

Abstract
Mycobacteriophages have provided numerous essential tools for mycobacterial genetics, including delivery systems for transposons, reporter genes, and allelic exchange substrates, and components for plasmid vectors and mutagenesis. Their genetically diverse genomes also reveal insights into the broader nature of the phage population and the evolutionary mechanisms that give rise to it. The substantial advances in our understanding of the biology of mycobacteriophages including a large collection of completely sequenced genomes indicates a rich potential for further contributions in tuberculosis genetics and beyond.
INTRODUCTION
Mycobacteriophages are viruses that infect mycobacterial hosts including Mycobacterium smegmatis and Mycobacterium tuberculosis. The first mycobacteriophages were isolated in the late 1940s using M. smegmatis as a host (1, 2), followed by isolation of phages that infect M. tuberculosis (3). The application of phages with distinct host preferences to typing clinical mycobacterial isolates was recognized, and numerous studies on mycobacteriophage typing were published over the subsequent 30 years (4–12). In the 1950s a variety of further investigations focusing on the biology of these phages and their potential applications were initiated including studies on generalized transduction (13), viral morphology (14, 15), lysogeny (16–20), transfection of phage DNA (21, 22), and other biochemical features (23–40). These early contributions provided a critical foundation for the further characterization and application of mycobacteriophages to tuberculosis research that emerged from them.
Prior to the mid-1990s, the lack of methods for efficient and reproducible introduction of DNA into mycobacteria—coupled with the lack of simple plasmid vectors—represented substantial impediments to the development of facile systems for genetic manipulation of M. tuberculosis and other mycobacteria (41). Bacteriophages played a critical role in overcoming these roadblocks, in part because of the ability to introduce phage DNA into M. smegmatis spheroplasts using strategies developed earlier for Streptomyces (42, 43) and because of the development of shuttle phasmids, which grow as phages in mycobacteria and as large plasmids in Escherichia coli (44, 45). These contributed to the development of methods for more efficient transfection and transformation, demonstration of genetically selectable systems, and general methods for gene transfer into mycobacteria (46–49).
In the early 1990s, the first complete genome sequence of a mycobacteriophage was described (50), followed by another dozen or so over the following decade (51–54). With the advancements in DNA sequence technologies and the development of integrated research and education programs in phage discovery and genomics (55–58), the number of sequenced mycobacteriophage genomes now exceeds 500. These show substantial degrees of both genetic diversity and genetic novelty, providing insights into viral evolution and greatly expanding the potential for developing additional tools for mycobacterial genetics and for gaining insights into the physiology of their hosts (59–61).
In this chapter I review our current understanding of mycobacteriophages including their genetic diversity and applications for tuberculosis genetics. The focus will primarily be on recent developments, and there are a number of other reviews that the reader may find useful (18, 59, 60, 62–78).
GENERAL ASPECTS OF MYCOBACTERIOPHAGES
Mycobacteriophage Isolation
Bacteriophages can typically be isolated from any environment in which their bacterial host—or close relatives of their host—is present. There is no obvious reservoir of M. tuberculosis outside of its human host, but mycobacteriophages have been isolated from various patient samples including stool (79–81). However, because there are numerous saprophytic mycobacterial relatives, mycobacteriophages can be readily isolated from environmental samples such as soil or compost, using M. smegmatis or other nonpathogenic mycobacteria as hosts; a subset of these phages also infect M. tuberculosis (discussed further below). Phages can also be isolated by release from lysogenic host bacteria (82, 83), and phage genomic sequences can be identified in sequenced mycobacterial genomes (83–88).
M. smegmatis has proven to be a useful surrogate for phage isolation, using either direct plating from environmental samplesorbyenrichment, in which the sample is first incubated with M. smegmatis to promote amplification of phages for that host. Typically, direct plating yields only a small number of plaques from ~10% of the samples that are tested, whereas enrichment generates plaques from a higher proportion of samples, and the phage titers can be greater than 109 plaque forming units (pfu)/ml. Although enrichment might be anticipated to reduce the diversity of the phages isolated due to the potential growth advantages that a subset of phages might enjoy under these conditions, there is little evidence to support this with the mycobacteriophages. However, it does appear to alter the prevalence with which different phage types are recovered. For example, phages with one particular genome type (Cluster A; see below for details) represent over 40% of the phages recovered after enrichment, whereas they are only 25% isolated by direct plating. However, phages of a second genome type (Cluster B) are more prevalently recovered by direct plating (27%) than by enrichment (13%). But both of these general phage types themselves encompass considerable diversity and can be divided into several subtypes (i.e., subclusters), and enrichment also influences the abundance of particular subclusters. For example, phages of Subcluster A4 are relatively abundant in enriched samples (25% of the Cluster Aphages, and 10% of the total) relative to direct plating (10% of Cluster A phages and 1% of the total). However, these biases in isolation should be interpreted cautiously, because there are numerous influences on which particular phages are selected for sequencing and further characterization. Additional hosts including M. tuberculosis could be used for enrichment, although plating directly on M. tuberculosis lawns is challenging because of its slow growth and the propensity for growth of contaminants that increase the difficulty of identifying plaques.
All mycobacteriophages examined to date carry double-stranded DNA (dsDNA). No phages with single-stranded DNA (ssDNA) or RNA genomes—or with virion morphologies other than the caudoviruses (see below)—have been described. It is plausible that they exist in these or other environments but that the isolation methods used to date bias against their growth and recovery. Phages with ssDNA or RNA genomes have been described for many other bacterial hosts, and it is somewhat surprising that none have been recovered for the mycobacteria. However, we note that this is also true for the phages described to date for all other species within the Actinomycetales.
Mycobacteriophage Virion Morphologies
The virion morphologies of many mycobacteriophages have been examined, and although three morphotypes of dsDNA tailed phages (Siphoviridae, Myoviridae, and Podoviridae) have been described for other hosts, only phages with siphoviral and myoviral morphologies (with long, flexible, noncontractile tails and contractile tails, respectively) have been identified for the mycobacteria (Fig. 1). No podoviral morphotypes (with short stubby tails) have been described, and with several hundred phages examined microscopically, it seems likely that these are truly excluded from the mycobacteriophage population. Although this could result from evolutionary exclusion, the rampant horizontal genetic exchange common to bacteriophages would suggest that this is unlikely, and it is plausible that a physical barrier, such as the complex mycobacterial cell wall, prevents them from accessing the cell membrane for successful infection. This may also account for why phages other than the tailed dsDNA phages have yet to be discovered.
FIGURE 1.
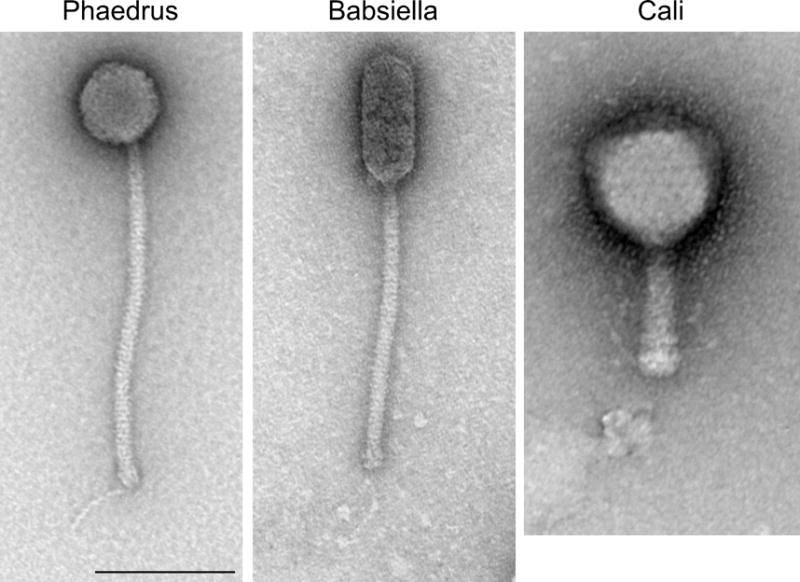
Mycobacteriophage morphologies. Three examples of virion morphologies are illustrated. Phaedrus and Babsiella exhibit siphoviral morphologies with long flexible tails; Phaedrus has an isometric head, whereas the Babsiella head is prolate. Cali is an example of myoviral morphology. Scale bar is 100 nm. 10.1128/microbiolspec.MGM2-0032-2013.f1
By far the majority (>90%) of mycobacteriophages have siphoviral morphologies with long flexible tails, and only ~8% have myoviral morphologies, all of which belong to a single genome type (Cluster C; see below). Most of the siphoviral morphotypes have isometric heads (varying in diameter from 50 to 80 nm), but a few have prolate heads with length:width ratio ranges from 2.5:1 to 4:1 (Fig. 1). There is substantial variation in tail length, ranging from 135 to 350 nm (89). The range in diameters of the isometric capsids—corresponding to differences in genome length—likely reflects different triangulation numbers in the capsid structures, but few have been determined experimentally. An electron micrograph–based reconstruction of mycobacteriophage Araucaria shows it has a T = 7 capsid symmetry (83), and a cryo-EM reconstruction of the phage BAKA capsid shows it has a T = 13 symmetry (90).
Phage Discovery and Genomics as an Integrated Research and Education Platform
Because phage genomes are relatively small, sequencing them is much less of a technological feat than it was 25 years ago. The chief limitation thus becomes the availability of phage isolates to sequence and characterize. In developing the Phage Hunters Integrating Research and Education (PHIRE) program at the University of Pittsburgh, this research mission (i.e., discovering and genomically characterizing new mycobacteriophages) is paired with a mission in science education, specifically to develop programs that introduce young students (high school and undergraduate students) to authentic scientific research (55–58). The success of this program fueled implementation of a nationwide research course for freshman undergraduate students in collaboration with the Science Education Alliance (SEA) program of the Howard Hughes Medical Institute. The SEA Phage Hunters Advancing Genomic and Evolutionary Science (SEA-PHAGES) initiative began in 2008 and has involved over 4,000 undergraduate students at over 70 institutions ranging from R1 research universities to primarily undergraduate teaching colleges. The research accomplishments of the PHIRE and SEA-PHAGES programs are reflected in the massive increase in the number of completely sequenced mycobacteriophage genomes, growing from fewer than 20 to more than 500 in a 10-year span (www.phagesdb.org). The impact on science education is indicated by numerous parameters including student surveys such as the Classroom Undergraduate Research Experience (CURE) instrument (91).
Successful implementation of the PHIRE and SEA-PHAGES programs is predicated on a suite of features that characterize phage discovery and genomics. The enormous diversity of the phage population stacks the deck in favor of individual phage hunters isolating a phage with novel features, in some instances being completely unrelated to previously sequenced phages. This, however, is only apparent from the genomic characterization, because little or nothing can be learned about phage diversity and evolution without the genomic sequence. Students name their phages following a non-systematic nomenclature that reflects the individualistic aspects of the phage genomes (see below). With over 500 completely sequenced genomes of phages isolated on M. smegmatis mc2155, the prospects of isolating a phage with a completely novel DNA sequence is greatly diminished, although it is still extremely rare to find two phages with identical or near-identical genomes. However, the diversity of phages isolated using other—but closely related—hosts is likely to be just as great, and it is easy to envisage that large collections of sequenced phage genomes using numerous hosts within the Actinomycetales are likely to substantially advance our understanding of viral evolution.
Seven additional attributes of the PHIRE program have been described that contribute to its productivity (55, 56). These are (i) technical simplicity in beginning the project, (ii) the lack of requirement for prior advanced conceptual understanding, (iii) flexibility in timing and implementation, (iv) multiple milestones for success, (v) a parallel project design with strong peer-mentoring opportunities and the potential to engage large numbers of students, (vi) authentic research leading to peer-reviewed publications, and (vii) project ownership that powerfully engages students in their own individual contributions. These attributes should be transferable to other research and education platforms, and there are several excellent examples (92–95).
MYCOBACTERIOPHAGE GENOMICS
At the time of writing, there are a total of 285 complete mycobacteriophage genomes available in GenBank, and we will focus on these in this chapter (Table 1). The total number of sequenced mycobacteriophages is larger—currently 531 (www.phagesdb.org)—and the outstanding genomes will be available in GenBank pending completion and review of the annotations. Of the 285, all except one were either isolated on M. smegmatis or are known to infect M. smegmatis. The single exception is DS6A (61), whose host preference is restricted to the M. tuberculosis complex (6, 24).
Table 1.
Sequenced mycobacteriophage genomes
| Phage | Cluster | Accession # | Length (bp) | GC % |
|---|---|---|---|---|
| Aeneas | A1 | JQ809703 | 53684 | 63.6 |
| Bethlehem | A1 | AY500153 | 52250 | 63.2 |
| BillKnuckles | A1 | JN699000 | 51821 | 63.4 |
| BPBiebs31 | A1 | JF957057 | 53171 | 63.4 |
| Bruns | A1 | JN698998 | 53003 | 63.6 |
| Bxb1 | A1 | AF271693 | 50550 | 63.6 |
| DD5 | A1 | EU744252 | 51621 | 63.4 |
| Doom | A1 | JN153085 | 51421 | 63.8 |
| Dreamboat | A1 | JN660814 | 51083 | 63.9 |
| Euphoria | A1 | JN153086 | 53597 | 63.7 |
| Jasper | A1 | EU744251 | 50968 | 63.7 |
| JC27 | A1 | JF937099 | 52169 | 63.6 |
| KBG | A1 | EU744248 | 53572 | 63.6 |
| KSSJEB | A1 | JF937110 | 51381 | 63.6 |
| Kugel | A1 | JN699016 | 52379 | 63.8 |
| Lesedi | A1 | JF937100 | 50486 | 63.8 |
| Lockley | A1 | EU744249 | 51478 | 63.4 |
| Marcell | A1 | JX307705 | 49186 | 64.0 |
| MrGordo | A1 | JN020140 | 50988 | 63.8 |
| Museum | A1 | JF937103 | 51426 | 63.6 |
| PattyP | A1 | KC661273 | 52057 | 63.6 |
| Perseus | A1 | JN572689 | 53142 | 63.7 |
| RidgeCB | A1 | JN398369 | 50844 | 64.0 |
| SarFire | A1 | KF024726 | 53701 | 63.8 |
| SkiPole | A1 | GU247132 | 53137 | 63.6 |
| Solon | A1 | EU826470 | 49487 | 63.8 |
| Switzer | A1 | JF937108 | 52298 | 63.8 |
| Trouble | A1 | KF024724 | 52102 | 63.6 |
