoters are all reduced by IE2, but not by mutant IE2 proteins that lack the HAT inhibition region. Furthermore, the p53 acetylation site mutant, K320/373/382R, retains both DNA binding and promoter transactivation activity in vivo and these effects are repressed by IE2 as well. Together with the finding that only wild-type IE2 exerts an antiapoptotic effect, our results suggest that HCMV IE2 downregulates p53-dependent gene activation by inhibiting p300/CBP-mediated local histone acetylation and that IE2 may have oncogenic activity.
Keywords: IE2, HAT, HCMV, p53
Introduction
Due to its ability to bind to specific DNA sequences and activate transcription, p53 is a critical regulator of cell proliferation and apoptosis in response to DNA damage (Wahl and Carr, 2001). The transcriptional activity of p53 is modulated in part by its post-translational acetylation (Brooks and Gu, 2003).
The binding of p53 to the coactivators, p300 and CREB-binding protein (CBP), which have histone acetyltransferase (HAT) activity (Bannister and Kouzarides, 1996; Ogryzko et al, 1996), results in synergistic enhancement of p53 transactivation activity (Avantaggiati et al, 1997; Gu et al, 1997). This is believed to be mediated by at least two different pathways, core histone acetylation and p53 acetylation. Hyperacetylation of histones correlates with enhanced transcription, presumably by increasing the accessibility of nucleosomal DNA to transcription factors (Brown et al, 2000). In response to DNA damage, p300/CBP also acetylate several lysine residues including K373 and K382 on p53, while another HAT, p300/CBP-associated factor (PCAF), acetylates K320 (Gu and Roeder, 1997; Sakaguchi et al, 1998; Liu et al, 1999). Whether p53 acetylation increases its affinity for DNA and thus results in activation of p53 function, as originally suggested by Gu et al (1997), is still controversial (Prives and Manley, 2001; Luo et al, 2004, and references therein).
Many DNA viruses, including the β-herpesvirus family member, human cytomegalovirus (HCMV), encode immediate-early gene products, which overcome the negative effects of p53 on cell cycle progression (Castillo and Kowalik, 2002). This is thought to promote cell growth and increase the available pool of deoxyribonucleotides for viral replication. However, loss of p53 activity often leads to serious disorders. Inactivation of p53 activity by the HCMV immediate-early 2 (IE2) protein can cause uncontrolled proliferation of vascular smooth muscle cells and endothelium cells surrounding blood vessels, possibly leading to clogging of the arteries (Speir et al, 1994). Uncontrolled cell growth caused by the loss of p53 activity is also linked to HCMV-associated cancer development (Castillo and Kowalik, 2002).
The gene encoding IE2 was first identified (Hermiston et al, 1987) as part of a region of the HCMV genome that could complement an adenovirus mutant defective in the E1A gene product, suggesting overlapping functions with E1A protein. By binding to specific DNA sequences or interacting with other transcription factors, IE2 can increase or decrease transcription (Castillo and Kowalik, 2002). Although IE2 is known to interact physically with p53 in vitro and downregulate p53-dependent gene activation (Tsai et al, 1996), the molecular mechanism underlying this inhibition and its physiological significance are still unclear. The aim of this study was to determine how HCMV IE2 downregulates p53 activity. Unexpectedly, we discovered that IE2 directly targets the p53 coactivators, p300 and CBP, and interferes with their acetylase activity.
Results
Since marked induction of p53 during HCMV infection is only seen after the expression of the viral IE genes (Castillo et al, 2000), in order to mimic this situation, throughout the present study, we used a plasmid encoding a temperature-sensitive mutant of p53, p53V143A, which adopts a nonfunctional conformation at 37°C, but folds into a functional conformation when the temperature is switched to 30°C (Zhang et al, 1994; Tsai et al, 1996). This allowed us to first express HCMV IE2, then induce p53 ‘wild-type' (WT) function by a temperature shift from 37 to 30°C.
HCMV IE2 downregulates p53-dependent gene activation
To determine whether HCMV IE2 downregulates p53 activity, we used a chloramphenicol acetyltransferase (CAT) reporter construct, p3PREcCAT, in which the reporter is driven by a promoter containing three copies of a consensus p53-binding site oligomer. Consistent with the previous report (Tsai et al, 1996), in p53-negative H1299 cells, we found that IE2 (Figure 1A, compare lane 4 to lane 2), but not IE1 (lane 3), a differentially spliced product from the same gene that encodes IE2, downregulated p53-dependent gene activation. Western blot analysis showed that IE2 did not decrease p53 protein expression (middle panel). To study the underlying mechanism, we examined whether IE2 suppresses p53 activity by recruiting corepressors, such as histone deacetylases (HDACs). It has been reported that class I and III HDACs play an important role in regulating p53 function (Juan et al, 2000; Luo et al, 2000; Vaziri et al, 2001; Langley et al, 2002). To examine this hypothesis, the effects of the class I HDAC inhibitor, trichostatin A (TSA), and the class III inhibitor, nicotinamide, were tested. We found that, alone or combined, they had no effect on the IE2-mediated inhibition of p53 activity (top panel, lanes 5–9). The inability of these HDAC inhibitors to recover p53 function was unlikely to be due to instability of the chemicals, at least in the case of TSA, since cells treated with a low dose of TSA (0.25 μM) showed greatly increased histone H3 acetylation (bottom panel). Thus, HDACs are unlikely to be involved in the IE2-mediated downregulation of p53 function. Similar results were obtained in p53-positive cells HEL299 (data not shown) and RKO (Figure 1B) when the IE2 effect on the p53-dependent p21 promoter (el-Deiry et al, 1993) was investigated. The use of IE2 deletion mutants (Figure 1C, bottom panel) in H1299 cells further showed that removal of the N-terminal 135 or 168 residues from IE2 resulted in a marked reduction in inhibitory ability (upper panel, compare lanes 5 and 6 to lane 4), although residual inhibition (about 20–30%) was still seen. These results suggest that the N-terminal 135 amino acids of IE2 are one of the important regions critical in repressing p53 activity.
Figure 1.