| U2 | A1 | AY500152 | 51277 | 63.7 |
| Violet | A1 | JN687951 | 52481 | 63.8 |
| Che12 | A2 | DQ398043 | 52047 | 62.9 |
| D29 | A2 | AF022214 | 49136 | 63.5 |
| L5 | A2 | Z18946 | 52297 | 62.3 |
| Odin | A2 | KF017927 | 52807 | 62.3 |
| Pukovnik | A2 | EU744250 | 52892 | 63.3 |
| RedRock | A2 | GU339467 | 53332 | 64.5 |
| Trixie | A2 | JN408461 | 53526 | 64.5 |
| Turbido | A2 | JN408460 | 53169 | 63.3 |
| Bxz2 | A3 | AY129332 | 50913 | 64.2 |
| HelDan | A3 | JF957058 | 50364 | 64.0 |
| JHC117 | A3 | JF704098 | 50877 | 64.0 |
| Jobu08 | A3 | KC661281 | 50679 | 64.0 |
| Methuselah | A3 | KC661272 | 50891 | 64.2 |
| Microwolf | A3 | JF704101 | 50864 | 64.0 |
| Rockstar | A3 | JF704111 | 47780 | 64.3 |
| Vix | A3 | JF704114 | 50963 | 64.0 |
| Arturo | A4 | JX307702 | 51500 | 64.1 |
| Backyardigan | A4 | JF704093 | 51308 | 63.7 |
| Dhanush | A4 | KC661271 | 51373 | 63.9 |
| Eagle | A4 | HM152766.1 | 51436 | 63.9 |
| Flux | A4 | JQ809701 | 51370 | 63.9 |
| ICleared | A4 | JQ896627 | 51440 | 63.9 |
| LHTSCC | A4 | JN699015 | 51813 | 63.9 |
| Medusa | A4 | KF024733 | 51384 | 63.9 |
| MeeZee | A4 | JN243856 | 51368 | 63.9 |
| Peaches | A4 | GQ303263.1 | 51376 | 63.9 |
| Sabertooth | A4 | JX307703 | 51377 | 63.9 |
| Shaka | A4 | JF792674 | 51369 | 63.9 |
| TiroTheta9 | A4 | JN561150 | 51367 | 63.9 |
| Wile | A4 | JN243857 | 51308 | 63.7 |
| Airmid | A5 | JN083853 | 51241 | 60.0 |
| Benedict | A5 | JN083852 | 51083 | 59.8 |
| Cuco | A5 | JN408459 | 50965 | 60.9 |
| ElTiger69 | A5 | JX042578 | 51505 | 59.8 |
| George | A5 | JF704107 | 51578 | 61.0 |
| LittleCherry | A5 | KF017001 | 50690 | 60.9 |
| Tiger | A5 | JQ684677 | 50332 | 60.7 |
| Blue7 | A6 | JN698999 | 52288 | 61.4 |
| DaVinci | A6 | JF937092 | 51547 | 61.5 |
| EricB | A6 | JN049605 | 51702 | 61.5 |
| Gladiator | A6 | JF704097 | 52213 | 61.4 |
| Hammer | A6 | JF937094 | 51889 | 61.3 |
| Jeffabunny | A6 | JN699019 | 48963 | 61.6 |
| HINdeR | A7 | KC661275 | 52617 | 62.8 |
| Timshel | A7 | JF957060 | 53278 | 63.1 |
| Astro | A8 | JX015524.1 | 52494 | 61.4 |
| Saintus | A8 | JN831654 | 49228 | 61.2 |
| Alma | A9 | JN699005 | 53177 | 62.5 |
| PackMan | A9 | JF704110 | 51339 | 62.6 |
| Goose | A10 | JX307704 | 50645 | 65.1 |
| Rebeuca | A10 | JX411619 | 51235 | 65.1 |
| Severus | A10 | KC661279 | 49894 | 64.4 |
| Twister | A10 | JQ512844 | 51094 | 65.0 |
| ABU | B1 | JF704091 | 68850 | 66.5 |
| Chah | B1 | FJ174694 | 68450 | 66.5 |
| Colbert | B1 | GQ303259.1 | 67774 | 66.5 |
| Fang | B1 | GU247133 | 68569 | 66.5 |
| Harvey | B1 | JF937095 | 68193 | 66.5 |
| Hertubise | B1 | JF937097 | 68675 | 66.4 |
| IsaacEli | B1 | JN698990 | 68839 | 66.5 |
| JacAttac | B1 | JN698989 | 68311 | 66.5 |
| Kikipoo | B1 | JN699017 | 68839 | 66.5 |
| KLucky39 | B1 | JF704099 | 68138 | 66.5 |
| Morgushi | B1 | JN638753 | 68307 | 66.4 |
| Murdoc | B1 | JN638752 | 68600 | 66.4 |
| Newman | B1 | KC691258 | 68598 | 66.5 |
| Oline | B1 | JN192463 | 68720 | 66.4 |
| Oosterbaan | B1 | JF704109 | 68735 | 66.5 |
| Orion | B1 | DQ398046 | 68427 | 66.5 |
| OSmaximus | B1 | JN006064 | 69118 | 66.3 |
| PG1 | B1 | AF547430 | 68999 | 66.5 |
| Phipps | B1 | JF704102 | 68293 | 66.5 |
| Puhltonio | B1 | GQ303264.1 | 68323 | 66.4 |
| Scoot17C | B1 | GU247134 | 68432 | 66.5 |
| SDcharge11 | B1 | KC661274 | 67702 | 66.5 |
| Serendipity | B1 | JN006063 | 68804 | 66.5 |
| ShiVal | B1 | KC576784 | 68355 | 66.5 |
| TallGrassMM | B1 | JN699010 | 68133 | 66.5 |
| Thora | B1 | JF957056 | 68839 | 66.5 |
| ThreeOh3D2 | B1 | JN699009 | 68992 | 66.5 |
| UncleHowie | B1 | GQ303266.1 | 68016 | 66.5 |
| Vista | B1 | JN699008 | 68494 | 66.5 |
| Vortex | B1 | JF704103 | 68346 | 66.5 |
| Yoshand | B1 | JF937109 | 68719 | 66.5 |
| Arbiter | B2 | JN618996 | 67169 | 68.9 |
| Ares | B2 | JN699004 | 67436 | 69.0 |
| Hedgerow | B2 | JN698991 | 67451 | 69.0 |
| Qyrzula | B2 | DQ398048 | 67188 | 68.9 |
| Rosebush | B2 | AY129334 | 67480 | 68.9 |
| Akoma | B3 | JN699006 | 68711 | 67.5 |
| Athena | B3 | JN699003 | 69409 | 67.5 |
| Daisy | B3 | JF704095 | 68245 | 67.6 |
| Gadjet | B3 | JN698992 | 67949 | 67.5 |
| Kamiyu | B3 | JN699018 | 68633 | 67.5 |
| Phaedrus | B3 | EU816589 | 68090 | 67.6 |
| Phlyer | B3 | FJ641182.1 | 69378 | 67.5 |
| Pipefish | B3 | DQ398049 | 69059 | 67.3 |
| ChrisnMich | B4 | JF704094 | 70428 | 69.1 |
| Cooper | B4 | DQ398044 | 70654 | 69.1 |
| KayaCho | B4 | KF024729 | 70838 | 70.0 |
| Nigel | B4 | EU770221 | 69904 | 68.3 |
| Stinger | B4 | JN699011 | 69641 | 68.6 |
| Zemanar | B4 | JF704104 | 71092 | 68.9 |
| Acadian | B5 | JN699007 | 69864 | 68.4 |
| Reprobate | B5 | KF024727 | 70120 | 68.3 |
| Alice | C1 | JF704092 | 153401 | 64.7 |
| ArcherS7 | C1 | KC748970 | 156558 | 64.7 |
| Astraea | C1 | KC691257 | 154872 | 64.7 |
| Ava3 | C1 | JQ911768 | 154466 | 64.8 |
| Breeniome | C1 | KF006817 | 154434 | 64.8 |
| Bxz1 | C1 | AY129337 | 156102 | 64.8 |
| Cali | C1 | EU826471 | 155372 | 64.7 |
| Catera | C1 | DQ398053 | 153766 | 64.7 |
| Dandelion | C1 | JN412588 | 157568 | 64.7 |
| Drazdys | C1 | JF704116 | 156281 | 64.7 |
| ET08 | C1 | GQ303260.1 | 155445 | 64.6 |
| Ghost | C1 | JF704096 | 155167 | 64.6 |
| Gizmo | C1 | KC748968 | 157482 | 64.6 |
| LinStu | C1 | JN412592 | 153882 | 64.8 |
| LRRHood | C1 | GQ303262.1 | 154349 | 64.7 |
| MoMoMixon | C1 | JN699626 | 154573 | 64.8 |
| Nappy | C1 | JN699627 | 156646 | 64.7 |
| Pio | C1 | JN699013 | 156758 | 64.8 |
| Pleione | C1 | JN624850 | 155586 | 64.7 |
| Rizal | C1 | EU826467 | 153894 | 64.7 |
| ScottMcG | C1 | EU826469 | 154017 | 64.8 |
| Sebata | C1 | JN204348 | 155286 | 64.8 |
| Shrimp | C1 | KF024734 | 155714 | 64.7 |
| Spud | C1 | EU826468 | 154906 | 64.8 |
| Wally | C1 | JN699625 | 155299 | 64.7 |
| Myrna | C2 | EU826466 | 164602 | 65.4 |
| Adjutor | D | EU676000 | 64511 | 59.9 |
| Butterscotch | D | FJ168660 | 64562 | 59.7 |
| Gumball | D | FJ168661 | 64807 | 59.6 |
| Nova | D | JN699014 | 65108 | 59.7 |
| PBI1 | D | DQ398047 | 64494 | 59.8 |
| PLot | D | DQ398051 | 64787 | 59.8 |
| SirHarley | D | JF937107 | 64791 | 59.6 |
| Troll4 | D | FJ168662 | 64618 | 59.6 |
| 244 | E | DQ398041 | 74483 | 63.4 |
| ABCat | E | KF188414 | 76131 | 63.0 |
| Bask21 | E | JF937091 | 74997 | 62.9 |
| Cjw1 | E | AY129331 | 75931 | 63.7 |
| Contagion | E | KF024732 | 74533 | 63.1 |
| Dumbo | E | KC691255 | 75736 | 63.0 |
| Elph10 | E | JN391441 | 74675 | 63.0 |
| Eureka | E | JN412590 | 76174 | 62.9 |
| Henry | E | JF937096 | 76049 | 63.0 |
| Kostya | E | EU816591 | 75811 | 63.5 |
| Lilac | E | JN382248 | 76260 | 63.0 |
| Murphy | E | KC748971 | 76179 | 62.9 |
| Phaux | E | KC748969 | 76479 | 62.9 |
| Phrux | E | KC661277 | 74711 | 63.1 |
| Porky | E | EU816588 | 76312 | 63.5 |
| Pumpkin | E | GQ303265.1 | 74491 | 63.0 |
| Rakim | E | JN006062 | 75706 | 62.9 |
| SirDuracell | E | JF937106 | 75793 | 62.9 |
| Toto | E | JN006061 | 75933 | 63.0 |
| Ardmore | F1 | GU060500 | 52141 | 61.5 |
| Bobi | F1 | KF114874 | 59179 | 61.7 |
| Boomer | F1 | EU816590 | 58037 | 61.1 |
| Che8 | F1 | AY129330 | 59471 | 61.3 |
| Daenerys | F1 | KF017005 | 58043 | 61.6 |
| DeadP | F1 | JN698996 | 56461 | 61.6 |
| DLane | F1 | JF937093 | 58899 | 61.9 |
| Dorothy | F1 | JX411620 | 58866 | 61.4 |
| DotProduct | F1 | JN859129 | 55363 | 61.8 |
| Drago | F1 | JN542517 | 54411 | 61.2 |
| Fruitloop | F1 | FJ174690 | 58471 | 61.8 |
| GUmbie | F1 | JN398368 | 57387 | 61.4 |
| Hamulus | F1 | KF024723 | 57155 | 61.8 |
| Ibhubesi | F1 | JF937098 | 55600 | 61.2 |
| Job42 | F1 | KC661280 | 59626 | 61.2 |
| Llij | F1 | DQ398045 | 56852 | 61.5 |
| Mozy | F1 | JF937102 | 57278 | 61.1 |
| Mutaforma13 | F1 | JN020142 | 57701 | 61.3 |
| Pacc40 | F1 | FJ174692 | 58554 | 61.3 |
| PMC | F1 | DQ398050 | 56692 | 61.4 |
| Ramsey | F1 | FJ174693 | 58578 | 61.2 |
| RockyHorror | F1 | JF704117 | 56719 | 61.1 |
| SG4 | F1 | JN699012 | 59016 | 61.9 |
| Shauna1 | F1 | JN020141 | 59315 | 61.7 |
| ShiLan | F1 | JN020143 | 59794 | 61.4 |
| SiSi | F1 | KC661278 | 56279 | 61.5 |
| Spartacus | F1 | JQ300538 | 61164 | 61.7 |
| Taj | F1 | JX121091 | 58550 | 61.9 |
| Tweety | F1 | EF536069 | 58692 | 61.7 |
| Velveteen | F1 | KF017004 | 54314 | 61.5 |
| Wee | F1 | HQ728524 | 59230 | 61.8 |
| Avani | F2 | JQ809702 | 54470 | 61.0 |
| Che9d | F2 | AY129336 | 56276 | 60.9 |
| Jabbawokkie | F2 | KF017003 | 55213 | 61.1 |
| Yoshi | F2 | JF704115 | 58714 | 61.0 |
| Angel | G | FJ973624 | 41441 | 66.7 |
| Avrafan | G | JN699002 | 41901 | 66.6 |
| BPs | G | EU568876 | 41901 | 66.6 |
| Halo | G | DQ398042 | 42289 | 66.7 |
| Hope | G | GQ303261.1 | 41901 | 66.6 |
| Liefie | G | JN412593 | 41650 | 66.8 |
| Konstantine | H1 | FJ174691 | 68952 | 57.4 |
| Predator | H1 | EU770222 | 70110 | 56.4 |
| Barnyard | H2 | AY129339 | 70797 | 57.5 |
| Babsiella | I1 | JN699001 | 48420 | 67.1 |
| Brujita | I1 | FJ168659 | 47057 | 66.8 |
| Island3 | I1 | HM152765 | 47287 | 66.8 |
| Che9c | I2 | AY129333 | 57050 | 65.4 |
| BAKA | J | JF937090 | 111688 | 60.7 |
| Courthouse | J | JN698997 | 110569 | 60.9 |
| LittleE | J | JF937101 | 109086 | 61.3 |
| Omega | J | AY129338 | 110865 | 61.4 |
| Optimus | J | JF957059 | 109270 | 60.8 |
| Redno2 | J | KF114875 | 108297 | 60.9 |
| Thibault | J | JN201525 | 106327 | 60.8 |
| Wanda | J | KF006818 | 109960 | 60.8 |
| Adephagia | K1 | JF704105 | 59646 | 66.6 |
| Anaya | K1 | JF704106 | 60835 | 66.4 |
| Angelica | K1 | HM152764 | 59598 | 66.4 |
| BarrelRoll | K1 | JN643714 | 59672 | 66.6 |
| CrimD | K1 | HM152767 | 59798 | 66.9 |
| JAWS | K1 | JN185608 | 59749 | 66.6 |
| TM4 | K2 | AF068845 | 52797 | 68.1 |
| MacnCheese | K3 | JX042579 | 61567 | 67.3 |
| Pixie | K3 | JF937104 | 61147 | 67.3 |
| Fionnbharth | K4 | JN831653 | 58076 | 68.0 |
| Larva | K5 | JN243855 | 62991 | 65.3 |
| JoeDirt | L1 | JF704108 | 74914 | 58.8 |
| LeBron | L1 | HM152763 | 73453 | 58.8 |
| UPIE | L1 | JF704113 | 73784 | 58.8 |
| Breezona | L2 | KC691254 | 76652 | 58.9 |
| Crossroads | L2 | KF024731 | 76129 | 58.9 |
| Faith1 | L2 | JF744988 | 75960 | 58.9 |
| Rumpelstiltskin | L2 | JN680858 | 69279 | 58.9 |
| Winky | L2 | KC661276 | 76653 | 58.9 |
| Whirlwind | L3 | KF024725 | 76050 | 59.3 |
| Bongo | M | JN699628 | 80228 | 61.6 |
| PegLeg | M | KC900379 | 80955 | 61.5 |
| Rey | M | JF937105 | 83724 | 60.9 |
| Butters | N | KC576783 | 41491 | 65.8 |
| Charlie | N | JN256079 | 43036 | 66.3 |
| Redi | N | JN624851 | 42594 | 66.1 |
| Catdawg | O | KF017002 | 72108 | 65.4 |
| Corndog | O | AY129335 | 69777 | 65.4 |
| Dylan | O | KF024730 | 69815 | 65.4 |
| Firecracker | O | JN698993 | 71341 | 65.5 |
| BigNuz | P | JN412591 | 48984 | 66.7 |
| Fishburne | P | KC691256 | 47109 | 67.3 |
| Jebeks | P | JN572061 | 45580 | 67.3 |
| Giles | Q | EU203571 | 53746 | 67.3 |
| Send513 | R | JF704112 | 71547 | 56.0 |
| Marvin | S | JF704100 | 65100 | 63.4 |
| Dori | Single | JN698995 | 64613 | 66.0 |
| DS6A | Single | JN698994 | 60588 | 68.4 |
| Muddy | Single | KF024728 | 48228 | 58.8 |
| Patience | Single | JN412589 | 70506 | 50.3 |
| Wildcat | Single | DQ398052 | 78296 | 57.2 |
Grouping of Mycobacteriophages into Clusters and Subclusters
A simple dotplot comparison of the 285 genomes reveals an obvious feature of these genomes: that there is substantial diversity (i.e., many different types) but that the diversity is heterogeneous, and some phages are more closely related to each other than to others (Fig. 2). To recognize this heterogeneity and to simplify discussion, analysis, and presentation, these phages are assorted into groups called clusters (Cluster A, Cluster B, Cluster C, etc.), with the main criterion being that grouping of phages within a cluster requires recognizable nucleotide sequence similarity spanning over 50% of genome lengths with another phage in that cluster (56). Phages without any close relatives are referred to as singletons. For most phages, assignment to a cluster is simple, and extensive DNA sequence similarity is clear and apparent (see Fig. 3). However, assignment of phages to clusters is a taxonomy of convenience and does not reflect well-defined distinctions based on phylogeny or evolutionary relationships. Closer examination of sequence relationships reveals that they are mosaic and are constructed from segments swapped horizontally across the phage population (54, 96). This mosaicism is evident at both the DNA and the gene product levels and imposes challenges to some cluster assignments.
FIGURE 2.

Dotplot comparison of 285 mycobacteriophage genomes. A concatenated file of 285 mycobacteriophage nucleotide sequences was compared against itself using the Gepard program (242) to generate the dotplot. The order of the genomes was arranged such that genomically related phages were adjacent to each other in this file, and the clusters of related phages (Clusters A, B, C, etc.) are shown above the plot. Five of the genomes are singletons with no closely related phages and are denoted collectively as Sin. 10.1128/microbiolspec.MGM2-0032-2013.f2
FIGURE 3.
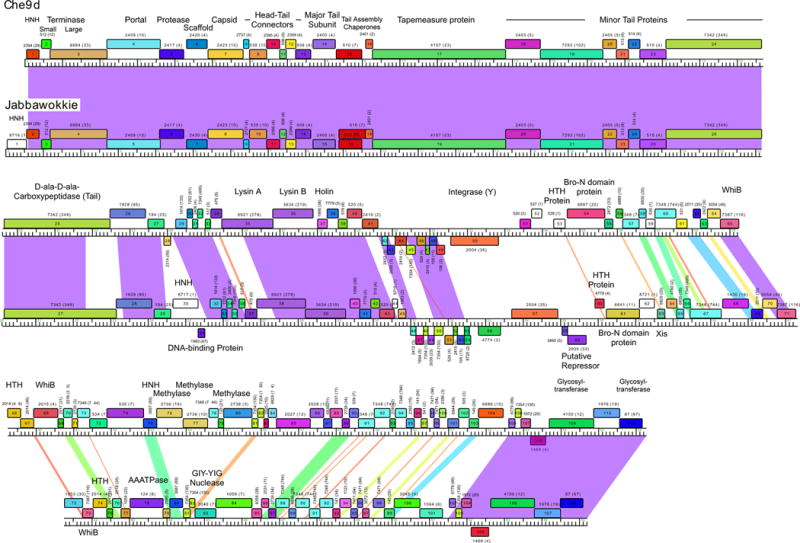
Comparison of mycobacteriophage Che9d and Jabbawokkie genome maps. Mycobacteriophages Che9d and Jabbawokkie are grouped into Subcluster F2, and their genome maps are shown as represented by the Phamerator program (100). Each genome is shown with markers, and the shading between the genomes reflects nucleotide sequence similarity determined by BLASTN, spectrum-colored with the greatest similarity in purple and the least in red. Protein-coding genes are shown as colored boxes above or below the genomes, reflecting rightward or leftward transcription, respectively. Each gene is assigned a phamily (Pham) designation based on amino acid sequence similarity (see text), as shown above or below each box, with the number of phamily members shown in parentheses; genes shown as white boxes are orphams and have no other phamily members. Putative gene functions are indicated. 10.1128/microbiolspec.MGM2-0032-2013.f3
Difficulties in cluster assignment have become more prevalent as the number of sequenced genomes has increased and generally fall into two categories. In the first, there are examples of genomes that are distantly related such that the nucleotide sequence similarity extends over substantial parts of the genomes but is sufficiently weak that it barely rises above the threshold levels of recognizable similarity, whether it is viewed by dotplot analysis or more quantitative methods. One example is phage Wildcat, which is currently classified as a singleton and although it has many similarities to the Cluster M phages, is not so closely related that it warrants inclusion in the cluster. A second conundrum arises from genomes that have segments of strong similarity to other phages, but over a span that either doesn’t convincingly meet the 50% threshold or spans more than 50% of one genome but not the other. An example is the inclusion of phage Che9c in Cluster I, where it belongs somewhat tenuously. Notwithstanding these caveats, the 285 genomes are grouped into 19 clusters (Clusters A to S) and five singleton genomes (Table 1).
Within some of these groupings, there is further heterogeneity in the degrees and extent of DNA sequence similarities. Thus, some of the clusters can be divided into subclusters, and the groupings are usually apparent by differences in the average nucleotide identity values (56, 89, 97, 98). However, the diversity varies from cluster to cluster, and thus the average nucleotide identity subcluster threshold values are not fixed and are relative to other members of the cluster. The subdivisions are usually apparent by dotplot comparison and, for example, the five subclusters within Cluster B can be easily seen in Fig. 2. Of the current 19 clusters, 9 are divided into subclusters, with the greatest division being the 10 subclusters within Cluster A (Table 1). The total number of different cluster-subcluster-singleton types is 47.
Assortment of Genes into Phamilies using Phamerator
An alternative representative of genome diversity is through gene content comparison (56, 89, 98, 99). This is accomplished with the program Phamerator (100), which sorts genes into phamilies (phams)—groups of genes in which each gene product has amino acid sequence similarity to at least one other phamily member above threshold levels (typically 32.5% amino acid identity or a BLASTP cutoff of less than 10−50) (100). Genomes can then be compared according to whether they do or do not have a member of each phamily. In a Phamerator database (Mycobacteriophage_285) generated with these 285 genomes there are a total of 3,435 phams, of which 1,322 (38%) are orphams, i.e., they contain only a single gene member. The relationships can be displayed using a network comparison in Splitstree (101) and in general show groupings of related phages that closely mirror the cluster designations from nucleotide sequence comparisons (56). It should be noted, however, that not all of the genes within the genomes necessarily have the same evolutionary histories—as a consequence of the mosaicism generated by horizontal genetic exchange—and thus these relationships only reflect the aggregate similarities and differences among the phages (56).
Comparative Genome Analysis
An especially informative representation of genome comparisons is by alignment of genome maps. These maps can be generated using Phamerator (100) and provide an overview of similarities at both the nucleotide and amino acid sequence levels (Fig. 3). Pairwise nucleotide sequence similarities are calculated using BLASTN, and values above a threshold level are displayed as colored shading between adjacent genomes in the display (Fig. 3); this is especially useful for identifying differences between genomes that have occurred in relatively recent evolutionary time. Because each gene is represented as a box colored according to its phamily membership, gene content comparisons and synteny also are displayed (Fig. 3).
Comparison of the genome maps of phages Che9d and Jabbawokkie—both members of Subcluster F2 (Table 1)—provides an informative illustration (Fig. 3). A large segment (~22 kbp) in the leftmost parts of the genomes (extending from the left ends through Che9d 25 and Jabbawokkie 27), encoding the virion structure and assembly genes, is extremely similar at the nucleotide sequence level (98% identity), as shown by the purple shading between the genomes in Fig. 3. The most obvious difference in this region is the insertion of an orpham (a gene that has no close mycobacteriophage relatives; i.e., it is the sole member of that pham) between the left physical end of the genome and a homing endonuclease (HNH) gene (Fig. 3), which is also predicted to encode an HNH endonuclease.
To the right of Che9d 25 and Jabbawokkie 27, there is another region of close similarity that extends to the left of the integrase genes and includes the lysis cassette (Fig. 3). Although there are several large interruptions in the alignment, the matching segments are closely related and vary between 90% and 100% nucleotide identity. Apart from the lysis genes and another HNH insertion in Jabbawokkie (gene 30), most of these genes are of unknown function. The entire right parts of the genomes (from ~31 kbp coordinates to the right physical ends) are much less closely related at the nucleotide sequence level, and many regions are sufficiently different that they are below the threshold for displaying any similarity; there are also many small segments of intermediate similarity and a block of closely related sequences (96% identity) at the right end. Another noteworthy feature is that some of the genes encode proteins of the same phamily even though there is little detectable nucleotide similarity. Perhaps the best example is the integrase genes (Che9d 50, Jabbawokkie 57; Fig. 3); the proteins share 45% amino acid identity but do not have recognizable DNA sequence similarity.
This comparison illustrates six general aspects of mycobacteriophage genomes. First, the genes are tightly packed and there is little noncoding space. Second, they are mosaic in their architecture, with different segments having different evolutionary histories (54, 96). Third, among those with siphoviral morphologies, the synteny of the virion structure and assembly genes is well conserved. Fourth, there are large numbers of genes of unknown function (56, 89, 98). Fifth, there is an abundance of small open reading frames, especially in the right parts of the genomes. Sixth, even where putative gene functions can be assigned, it is often unclear what their specific roles are (for example, why are there two whiB genes in Che9d, distantly related and sharing only ~25% amino acid sequence identity?).
Virion Structure and Assembly Genes
Phages with siphoviral morphologies (i.e., with a long flexible noncontractile tail)—including all mycobacteriophages except those in Cluster C—have a well-defined operon of virion structure and assembly genes in a syntenically well-conserved order. The genomes are typically represented, following the Lambda precedent, with these genes at the left end of the genome and transcribed rightward (see Fig. 3, 4, and 5). Sometimes this structural operon is situated such that the terminase genes at its left end are close to the physical ends of the genome, but there are many examples (e.g., Cluster A phages) where other genes are situated in this interval. Large subunit terminase genes can be readily identified in most of these phages, and many—but not all—have an identifiable small terminase subunit. The other virion structural genes typically follow in the order: terminase, portal, capsid maturation protease, scaffolding, capsid subunit, 4 to 6 head-tail connector proteins, major tail subunit, tail assembly chaperones, Tapemeasure protein, and 5 to 10 minor tail proteins.
FIGURE 4.
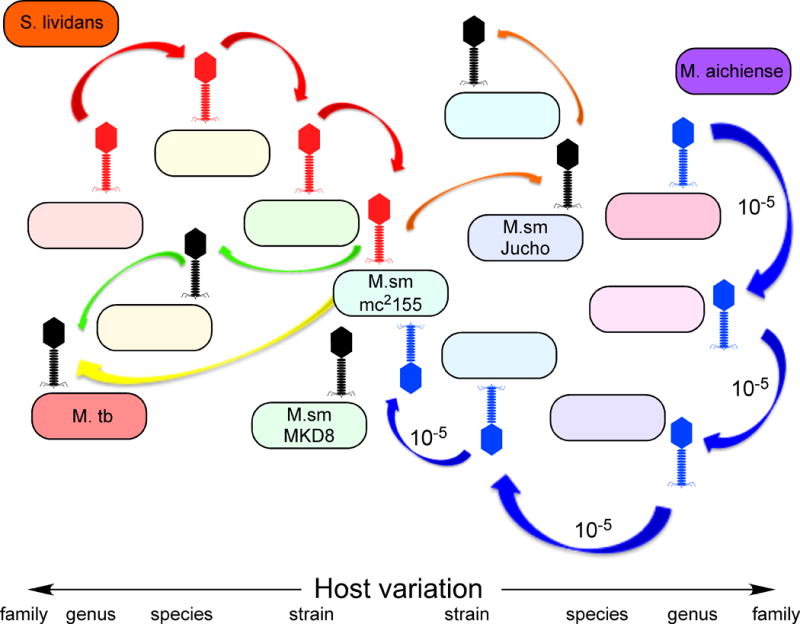
Functional genomics of mycobacteriophage Giles. A map of the mycobacteriophage Giles was generated using Phamerator and annotated as described for Fig. 3. Boxes below the genome indicate whether the gene is nonessential for lytic growth (yellow), likely essential (blue), or essential (green). Arrows indicate genes expressed in lysogeny (red) or early (green) or late (purple) lytic growth, with line thickness reflecting transcription strength. Reproduced with permission from Dedrick et al. (107). 10.1128/microbiolspec.MGM2-0032-2013.f4
FIGURE 5.
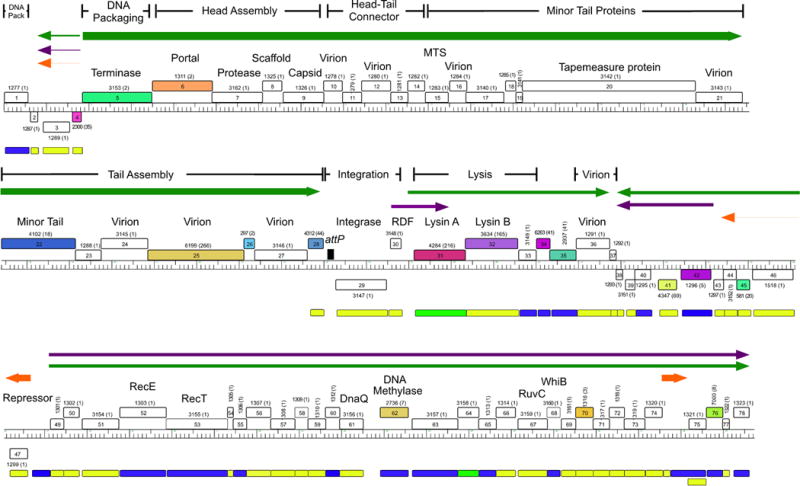
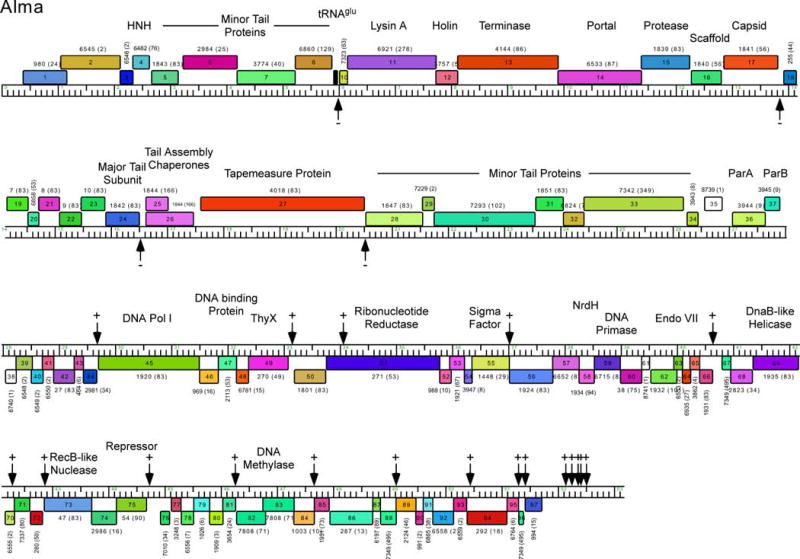
Genome map of mycobacteriophage Alma. The genome map of mycobacteriophage Alma was generated using Phamerator and is illustrated as described for Fig. 3. Alma is a Subcluster A6 phage and shares the features of other Cluster A phages in having multiple binding sites for its repressor protein (gp75). These stoperator sites are indicated by vertical arrows, and the orientation of the asymmetric sites relative to genome orientation are shown as (−) or (+). Stoperators were identified as sequences corresponding to the consensus sequence 5′-GATGAGTGTCAAG with no more than a single mismatch. Note that the stoperator consensus sequences can differ for different Subcluster A phages (98). 10.1128/microbiolspec.MGM2-0032-2013.f5
Although the gene order is very well conserved, there is substantial sequence diversity, to the extent that genes conferring some of the specific functions cannot be predicted from their sequence alone, although their position is also informative. However, because it is relatively simple to characterize the virions themselves, by SDS-PAGE, N-terminal sequencing, or mass spectrometry, correlations between the structural genes and proteins can be readily determined (50, 53, 90, 102, 103). In general, these studies show that the major capsid and tail subunits are the most abundant and that in some phages (e.g., L5) the capsid subunit is extensively covalently cross-linked, similar to the well-studied phage HK97 (50, 104). A scaffold protein gene involved in head assembly is absent from some mycobacteriophages, although its function may be provided by a domain of the capsid subunit.
The Tapemeasure protein is simple to identify because it is typically encoded by the longest gene in the genome, and the two reading frames immediately upstream are expressed via a programmed translational frameshift (105). Although the sequences of the Tapemeasure proteins are very diverse, they often contain small conserved domains corresponding to peptidoglycan hydrolysis motifs (54). The precise roles for these is not clear, but in TM4 it has been shown that the motif is not required for phage assembly or growth, and removal of it results in a predictably shorter tail but also a reduction of its efficiency of infection of stationary phase cells (106). Following the tapemeasure gene are the minor tail protein genes encoding the proteins that constitute the tail tip structure. These are among some of the most diverse sequences in the mycobacteriophages, with many complex relationships, reflecting recombination and a mutational bias likely associated with host resistance. Many of the phages have a tail protein containing a D-Ala-D-Ala carboxypeptidase motif that presumably promotes infection through enzymatic modification or remodeling of the peptidoglycan. As described below, mutations conferring expanded host range also map to these minor tail protein genes (99).
Phages such as BPs and its Cluster G relatives have a compact virion structure and assembly operons with a total of 26 genes in a ~24-kbp span (103). At the opposite extreme, the Cluster J phages have a similar number of structural genes, but they span >30 kbp as a result of insertions of HNH endonucleases, introns, inteins (see below), and an assortment of other genes including those coding for methyltransferases and glycosyltransferases, whose roles are unknown. In Marvin, there is an unusual genome rearrangement in which a segment encoding several of the minor tail protein genes is displaced and sits among the nonstructural genes in the right part of the genome (102). The organizations of structural genes in the Cluster C phages with myoviral morphologies are much less well characterized.
Nonstructural Genes
The right parts of the genomes encoding nonstructural functions are characterized by an abundance of small open reading frames of unknown functions. They usually include a subset of genes whose functions can be bioinformatically predicted, and these are often associated with nucleotide metabolism or DNA replication. For example, some phages encode a DNA polymerase similar to E. coli DNA Polymerase I, whereas others encode alpha subunits of bacterial DNA Polymerase III. While it is simple to reason that these are involved in phage DNA replication, this has not been shown directly, and there are many mycobacteriophages that do not encode their own DNA polymerase at all. So why it would be needed in some phages but not in others is not clear. It is also common to find helicase and primase-like proteins, and sometimes recombination functions, including both recA and recET-like genes. In phage Giles, proteins of previously unknown function are implicated in DNA replication from mutagenesis studies (see Fig. 4) (107); D29 gp65—predicted to be part of the RecA/DnaB helicase superfamily—has been demonstrated to be an exonuclease (108). But overall, very little is known about the mechanics of regulation of DNA replication in mycobacteriophages or many of the possible genes that are involved.
Some of the nonstructural genes are cytotoxic and kill the host when expressed or overexpressed. This has been extensively characterized in staphylococcal phages (109), where it has been developed as part of a drug development pipeline, but several mycobacteriophage-encoded cytotoxic proteins have also been identified. Initially, segments of the phage L5 genome were shown to be not tolerated on plasmid vectors and could not be transformed into M. smegmatis (110). Further dissection showed that L5 genes 77, 78, and 79 all have cytotoxic properties, with 77 being the most potent (111). Interestingly, expression of L5 gp79 seems to specifically inhibit cell division of M. smegmatis and promote filamentation (111). L5 gp77 appears to act by interacting directly with Msmeg_3532, a pyridoxal- 5′-phosphate-dependent L-serine dehydratase that converts L-serine to pyruvate (112), although it is unclear what the consequences of the interaction are. The role of such an interaction in the growth of L5—or the many other Cluster A phages encoding homologues of L5 gp77—is unclear.
tRNA and tmRNA Genes
A considerable variety of tRNA gene repertoires are seen among the mycobacteriophages. Many of them do not appear to code for any tRNA genes at all, whereas others have dozens, close to a nearly complete coding set (54). But there are many intermediate variations, with some carrying just a single tRNA gene, and others with five or six. For example, many of the Cluster A phages carry one or more tRNA genes, but their specificities are quite varied. For example, just among the Subcluster A2 phages, L5 has three tRNA genes (tRNAAsn, tRNATrp, and tRNAGln) (50), and D29 contains these plus two more (tRNAGlu and tRNATyr) (52). But Turbido and Pukovnik have a single tRNAGln gene, Redrock has a single tRNATrp gene, and Trixie has both tRNATrp and tRNAGln genes. So while there have been several efforts to account for the specific roles of these genes in accommodating phage codon usage requirements (113–116), this variation suggests considerable complexity. It is plausible, for example, that many of them are “legacy genes” that may have been required for growth in a particular unknown host in their recent evolutionary past but play no role in the current host (see Fig. 6). Furthermore, there are examples of genes that appear to encode nonsense tRNAs as well as potential frame-shifting tRNAs, suggesting regulatory roles in gene expression. The Subcluster K1 phages all encode a single tRNA gene (tRNATrp) but also carry genes coding for a putative RNA ligase (RtcB), which could a play a role in repair of host-mediated attack of the tRNA (97).
FIGURE 6.