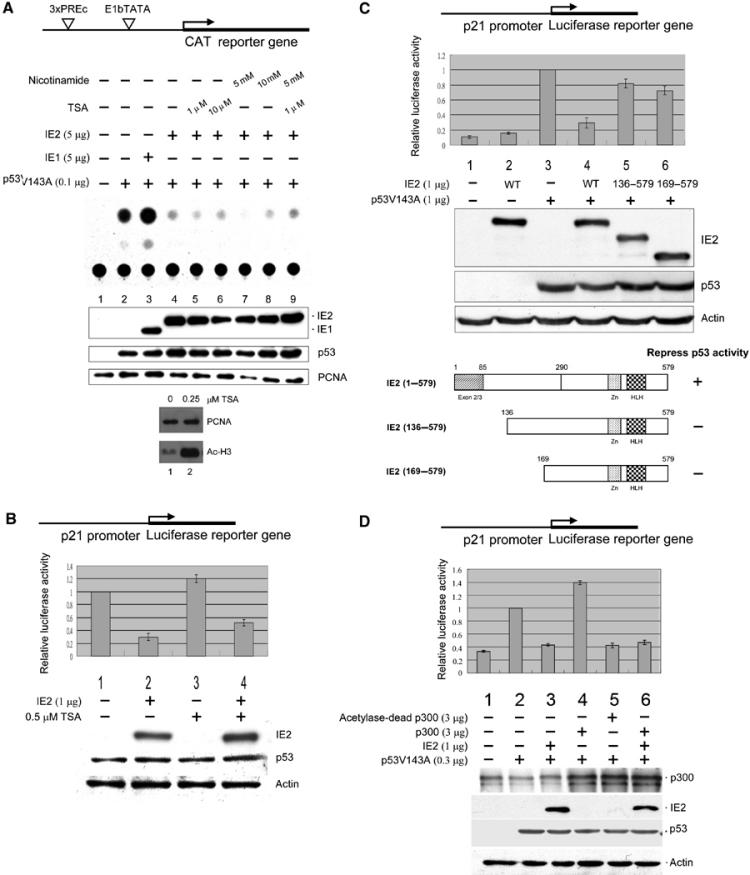
HCMV IE2-mediated suppression of p53-dependent gene activity. Cells were transfected with indicated expression plasmids, followed by CAT reporter assays (A) or luciferase assays (B–D) and Western analyses. The reporter constructs used are shown at the top of each figure. (A) IE2 inhibits p53-specific gene activation by a TSA-insensitive mechanism. Upper panel: Autoradiogram of the TLC separation. The experiment was repeated three times giving means and standard errors of 0.13±0.02, 1.00±0, 1.60±0.16, 0.46±0.06, 0.32±0.02, 0.39±0.02, 0.35±0.02, 0.4±0.03, and 0.32±0.06 for lanes 1–9, respectively. Bottom panel: Cells were incubated for 12 h with or without 0.25 μM TSA, followed by Western analyses. PCNA: internal control. (B) IE2 downregulates endogenous p53-mediated activation of the p21 promoter. Upper panel: Firefly luciferase activity (p21-Luc) normalized to that of the Renilla luciferase (pRL-SV40) control. Bottom panel: Western blots, with actin as the internal control. (C) IE2 deletion mutants lacking the N-terminal 135 amino acids lost p53 inhibition activity. The WT or IE2 deletion mutants are shown in the bottom panel. IE2 (1–579) is full-length IE2, while IE2 (136–579) and IE2 (169–579) lack the N-terminal 135 or 165 residues, respectively. Residues 1–85 are the common region shared by IE1 and IE2. Zn: Zn finger domain; HLH: helix–loop–helix motif. (D) IE2 inhibits p300-mediated induction of p53 activity. The reporters used in (B–D) were 10 ng of p21-Luc and 10 ng of pRL-SV40, as an internal control. The antibodies for the Western blots were directed against the N-terminal 85 amino acids common to HCMV IE1 and IE2 (MAb810, Chemicon) (A), IE2 (polyclonal, see Materials and methods) (B–D), p53 (Ab-6, Oncogene), proliferation cell nuclear antigen (PCNA) (SC56, Santa Cruz), acetylated histone H3 (06-599, Upstate), actin (MAB1501, Chemicon), and p300 (NA-46, Oncogene).
Given that p53 activity depends, in part, on the recruitment of its coactivators, p300/CBP (see Introduction), we next investigated if IE2 directly reduces the p300-mediated stimulation of p53 activity. As shown in Figure 1D, overexpression of WT p300 resulted in a moderate increase in p53-dependent gene activation (lane 4), whereas expression of an acetyltransferase-dead p300 mutant, p300D1399Y, not only failed to activate p53 but significantly repressed its effect (lane 5); this phenomenon was probably due to the dominant-negative effect caused by the mutant p300, which competed with the WT endogenous p300 for binding to p53. Importantly, p300-mediated p53 activation was also reduced by IE2 (lane 6). These data imply that IE2 may downregulate p53 function either by dissociating the coactivators p300/CBP from p53 or by directly interfering with their enzymatic activity.
p53 binding to p300/CBP is not affected by IE2
We then used immunoprecipitation (IP), followed by Western blotting, to determine whether binding of IE2 to p53 dissociates p300/CBP from p53. In H1299 cells, in the absence of IE2, anti-p53 antibody pulled down endogenous p300 and CBP (Figure 2A, lane 2) and this was unaffected by coexpression of IE2 (lane 3). In agreement with the previous GST pull-down experiment (Tsai et al, 1996), we also found that p53 associated with IE2 in vivo (lane 3). Furthermore, when anti-IE2 antibody was used, p300 and p53 were only precipitated in the presence of IE2 (Figure 2B, lane 4). Consistent with the results obtained in the overexpressing system, neither IE2 nor IE2 (169–579) that lacks the p53 inhibition activity reduced the endogenous p53/p300 binding in RKO cells (Figure 2C) and anti-IE2, but not the preimmune serum, precipitated the endogenous p300 (Figure 2D). These results suggest that IE2 downregulates p53 activity by a mechanism not involving the dissociation of p300/CBP from p53.
Figure 2.
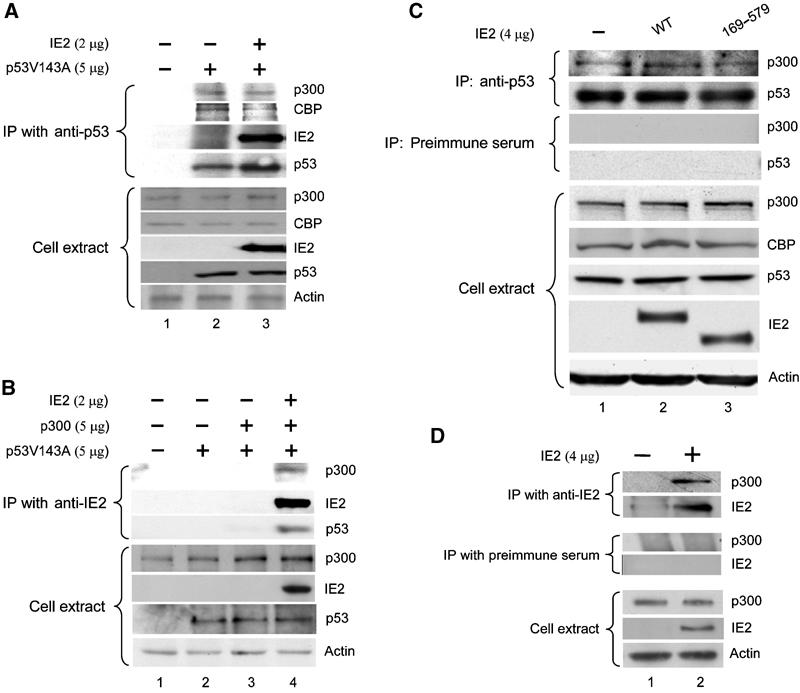
In vivo binding of IE2, p53, and p300/CBP. H1299 cells (A, B) or RKO cells (C, D) were transfected with the indicated expression plasmid(s), then lysed and the cell extract was immunoprecipitated (IPed) with anti-p53 antibody (FL-393-G, Santa Cruz) (A, C) or rabbit polyclonal anti-IE2 antibody (B, D). The precipitated proteins were electrophoresed and tested on Western blots with the indicated antibodies. The antibodies used were against p300 (05-267, Upstate in (A, B); Ab-1, Oncogene in (C, D)), CBP (Ab-22, Santa Cruz), IE2 (MAB810, Chemicon) in (A, B, D), IE2 (polyclonal) in (C), p53 (Ab-6, Oncogene), or actin (MAB1501, Chemicon).
IE2 inhibits histone and p53 acetylation by p300/CBP in vitro
We next examined if IE2 directly affects the HAT activity of p300/CBP using an in vitro acetylase assay. As shown in Figure 3A (top), histone H3 was acetylated in the presence (lane 2), but not in the absence (lane 1), of the p300 HAT domain. Importantly, full-length IE2 markedly reduced H3 acetylation (lane 3) in a dose-dependent manner (data not shown). IE1 also inhibited acetylation (lane 4), suggesting that this activity might, in part, reside in the N-terminal 85 residues shared by IE1 and IE2. The use of IE2 deletion mutants revealed that, although not as effective as full-length IE2, residues 1–98 (lane 5) and 1–290 (lane 6) caused significant inhibition of H3 acetylation, whereas the N-terminal-deleted mutant (residues 169–579) did not (lane 7). The controls, BSA (lane 8) and GST (lane 9), had no significant inhibitory effect. The relative histone acetylation inhibitory activities (middle panel), the constructs used, and the results of the experiments using them (bottom panel) are shown. Our results suggest that the N-terminal 98 residues of IE2 might be directly involved in interfering with the HAT activity of p300 and are in agreement with the data showing that deletion mutants lacking this region display a markedly reduced ability to suppress p53 activity (Figure 1C).
Figure 3.
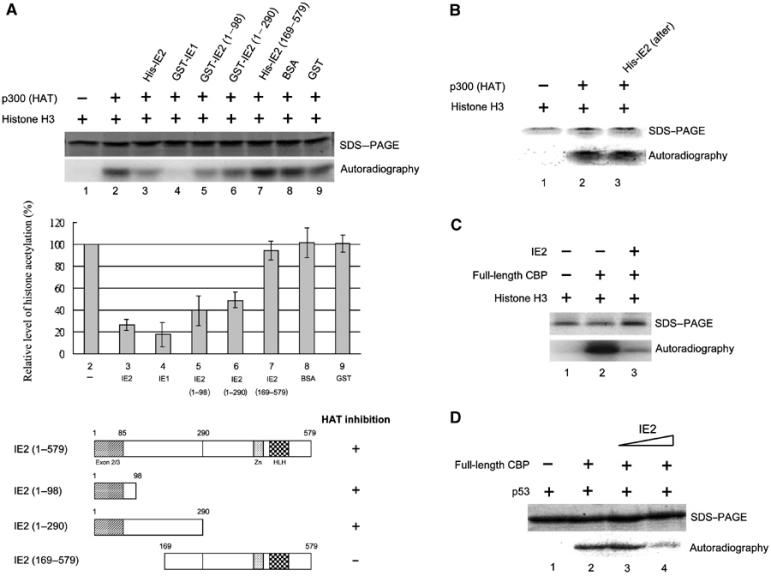
IE2 inhibits histone and p53 acetylation by p300/CBP in vitro. (A) Mapping of the HAT inhibitory domain of IE2. Indicated proteins were mixed with [3H]acetyl CoA in in vitro acetylase assays, then the reaction products were separated by 15% SDS–PAGE (top panel) and analyzed by phosphoimaging. The relative levels of histone acetylation were measured by normalization to histone H3 acetylation in the absence of IE2 (lane 2) (middle panel). The bottom panel is a schematic diagram of the IE2 deletion mutants and their HAT inhibitory activity. (B) IE2 added after incubation of histones, the p300 HAT domain, and [3H]acetyl CoA has no effect on H3 acetylation. The experiments were performed as in (A) except that IE2 was added 30 min after incubation of all other reagents. (C, D) IE2 suppresses acetyltransferase activity of full-length CBP in vitro. (C) As in (A) but using baculovirus-expressed full-length CBP, instead of the p300 HAT domain. (D) As in (C) but using p53 as substrate.
The alternative explanation that IE2 might act as a deacetylase, rather than inhibit HAT activity, was unlikely, since sequence alignment showed no homology between IE2 and known HDACs (data not shown) and the HDAC inhibitor, TSA or nicotinamide, had no effect on the IE2-mediated inhibition of p53 function (Figure 1A). To exclude formally this possibility, an alternative HAT assay was used in which histones were first incubated for 30 min with [3H]acetyl CoA and the p300 HAT domain, then IE2 was added for 15 min; using this system, histone acetylation would be reduced in the presence of IE2 if it did act as a histone deacetylase. However, this was not the case (Figure 3B). The inhibitory effect of IE2 on H3 acetylation was also evident when full-length CBP was used instead of the p300 HAT domain (Figure 3C). Furthermore, the acetylation of p53, a non-histone substrate, was also suppressed by IE2 (Figure 3D). Thus, like E1A (Chakravarti et al, 1999), IE2 can inhibit the HAT activity of p300/CBP.
IE2 binds to p300/CBP
Since IE2 inhibited p300/CBP acetylase activity in vitro (Figure 3) and IE2 could be co-immunoprecipitated with p300 in cells (Figure 2), we tested if it directly binds to p300/CBP. As expected, GST-full-length IE2 bound to CBP fragments consisting of amino acids 737–1626 (Figure 4A, CBP3; containing the HAT domain) or 1626–2260 (CBP4, containing the CH3 domain), but not those containing residues 117–737 (CBP2; containing the C/H1 and CREB-binding domains) or 2260–2389 (CBP5). GST or beads alone did not pull down any CBP fragment. Consistently, p53 bound to GST-IE2 beads (Figure 4B, lane 4). However, IE1, although exhibiting HAT inhibition activity (Figure 3A, lane 4), failed to interact with p53 (Figure 4B, lane 3).
Figure 4.
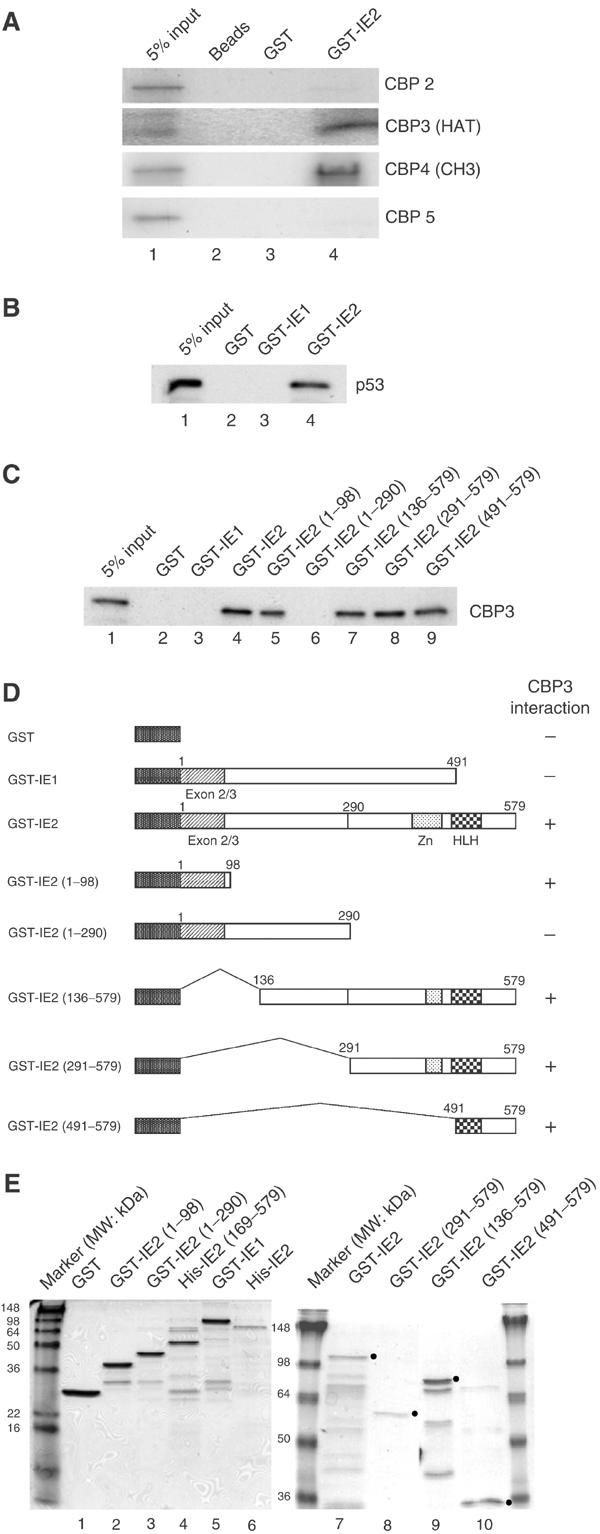
In vitro binding of IE2 and CBP. (A) Binding of GST-IE2 fusion proteins to in vitro-translated 35S-labeled CBP fragments. Beads (lane 2), GST beads (lane 3), or GST-IE2 beads (lane 4) were incubated with CBP2, CBP3, CBP4, or CBP5 fragments, then the bound complexes were separated by 10% SDS–PAGE and the gels subjected to autoradiography. (B) GST-IE2, but not GST-IE1, pulls down in vitro-translated p53. (C) Mapping of the CBP3-binding domain of IE2. Similar experiments were performed except that in vitro-translated 35S-methionine-labeled CBP3 was incubated with various GST-IE2 fragments. (D) Schematic diagram of the IE2 deletion mutants used in (C). (E) Purity of IE1 and IE2 derivatives used in Figures 3 and 4 shown by Coomassie blue staining. The molecular weight markers are indicated. The dots represent the expected position of the band corresponding to each protein.