A model for mycobacteriophage diversity. The large number of different types of mycobacteriophages isolated on M. smegmatis mc2155 can be explained by a model in which phages can readily infect new bacterial hosts—either by a switch or an expansion of host range—using a highly diverse bacterial population that includes many closely related strains. As such, phages with distinctly different genome sequences and GC% contents infecting distantly related bacterial hosts, such as those to the left (red) or right (blue) extremes of a spectrum of hosts, can migrate across a microbial landscape through multiple steps. Each host switch occurs at a relatively high frequency (~1 in 105 particles, or an average of about one every 103 bursts of lytic growth) and much faster than either amelioration of phage GC% to its new host or genetic recombination. Two phages (such as those shown in red and blue) can thus “arrive” at a common host (M. smegmatis mc2155) but be of distinctly different types (clusters, subclusters, and singletons). Reproduced with permission from Jacobs-Sera et al. (99). 10.1128/microbiolspec.MGM2-0032-2013.f6
Some phages (e.g., Cluster C) also encode a tmRNA gene, similar to host-encoded tmRNA genes that play a role in the release of ribosomes from broken mRNA. The specific roles of the phage-encoded tmRNAs are unclear, but it is plausible that they enhance the pool of free ribosomes for late gene expression by releasing them from early transcripts when they are no longer required.
Mobile Elements: Transposons, Introns, Inteins, and HNH Endonucleases
The key architectural feature of phage genomes—pervasive mosaicism—is likely generated by illegitimate recombination events that occur at regions of DNA sharing little or no DNA sequence similarity (96). Although most of these may result from replication or repair “accidents”, there are several active processes that could contribute, including transposition, intron and intein mobility, and HNH endonuclease activity.
A number of different transposable elements in mycobacterial genomes have been described, although these are generally not present in mycobacteriophage genomes. However, both active transposons and residual segments of transposons have been identified in the phages. Comparative analysis of the Cluster G phages provides compelling evidence for identification of novel ultra-small mycobacteriophage mobile elements (MPMEs), with two closely related subtypes, MPME1 and MPME2 (117). There are at least three instances in which an MPME insertion is present in one genome but absent from others, and because the Cluster G genomes are extremely similar to each other, the preintegration site and the precise insertion can be readily interpreted. The 439-bp MPME1 elements contain 11-bp imperfect inverted repeats (IRs) near their ends and a small (125-codon) open reading frame encoding a putative transposase. The MPME1 elements in phages BPs and Hope are 100% identical to each other (from IR-L to IR-R), but the insertions differ in a 6-bp segment between IR-L and the preintegration site. The origin of this 6-bp sequence is unclear but does not correspond to a target duplication (117).
MPME1 elements are found in a variety of other mycobacteriophage genomes including those in Clusters F and I and are either identical or have no more than a single base pair difference; there are also truncated versions of the element in the Cluster O phages (117). MPME2 elements are 1 base pair longer and share 79% nucleotide sequence identity to MPME1. They are present in the Cluster G phage Halo and phages within Clusters F, I, and N, being either identical to each other or having no more than a single base pair difference. As yet there is no direct evidence for the mobility of the MPME elements, the frequency of movement, or the mechanism involved, although the comparative genomics suggests that these are active and moving at a respectable rate. No MPME elements have been identified outside of the mycobacteriophages.
Comparative genomics also reveals an IS110-like element in phage Omega, which is absent from phage LittleE, and the genomes are sufficiently similar in these regions to indicate the preintegration site and a 5-bp target duplication (90). There are no closely related copies in other mycobacteriophages, but there are some more distantly related segments in some Subcluster A1 phages (90). It is unclear if this is a remnant or an active transposon.
There are few examples of self-splicing introns in the mycobacteriophages, but two have been recently described, both in Cluster J phages (90) and both within virion structural protein genes. In phage BAKA the intron is small (265 bp) and is within a putative tail protein gene; in phage LittleE the intron is within the capsid subunit gene and is larger (819 bp) due to inclusion of a small open reading frame on the opposite strand with similarities to homing endonucleases. Because both these proteins are required for virion assembly, and the capsid must be expressed at very high levels, the splicing events are expected to occur extremely efficiently. They are probably variants of group I introns, but little is known about their mechanisms of splicing (90). Other introns may be present in other genomes but have escaped detection because of insertion in genes of unknown function.
The rarity of self-splicing RNA introns contrasts with the relative abundance of inteins that splice out at the protein level. These are typically identifiable through conserved domains associated with intein splicing and are often apparent by comparative genomics. Phage ET08 has a total of five inteins—the most in any single mycobacteriophage genome to date (98). These are located within gp3, gp79, gp202, gp239, and gp248, but the functions of these are largely unknown, except for gp3, which is predicted to be a nucleotidyltransferase. For all five, there are examples of intein-less homologues in other phages. Most of the inteins contain an endonuclease domain that is predicted to promote DNA cleavage in intein-less genes followed by repair with the intein-containing DNA copy. The intein in Bethlehem gp51 has been dissected biochemically and shown to represent a new type (type III) of splicing mechanism (118), of which the Omega gp206 intein is an unusual variant (119).
It is common for mycobacteriophage genomes to have one or more putative HNH homing endonucleases encoded as freestanding genes (rather than as part of an intein or coded within an intron). These are usually recognized from the presence of conserved motifs, and none have yet been shown to have nuclease activity. However, a comparison of the freestanding HNH encoded by Courthouse gene 51 and phage Thibault, which lacks it, shows a precise insertion and the presumed location of endonucleolytic cleavage (90). A second example is an HNH insertion in Omega that is somewhat messier and when compared to phage Optimus, lacking the insertion, is associated with a loss of 35 bp, presumably during the process of HNH acquisition (90). While the specific mechanisms generating these events are not clear, it is not difficult to see how these could play important roles in generating genomic mosaicism during phage evolution.
MYCOBACTERIOPHAGE-HOST INTERACTIONS
Mycobacteriophage Host Range and Host Range Expansion
Considerable efforts in using mycobacteriophages for phage typing illustrate that they readily discern between different hosts and that by using panels of phages with defined host ranges, the identities of unknown hosts can be predicted (6). Unfortunately, for the most part the typing phages are no longer available, and the genomic information is lacking. However, the host ranges of a few sequenced phages have been examined and are informative in regard to understanding the genetic diversity of the phages. For example, Rybniker et al. (120) tested the host ranges of 14 mycobacteriophages and showed that L5 and D29 (both Subcluster A2 phages) as well as Bxz2 (Subcluster A3) have broad host ranges and infect M. tuberculosis, BCG, Mycobacterium scrofulaceum, Mycobacterium fortuitum, Mycobacterium chelonae, and some strains of both Mycobacterium ulcerans and Mycobacterium avium, in addition to M. smegmatis. Other phages have intermediate host preferences, such as phage Wildcat, which infects M. scrofulaceum, M. fortuitum, and M. chelonae, and some (e.g., Barnyard, Che8, Rosebush) infect only M. smegmatis or its substrains, out of the strains tested (120).
Jacobs-Sera et al. (99) determined whether a collection of 220 of the sequenced phages are able to infect M. tuberculosis. In general, there is a close correlation between the ability to infect M. tuberculosis and the cluster or subcluster type. For example, all of the Cluster K phages—regardless of subcluster—efficiently infect M. tuberculosis; among the Cluster A phages, only phages within specific subclusters can infect M. tuberculosis, including Subclusters A2 and A3. Interestingly, the Cluster G phages (e.g., BPs) do not efficiently infect M. tuberculosis, although mutants that do can be isolated at a frequency of ~10−5, and the mutants maintain the ability to infect M. smegmatis. Several of these mutants have been mapped, and all have single amino acid substitutions in gene 22, encoding a putative tail fiber protein (107, 117). A simple interpretation is that the mutants overcome the need for a specific interaction with receptors on the M. tuberculosis cell surface. However, adsorption assays suggest there is a more complex explanation, because the mutants do not appear to adsorb to M. tuberculosis more efficiently than the wild-type parent phage, and surprisingly, they adsorb substantially better to M. smegmatis, in some cases, dramatically so (99). This is not easy to explain, because the mutants appear to infect M. smegmatis quite normally and were selected for infection of M. tuberculosis.
The same set of phages was also tested for their ability to infect two other strains of M. smegmatis (99). Many, but not all, of the phages efficiently infect these strains, and although there is a correlation between infection and cluster/subcluster type, it is statically weaker than with M. tuberculosis infection. But similarly, there are instances where phages are able to overcome the host barrier at moderate frequencies (10−5), and examination of phage Rosebush mutants capable of infecting M. smegmatis Jucho (99) reveals amino acid substitutions in putative tail fibers, reminiscent of the Cluster G mutants that infect M. tuberculosis.
Mechanisms of Phage Resistance
Resistance to phage infection can occur by a variety of mechanisms including surface changes, and phages can presumably coevolve to overcome this resistance, reflecting the processes giving rise to the expanded-host-range mutants described above. However, in general, little is known about mycobacterial receptors for phage recognition or the determinants of host specificity. For phage I3 (which is not completely sequenced but is a Cluster C–like phage) resistance is accompanied by loss of cell-wall-associated glycopeptidolipids (121), and an M. smegmatis peptidoglycolipid, mycoside C(sm), has been implicated in the binding of phage D4 (which is also genomically uncharacterized) (122). Lyxose-containing molecules have also been proposed as possible receptors for the unsequenced phage Phlei (123, 124). There is clearly much to be learned about mycobacteriophage receptors and how phage tail structures specifically recognize them.
However, there are many other determinants of host range beyond the availability of cell wall receptors, including restriction/modification systems, lysogenic immunity, CRISPR elements (125), and various abortive infection systems including those mediated by toxin-antitoxin systems (126). M. smegmatis mc2155 has no known prophages, restriction modification systems, or CRISPR arrays, which may contribute to its suitability as a permissive host for phage isolation. However, M. tuberculosis H37Rv contains a type III-A family CRISPR array (127) and more than 80 putative toxin/antitoxin systems (128), perhaps suggesting development within a phage-rich environment in its recent evolutionary history. None of the spacer elements within the M. tuberculosis CRISPR array (which are used for spoligotyping M. tuberculosis isolates) closely match known phage sequences, and it is unclear whether the CRISPR arrays are currently active or contribute in any way to phage resistance (129). M. tuberculosis H37Rv has two small (~10 kbp) prophage-like elements in its genome (φRv1 and φRv2), but it is unlikely that these confer immunity to other phages (96). However, a likely intact 55-kbp prophage has been identified in a Mycobacterium canettii strain (88), and other prophages are present in the genomes of M. avium 104 (130), Mycobacterium marinum (87), M. ulcerans (85), and Mycobacterium abscessus (83, 84). Resistance of M. smegmatis to D29 infection can result from overexpression of the host multicopy phage resistance (mpr) gene, perhaps by alteration of the cell surface, although the specific mechanism is not known (131, 132).
Because phages can easily replicate from a single particle to vast numbers (there are 106 to 108 particles in a typical plaque), and mutants arise at moderate frequencies, host range mutants can readily arise within environmental populations. As such, phages are expected to move from one host to another at frequencies that are vastly greater than the time required to ameliorate their genomic features, such as coding biases and GC%, to a specific host. This leads to a model (Fig. 6) in which two key parameters contribute to the diversity of phages: the ability of phages to rapidly switch hosts, and the availability of a broad spectrum of closely related hosts in the environment from which the phages are isolated (99). This model can therefore account for the nature of mycobacteriophage genomes and predicts that similar phage diversity will be seen using other hosts and similar sampling (99).
Lysis Systems
Host cell lysis is a critical step in phage lytic growth, and in the prototype lambda system is both efficient and precisely timed (133, 134). Understanding the lysis systems of mycobacteriophages is of particular interest in that these can illuminate features of mycobacterial cell walls. Mycobacteriophage lysis systems are described in detail in reference 243 and will be discussed only briefly here.
Most mycobacteriophages encode at least three proteins required for lysis: a peptidoglycan-cleaving endolysin (also called Lysin A), Lysin B, and a holin; a few lack Lysin B, some either lack a holin or it is difficult to identify bioinformatically, but all encode an endolysin (135–138). Phage Ms6 and its relatives encode an additional protein that acts as a chaperone for delivery of the endolysin to its peptidoglycan target (139). The endolysins are diverse and modular in nature, reflecting an intragenic mosaicism that is a microcosm of the generally mosaic nature of the phage genomes (138). Many are composed of three segments: an N-terminal domain with predicted peptidase activity, a central domain that cleaves the peptidoglycan sugar backbone, and a C-terminal cell wall binding domain; there are, however, numerous departures from this general organization (138). In total, there are at least 25 different organizations (Org-A to Org-Y) with unique combinations of the constituent domains. Interestingly, the Ms6 lysA (Org-J) gene encodes a second lysis gene that is wholly embedded within lysA and is expressed by translation initiation from an internal start codon; phage mutants that express only the longer (Lysin384) or the shorter (Lysin241) endolysin are viable (140). Lysin B encodes an esterase that cleaves the linkage of the mycolylarabinogalactan to the peptidoglycan (136, 137) and, unlike lysin A, is dispensable for lytic growth (137). However, it is required for optimal lysis and efficient phage reproduction (137) and likely plays an analogous role to the spanins that facilitate fusion of the inner and outer membranes of Gram-negative bacteria during phage lysis (134, 141). Analysis of the Ms6 gp4 shows that it is a likely holin, with a signal-arrest-release (SAR) domain followed by a transmembrane domain (142). However, Ms6 gp5 also has a transmembrane domain, and gp4 and gp5 interact to facilitate lysis (142).
INTEGRATION SYSTEMS
Phages within more than one-half of the different clusters encode a phage integrase, and there are numerous examples of both tyrosine integrases (Int-Y) and serine integrases (Int-S); these include Clusters A, E, F, G, I, J, K, L, M, and P and the singletons Dori and DS6A. But the distribution of Int-Y and Int-S types within different clusters is nonrandom. For example, all of the phages within Clusters E, F, G, I, J, K, L, and P (as well as Dori and DS6A) encode tyrosine integrases, and all of the Cluster M phages encode serine integrases. However, the distribution of different integrase types varies among the various subclusters within Cluster A. For example, all of the A1, A5, A7, and A10 phages encode an Int-S, as well as 2 of the 8 A3 phages, 11 of the 14 A4 phages, and 1 of the 2 A8 phages (the other has a deletion in this region of the genome); 7 of the 9 A2 phages, 6 A3 phages, and 3 A4 phages encode Int-Ys. These systems thus may evolve quickly relative to the rest of the genomes, presumably by promoting site-specific or quasisite-specific recombination events.
Interestingly, two of the A2 phages, the one A9 phage, and all six A6 phages have a parA/B partitioning system at the location where the integrase is in the related phages (67). Presumably, these genomes replicate extrachromosomally during lysogeny, and the parA/B systems ensure their accurate segregation at cell division and conferring prophage maintenance—essentially the same functionality provided by the integration systems (143). How these phages are able to replicate their genomes during lysogeny is unclear, especially as their related genomes that integrate presumably do not.
For most of the genomes encoding a tyrosine integrase, the location of attP can be bioinformatically predicted. Typically, these use a bacterial attachment site (attB) overlapping the 3′ end of a tRNA gene, and to preserve the integrity of the tRNA following integration, attP and attB typically share 30- to 40-bp sequence identity. Thus, a BLASTN search of the phage genome against the M. smegmatis chromosome usually identifies the putative attB site. At least 12 attB sites have been identified this way, and usage of the sites has been shown for at least 8 of these (67, 90, 97, 103, 144–146). For the Cluster E and Cluster L phages, bioinformatic analysis fails to identify putative attB sites, and these await experimental determination.
Phages encoding serine integrases do not integrate at host tRNA genes and often use attB sites located within open reading frames (147, 148). The attP and attB sites typically share only minimal segments of sequence identity (3 to 10 bp) and thus cannot be readily predicted bioinformatically and require experimental analysis. The attB sites for two different mycobacterial Int-S systems have been described: Bxb1, which integrates into the groEL1 gene of M. smegmatis, and Bxz2, which integrates into the gene Msmeg_5156 (145, 147). Bxb1 integration has been especially useful for gaining insights into mycobacterial physiology because Bxb1 lysogens are defective in the formation of mature biofilms (149). This results specifically from inactivation of the groEL1 gene by integrative disruption and led to the demonstration that GroEL1 plays a role as a dedicated chaperone for mycolic acid biosynthesis (149, 150). It is not known if disruption of Msmeg_5156 or any other host gene as a consequence of phage integration via a serine integrase has specific physiological consequences.
Phage integration systems typically are carefully regulated in their recombination directionality, such that integrase catalyzes integrative recombination using attP and attB sites (252 bp and 29 bp, respectively [151, 152]) but in the absence of accessory factors does not mediate excisive recombination utilizing attL and attR sites (which are themselves the products of integration) (153). Directional control is enabled by a recombinational directionality factor (RDF), a phage-encoded accessory actor that determines which pairs of attachment sites can undergo recombination. The L5 RDF has been identified (gp36) and acts by binding to specific DNA sites within attP and attR to bend DNA and alter the nature of higher-order protein-DNA architectures that can be formed by the Int-Y, the RDF, and the host integration factor, mIHF (154–157). Directional control is determined by a different mechanism for the serine integrases, as illustrated for Bxb1. The attachment sites are relatively small (<50 bp), and Int binds as a dimer to each site, and the choice of pairs of sites competent for recombination is determined by which protein-DNA complexes can undergo synapsis, presumably predicated on compatible conformations of Int bound to specific sites (158, 159). A phage-encoded RDF, Bxb1 gp47, associates with Int-DNA complexes not through DNA binding, but by direct protein-protein interactions and presumably alters the conformations such that attL and attR can undergo synapsis, but attP and attB cannot (160, 161). A curious feature of Bxb1 is that the RDF is encoded by gene 47, which is situated among DNA replication genes, and is not closely linked to int (gene 35). Numerous mycobacteriophages have homologues of Bxb1 47, including many (such as L5) that utilize tyrosine integrases and for which there is no obvious role in recombination. Presumably, these genes play alternative roles in phage growth, most likely in DNA replication, and have been coopted by the integration systems for directional regulation (162).