Since CBP3 contains the HAT domain, we reasoned that IE2 might affect HAT activity by binding to this region. We therefore examined the binding of different GST-IE2 fragments to CBP3. As shown in Figure 4C, full-length IE2, the construct containing only the N-terminal 98 amino acids, and all constructs containing the C-terminal 89 residues (136–579, 291–579, and 491–579) of IE2 were able to bind (lanes 4, 5, and 7–9). However, since GST-IE2 (1–290) did not bind (lane 6), it is not clear whether the binding seen with the N-terminal 98 residues is meaningful. Similarly, if residues 1–98 were important, we might expect to observe binding of CBP3 to GST-IE1, which has the same N-terminal 85 residues as IE2; however, this was not the case (lane 3). Thus, we conclude that IE2 might interact with the HAT domain of CBP through its C-terminal 89 residues. The constructs used and the results obtained with them are shown in Figure 4D. Figure 4E shows sodium dodecyl sulfate (SDS) gels demonstrating the purity of the Escherichia coli-expressed proteins used in Figures 3 and 4. These results demonstrate that IE2 specifically targets the acetyltransferases, p300/CBP, and it is therefore highly likely that this plays an important role in the IE2-mediated downregulation of p53 function.
IE2 inhibits p300/CBP-induced p53 acetylation and p21 expression in vivo
These in vitro results prompted us to ask whether IE2 regulates the acetylation status of p53 in vivo. As shown in Figure 5, transfection of H1299 cells with p300 resulted in a clear increase of acetylated p53 (Ace-p53 (K373), compare lane 5 to 2) and this effect was inhibited by expression of WT IE2 (compare lane 6 to 5) but not of IE2 (169–579), which lacks both HAT and p53 inhibitory activities (compare lane 7 to 6). It should be noted that total p53 protein levels were similar in all experiments. Together with the previous in vitro data, these results show that IE2 inhibits p53 acetylation by p300/CBP, suggesting that the expression of genes downstream of p53 might also be affected by IE2. Consistently, p21 protein levels were increased in cells transfected with p53 or p53 and p300 (p21, lanes 2 and 5) and this effect was blocked by IE2 (compare lane 3 to 2 and lane 6 to 5), but not by IE2 (169–579) (compare lane 4 to 3 and lane 7 to 6). These studies imply that acetylation of p53, at least by p300/CBP, is highly correlated with p21 gene activation.
Figure 5.
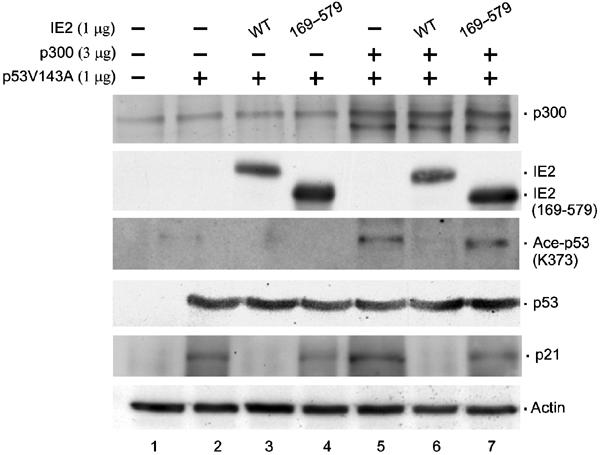
In vivo suppression of p300-dependent p53 acetylation and p21 expression by IE2. H1299 cells were transfected with the indicated expression plasmids, followed by Westerns using antibodies against p300 (Ab-1, Oncogene), IE2 (polyclonal), acetylated p53 (K373) (06-916, Upstate), p53 (FL-393, Santa Cruz), p21 (C-19, Santa Cruz), or actin (MAB1501, Chemicon). Actin: loading control.
IE2 represses p53 acetylation site mutant-induced differential effects in both transactivation and in vivo DNA-binding abilities
To determine whether p53 acetylation plays a role in its transactivation activity, we constructed three p300/CBP acetylation site mutants of p53 (3A, 3R, and 3D) in which all three lysines, K320, K373, and K382, were replaced by alanine (A), arginine (R), or aspartic acid (D). These mutants were expressed in H1299 cells to assay their effect on p21 promoter (Figure 6A). To our surprise, we found that these three mutants displayed differential ability to activate the p21 promoter, with mutants 3D and 3A having only about 20 and 40%, respectively, of the activity of WT p53, whereas mutant 3R retained most of the activity (gray bars). Importantly, the transactivation activities induced by either WT p53 or the p53 acetylation site mutants were all repressed by IE2 (dark bars), indicating that the IE2-mediated suppression of p53 function is independent of p53 acetylation. As revealed by Western blots (bottom panel), the differences in luciferase activity caused by the WT or mutant p53 were not due to differences in the levels of expressed proteins. Figure 6B shows that the three mutants were not recognized by antibodies specific for p53 acetylated at either K373/382 or K320. In addition, less p21 protein level was found induced by mutants 3A and 3D, whereas similar induction of the protein was seen with the 3R mutant (p21, compare lanes 3, 4, and 5 to lane 2).
Figure 6.
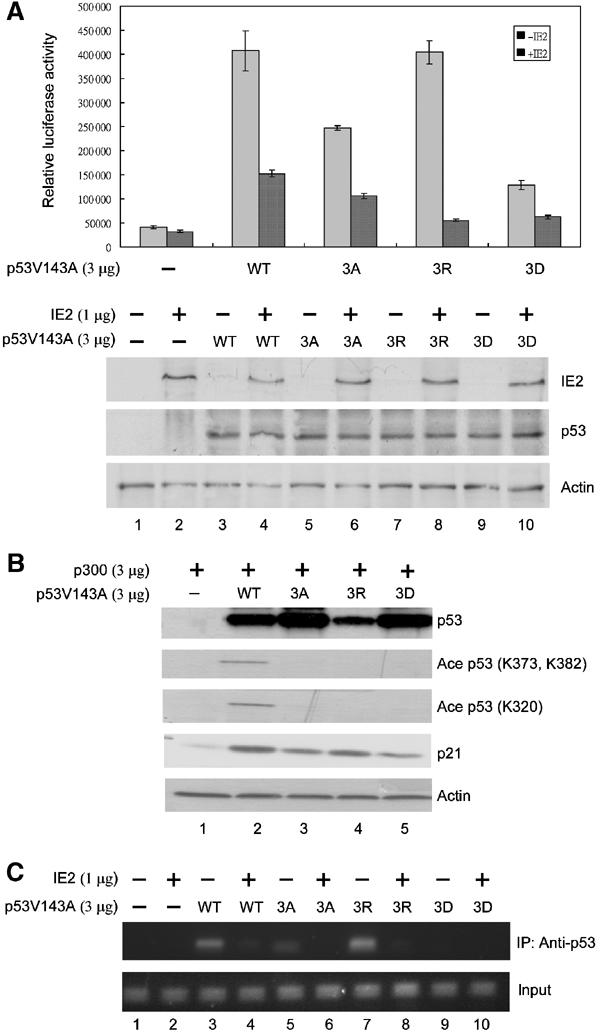
Effects of p53 acetylation site mutants. H1299 cells were transfected with the indicated expression vectors, followed by luciferase assays (A), Western analyses (B), and ChIP assays (C). (A) Differential effect of p53 acetylation mutants on the p21 promoter. Lysines 320, 373, and 382 of p53 were simultaneously substituted with alanine (3A), arginine (3R), or aspartic acid (3D). The promoter activity of WT p53 or mutant 3A, 3R, or 3D was assayed in the absence (gray bar) or presence (dark bar) of IE2. Bottom panel: Western blots of the expression levels of p53 (Ab-6, Oncogene) and IE2 (polyclonal). Actin: loading control. (B) Western blots using antibodies against p53 (Ab-6, Oncogene), p53 acetylated at K373 and K382 (06-758, Upstate), p53 acetylated at K320 (05-915, Upstate), p21 (Ab-397, Santa Cruz), or PCNA (SC56, Santa Cruz). (C) ChIP assays showing binding of the p53 mutants to the endogenous p21 promoter in the absence (lanes 1, 3, 5, 7, and 9) or presence (2, 4, 6, 8, and 10) of IE2. The upper panel shows the PCR products from the immunoprecipitates using anti-p53 antibodies (FL-393, Santa Cruz), while the bottom panel is the input control that shows the PCR products from the cell extract before IP. The transfections in (B, C) were similar to those in (A), except that the p21-Luc reporter construct was omitted and p300 expression plasmid was included in (B).
A similar effect was observed in the chromatin immunoprecipitation (ChIP) assays, which can detect the in vivo binding of p53 to DNA by precipitating the p53-associated chromatin, followed by PCR amplification of the p53 binding sequences (Figure 6C). We found that 3A (lane 5) and 3D (lane 9), but not 3R (lane 7), showed reduced DNA binding to the endogenous p21 promoter (compare to lane 3). In the presence of IE2, the binding of all four forms was significantly suppressed (compare lane 4 to 3, 6 to 5, 8 to 7, and 10 to 9). Together, since mutant 3R retained most of the functions of p53, we believe that, at least in the case of p21 promoter, acetylation of p53 may not directly affect its binding to DNA and the expression of the downstream genes and that the IE2-mediated downregulation of p53 activity is very likely due to reduced local histone acetylation.
IE2 decreases in vivo, but not in vitro, DNA binding of p53 and p53-dependent local histone acetylation on responsive promoters
We next determined whether IE2 affects the local histone acetylation on endogenous p53-dependent promoters. Before we answered this question, we used ChIP and in vitro gel shift assays to explore the mechanisms underlying IE2-mediated inhibition of p53 binding to DNA. Consistent to what we observed in Figure 6C, in H1299 cells, ChIP assays detected that IE2 blocked the in vivo binding of p53 to the p21 promoter dose-dependently (Figure 7A, upper panel). Furthermore, IE2 binding to the promoter was only clearly observed when p53 was bound to the promoter. At the higher concentration of IE2, the binding of both p53 and IE2 to the promoter was decreased (lane 4). These experiments clearly demonstrated that IE2 could suppress the binding of p53 to the p21 promoter in vivo and that the interaction of IE2 with this promoter was dependent on p53. Similar results were obtained when the IE2 effect on the PIG3 promoter, another p53-responsive gene that plays an important role in cell apoptosis (el-Deiry, 1998), was examined (lower panel). However, the disruption of p53/DNA complex by IE2 was not observed in in vitro gel shift assays with procaryotically purified proteins. As shown in Figure 7B, p53 was bound to WT (lanes 1–5), but not mutant (lanes 7–10), p21 oligo in a dose-dependent manner. Inclusion of a 2.5-fold molar excess (for monomeric IE2 and p53) of IE2 in the reaction did not significantly block the formation of p53/DNA complex (compare lane 5 to 4). And as expected, IE2, although capable of binding to its DNA-binding sequences on the RSV LTR (Tsai et al, 1997) (data not shown), did not bind to p21 oligo (lane 6). The observation that IE2 interfered with p53 binding to DNA only in vivo, not in vitro, strongly suggests that, rather than simply block p53's DNA-binding domain, IE2 may utilize a more complex mechanism such as alteration of the chromatin structure to regulate p53's function.
Figure 7.

IE2 decreases binding of p53 to, and histone acetylation on, p53-responsive promoters in vivo but does not impair p53 binding to p21 oligo in vitro. H1299 cells (A, C, D) or RKO cells (E, F) were transfected with the indicated expression vectors, followed by ChIP assays. (A) IE2 suppresses p53 binding to endogenous p21 (upper panel) and PIG3 (lower panel) promoters. (B) A gel shift assay shows that the DNA-binding activity of p53 is not impeded by IE2. The 32P end-labeled double-stranded oligonucleotide containing the WT (lanes 1–6) or mutant (lanes 7–10) p53-binding site on the p21 gene promoter (−2337 to −2358, relative to the transcription start site) was incubated with indicated proteins, followed by electrophoresis in a 4% nondenaturing polyacrylamide gel. (C) IE2 reduces p53-dependent histone H3 acetylation on the endogenous p21 promoter. (D) The acetyltransferase activity of p300 enhances the binding of p53. (E) The HAT inhibitory domain of IE2 is required to inhibit endogenous p53 binding to DNA and local histone acetylation in a TSA-insensitive manner. (F) The HAT inhibition mutant of IE2 binds to the endogenous p53/p300 complex on the p21 promoter. The antibodies used were directed against p53 (FL-393, Santa Cruz), IE2 (MAB810 from Chemicon) in (A, C), IE2 (polyclonal) in (F), Ace H3 (06-599, Upstate), Ace H4 (06-866, Upstate), or p300 (NA-46, Oncogene).
The ChIP assays were then applied to test whether IE2 influences p53-stimulated histone acetylation on p21 promoter (Figure 7C). IE2, when coexpressed with p53 in H1299 cells, abolished the acetylation of endogenous H3 (lane 3). Consistently, no binding of IE2 to the p21 promoter was seen in the absence of p53 (lane 4). Furthermore, we found that binding of p53 to its cognate sites was increased by exogenous WT p300, but not by acetylase-dead p300 (Figure 7D, lanes 3 and 4, upper panel). In agreement with the results shown in Figure 1D, we demonstrated that p53 binding in the presence of acetylase-dead p300 was even lower than that mediated by the endogenous p300 (compare lane 4 to lane 2). Again, we believe that this phenomenon was due to the dominant-negative effect caused by the mutant p300. As expected, acetylation of histones on the promoter only increased in the presence of both exogenous WT p300 and p53 (Figure 7D, middle panel). These results clearly indicate that p300 acetylase activity plays an important role in the binding of p53 to its target promoter and imply that p53 binding to DNA may be increased by local histone acetylation.
Similar results were obtained in RKO cells. We found that TSA slightly increased both the binding of endogenous p53 to the p21 promoter and local histone acetylation (Figure 7E, compare lane 5 to lane 1). However, both activities were suppressed by WT IE2, but not by the N-terminal deleted mutants (136–579 and 169–579) lacking the HAT inhibition activity (compare lanes 7 and 8 to lane 6). These results reinforce our earlier hypothesis that HDACs are unlikely to be involved in IE2-mediated inhibition of p53 function (Figure 1A). Furthermore, only WT IE2 reduced the association of p300 with the promoter (Figure 7F, lane 2), suggesting that, similar to IE2, the interaction of p300 with this promoter is dependent on p53. Similar results were observed in the apoptosis-associated promoters, PIG3 and BID (Sax et al, 2002) (data not shown). Taken together, these studies further support the notion that IE2 may reduce the local histone acetylation and therefore downregulate p53 binding to chromatinized DNA in vivo.
WT IE2, but not the HAT inhibition mutants, exhibits antiapoptosis activity
Thus far, we had demonstrated that IE2 could downregulate p53 function and inhibit the acetylase activity of the p53 coactivators, p300/CBP, both in vitro and in vivo. We had also shown that both overexpressed and endogenous p53 were affected by IE2. We then asked if any of the physiological functions of p53 is influenced by IE2. To this end, DNA fragmentation assays were performed in RKO cells to examine if IE2 suppresses p53-mediated cell apoptosis. As shown in Figure 8, DNA laddering induced by UV (lane 2) was reduced by expression of WT IE2 (lane 3), but not of the HAT inhibition mutants of IE2, IE2 (136–579) (lane 4) and IE2 (169–579) (lane 5). The lower panel shows the expression level of IE2 and its derivatives in RKO cells. To evaluate the number of apoptotic cells, we then applied terminal deoxyribonucleotide transferase-mediated dUTP nick end labeling (TUNEL) assays that also detect the fragmented DNA in individual cells. As revealed in Figure 9, in the absence of IE2, treatment of RKO cells with UV dramatically increased the number of TUNEL-positive cells (compare F to E). When WT IE2 was added, the UV-induced apoptosis was greatly inhibited (compare G to F). As expected, IE2 (169–579) exhibited limited ability to protect cells from programmed cell death (compare H to F). The relative percentage of TUNEL-positive cells was shown (Q). Importantly, under × 400 magnification, we observed that the UV-induced changes in the cell morphology (compare I, J, K, and L), such as shrinkage, round-up, and blebbing, and nuclear fragmentation (compare M, N, O, and P, arrowhead) were all significantly prevented by WT IE2, but not by IE2 (169–579). The proportion of dead cells as judged by cell morphology changes was estimated and shown (R). In addition to TUNEL assays, consistent results were obtained by annexin V staining (data not shown). Annexin V binds to phosphatidylserine exposed at the outer leaflet of the cytoplasmic membrane at the early stage of the apoptotic process. Based on these data, we strongly believe that regulation of p53 function by HCMV IE2 through inhibition of HAT activity is biologically significant.