MYCOBACTERIOPHAGE GENE EXPRESSION AND ITS REGULATION
None of the sequenced mycobacteriophage genomes encode single-subunit RNA polymerases, and it is likely that all phage transcription utilizes the host RNA polymerase. In some examples, phage growth has specifically been shown to be sensitive to the transcription inhibitor, rifampin (33, 50). Promoters corresponding to those used by the host major sigma factor (SigA) have also been identified in several mycobacteriophages and can be predicted in many others. For example, the strong Pleft promoter in phage L5 closely corresponds to −10 and −35 SigA sequences (163), as do both the early lytic promoter (PR) and the repressor promoter (Prep) of BPs (164), and in all three examples, the transcription start site has been mapped. Interestingly, in both the BPs promoters, these transcripts provide a leaderless mRNA for the first gene in the operon, with the first base of the translation initiation codon corresponding to the 5′ end of the mRNA. The use of leaderless mRNA transcripts has been shown previously in expression of the firefly luciferase reporter gene in an L5 recombinant phage (165). A SigA-like promoter has also been described for expression of the Ms6 lysis cassette, but this does include a leader between the transcription initiation site and the predicted translational start codon (166).
The use of host SigA-like promoters does not, however, appear to be universal for mycobacteriophage transcription. For example, a search for SigA-like promoters in mycobacteriophage Rey reveals no close matches and none that closely correlate with positions where promoters are anticipated to be. In phage Giles, where the transcripts in lysogeny as well as early and late lytic growth have been mapped by RNAseq (Fig. 4), there are no evident SigA-like promoter sequences upstream of where transcription starts. Although late transcripts are likely to utilize phage-encoded activators, it is unclear whether the early promoters in Giles use an alternative host sigma factor or if these also are regulated by phage-encoded functions. There is clearly much to be learned about how transcription initiation is regulated in mycobacteriophage growth. The Cluster A phages all encode a potential sigma factor (Phams 1448, 1922, 2982 in the Phamerator database Mycobacteriophage_285), as indicated by HHPred searches with the closest similarity to SigK of M. tuberculosis, which could regulate phage gene expression. Many phages also encode one or more WhiB proteins, but whether these regulate host or phage expression is not clear. In TM4, it has been shown that the phage-encoded WhiB protein is not required for phage growth, although it contains an Fe-S cluster, is a dominant negative regulator of the host whiB2 gene, and promotes superinfection exclusion (167).
Transcriptomics
RNAseq analysis of transcription in phage Giles is quite informative. In lysogeny, few regions are expressed—as expected—and these include gene 47, which encodes the phage repressor. Somewhat surprisingly, the repressor is expressed at high levels, equivalent to the 0.5 percentile of highest expressed genes in M. smegmatis (107), in noted contrast to the lower expression of other phage repressors (168). Presumably, the Giles repressor binds with relatively low affinity to its binding site(s), although these have yet to be identified. Lysogenic transcription proceeds through the four downstream genes, although at a much lower level (Fig. 4). The leftward-transcribed genes at the left end of the genome (3–4) are also expressed at a low level in lysogeny. Perhaps most surprising from this RNAseq analysis is the expression of an apparent noncoding RNA near the right end of the genome (Fig. 4), which is made during both lysogenic and early lytic growth. It is expressed at a high level, but its function is unclear, and the DNA segment encoding it is not required for phage growth or lysogeny.
Immunity Regulation in Cluster A Phages
An unusual system for gene expression is found in the temperate Cluster A phages but has been studied in detail in just a few examples: L1, L5, and Bxb1 (163, 169–177). The repressor is encoded in the right part of the genome (gene 71 in phage L5) and codes for a small protein (183 amino acids) containing a putative helix-turn-helix DNA binding motif near its N-terminus; deletion of the gene interferes with lysogeny and generates a clear plaque phenotype. L5 gene 71 is sufficient to confer immunity to superinfection, and thermo-inducible mutations map in the repressor gene (110, 176, 178). The repressor sequence of the closely related L1 phage is identical to that of L5 (176). The repressor functions by binding to a 13-bp operator site that overlaps the early lytic promoter, Pleft, situated near the right end of the genome and transcribing leftward (163, 169). It is a two-domain protein with an N-terminal domain (residues 1 to 64) and a C-terminal domain (residues 64 to 183) separated by a hinge region (172), and mutations in either domain influence DNA binding (172, 176). The repressor binds as a monomer and imparts a modest DNA bend (30°) at its binding site (171).
Surprisingly, there are many additional repressor-binding sites situated throughout the L5 genome, and the repressor has been shown to bind to 23 of these in addition to its operator at Pleft (169). These sites correspond to a tightly conserved asymmetric consensus sequence, 5′-GGTGGc/aTGTCAAG, and all or most of the base positions are critical for repressor binding (169, 171). A clue to the role of these additional binding sites emerges from their genomic locations and orientation. With few exceptions, they are located within short intergenic regions or overlapping translation initiation or termination codons and are oriented in one direction relative to the direction of transcription (169). Thus, in the left arm of L5, in which the virion structure and assembly genes are transcribed rightward, there are four sites in the “−” orientation (and one at the extreme left end in the nontranscribed region in the “+” orientation, where “+” and “−” refer to whether the sequence corresponds to the top or bottom strand of the genome), and in the leftward transcribed right arm there are 17 sites in the “+” orientation (and one at the extreme right end in the nontranscribed region in the “−” orientation). There are no apparent promoters at each of these sites, suggesting a role different from that of the true operator site at Pleft (169). Insertion of one or more sites between a nonphage promoter (hsp60) and a reporter gene results in a reduction of reporter activity, in a manner that is dependent on both the repressor and the orientation of the site, which is magnified with larger numbers of binding sites (169). This suggests a model in which repressor binding to these sites acts to promote cessation of transcription and facilitate silencing of the L5 genome in the lysogenic state. These sites are thus referred to as “stoperators” (169). An example of stoperator location and orientation in mycobacteriophage Alma is illustrated in Fig. 5.
Mycobacteriophage Bxb1 is heteroimmune to L5 but contains a similar regulatory system (51, 175). The Bxb1 repressor (gp69) shares only 41% amino acid sequence identity to L5 gp71, and there are a total of 34 putative operator/stoperator sites in the Bxb1 genome. As in L5, these sites correspond to a tightly conserved asymmetric consensus sequence, 5′-GTTACGt/ag/aTCAAG, and are located in short intergenic regions in one orientation relative to transcription. The consensus sequences of the L5 and Bxb1 stoperators are closely related but with differences that likely contribute to heteroimmunity of the two phages (175). Genomic analysis of the large number of Cluster A phages shows that they all share this unusual repressor/stoperator system, with variations of the stoperator consensus sequence that correlate with subcluster designation (98). However, the variation typically occurs within positions 2 to 8 of the consensus sequence, and positions 1 (G) and 9 to 13 (5′-TCAAG) are invariant. How heteroimmunity evolves in this system is unclear, because although amino substitutions within the repressor could readily lead to altered recognition of positions 2 to 8 in the stoperator sites, it is unclear what selection pressure leads to coordinate evolution of over 30 genomically dispersed binding sites to new specificities (see Fig. 6). Interestingly, although the repressor/stoperator system is restricted to the Cluster A phages within the collection of sequenced mycobacteriophages, it appears to be shared with phages of some other Actinomycetales hosts. For example, there are multiple stoperator-like sites in Streptomyces phage R4 and its relatives (244), as well as in Rhodococcus phage RER2 (179); in the latter phage the invariant positions (1, 9 to 13) are the same as in the Cluster A mycobacteriophages, but the R4-like Streptomyces phage stoperators are substantially different. The prevalence of this regulatory system among the broader Actinomycetales phage population remains to be explored.
Although this particular regulatory system is confined to Cluster A phages, within the mycobacteriophages, other phages contain repeated sequences that are likely involved in the regulation of gene expression. One example is the Cluster K phages that contain a conserved sequence (5′-GGGATAGGAGCCC) positioned 2 to 8 bp upstream of putative translation start sites and are thus referred to as start-associated sequences (SASs); there are 10 to 19 copies per genome (97). The sequence is asymmetric and is situated in the expected location of the ribosome binding site, and eight of the conserved positions can pair with the 3′ end of the 16S rRNA. However, there are well-conserved positions at the edges of the site that cannot pair with the 16SrRNA, suggesting that it is not just a common variation of a ribosome binding site. Moreover, the sites are situated exclusively next to nonvirion structural genes, and the consensus site is present neither in other mycobacteriophages nor in the M. smegmatis genome (97). So if these SASs are involved in translation initiation, they likely also require a phageencoded regulator. An intriguing hypothesis is that the SAS-associated genes are highly expressed in early lytic growth and that the SASs play a role in releasing ribosomes from these transcripts during late lytic growth to optimize ribosome availability for late gene expression. Curiously, a subset of the SAS sites has a second conserved sequence composed of imperfect 17-bp IRs separated by a variable spacer. Because of their tight association with SASs—typically located with a few base pairs upstream—these are referred to as extended start-associated sequences, and their roles are unknown (97).
Integration-Dependent Immunity
A novel system of immunity regulation has been described in several different types of mycobacteriophages, including BPs (Cluster G), Brujita (Cluster I), Charlie (Cluster N) and BigNuz (Cluster P) (164, 180). The characteristic feature—readily recognizable bioinformatically—is that the phage attachment site for integration (attP) is located within the coding sequence for the phage repressor (e.g., BPs gp33). In these systems, the repressor is located immediately upstream of the integrase gene (int), in contrast to the immunity system of Cluster A phages described above, in which int and the repressor are separated by ~20 kbp (see Fig. 6). The immunity functions are thus compactly organized, with all of the required functions situated within ~2 kbp of the genomes. Because of the specific location of attP with the repressor, integration of the phage genome plays a central role in the lytic-lysogenic decision, generating two alternative forms of the repressor: a longer viral form (e.g., BPs gp33136) and a shorter prophage form (BPs gp33103) that lacks the C-terminal 33 residues of the viral form. As expected, the shorter prophage form is active in conferring superinfection immunity, but interestingly, the longer viral form is not. The reason for this is that the extreme C-terminus of the viral form contains an ssrA-like tag that targets it for degradation, presumably by the ClpXP protease (164). This is a functionally important distinction in the two forms of the repressor and directly determines the frequency at which lysogeny is established; a mutant phage encoding a stabilized viral repressor form has a greatly elevated frequency of lysogenization (164). Nonetheless, establishment of immunity is dependent on phage integration, because otherwise the active prophage form of the repressor cannot be expressed (164). The activity of the integration system thus must also play a central role in the lytic-lysogenic decision, because if Integrase (Int) was always expressed and fully active, then lytic growth would not occur.
The resolution to this conundrum is that the integrase also contains a C-terminal ssrA-like tag, such that degradation of Int is anticipated to also determine the lysogenization frequency. This feature is illustrated by the behavior of integration-proficient plasmid vectors (see below) that transform at unexpectedly low frequencies due to Int degradation; stabilization of Int leads to large increases in the transformation frequencies (164). The relative simplicity of these immunity systems with a few genes and DNA sites serving multiple functions suggests that these may have evolved relatively early in the development of phage immunity systems (180).
GENETIC MANIPULATION OF MYCOBACTERIOPHAGES
Four approaches have been described for manipulation of mycobacteriophage genomes. The first is a simple genetic cross in which a phage can acquire a DNA segment by homologous recombination from a plasmid containing homologous phage sequences. Although this should be a generalizable technique, there is only a single example, in which the firefly luciferase gene was crossed onto the L5 genome and recombinants were identified by hybridization (165). A second approach was through direct cloning into the TM4 genome, replacing a small restriction fragment with a PCR-generated substitute containing deletion of gene 49 (167). A third approach is the construction of shuttle phasmids, which is discussed in detail in reference 245. Shuttle phasmids provide information about which phage genome segments are likely to be nonessential for lytic growth and are instrumental for delivery of reporter genes, transposons, and allelic exchange substrates to mycobacteria. However, they are of more limited use for constructing specific mutations in phage genes.
A general method for genome manipulation is the bacteriophage recombineering of electroporated DNA (BRED) technique, which can be used to efficiently construct precise gene deletions (minimizing genetic polarity) and to introduce point mutations (181–183). The technique takes advantage of a mycobacterial recombineering system in which recombinase genes from mycobacteriophage Che9c are inducibly expressed in M. smegmatis (184–186). Following coelectroporation of phage genomic DNA and a dsDNA substrate containing the desired mutation (typically ~200 bp), plaques are recovered using an infectious center assay, and individual plaques are screened by PCR for the presence of wild-type and mutant alleles at the site of interest (182). Because the recombination frequencies are high, mutant plaques are typically present in at least 10% of the plaques tested; however, all plaques contain a wild-type signal, likely reflecting the use of replicating genomes as substrates for recombination. If the mutant is viable, it can be recovered from replating of single plaques. If the mutant is not viable, it can usually be identified among the primary plaques due to the presence of wild-type helper phage within each plaque but then is not recoverable from a secondary plating (182).
The BRED approach is applicable to many different types of phage genomes but perhaps not all. For example, it is not possible to recover plaques of Omega by electroporation of phage DNA into M. smegmatis (90). The reason for this is not clear. An intriguing possibility is that a capsid-enclosed protein is required for recircularization of the phage DNA upon injection, which is then absent during electroporation. Phage Omega is unusual in that it has defined genome termini with very short (4-base) single-stranded DNA extension and requires the host nonhomologous end joining (NHEJ) system for efficient infection (187). Omega encodes its own Ku-like protein, which could be required for recircularization, but mass spectrometry of Omega virions failed to identify any such components (90). Some phage genomes are readily engineered with BRED, and Dedrick et al. performed a whole-genome analysis for essential genes in phage Giles (see Fig. 4) (107); the introduction of point mutations into the BPs genome was critical for establishing the mechanisms of integration-dependent immunity (164).
APPLICATIONS FOR MYCOBACTERIAL GENETICS
Mycobacteriophages have proven to be essential tools for the development of mycobacterial genetics and to provide potential clinical tools for tuberculosis control. Given the well-established advantages of viral approaches to a variety of biological problems, mycobacteriophages would seem to offer considerable promise for genetic and clinical applications. Like most viruses, mycobacteriophages show host specificity, efficiently introduce their DNA into the host, replicate efficiently, express genes at high levels, utilize a variety of regulatory strategies, and are simple to grow. Mycobacteriophages offer an additional advantage in that their rapid growth contrasts to the extremely slow growth rate of M. tuberculosis. The tools for mycobacterial genetics are typically of two general classes. First, there are those that take advantage of the phages themselves, such as for efficient delivery of foreign DNA to the hosts. The second is the exploitation of phage components such as plasmid vectors or selectable markers. Applications in clinical microbiology have largely focused on simple and rapid tools for tuberculosis diagnosis.
Generalized Transduction
Generalized transduction of genetic markers in M. smegmatis was one of the earliest applications of mycobacteriophages (13). Although transduction was initially demonstrated using phage I3—which has yet to be fully genomically characterized but liklely is grouped within Cluster C—it has also been shown for other Cluster C phages such as Bxz1 (246). Transduction using these phages typically generates relatively few transductants, depending on the locus, but a number of markers have been moved between M. smegmatis strains. Unfortunately, the Cluster C phages do not infect M. tuberculosis and there currently is no generalized transducing phage for slow-growing mycobacteria. Moreover, all of the phages that infect M. tuberculosis contain genomic cohesive ends and are thus not good candidates for generalized transducers. Presumably phages of M. tuberculosis that have terminally redundant genomes—and are thus candidates for generalized transducers—exist in nature, but have yet to be isolated.
Mycobacteriophages for Efficient Gene Delivery
A key feature of mycobacteriophages is that when added to host cells at a multiplicity of infection (moi) greater than about 3, every cell in the culture is infected. This is vastly more efficient than introducing DNA by electroporation and when using recombinant phages provides a simple means of introducing foreign DNA. The first strategy to exploit this was the construction of shuttle phasmids that replicate as large (~50 kbp) cosmids in E. coli and as phages in mycobacteria (44), and these continue to be indispensible tools of mycobacterial genetics; see reference 245 for a detailed discussion. Shuttle phasmids are typically constructed by ligation of phage DNA to a cosmid vector and packaging into phage lambda particles in vitro. These are used to infect E. coli, the library of colonies is harvested, and DNA is isolated and then used to electroporate M. smegmatis. The plaques recovered are purified and demonstrated to retain the cosmid vector (76). The strategy relies on the use of mycobacteriophages with appropriately sized genomes, as the packaging constraints for phage lambda are ~53 kbp and there must be a region of nonessential genes that can be replaced by the cosmid vector. Shuttle phasmids have been constructed from phages TM4 and D29, both of which satisfy these constraints, as well as having the ability to efficiently infect both M. smegmatis and M. tuberculosis. TM4 shuttle phasmids are the most widely used, and conditionally replicating temperature-sensitive mutants have been developed that facilitate the recovery of survivors following shuttle phasmid infection (49); these mutations have been mapped through analysis of temperature-resistant revertants (97).
Shuttle phasmids can be readily manipulated in E. coli by replacing the cosmid moiety with another cosmid carrying desired genes. The three primary applications have been in the construction of reporter phages for the delivery of luciferase or fluorescent genes (188–190), for the delivery of allelic exchange substrates for constructing M. tuberculosis mutants (48), and for transposon delivery (49). Because transposon insertion is typically a low-frequency event, the high efficiency of shuttle phasmid infection is critical and has enabled the construction of high-density transposon libraries and their use for determining gene requirements under specific conditions (191–194).
Recombinant phages have been constructed for the delivery of reporter genes by two other methods. First, a derivative of phage L5 that carries the firefly luciferase gene was constructed by crossing over from a plasmid and identifying the recombinants by hybridization (165); these infect both fast- and slow-growing mycobacteria (195). Second, a reporter phage derivative of D29 has been constructed carrying green fluorescent protein (GFP) using recombineering (196). These approaches are more directed than using shuttle phasmids but contribute toward a suite of strategies for using phages to deliver DNA efficiently to mycobacterial hosts.