Figure 8.
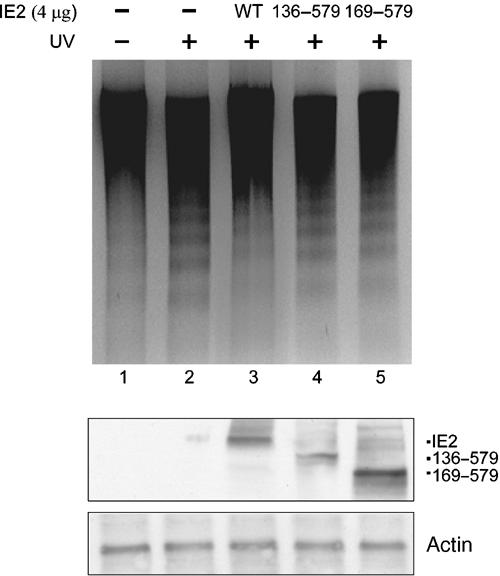
WT IE2, but not the HAT inhibition mutants, inhibits UV-induced DNA fragmentation in RKO cells. The fragmented DNAs were extracted, recovered, and analyzed on a 2% agarose gel containing ethidium bromide. Bottom panel: Western blots that show the expression of IE2 proteins (polyclonal) and p53 (FL-393, Santa Cruz). Actin: loading control.
Figure 9.
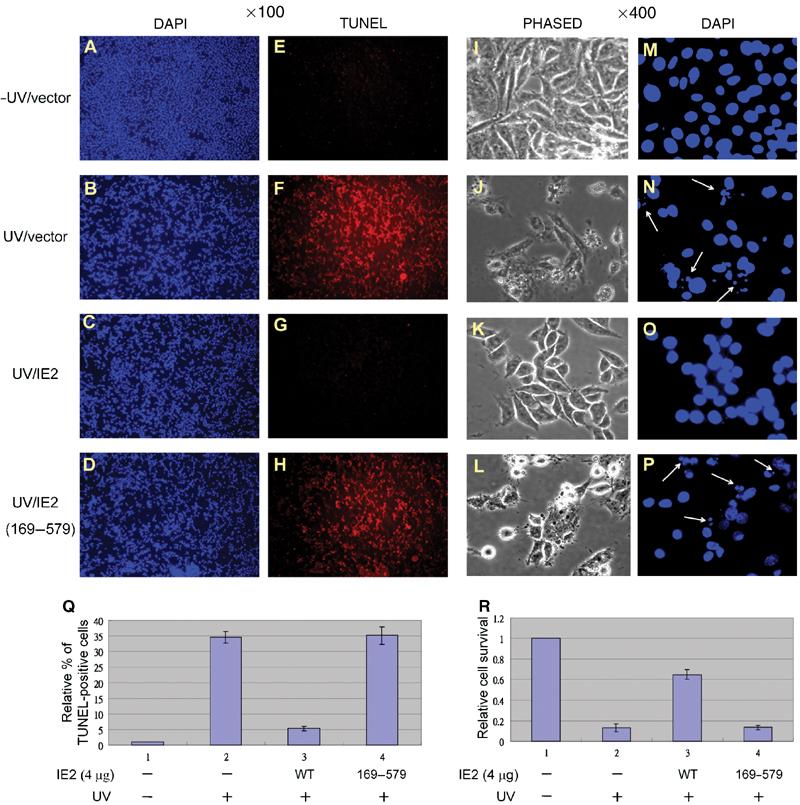
Apoptosis determined by TUNEL. RKO Cells were stained doubly with DAPI (blue dots, A–D, M–P) and TUNEL (red dots, E–H) and visualized under a fluorescence microscope with either × 100 magnification (A–H) or × 400 magnification (I–P). The relative % of the TUNEL-positive cells (Q) and the cell survival rate (R) were each determined in four fields ( × 100 magnification for TUNEL-positive cells, × 400 magnification for cell survival) for each group and normalized to that of the group of cells without UV treatment and IE2 transfection. The arrowheads point to the fragmented nucleus.
Discussion
Loss of p53 activity is a common event in tumor progression. Many DNA tumor viruses interfere with p53 functions through various mechanisms (Mauser et al, 2002, and references therein). The present data reveal a novel p53 regulatory pathway controlled by HCMV IE2. We found that IE2 was able to suppress the HAT activity of the p53 transcriptional coactivators, p300/CBP (Figures 3, 5, and 7), and that IE2 deletion mutants (aa 136–579 and 169–579) lacking the minimal N-terminal HAT inhibitory domain failed to repress the in vivo DNA binding of p53 and local histone acetylation (Figures 1C, 5, 7E, F, 8, and 9). Since p53 acetylation mutant K320/373/382R still activated p21 promoter and this activation could be repressed by IE2, we conclude that the in vivo impairment of p53 function by IE2 is most likely due to decreased p53 binding to chromatinized DNA, a result of IE2-mediated inhibition of local histone acetylation.
IE2 inhibits endogenous p53
Importantly, the IE2-mediated inhibition of p53 function was also prominent in p53-positive cells such as RKO. In RKO cells, we demonstrated that IE2 inhibited (1) p53-dependent p21 promoter activity (Figure 1B), (2) the in vivo p53 binding to and (3) local histone acetylation on p53-responsive promoters (Figure 7E and F), and (4) UV-induced apoptosis (Figures 8 and 9). Moreover, we showed that inhibition of p53 activity mediated by IE2 unlikely involved disruption of p300/p53 complex (Figure 2C). However, we cannot completely eliminate this possibility due to 30–40% transfection efficiency in RKO cells, as determined by the expression of the green fluorescent protein (data not shown). But since the ability of p300 to interact with p53 was not reduced at all even though 40% of the cells contained IE2 (Figure 2C, compare lane 2 to lane 1), it is most likely that disruption of p300/p53 complex is not the main mechanism that contributes to the IE2 effect on p53 activity.
IE2 and the coactivators p300/CBP
Like p53, the HATs and HDACs have recently been found to be major targets of viral proteins (Caron et al, 2003). Interestingly, IE2 is reported to recruit coactivators, such as p300/CBP (Schwartz et al, 1996) or P/CAF (Bryant et al, 2000), to potentiate synergistically the expression of the target genes. These results seem to contradict directly our present findings, since, if the HAT activity of p300/CBP is inhibited by IE2, these factors would not be expected to enhance IE2-mediated gene activation. This discrepancy may be explained, in part, by the observation from Western blots, which showed that IE2 decreased acetylation of the bulk p53 (Figure 5, compare lane 6 to lane 5), but not of the bulk histones (data not shown). However, since IE2 suppressed local histone acetylation around the p53-binding sites on the p21 promoter (Figure 7; ChIP assays), we believe that the effect of IE2 inhibition of the acetyltransferase activity of p300/CBP might be p53-specific or promoter-dependent. This hypothesis remains to be investigated. In addition, a recent report indicates that p300 not only acetylates p53 but also ubiquitinizes p53 (Grossman et al, 2003). Whether IE2 is involved in this process is currently unknown.