Integration-Proficient Plasmid Vectors
Some of the earliest tools developed by exploiting parts of the phage genomes are the integration-proficient plasmids derived from phage L5 (144). The plasmids were generated by inserting a segment of the phage genome carrying the integrase gene and attP site (~2 kbp) into a plasmid backbone that has an origin of replication for E. coli and a selectable marker for mycobacteria. These plasmids are not capable of extrachromosomal replication in mycobacteria and can only transform mycobacterial cells if the integrase gene is expressed and can mediate site-specific recombination between the plasmid attP and host attB site (197). In practice, these plasmids transform both M. smegmatis mc2155 and M. tuberculosis H36Rv efficiently (~106 transformants/μg of DNA) (144) as a result of a single well-defined predictable plasmid integration event into the host chromosome. These integration-proficient plasmids offer a number of useful attributes for mycobacterial genetic manipulation. Perhaps most importantly, they provide a simple method for inserting genes into single copy on the chromosome, in contrast to the commonly used extrachromosomal plasmids that have copy numbers in excess of 20 (198). This is specifically useful in complementation experiments, where extrachromosomal vectors can present misleading behaviors (199–201). Furthermore, as integration results in reconstruction of the tRNA gene at the site of insertion, there is little or no disruption to the behavior of the host cell.
An additional attribute is that the integrated vectors are stably maintained in the absence of selection (144), although if the plasmid is expressing genes that disfavor bacterial growth and there is a selection for plasmid loss, then a low level of Xis-independent recombination of attL and attR gives rise to plasmid loss (202). This issue can be readily addressed by using a two-plasmid system in which the integrase gene is provided on a second, nonreplicating, nonintegrating vector, such that after co-electroporation only the attP-containing vector integrates (151). This eliminates the possibility of integrase-mediated events conferring instability, although homologous recombination can still occur between the ~40 bp of homology between attL and attR.
Because of the large number of sequenced mycobacteriophage genomes and their overall diversity, there are numerous alternative types of integration-proficient vectors that can be constructed and, if they use different attB sites, should be fully compatible with each other and could be used in combination to construct complex recombinants. To date, integrating vectors have been constructed from phages Tweety (145), Giles (103), Omega (90), Adephagia (97), Ms6 (146), BPs (164), Charlie (164), Brujita (164), and Streptomyces pSAM2 (203) and phage phiC31 (204), all of which use tyrosine integrases and insert into different attB sites overlapping tRNA genes. Because there is generally excellent conservation of tRNA genes between M. smegmatis and M. tuberculosis, these vectors usually work well in both hosts, even though the parent phage may not necessarily infect M. tuberculosis. It is important to note that for those systems derived from phages that use integration-dependent immunity systems (e.g., BPs, Charlie, Brujita [164]), transformation frequencies are extremely low unless a stabilized mutant form of the integrase is used (as discussed above). Additional vectors have been constructed from serine-integrase systems, including Bxb1 (147) and Bxz2 (145), although because the attB sites are located within open reading frames, the recombinants may have altered physiological features (149).
Further refinements to these vector systems broaden their utilities. For example, Huff et al. (198) constructed a series of related vector systems that enable use of different integration systems and easy subsequent removal of the integrase gene, and Saviola and Bishai (205) described a system for multiple integration events by introducing a new attB copy on the integrated vector itself. Taking advantage of the ability to promote excision of the plasmids in the presence of the L5 RDF (gp36), Parish and colleagues developed systems to test for gene essentiality (206) and for doing integrated plasmid switching (207).
Other Applications for Mycobacteriophage Site-Specific Recombination Systems
The serine-integrase systems have considerable potential for broad use in molecular biology and genetics. Although only a few have been studied in biochemical detail (including phiC31 and Bxb1 [148, 158, 208]), they generally share the features of using small attachments sites (<50 bp), lack requirements for host-encoded accessory factors, and have tightly regulated directionality. As such, they typically function efficiently in other biological contexts, and the mycobacteriophage Bxb1 system has been used in Plasmodium (209), Drosophila (210), Arabidopsis (211), and mammalian cells (212).
The Bxb1 system has also been used to construct microbial data storage and computing systems (213, 214). These take advantage of fine-tuning expression of both the integrase and the RDF (gp47), so as to be able to reproducibly flip chromosomal segments in E. coli. Generation of greater complexity to these systems will require exploitation of additional but compatible serine integrases, and phage diversity should more than satisfy this requirement (213).
Selectable Markers
Although they have yet to find widespread use, phage immunity systems provide effective selectable markers that avoid the use of antibiotics and antibiotic resistance genes. These have advantages both for use in combination with other antibiotic resistance genes when constructing complex recombinants and few antibiotic resistance markers are available, as well as for constructing recombinants of virulent strains where introducing additional antibiotic resistance genes can enhance the biosafety concerns. The first example of immunity selection was demonstrated using the L5 repressor, which confers immunity to both L5 and D29 (110), but the strategy is adaptable for any phage in which the repressor has been identified and shown to confer immunity to superinfection (97, 107, 164). Patterns of phage heteroimmunity indicate which of these can be used in combination with each other. In principle, there are dozens of additional immunity systems and thus the potential to create a large suite of compatible selectable systems.
Selection for transformants carrying the immunity marker typically uses a clear-plaque derivative of the parent phage from which the repressor gene was derived (110). Such mutants can usually be isolated easily and efficiently kill nontransformed cells, although phage-resistant mutants can arise at low frequency. Selection can be accomplished by first spreading 108 to 109 virion particles onto the surface of solid media, followed by plating of the electroporated cells, or by first adding the phage to the recovering electroporated cells and plating the mixture onto solid media (164). When using such selectable schemes, it is helpful to be mindful of the nature of the recombinants recovered. If the phage is competent for integration (the more usual circumstance), then the recombinants recovered will also harbor an integrated complete prophage. For many experimental purposes this may not matter and has the potential advantage of providing a system for complete maintenance of the repressor plasmid, even when replicating extrachromosomally, because should plasmid loss occur, the phage immediately begins lytic growth leading to phage propagation and cell death. If this is undesirable, then phages defective in integration can be used, allowing the recovery of transformants that are completely phage free. The availability of the BRED engineering approach (182) facilitates the construction of such integration-defective mutants (164; G. W. Broussard and G. F. Hatfull, unpublished observations).
Mycobacterial Recombineering
Recombineering—genetic engineering by recombination (215)—is a method developed originally for manipulation of E. coli, taking advantage of the ability of phageencoded recombination systems to greatly stimulate the levels of recombination, such that introduction of DNA substrates containing homology to the chromosome are efficiently incorporated (215, 216). The commonly used recombination systems are the Red function of phage Lambda, which contains three components, an exonuclease (Exo), a DNA pairing enzyme (Bet), and a RecBCD inhibitor (Gam), or the rac prophage-derived RecET system (215, 217). There are anecdotal reports of attempts to use the Lambda Red system in mycobacteria, all without success, although this is perhaps not surprising given the species specificities seen in similar recombineering systems (218).
A mycobacterial recombineering system was developed using recombinases encoded by mycobacteriophages (184–186), and this strategy of using species-cognate recombination systems has been used for other bacteria (219). However, not all phages encode recombination systems that are bioinformatically identifiable, and it was not until a substantial collection of sequenced genomes was available (56) that recET-like genes were identified in mycobacteriophage Che9c (184). Induced expression of Che9c 60 and 61 promotes recombination levels such that recombinants can be recovered following electroporation of a dsDNA substrate with homology to the chromosome, both in M. smegmatis and in M. tuberculosis (184). The requirements for homology seem less permissive than in E. coli, and the frequency of recombination drops substantially once the length falls below about 500 bp. Recombineering can also use ssDNA substrates, and then only Che9c gp61 is required (185). This enables the introduction of point mutations, especially when they can be selected for directly, such as those that confer antibiotic resistance, where the total number of recombinants recovered can be greater than 105 (185). A mutant that cannot be selected for directly may also be constructed by co-electroporation with one that does, because a high proportion of the selected transformants also carry the unselected mutation and can be screened for using PCR (185). This approach mirrors that developed for manipulation of phage genomes (182), as described above.
Tools for Diagnosis of Tuberculosis
Several approaches have been described that use phagebased systems for diagnosis of tuberculosis and assessment of drug susceptibility profiles. There is a strong motivation to develop such systems because traditional methodologies are typically very slow and more modern ones are often expensive. Phage-based diagnostics are thus typically aimed toward a low-cost point-of-care application (220). The phage amplified biologically (PhaB) assay (221) takes advantage of the ability of M. tuberculosis cells present in a clinical sample to support replication of phage D29, efficient elimination of excess input phage particles, and scoring phage amplification on lawns of M. smegmatis. The test requires simple microbiological methods, has reasonable sensitivity, and can be used to distinguish between rifampin-resistant and -sensitive strains (74, 222, 223). More than a dozen evaluations of specificity and sensitivity have been conducted, which have been summarized in meta-analyses reported in 2005 (224) and 2010 (225). The more recent analysis shows that the commercially available FastPlaque assay has rather variable estimates of both sensitivity (81% to 100%) and specificity (73% to 100%) (225).
An alternative approach uses reporter mycobacteriophages to deliver a readily assayable gene such as the firefly luciferase gene or a fluorescent reporter such as GFP (188, 190). Reporter phages have been built using a variety of phage genomes including TM4, D29, and L5, all of which can infect both M. smegmatis and M. tuberculosis (165, 188, 196, 226); the TM4 platform may offer some advantages because of its ability to infect stationary-phase cells, a phenomenon that is dependent on the peptidoglycan hydrolytic motif embedded within the phage Tapemeasure protein (106). The main difference in the choice of the reporter is the detection method used. For luciferase reporter phages, light emission from infected cells can be detected using either a luminometer or photographically (188, 227), whereas fluorescent reporter phage infection can be detected using either microscopy or flow cytometry (189, 190). For optimal signal generation, it is important to prevent phagemediated lysis, which can be accomplished using conditionally replicating mutants (49, 228, 229). Several evaluations of both luciferase and fluorescent phages have been conducted (230–235), and a meta-analysis of the luciferase studies shows consistent estimates of diagnostic accuracy (with seven of eight studies reporting 100% sensitivity) and specificity (with four of eight reporting 100% specificity) (225). Addition of a tag to the capsid protein provides an approach for the simple recovery of phage and phage-bacterial complexes (236).
Ideally, both PhaB and reporter phage assays could be applied directly to sputum, without the requirement for complex processing methods. Although it has proven tricky to develop methods for efficient sputum processing that provide good recovery of viable mycobacterial cells and inhibition of contaminant growth and that facilitate good phage infection, recent studies show that this is feasible (189), and it now awaits evaluation in larger collections of patient samples.
Phage Therapy?
No discussion of mycobacteriophages is complete without some consideration of their use for direct therapeutic control of mycobacterial infections including tuberculosis. The general concept of phage therapy is an old one and was championed by Felix D’Herelle for several decades following his discovery of bacteriophages (237). Phage therapy has been widely employed in the former Soviet Union but has been slow to gain acceptance elsewhere, although recently, phages have been approved for control of both Listeria and E. coli contaminations in meat. The use of mycobacteriophages for control of tuberculosis has considerable appeal given the predominance of multidrug-resistant (MDR) strains and the appearance of extensively drug-resistant (XDR) strains (238). Although there has been some laboratory assessment of phage therapy in M. tuberculosis–infected guinea pigs (239), and the possible use of surrogate systems for phage delivery to infected macrophages (240, 241), there have been no human trials.
There are two obvious impediments to phage therapy of tuberculosis. The first is the concern that during a pulmonary infection with M. tuberculosis, it would prove difficult for phages to efficiently access the bacteria, particularly where they are intracellular or contained with granulomas. This is a serious concern, but one that could be alleviated if there is any significant dynamic exchange of bacteria within an infection and if surrogates were used for phage delivery (240, 241). The second concern is the rapid development of phage resistance, which is expected to occur at frequencies equivalent to that of antibiotic resistance. This could be alleviated by use of multiple phages for which there are different resistance mechanisms, or inclusion of phage mutants isolated in the laboratory that promote escape from host resistance. Although these two problems may be tough, they would not seem insurmountable.
An alternative application might be to use mycobacteriophages prophylactically rather than therapeutically. For example, family members or coworkers of patients recently diagnosed with pulmonary tuberculosis could use aspirated phages to interfere with transmission and acquisition of the disease. This is appealing because the bacteria involved in disease acquisition are more likely to be accessible to phage attack, and typically only relatively small numbers of bacteria are inhaled, so that not only could relatively high multiplicities of infection be achieved, but the incidence of phage resistance would be low due to the relatively small size of the bacterial population under control. Although this would seem like an effective way to disrupt transmission that is both cheap and safe—without interfering with ongoing treatment of infected patients—to our knowledge it has not yet been evaluated.
PERSPECTIVES: PAST, PRESENT, AND FUTURE
In closing a comprehensive review of mycobacteriophages in 1984, Mizugichi concluded, “Studies on mycobacteriophages have contributed little to the knowledge and understanding of general virology especially in the fields of molecular biology and genetics. The future use of mycobacteriophages in studies of molecular biology would appear to be nonprofitable because of the paucity of necessary basic information. It would seem that the thrust of current mycobacteriophage studies should be to clarify the host-virus interaction in mycobacteria. A profound analysis of the relationship between mycobacteria and their phages would certainly enhance our knowledge of genetics, pathogenicity, drug resistance, variation and evolution of mycobacteria” (18). These words were quite prescient, especially with the emphasis on the need for a stronger basic understanding of mycobacteriophages and the ways in which this would be useful. The discussion of mycobacteriophages in the 2000 first edition of Molecular Genetics of Mycobacteria (64) reflected the considerable advances in both basic and applied studies over the subsequent dozen years.
However, the 13 years since the first edition in 2000 have quite dramatically advanced our understanding of mycobacteriophages and ways in which they can be exploited for tuberculosis genetics. In 2000, the complete genome sequences of only four mycobacteriophages (L5, D29, Bxb1, TM4) had been determined, dwarfed now by the ~285 complete genome sequences in GenBank (61, 90, 97, 98) and the total of over 500 (http://www.phagesdb.com). This wealth of genomic data has changed our perspectives of these phages and our expectations for exploiting them for mycobacterial genetics. The diversity is clearly enormous, and although we may now be closer to saturating the isolation of the vast majority of genome types that infect M. smegmatis mc2155, the use of other mycobacterial hosts is likely to reveal further expansion of this diversity.
The concluding sentence of the chapter on mycobacteriophages in the first edition stated, “We can therefore expect a rich and exciting future in elucidating mycobacteriophage evolution, novel systems of genetic regulation, tools for genetic and clinical applications, and the role of phages in mycobacterial physiology and virulence” (64). The ensuing 13 years more than adequately met these expectations; phage genomics has elucidated evolutionary mechanisms, integration-dependent immunity is an excellent example of novel regulation, recombineering is an effective genetic application, and Bxb1 integration has revealed aspects of host physiology and biofilm formation.
What will the next period of mycobacteriophage research bring? There are numerous areas in which advances might be anticipated. Currently, we have only limited insights into the host ranges of the currently characterized phages, know very little about what receptors are used and what determines specificity, and have only limited information on the rates and mechanisms by which phages switch host ranges. It is also reasonable to expect substantial advances in determining the functions of mycobacteriophage genes. There are a vast number of genes (>40,000) with unknown functions, and with new mutagenesis approaches we should be poised to find out what these do and why they are carried in phage genomes. Likewise, although we’re learning more about phage gene expression and the regulation of gene expression, this is mostly confined to small numbers of phages, and there is a great deal more to learn. The genomic information also suggests numerous ways in which these phages can be exploited for mycobacterial genetics and further insights into mycobacterial physiology.
Finally, the disseminated use of mycobacteriophage research for science education and research training has engaged hundreds of faculty and thousands of undergraduate students. Their involvement has had a huge impact over the past 10 years, and the continued engagement of these students and researchers will secure an active future for mycobacteriophage investigations.
Acknowledgments
I am grateful to Roger Hendrix, Craig Peebles, and Jeffrey Lawrence for discussions; to all members of my laboratory past and present for their insights and contributions; to Welkin Pope, Bekah Dedrick, Greg Broussard, and Debbie Jacobs-Sera for comments on the manuscript; and to all student phage hunters everywhere.
Studies in the Hatfull laboratory were supported by National Institutes of Health grant GM51975 and by the Howard Hughes Medical Institute through its Professorship grant to GFH.
References
- 1.Gardner GM, Weiser RS. A bacteriophage for Mycobacterium smegmatis. Proc Soc Exp Biol Med. 1947;66:205–206. doi: 10.3181/00379727-66-16037. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker E. Two bacteriophages for Mycobacterium smegmatis. Can J Public Health. 1950;41:431–436. [PubMed] [Google Scholar]
- 3.Froman S, Will DW, Bogen E. Bacteriophage active against Mycobacterium tuberculosis. I. Isolation and activity. Am J Pub Health. 1954;44:1326–1333. doi: 10.2105/ajph.44.10.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grange JM. Proceedings: bacteriophage typing of strains of Mycobacterium tuberculosis isolated in south-east England. J Med Microbiol. 1975;8:ix(2). [PubMed] [Google Scholar]
- 5.Grange JM. The genetics of mycobacteria and mycobacteriophages: a review. Tubercle. 1975;56:227–238. doi: 10.1016/0041-3879(75)90056-2. [DOI] [PubMed] [Google Scholar]
- 6.Jones WD., Jr Phage typing report of 125 strains of “Mycobacterium tuberculosis”. Ann Sclavo. 1975;17:599–604. [PubMed] [Google Scholar]
- 7.Jones WD., Jr Bacteriophage typing of Mycobacterium tuberculosis cultures from incidents of suspected laboratory cross-contamination. Tubercle. 1988;69:43–46. doi: 10.1016/0041-3879(88)90039-6. [DOI] [PubMed] [Google Scholar]
- 8.Snider DE, Jr, Jones WD, Good RC. The usefulness of phage typing Mycobacterium tuberculosis isolates. Am Rev Respir Dis. 1984;130:1095–1099. doi: 10.1164/arrd.1984.130.6.1095. [DOI] [PubMed] [Google Scholar]
- 9.Goode D. Bacteriophage typing of strains of Mycobacterium tuberculosis from Nepal. Tubercle. 1983;64:15–21. doi: 10.1016/0041-3879(83)90045-4. [DOI] [PubMed] [Google Scholar]
- 10.Murohashi T, Tokunaga T, Mizuguchi Y, Maruyama Y. Phage typing of slow-growing mycobacteria. Am Rev Respir Dis. 1963;88:664–669. doi: 10.1164/arrd.1963.88.5.664. [DOI] [PubMed] [Google Scholar]
- 11.Baess I. Subdivision of M. tuberculosis by means of bacteriophages. With special reference to epidemiological studies. Acta Pathol Microbiol Scand. 1969;76:464–474. [PubMed] [Google Scholar]
- 12.Gangadharam PR, Simmons CS, Stager CE. Phage typing of mycobacteria using paper discs. Am Rev Respir Dis. 1978;118:148–150. doi: 10.1164/arrd.1978.118.1.148. [DOI] [PubMed] [Google Scholar]
- 13.Raj CV, Ramakrishnan T. Transduction in Mycobacterium smegmatis. Nature. 1970;228:280–281. doi: 10.1038/228280b0. [DOI] [PubMed] [Google Scholar]
- 14.Sellers MI, Tokuyasu K, Price Z, Froman S. Electron microscopic studies of mycobacteriophages. Am Rev Tuberc. 1957;76:964–969. doi: 10.1164/artpd.1957.76.6.964. [DOI] [PubMed] [Google Scholar]
- 15.Buraczewska M, Kwiatkowski B, Manowska W, Rdultowska H. Ultrastructure of some mycobacteriophages. Am Rev Respir Dis. 1972;105:22–29. doi: 10.1164/arrd.1972.105.1.22. [DOI] [PubMed] [Google Scholar]
- 16.Jones W, White A. Lysogeny in mycobacteria. I. Conversion of colony morphology, nitrate reductase activity, and tween 80 hydrolysis of Mycobacterium sp. ATCC 607 associated with lysogeny. Can J Microbiol. 1968;14:551–555. doi: 10.1139/m68-093. [DOI] [PubMed] [Google Scholar]
- 17.Mankiewicz E, Liivak M, Dernuet S. Lysogenic mycobacteria: phage variations and changes in host cells. J Gen Microbiol. 1969;55:409–416. doi: 10.1099/00221287-55-3-409. [DOI] [PubMed] [Google Scholar]
- 18.Mizuguchi Y. Mycobacteriophages. In: Kubica GP, Wayne LG, editors. The Mycobacteria: A Sourcebook, part A. Marcel Dekker; New York: 1984. pp. 641–662. [Google Scholar]
- 19.Grange JM, Bird RG. The nature and incidence of lysogeny in Mycobacterium fortuitum. J Med Microbiol. 1975;8:215–223. doi: 10.1099/00222615-8-2-215. [DOI] [PubMed] [Google Scholar]
- 20.Grange JM, Bird RG. Lysogeny associated with mucoid variation in Mycobacterium kansasii. J Med Microbiol. 1978;11:1–6. doi: 10.1099/00222615-11-1-1. [DOI] [PubMed] [Google Scholar]
- 21.Karnik SS, Gopinathan KP. Transfection of Mycobacterium smegmatis SN2 with mycobacteriophage I3 DNA. Arch Microbiol. 1983;136:275–280. doi: 10.1007/BF00425216. [DOI] [PubMed] [Google Scholar]
- 22.Tokunaga T, Nakamura RM. Infection of competent Mycobacterium smegmatis with deoxyribonucleic acid extracted from bacteriophage B1. J Virol. 1968;2:110–117. doi: 10.1128/jvi.2.2.110-117.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tokunaga T, Mizuguchi Y, Murohashi T. Deoxyribonucleic acid base composition of a mycobacteriophage. J Bacteriol. 1963;86:608–609. doi: 10.1128/jb.86.3.608-609.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowman BU. Properties of mycobacteriophage DS6A. I. Immunogenicity in rabbits. Proc Soc Exp Biol Med. 1969;131:196–200. doi: 10.3181/00379727-131-33838. [DOI] [PubMed] [Google Scholar]
- 25.Buraczewska M, Manowska W, Rdultowska H. Studies on mycobacteriophages. Adsorption of mycobacteriophages on bacillary cells. Pol Med J. 1969;8:1337–1341. [PubMed] [Google Scholar]
- 26.Castelnuovo G, Giuliani HI, Luchini de Giuliani E, Arancia G. Protein components of a mycobacteriophage. J Gen Virol. 1969;4:253–257. doi: 10.1099/0022-1317-4-2-253. [DOI] [PubMed] [Google Scholar]
- 27.Juhasz SE, Gelbart S, Harize M. Phage-induced alteration of enzymic activity in lysogenic Mycobacterium smegmatis strains. J Gen Microbiol. 1969;56:251–255. doi: 10.1099/00221287-56-2-251. [DOI] [PubMed] [Google Scholar]
- 28.Menezes J, Pavilanis V. Properties of mycobacteriophage C2. Experientia. 1969;25:1112–1113. doi: 10.1007/BF01901466. [DOI] [PubMed] [Google Scholar]
- 29.Bonicke R, Saito H. Phage conversion of biochemical properties in the genus Mycobacterium. Bull Int Union Tuberc. 1970;43:217–225. [PubMed] [Google Scholar]
- 30.David HL, Jones WD., Jr Biosynthesis of a lipase by Mycobacterium smegmatis ATCC 607 infected by mycobacteriophage D29. Am Rev Respir Dis. 1970;102:818–820. doi: 10.1164/arrd.1970.102.5.818. [DOI] [PubMed] [Google Scholar]
- 31.Jones WD, David HL, Beam RE. The occurrence of lipids in mycobacteriophage D29 propagated in Mycobacterium smegmatis ATCC 607. Am Rev Respir Dis. 1970;102:814–817. doi: 10.1164/arrd.1970.102.5.814. [DOI] [PubMed] [Google Scholar]
- 32.Tokunaga T, Kataoka T, Suga K. Phage inactivation by an ethanol-ether extract of Mycobacterium smegmatis. Am Rev Respir Dis. 1970;101:309–313. doi: 10.1164/arrd.1970.101.2.309. [DOI] [PubMed] [Google Scholar]
- 33.Jones WD, Jr, David HL. Inhibition by fifampin of mycobacteriophage D29 replication in its drug-resistant host, Mycobacterium smegmatis ATCC 607. Am Rev Respir Dis. 1971;103:618–624. doi: 10.1164/arrd.1971.103.5.618. [DOI] [PubMed] [Google Scholar]
- 34.Bowman BU, Newman HA, Moritz JM, Koehler RM. Properties of mycobacteriophage DS6A. II. Lipid composition. Am Rev Respir Dis. 1973;107:42–49. doi: 10.1164/arrd.1973.107.1.42. [DOI] [PubMed] [Google Scholar]
- 35.Bowman BU, Jr, Fisher LJ, Witiak DT, Newman HA. Inactivation of phages DS6A and D29 by acetone extracts of Mycobacterium tuberculosis and Mycobacterium bovis. Am Rev Respir Dis. 1975;112:17–22. doi: 10.1164/arrd.1975.112.1.17. [DOI] [PubMed] [Google Scholar]
- 36.Wisingerova E, Sulova J, Sassmannova E. Inactivation of the mycophage activity by lipid solvents. Ann Sclavo. 1975;17:634–640. [PubMed] [Google Scholar]
- 37.Schafer R, Huber U, Franklin RM. Chemical and physical properties of mycobacteriophage D29. Eur J Biochem. 1977;73:239–246. doi: 10.1111/j.1432-1033.1977.tb11312.x. [DOI] [PubMed] [Google Scholar]
- 38.Soloff BL, Rado TA, Henry BE, 2nd, Bates JH. Biochemical and morphological characterization of mycobacteriophage R1. J Virol. 1978;25:253–262. doi: 10.1128/jvi.25.1.253-262.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somogyi PA, Maso Bel M, Foldes I. Methylated nucleic acid bases in mycobacterium and mycobacteriophage DNA. Acta Microbiol Acad Sci Hung. 1982;29:181–185. [PubMed] [Google Scholar]
- 40.Reddy AB, Gopinathan KP. Presence of random single-strand gaps in mycobacteriophage I3 DNA. Gene. 1986;44:227–234. doi: 10.1016/0378-1119(86)90186-1. [DOI] [PubMed] [Google Scholar]
- 41.Jacobs WR., Jr . Mycobacterium tuberculosis: a once genetically intractable organism. In: Hatfull GF, Jacobs WR Jr, editors. Molecular Genetics of the Mycobacteria. ASM Press; Washington, DC: 2000. pp. 1–16. [Google Scholar]
- 42.Hopwood DA, Wright HM. Bacterial protoplast fusion: recombination in fused protoplasts of Streptomyces coelicolor. Mol Gen Genet. 1978;162:307–317. doi: 10.1007/BF00268856. [DOI] [PubMed] [Google Scholar]
- 43.Okanishi M, Suzuki K, Umezawa H. Formation and reversion of streptomycete protoplasts: cultural condition and morphological study. J Gen Microbiol. 1974;80:389–400. doi: 10.1099/00221287-80-2-389. [DOI] [PubMed] [Google Scholar]
- 44.Jacobs WR, Jr, Tuckman M, Bloom BR. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature. 1987;327:532–535. doi: 10.1038/327532a0. [DOI] [PubMed] [Google Scholar]
- 45.Jacobs WR, Jr, Kalpana GV, Cirillo JD, Pascopella L, Snapper SB, Udani RA, Jones W, Barletta RG, Bloom BR. Genetic systems for mycobacteria. Methods Enzymol. 1991;204:537–555. doi: 10.1016/0076-6879(91)04027-l. [DOI] [PubMed] [Google Scholar]
- 46.Snapper SB, Lugosi L, Jekkel A, Melton RE, Kieser T, Bloom BR, Jacobs WR., Jr Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc Natl Acad Sci USA. 1988;85:6987–6991. doi: 10.1073/pnas.85.18.6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 48.Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 49.Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR., Jr Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 1997;94:10961–10966. doi: 10.1073/pnas.94.20.10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hatfull GF, Sarkis GJ. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol. 1993;7:395–405. doi: 10.1111/j.1365-2958.1993.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 51.Mediavilla J, Jain S, Kriakov J, Ford ME, Duda RL, Jacobs WR, Jr, Hendrix RW, Hatfull GF. Genome organization and characterization of mycobacteriophage Bxb1. Mol Microbiol. 2000;38:955–970. doi: 10.1046/j.1365-2958.2000.02183.x. [DOI] [PubMed] [Google Scholar]
- 52.Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF. Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol. 1998;279:143–164. doi: 10.1006/jmbi.1997.1610. [DOI] [PubMed] [Google Scholar]
- 53.Ford ME, Stenstrom C, Hendrix RW, Hatfull GF. Mycobacteriophage TM4: genome structure and gene expression. Tuber Lung Dis. 1998;79:63–73. doi: 10.1054/tuld.1998.0007. [DOI] [PubMed] [Google Scholar]
- 54.Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Hendrix RW, Hatfull GF. Origins of highly mosaic mycobacteriophage genomes. Cell. 2003;113:171–182. doi: 10.1016/s0092-8674(03)00233-2. [DOI] [PubMed] [Google Scholar]
- 55.Hatfull GF. Bacteriophage research: gateway to learning science. Microbe. 2010;5:243–250. [Google Scholar]
- 56.Hatfull GF, Pedulla ML, Jacobs-Sera D, Cichon PM, Foley A, Ford ME, Gonda RM, Houtz JM, Hryckowian AJ, Kelchner VA, Namburi S, Pajcini KV, Popovich MG, Schleicher DT, Simanek BZ, Smith AL, Zdanowicz GM, Kumar V, Peebles CL, Jacobs WR, Jr, Lawrence JG, Hendrix RW. Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet. 2006;2:e92. doi: 10.1371/journal.pgen.0020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanauer D, Hatfull GF, Jacobs-Sera D. Active Assessment: Assessing Scientific Inquiry. Springer; New York, NY: 2009. [Google Scholar]
- 58.Hanauer DI, Jacobs-Sera D, Pedulla ML, Cresawn SG, Hendrix RW, Hatfull GF. Inquiry learning. Teaching scientific inquiry. Science. 2006;314:1880–1881. doi: 10.1126/science.1136796. [DOI] [PubMed] [Google Scholar]
- 59.Hatfull GF. Bacteriophage genomics. Curr Opin Microbiol. 2008;11:447–453. doi: 10.1016/j.mib.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hatfull GF. Mycobacteriophages: genes and genomes. Annu Rev Microbiol. 2010;64:331–356. doi: 10.1146/annurev.micro.112408.134233. [DOI] [PubMed] [Google Scholar]
- 61.Hatfull GF. Complete genome sequences of 138 mycobacteriophages. J Virol. 2012;86:2382–2384. doi: 10.1128/JVI.06870-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hatfull GF. Mycobacteriophage L5: a toolbox for tuberculosis. ASM News. 1994;60:255–260. [Google Scholar]
- 63.Hatfull GF. Mycobacteriophages. In: Ratledge C, Dale J, editors. Mycobacteria: Molecular Biology and Virulence. Chapman and Hall; London: 1999. pp. 38–58. [Google Scholar]
- 64.Hatfull GF. Molecular genetics of mycobacteriophages. In: Hatfull GF, Jacobs WR Jr, editors. Molecular Genetics of Mycobacteria. ASM Press; Washington, DC: 2000. pp. 37–54. [Google Scholar]
- 65.Hatfull GF. Mycobacteriophages and tuberculosis. In: Eisenach K, Cole ST, Jacobs WR Jr, McMurray D, editors. Tuberculosis. ASM Press; Washington, DC: 2004. pp. 203–218. [Google Scholar]
- 66.Hatfull GF. Mycobacteriophages. In: Calendar R, editor. The Bacteriophages. Oxford University Press; New York, NY: 2006. pp. 602–620. [Google Scholar]
- 67.Hatfull GF. The secret lives of mycobacteriophages. Adv Virus Res. 2012;82:179–288. doi: 10.1016/B978-0-12-394621-8.00015-7. [DOI] [PubMed] [Google Scholar]
- 68.Hatfull GF, Barsom L, Chang L, Donnelly-Wu M, Lee MH, Levin M, Nesbit C, Sarkis GJ. Bacteriophages as tools for vaccine development. Dev Biol Stand. 1994;82:43–47. [PubMed] [Google Scholar]
- 69.Hatfull GF, Cresawn SG, Hendrix RW. Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res Microbiol. 2008;159:332–339. doi: 10.1016/j.resmic.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hatfull GF, Hendrix RW. Bacteriophages and their genomes. Curr Opin Virol. 2011;1:298–303. doi: 10.1016/j.coviro.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hatfull GF, Jacobs WR., Jr . Mycobacteriophages: cornerstones of mycobacterial research. In: Bloom BR, editor. Tuberculosis: Pathogenesis, Protection and Control. ASM Press; Washington, DC: 1994. pp. 165–183. [Google Scholar]
- 72.Stella EJ, de la Iglesia AI, Morbidoni HR. Mycobacteriophages as versatile tools for genetic manipulation of mycobacteria and development of simple methods for diagnosis of mycobacterial diseases. Rev Argent Microbiol. 2009;41:45–55. [PubMed] [Google Scholar]
- 73.McNerney R. TB: the return of the phage. A review of fifty years of mycobacteriophage research. Int J Tuberc Lung Dis. 1999;3:179–184. [PubMed] [Google Scholar]
- 74.McNerney R. Phage tests for diagnosis and drug susceptibility testing. Int J Tuberc Lung Dis. 2002;6:1129–1130. author reply 1130. [PubMed] [Google Scholar]
- 75.McNerney R, Traore H. Mycobacteriophage and their application to disease control. J Appl Microbiol. 2005;99:223–233. doi: 10.1111/j.1365-2672.2005.02596.x. [DOI] [PubMed] [Google Scholar]
- 76.Jacobs WR, Jr, Snapper SB, Tuckman M, Bloom BR. Mycobacteriophage vector systems. Rev Infect Dis. 1989;11(Suppl 2):S404–S410. doi: 10.1093/clinids/11.supplement_2.s404. [DOI] [PubMed] [Google Scholar]
- 77.Redmond WB. Bacteriophages of mycobacteria: a review. Adv Tuberc Rev. 1963;12:191–229. [Google Scholar]
- 78.Sarkis GJ, Hatfull GF. Mycobacteriophages. Methods Mol Biol. 1998;101:145–173. doi: 10.1385/0-89603-471-2:145. [DOI] [PubMed] [Google Scholar]
- 79.Cater JC, Redmond WB. Isolation of and studies on bacteriophage active against mycobacteria. Can J Microbiol. 1961;7:697–703. doi: 10.1139/m61-083. [DOI] [PubMed] [Google Scholar]
- 80.Cater JC, Redmond WB. Mycobacterial phages isolated from stool specimens of patients with pulmonary disease. Am Rev Respir Dis. 1963;87:726–729. doi: 10.1164/arrd.1963.87.5.726. [DOI] [PubMed] [Google Scholar]
- 81.Redmond WB, Carter JC. A bacteriophage specific to Mycobacterium tuberculosis varieties hominis and bovis. Am Rev Respir Dis. 1960;82:781–786. doi: 10.1164/arrd.1960.82.6.781. [DOI] [PubMed] [Google Scholar]