IE1 and IE2
HCMV IE1 and IE2 proteins are two of the alternatively spliced products encoded by the same gene under the control of the major IE promoter. These two proteins have the first 85 amino acids in common, but otherwise have totally different amino-acid sequences and have distinct functions (Castillo and Kowalik, 2002). In addition to IE2, in our present study, IE1 was shown to have HAT inhibitory activity in vitro (Figure 3A). However, the effect in vivo remains to be further studied. Nevertheless, this finding poses the interesting question of why IE1 does not affect p53 function. One possible explanation is based on the observations that IE1 does not bind to p53 (Figure 4B) or form a stable complex with CBP, at least within the HAT domain (Figure 4C, lane 3). Thus, we believe that, even if IE1 could repress HAT activity, it might not affect p53-specific gene activation because it could not be recruited by p53.
p53 acetylation and its functions
As mentioned in Introduction, the direct consequences of p53 acetylation are still a matter of debate. In our study, although p53 acetylation seemed highly correlated with its transactivation activity (Figure 5), one cannot rule out the possibility that the stimulated p53 activity seen when coactivators are recruited is due to local histone acetylation. To answer this question, lysines 320, 373, and 382 of p53 were simultaneously substituted with alanine (3A), arginine (3R), or aspartic acid (3D) (Figure 6A). Acetylation of lysine residues neutralizes their positive charge; so, if charge were the only important factor, we would expect to see a similar activity for the WT and 3A mutant, a decreased activity for 3R, and possibly an increased activity for 3D. However, if the positions of these lysines, rather than their acetylation, were important, we would expect 3A and 3D, but not 3R, to show decreased activity, because, in theory, substitution of lysines with arginines should cause the least change in overall conformation. In addition, if the lysine residues were important for p53 degradation, for example, as sites for modifications, such as ubiquitination, the three mutants would not be degraded and therefore retain p53 activity, as reported by Nakamura et al (2000). Interestingly, our results matched the second hypothesis. We found that 3R retained most of the DNA-binding and transactivation activity of the WT protein, whereas 3A and 3D were much less active in both assays (Figure 6). Together with the finding that these three p53 mutants were all affected by IE2 (Figure 6), we strongly believe that other acetylation targets, such as histones, are important in IE2-mediated regulation of p53 function.
Physiological significance of IE2 inhibition of p53 functions
Cell apoptosis and cell cycle arrest are the two most studied downstream functions of p53. Many viruses have evolved specific proteins to stimulate cell cycle progression by antagonizing p53 function (see Introduction). In addition to cell cycle control regulation, viruses, including HCMV, have developed several antiapoptotic pathways (Skaletskaya et al, 2001). Our present study further demonstrated that HCMV IE2 inhibits UV-induced DNA fragmentation (Figures 8 and 9), a marker of cell apoptosis. These data provide evidence for an additional role of IE2 in the regulation of p53 function. Since both p53 and p300/CBP are tumor suppressor proteins, IE2, by inhibiting their activities, could potentially increase the incidence of tumor formation. The physiological significance of the IE2-mediated inhibition of both p53 and p300/CBP function is currently under extensive investigation.
Materials and methods
Plasmids and mutagenesis
The following plasmids have been described previously: pSVp53V143A, which expresses the temperature-sensitive p53 derivative, p53V143A; p3PREcCAT, the p53 reporter construct; pSIE1 and pSIE2, which encode IE1 and IE2 proteins under the control of the SV40 promoter (Tsai et al, 1996); pRL-SV40 (Dual-Luciferase Reporter Assay System, Promega); p21-Luc (el-Deiry et al, 1993); pSK-p300-myc and pSK-p300D1399Y-myc, which, respectively, encode WT p300 and the acetylase-dead mutant (Ito et al, 2001); pSKII-CBP2, pSKII-CBP3, pSKII-CBP4, and pSKII-CBP5, which produce in vitro-translated CBP fragments (Asahara et al, 2002); pGEM4-p53, which produces in vitro-translated WT p53 (Chen et al, 1993); pGEX-3X-IE1 and pGEX-3X-IE2 series, which express GST-fusion IE1 and IE2 derivatives in bacteria (Wang et al, 1997); and pET11d-IE2 and pET11d-IE2 (169–579), which express His-tagged IE2 derivatives in E. coli (Wang et al, 2000). pSEP7IE1 and pSEP7IE2, which express IE1 and IE2 proteins in mammalian cells (Figure 1A), were constructed by insertion of the corresponding cDNA into the HindIII site of plasmid pSEP7, which was derived from pSEP4 by replacing the CMV promoter with the SV40 promoter. pSIE2 (136–579) was constructed by the digestion of pSIE2 with SmaI to remove the DNA sequence corresponding to amino-acid residues 1–135. pSIE2 (169–579) was generated by insertion of the corresponding cDNA into pSG424IE2 (Tsai et al, 1996) between HindIII and BamHI sites. pZeoSV2(−)-p300 (Figures 1D and 5), which expresses p300 under the control of the SV40 promoter, was constructed by insertion of the p300 cDNA into pZeoSV2(−) (Invitrogen) between NotI and HindIII sites. Point mutations in the C-terminal domain of p53V143A were introduced using an in vitro site-directed mutagenesis system (Stratagene) according to the manufacturer's instructions.
Cell culture, CAT assay, and luciferase assay
The human lung carcinoma cell line H1299, the human colon cancer cell line RKO, and human fibroblast HEL299 cells were all obtained from, and maintained as instructed by, the ATCC. Temperature shift assays, CAT assays, and luciferase assays were performed as described previously (Juan et al, 2000). With the exception of the experiment shown in Figure 1A, in which calcium phosphate-mediated DNA transfection was used as described in Juan et al (2000), cells were transfected using lipofectamine™ 2000 (Invitrogen) in serum-free medium. Normally, 5 × 105 or 1 × 106 H1299 cells, or 1 × 106 or 2–3 × 106 RKO cells were seeded onto six-well plates or 10 cm plates (for IP or ChIP assays, respectively), and transfections carried out at 80% confluency for H1299 or 60% confluency for RKO.
Western, IP, and GST pull-down
Western analyses, GST pull-down, and IP assays were performed as described in Juan et al (2000) except that the cells were lysed on ice in ice-cold RIPA lysis buffer (50 mM Tris–HCl, pH 8.0, 5 mM EDTA, 150 mM NaCl, 1% NP-40, 0.1% SDS, and 0.5% deoxycholate) containing a mixture of 13 protease inhibitors (Complete TM, Roche Molecular Biochemicals). Equal amounts (approximately 40–150 μg) of total extract protein were separated on a 10 or 15% SDS–polyacrylamide gel. For simultaneous detection of p300 and histone protein, a discontinuous gel (upper gel: 6%; lower gel: 15%) was used. The polyclonal anti-IE2 antibody was raised by immunizing rabbits with IE2 (aa 169–579), purified from E. coli XA-90, according to standard protocols. For IP assays, 0.6 mg (H1299) or 1 mg (RKO) of cell lysate protein was used.
In vitro acetylase assays
A 3 μg portion of histone H3 (Upstate) or 5 μg of p53, made from baculovirus (Wang et al, 2003), was mixed with various IE2 fragments and/or the p300 HAT domain (residues 1195–1673), purified from bacteria (Ogryzko et al, 1996), and/or full-length CBP, purified from baculovirus (Chen et al, 2001). Acetylation reactions were performed for 0.5–1 h at 30°C in the presence of [3H]acetyl-CoA (Amersham) in 50 mM Tris–HCl, pH 8.0, 10% glycerol, 1 mM DTT, 1 mM PMSF, and 0.1 mM EDTA. Then the reaction mixtures were separated by 15% SDS—polyacrylamide gel electrophoresis (PAGE) and the gels were stained with Coomassie blue, dried, and subjected to autoradiography.
ChIP and gel shift assays
ChIP assays were performed as described (Ishizuka and Lazar, 2003) with minor modifications. The final DNA samples were analyzed with 25–35 cycles of PCR to amplify indicated promoter sequences. Each cycle consisted of denaturation at 94°C for 30 s, annealing at 63°C (PIG3) or 65°C (p21) for 30 s, and extension at 72°C for 25 s (PIG3) or 2 min (p21). The primers used for amplifying PIG3 and p21 promoters were described previously (Szak et al, 2001; Jin et al, 2002). The gel mobility shift assays were conducted as described in Shieh et al (1997). The oligonucleotide containing the WT p53 consensus site in the p21 gene promoter (Shieh et al, 1997) and the mutant sequences 5′-AATTCTCGAGGAACACACAAACCACTGTTGCTCGAG-3′ (sense) and 5′-AATTCTCGAGCAACAGTGGTTTGTGTGTTCCTCGAG-3′ (antisense) were used. These sequences exactly match the p21 gene region analyzed in the ChIP assays. Binding of IE2 to the CRS element of RSV-LTR was performed as described previously (Tsai et al, 1997).
DNA fragmentation, TUNEL, and annexin V assays
RKO cells were transfected as described above except that, after 24 h, the cells were exposed to UV-C (25 J/m2, 72 h of recovery for DNA fragmentation; 25 J/m2, 24 h of recovery for annexin V staining; 50 J/m2, 12 h of recovery for TUNEL) and harvested. Fragmented DNAs were extracted using a Wizard® Genomic DNA Purification Kit (Promega) and analyzed on a 2% agarose gel containing ethidium bromide. The TUNEL and annexin V experiments were performed with the In situ Cell Death Detection kit, Fluorescein (Roche Molecular Biochemicals) and Annexin V-Cy3 Reagent Kit (BioVision), respectively, according to the manufacturer's instructions.
Acknowledgments
We thank Dr S-C Chen for constructing plasmids pSEP7IE1 and pSEP7IE2, Dr S-Y Shieh for providing bacterially purified His-tagged p53, Drs Y-S Lin, KW Kinzler, B Vogelstein, Y Nakatani, T-P Yao, M Montminy, and J-Y Chen for supplying plasmids. We also gratefully acknowledge Dr T Barkas for carefully editing the manuscript and Drs JL Workman, T-P Yao, Y-HW Lee, W-Y Tarn, and H-M Shih for critical comments. This work was supported by grants from the NHRI to L-JJ, from the NHRI, the IBMS, and the NSC to C-WW, and from the NSC and MOE to MD-TC.
References
- Asahara H, Tartare-Deckert S, Nakagawa T, Ikehara T, Hirose F, Hunter T, Ito T, Montminy M (2002) Dual roles of p300 in chromatin assembly and transcriptional activation in cooperation with nucleosome assembly protein 1 in vitro. Mol Cell Biol 22: 2974–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89: 1175–1184 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T (1996) The CBP co-activator is a histone acetyltransferase. Nature 384: 641–643 [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W (2003) Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 15: 164–171 [DOI] [PubMed] [Google Scholar]
- Brown CE, Lechner T, Howe L, Workman JL (2000) The many HATs of transcription coactivators. Trends Biochem Sci 25: 15–19 [DOI] [PubMed] [Google Scholar]
- Bryant LA, Mixon P, Davidson M, Bannister AJ, Kouzarides T, Sinclair JH (2000) The human cytomegalovirus 86-kilodalton major immediate-early protein interacts physically and functionally with histone acetyltransferase P/CAF. J Virol 74: 7230–7237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron C, Col E, Khochbin S (2003) The viral control of cellular acetylation signaling. BioEssays 25: 58–65 [DOI] [PubMed] [Google Scholar]
- Castillo JP, Kowalik TF (2002) Human cytomegalovirus immediate early proteins and cell growth control. Gene 290: 19–34 [DOI] [PubMed] [Google Scholar]
- Castillo JP, Yurochko AD, Kowalik TF (2000) Role of human cytomegalovirus immediate-early proteins in cell growth control. J Virol 74: 8028–8037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti D, Ogryzko V, Kao HY, Nash A, Chen H, Nakatani Y, Evans RM (1999) A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 96: 393–403 [DOI] [PubMed] [Google Scholar]
- Chen CJ, Deng Z, Kim AY, Blobel GA, Lieberman PM (2001) Stimulation of CREB binding protein nucleosomal histone acetyltransferase activity by a class of transcriptional activators. Mol Cell Biol 21: 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Funk WD, Wright WE, Shay JW, Minna JD (1993) Heterogeneity of transcriptional activity of mutant p53 proteins and p53 DNA target sequences. Oncogene 8: 2159–2166 [PubMed] [Google Scholar]
- el-Deiry WS (1998) Regulation of p53 downstream genes. Semin Cancer Biol 8: 345–357 [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, Nakatani Y, Livingston DM (2003) Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 300: 342–344 [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90: 595–606 [DOI] [PubMed] [Google Scholar]
- Gu W, Shi X-L, Roeder RG (1997) Synergistic activation of transcription by CBP and p53. Nature 387: 819–827 [DOI] [PubMed] [Google Scholar]
- Hermiston TW, Malone CL, Witte PR, Stinski MF (1987) Identification and characterization of the human cytomegalovirus immediate-early region 2 gene that stimulates gene expression from an inducible promoter. J Virol 61: 3214–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Lazar MA (2003) The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Mol Cell Biol 23: 5122–5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 20: 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zeng SX, Dai MS, Yang XJ, Lu H (2002) MDM2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. J Biol Chem 277: 30838–30843 [DOI] [PubMed] [Google Scholar]
- Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, Wu CW (2000) Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem 275: 20436–20443 [DOI] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 21: 2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol 19: 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W (2004) Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci USA 101: 2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408: 377–381 [DOI] [PubMed] [Google Scholar]
- Mauser A, Saito S, Appella E, Anderson CW, Seaman WT, Kenney S (2002) The Epstein–Barr virus immediate-early protein BZLF1 regulates p53 function through multiple mechanisms. J Virol 76: 12503–12512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Roth JA, Mukhopadhyay T (2000) Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol Cell Biol 20: 9391–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87: 953–959 [DOI] [PubMed] [Google Scholar]
- Prives C, Manley JL (2001) Why is p53 acetylated? Cell 107: 815–818 [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E (1998) DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev 12: 2831–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS (2002) BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 4: 842–849 [DOI] [PubMed] [Google Scholar]
- Schwartz R, Helmich B, Spector DH (1996) CREB and CREB-binding proteins play an important role in the IE2 86-kilodalton protein-mediated transactivation of the human cytomegalovirus 2.2-kilobase RNA promoter. J Virol 70: 6955–6966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh S-Y, Ikeda M, Taya Y, Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334 [DOI] [PubMed] [Google Scholar]
- Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS (2001) A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci USA 98: 7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE (1994) Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 265: 391–394 [DOI] [PubMed] [Google Scholar]
- Szak ST, Mays D, Pietenpol JA (2001) Kinetics of p53 binding to promoter sites in vivo. Mol Cell Biol 21: 3375–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HL, Kou GH, Chen SC, Wu CW, Lin YS (1996) Human cytomegalovirus immediate-early protein IE2 tethers a transcriptional repression domain to p53. J Biol Chem 271: 3534–3540 [PubMed] [Google Scholar]
- Tsai HL, Kou GH, Tang F-M, Wu CW, Lin YS (1997) Negative regulation of a heterologous promoter by human cytomegalovirus immediate-early protein IE2. Virology 238: 372–379 [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107: 149–159 [DOI] [PubMed] [Google Scholar]
- Wahl GM, Carr AM (2001) The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol 3: E277–E286 [DOI] [PubMed] [Google Scholar]
- Wang YC, Huang CF, Tung SF, Lin YS (2000) Competition with TATA box-binding protein for binding to the TATA box implicated in human cytomegalovirus IE2-mediated transcriptional repression of cellular promoters. DNA Cell Biol 19: 613–619 [DOI] [PubMed] [Google Scholar]
- Wang YF, Chen SC, Wu FY, Wu CW (1997) The interaction between human cytomegalovirus immediate-early gene 2 (IE2) protein and heterogeneous ribonucleoprotein A1. Biochem Biophys Res Commun 232: 590–594 [DOI] [PubMed] [Google Scholar]
- Wang YH, Tsay YG, Tan BC, Lo WY, Lee SC (2003) Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem 278: 25568–25576 [DOI] [PubMed] [Google Scholar]
- Zhang W, Guo XY, Hu GY, Liu WB, Shay JW, Deisseroth AB (1994) A temperature-sensitive mutant of human p53. EMBO J 13: 2535–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]