- 82.Timme TL, Brennan PJ. Induction of bacteriophage from members of the Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium scrofulaceum serocomplex. J Gen Microbiol. 1984;130:2059–2066. doi: 10.1099/00221287-130-8-2059. [DOI] [PubMed] [Google Scholar]
- 83.Sassi M, Bebeacua C, Drancourt M, Cambillau C. The first structure of a mycobacteriophage, the Mycobacterium abscessus subsp. bolletii phage Araucaria. J Virol. 2013;87:8099–8109. doi: 10.1128/JVI.01209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choo SW, Yusoff AM, Wong YL, Wee WY, Ong CS, Ng KP, Ngeow YF. Genome analysis of Mycobacterium massiliense strain M172, which contains a putative mycobacteriophage. J Bacteriol. 2012;194:5128. doi: 10.1128/JB.01096-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tobias NJ, Doig KD, Medema MH, Chen H, Haring V, Moore R, Seemann T, Stinear TP. Complete genome sequence of the frog pathogen Mycobacterium ulcerans ecovar Liflandii. J Bacteriol. 2013;195:556–564. doi: 10.1128/JB.02132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 87.Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, Abdellah Z, Arrowsmith C, Chillingworth T, Churcher C, Clarke K, Cronin A, Davis P, Goodhead I, Holroyd N, Jagels K, Lord A, Moule S, Mungall K, Norbertczak H, Quail MA, Rabbinowitsch E, Walker D, White B, Whitehead S, Small PL, Brosch R, Ramakrishnan L, Fischbach MA, Parkhill J, Cole ST. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008;18:729–741. doi: 10.1101/gr.075069.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, Khanna V, Majlessi L, Criscuolo A, Tap J, Pawlik A, Fiette L, Orgeur M, Fabre M, Parmentier C, Frigui W, Simeone R, Boritsch EC, Debrie AS, Willery E, Walker D, Quail MA, Ma L, Bouchier C, Salvignol G, Sayes F, Cascioferro A, Seemann T, Barbe V, Locht C, Gutierrez MC, Leclerc C, Bentley SD, Stinear TP, Brisse S, Medigue C, Parkhill J, Cruveiller S, Brosch R. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet. 2013;45:172–179. doi: 10.1038/ng.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hatfull GF, Jacobs-Sera D, Lawrence JG, Pope WH, Russell DA, Ko CC, Weber RJ, Patel MC, Germane KL, Edgar RH, Hoyte NN, Bowman CA, Tantoco AT, Paladin EC, Myers MS, Smith AL, Grace MS, Pham TT, O’Brien MB, Vogelsberger AM, Hryckowian AJ, Wynalek JL, Donis-Keller H, Bogel MW, Peebles CL, Cresawn SG, Hendrix RW. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J Mol Biol. 2010;397:119–143. doi: 10.1016/j.jmb.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pope WH, Jacobs-Sera D, Best AA, Broussard GW, Connerly PL, Dedrick RM, Kremer TA, Offner S, Ogiefo AH, Pizzorno MC, Rockenbach K, Russell DA, Stowe EL, Stukey J, Thibault SA, Conway JF, Hendrix RW, Hatfull GF. Cluster J mycobacteriophages: intron splicing in capsid and tail genes. PLoS One. 2013;8:e69273. doi: 10.1371/journal.pone.0069273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harrison M, Dunbar D, Ratmansky L, Boyd K, Lopatto D. Classroom-based science research at the introductory level: changes in career choices and attitude. CBE Life Sci Educ. 2011;10:279–286. doi: 10.1187/cbe.10-12-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shaffer CD, Alvarez C, Bailey C, Barnard D, Bhalla S, Chandrasekaran C, Chandrasekaran V, Chung HM, Dorer DR, Du C, Eckdahl TT, Poet JL, Frohlich D, Goodman AL, Gosser Y, Hauser C, Hoopes LL, Johnson D, Jones CJ, Kaehler M, Kokan N, Kopp OR, Kuleck GA, McNeil G, Moss R, Myka JL, Nagengast A, Morris R, Overvoorde PJ, Shoop E, Parrish S, Reed K, Regisford EG, Revie D, Rosenwald AG, Saville K, Schroeder S, Shaw M, Skuse G, Smith C, Smith M, Spana EP, Spratt M, Stamm J, Thompson JS, Wawersik M, Wilson BA, Youngblom J, Leung W, Buhler J, Mardis ER, Lopatto D, Elgin SC. The genomics education partnership: successful integration of research into laboratory classes at a diverse group of undergraduate institutions. CBE Life Sci Educ. 2010;9:55–69. doi: 10.1187/09-11-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lopatto D, Alvarez C, Barnard D, Chandrasekaran C, Chung HM, Du C, Eckdahl T, Goodman AL, Hauser C, Jones CJ, Kopp OR, Kuleck GA, McNeil G, Morris R, Myka JL, Nagengast A, Overvoorde PJ, Poet JL, Reed K, Regisford G, Revie D, Rosenwald A, Saville K, Shaw M, Skuse GR, Smith C, Smith M, Spratt M, Stamm J, Thompson JS, Wilson BA, Witkowski C, Youngblom J, Leung W, Shaffer CD, Buhler J, Mardis E, Elgin SC. Undergraduate research. Genom Educ Partnership Sci. 2008;322:684–685. doi: 10.1126/science.1165351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smith SA, Tank DC, Boulanger LA, Bascom-Slack CA, Eisenman K, Kingery D, Babbs B, Fenn K, Greene JS, Hann BD, Keehner J, Kelley-Swift EG, Kembaiyan V, Lee SJ, Li P, Light DY, Lin EH, Ma C, Moore E, Schorn MA, Vekhter D, Nunez PV, Strobel GA, Donoghue MJ, Strobel SA. Bioactive endophytes warrant intensified exploration and conservation. PLoS One. 2008;3:e3052. doi: 10.1371/journal.pone.0003052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hanauer DI, Frederick J, Fotinakes B, Strobel SA. Linguistic analysis of project ownership for undergraduate research experiences. CBE Life Sci Educ. 2012;11:378–385. doi: 10.1187/cbe.12-04-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc Natl Acad Sci USA. 1999;96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pope WH, Ferreira CM, Jacobs-Sera D, Benjamin RC, Davis AJ, DeJong RJ, Elgin SCR, Guilfoile FR, Forsyth MH, Harris AD, Harvey SE, Hughes LE, Hynes PM, Jackson AS, Jalal MD, MacMurray EA, Manley CM, McDonough MJ, Mosier JL, Osterbann LJ, Rabinowitz HS, Rhyan CN, Russell DA, Saha MS, Shaffer CD, Simon SE, Sims EF, Tovar IG, Weisser EG, Wertz JT, Weston-Hafer KA, Williamson KE, Zhang B, Cresawn SG, Jain P, Piuri M, Jacobs WR, Jr, Hendrix RW, Hatfull GF. Cluster K mycobacteriophages: insights into the evolutionary origins of mycobacteriophage TM4. PLoS One. 2011;6:e26750. doi: 10.1371/journal.pone.0026750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pope WH, Jacobs-Sera D, Russell DA, Peebles CL, Al-Atrache Z, Alcoser TA, Alexander LM, Alfano MB, Alford ST, Amy NE, Anderson MD, Anderson AG, Ang AAS, Ares M, Jr, Barber AJ, Barker LP, Barrett JM, Barshop WD, Bauerle CM, Bayles IM, Belfield KL, Best AA, Borjon A, Jr, Bowman CA, Boyer CA, Bradley KW, Bradley VA, Broadway LN, Budwal K, Busby KN, Campbell IW, Campbell AM, Carey A, Caruso SM, Chew RD, Cockburn CL, Cohen LB, Corajod JM, Cresawn SG, Davis KR, Deng L, Denver DR, Dixon BR, Ekram S, Elgin SCR, Engelsen AE, English BEV, Erb ML, Estrada C, Filliger LZ, Findley AM, Forbes L, Forsyth MH, Fox TM, Fritz MJ, Garcia R, George ZD, Georges AE, Gissendanner CR, Goff S, Goldstein R, Gordon KC, Green RD, Guerra SL, Guiney-Olsen KR, Guiza BG, Haghighat L, Hagopian GV, Harmon CJ, Harmson JS, Hartzog GA, Harvey SE, He S, He KJ, Healy KE, Higinbotham ER, Hildebrandt EN, Ho JH, Hogan GM, Hohenstein VG, Holz NA, Huang VJ, Hufford EL, Hynes PM, Jackson AS, Jansen EC, Jarvik J, Jasinto PG, Jordan TC, Kasza T, Katelyn MA, Kelsey JS, Kerrigan LA, Khaw D, Kim J, Knutter JZ, Ko C-C, Larkin GV, Laroche JR, Latif A, Leuba KD, Leuba SI, Lewis LO, Loesser-Casey KE, Long CA, Lopez AJ, Lowery N, Lu TQ, Mac V, Masters IR, McCloud JJ, McDonough MJ, Medenbach AJ, Menon A, Miller R, Morgan BK, Ng PC, Nguyen E, Nguyen KT, Nguyen ET, Nicholson KM, Parnell LA, Peirce CE, Perz AM, Peterson LJ, Pferdehirt RE, Philip SV, Pogliano K, Pogliano J, Polley T, Puopolo EJ, Rabinowitz HS, Resiss MJ, Rhyan CN, Robinson YM, Rodriguez LL, Rose AC, Rubin JD, Ruby JA, Saha MS, Sandoz JW, Savitskaya J, Schipper DJ, Schnitzler CE, Schott AR, Segal JB, Shaffer CD, Sheldon KE, Shepard EM, Shepardson JW, Shroff MK, Simmons JM, Simms EF, Simpson BM, Sinclair KM, Sjoholm RL, Slette IJ, Spaulding BC, Straub CL, Stukey J, Sughrue T, Tang T-Y, Tatyana LM, Taylor SB, Taylor BJ, Temple LM, Thompson JV, Tokarz MP, Trapani SE, Troum AP, Tsay J, Tubbs AT, Walton JM, Wang DH, Wang H, Warner JR, Weisser EG, Wendler SC, Weston-Hafer KA, Whelan HM, Williamson KE, Willis AN, Wirtshafter HS, Wong TW, Wu P, Yang YJ, Yee BC, Zaidins DA, Zhang B, Zúniga MY, Hendrix RW, Hatfull GF. Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution. PLoS One. 2011;6:e16329. doi: 10.1371/journal.pone.0016329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science Sea-Phages Program. Modlin RL, Hendrix RW, Hatfull GF. On the nature of mycobacteriophage diversity and host preference. Virology. 2012;434:187–201. doi: 10.1016/j.virol.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, Hatfull GF. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics. 2011;12:395. doi: 10.1186/1471-2105-12-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 102.Mageeney C, Pope WH, Harrison M, Moran D, Cross T, Jacobs-Sera D, Hendrix RW, Dunbar D, Hatfull GF. Mycobacteriophage Marvin: a new singleton phage with an unusual genome organization. J Virol. 2012;86:4762–4775. doi: 10.1128/JVI.00075-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morris P, Marinelli LJ, Jacobs-Sera D, Hendrix RW, Hatfull GF. Genomic characterization of mycobacteriophage Giles: evidence for phage acquisition of host DNA by illegitimate recombination. J Bacteriol. 2008;190:2172–2182. doi: 10.1128/JB.01657-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duda RL, Hempel J, Michel H, Shabanowitz J, Hunt D, Hendrix RW. Structural transitions during bacteriophage HK97 head assembly. J Mol Biol. 1995;247:618–635. doi: 10.1006/jmbi.1995.0168. [DOI] [PubMed] [Google Scholar]
- 105.Xu J, Hendrix RW, Duda RL. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell. 2004;16:11–21. doi: 10.1016/j.molcel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 106.Piuri M, Hatfull GF. A peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol Microbiol. 2006;62:1569–1585. doi: 10.1111/j.1365-2958.2006.05473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dedrick RM, Marinelli LJ, Newton GL, Pogliano K, Pogliano J, Hatfull GF. Functional requirements for bacteriophage growth: gene essentiality and expression in mycobacteriophage Giles. Mol Microbiol. 2013;88:577–589. doi: 10.1111/mmi.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giri N, Bhowmik P, Bhattacharya B, Mitra M, Das Gupta SK. The mycobacteriophage D29 gene 65 encodes an early-expressed protein that functions as a structure-specific nuclease. J Bacteriol. 2009;191:959–967. doi: 10.1128/JB.00960-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu J, Dehbi M, Moeck G, Arhin F, Bauda P, Bergeron D, Callejo M, Ferretti V, Ha N, Kwan T, McCarty J, Srikumar R, Williams D, Wu JJ, Gros P, Pelletier J, DuBow M. Antimicrobial drug discovery through bacteriophage genomics. Nat Biotechnol. 2004;22:185–191. doi: 10.1038/nbt932. [DOI] [PubMed] [Google Scholar]
- 110.Donnelly-Wu MK, Jacobs WR, Jr, Hatfull GF. Superinfection immunity of mycobacteriophage L5: applications for genetic transformation of mycobacteria. Mol Microbiol. 1993;7:407–417. doi: 10.1111/j.1365-2958.1993.tb01132.x. [DOI] [PubMed] [Google Scholar]
- 111.Rybniker J, Plum G, Robinson N, Small PL, Hartmann P. Identification of three cytotoxic early proteins of mycobacteriophage L5 leading to growth inhibition in Mycobacterium smegmatis. Microbiology. 2008;154:2304–2314. doi: 10.1099/mic.0.2008/017004-0. [DOI] [PubMed] [Google Scholar]
- 112.Rybniker J, Krumbach K, van Gumpel E, Plum G, Eggeling L, Hartmann P. The cytotoxic early protein 77 of mycobacteriophage L5 interacts with MSMEG_3532, an L-serine dehydratase of Mycobacterium smegmatis. J Basic Microbiol. 2011;51:515–522. doi: 10.1002/jobm.201000446. [DOI] [PubMed] [Google Scholar]
- 113.Hassan S, Mahalingam V, Kumar V. Synonymous codon usage analysis of thirty two mycobacteriophage genomes. Adv Bioinformatics. 2009;2009:1–11. doi: 10.1155/2009/316936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kunisawa T. Functional role of mycobacteriophage transfer RNAs. J Theor Biol. 2000;205:167–170. doi: 10.1006/jtbi.2000.2057. [DOI] [PubMed] [Google Scholar]
- 115.Kunisawa T, Kanaya S, Kutter E. Comparison of synonymous codon distribution patterns of bacteriophage and host genomes. DNA Res. 1998;5:319–326. doi: 10.1093/dnares/5.6.319. [DOI] [PubMed] [Google Scholar]
- 116.Sahu K, Gupta SK, Ghosh TC, Sau S. Synonymous codon usage analysis of the mycobacteriophage Bxz1 and its plating bacteria M. smegmatis: identification of highly and lowly expressed genes of Bxz1 and the possible function of its tRNA species. J Biochem Mol Biol. 2004;37:487–492. doi: 10.5483/bmbrep.2004.37.4.487. [DOI] [PubMed] [Google Scholar]
- 117.Sampson T, Broussard GW, Marinelli LJ, Jacobs-Sera D, Ray M, Ko CC, Russell D, Hendrix RW, Hatfull GF. Mycobacteriophages BPs, Angel and Halo: comparative genomics reveals a novel class of ultra-small mobile genetic elements. Microbiology. 2009;155:2962–2977. doi: 10.1099/mic.0.030486-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tori K, Dassa B, Johnson MA, Southworth MW, Brace LE, Ishino Y, Pietrokovski S, Perler FB. Splicing of the mycobacteriophage Bethlehem DnaB intein: identification of a new mechanistic class of inteins that contain an obligate block F nucleophile. J Biol Chem. 2009;285:2515–2526. doi: 10.1074/jbc.M109.069567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tori K, Perler FB. Expanding the definition of class 3 inteins and their proposed phage origin. J Bacteriol. 2011;193:2035–2041. doi: 10.1128/JB.01407-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rybniker J, Kramme S, Small PL. Host range of 14 mycobacteriophages in Mycobacterium ulcerans and seven other mycobacteria including Mycobacterium tuberculosis: application for identification and susceptibility testing. J Med Microbiol. 2006;55:37–42. doi: 10.1099/jmm.0.46238-0. [DOI] [PubMed] [Google Scholar]
- 121.Chen J, Kriakov J, Singh A, Jacobs WR, Jr, Besra GS, Bhatt A. Defects in glycopeptidolipid biosynthesis confer phage I3 resistance in Mycobacterium smegmatis. Microbiology. 2009;155:4050–4057. doi: 10.1099/mic.0.033209-0. [DOI] [PubMed] [Google Scholar]
- 122.Furuchi A, Tokunaga T. Nature of the receptor substance of Mycobacterium smegmatis for D4 bacteriophage adsorption. J Bacteriol. 1972;111:404–411. doi: 10.1128/jb.111.2.404-411.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bisso G, Castelnuovo G, Nardelli MG, Orefici G, Arancia G, Laneelle G, Asselineau C, Asselineau J. A study on the receptor for a mycobacteriophage: phage phlei. Biochimie. 1976;58:87–97. doi: 10.1016/s0300-9084(76)80359-8. [DOI] [PubMed] [Google Scholar]
- 124.Khoo KH, Suzuki R, Dell A, Morris HR, McNeil MR, Brennan PJ, Besra GS. Chemistry of the lyxose-containing mycobacteriophage receptors of Mycobacterium phlei/Mycobacterium smegmatis. Biochemistry. 1996;35:11812–11819. doi: 10.1021/bi961055+. [DOI] [PubMed] [Google Scholar]
- 125.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 126.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci USA. 2009;106:894–899. doi: 10.1073/pnas.0808832106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Allix C, Aristimuno L, Arora J, Baumanis V, Binder L, Cafrune P, Cataldi A, Cheong S, Diel R, Ellermeier C, Evans JT, Fauville-Dufaux M, Ferdinand S, Garcia de Viedma D, Garzelli C, Gazzola L, Gomes HM, Guttierez MC, Hawkey PM, van Helden PD, Kadival GV, Kreiswirth BN, Kremer K, Kubin M, Kulkarni SP, Liens B, Lillebaek T, Ho ML, Martin C, Martin C, Mokrousov I, Narvskaia O, Ngeow YF, Naumann L, Niemann S, Parwati I, Rahim Z, Rasolofo-Razanamparany V, Rasolonavalona T, Rossetti ML, Rusch-Gerdes S, Sajduda A, Samper S, Shemyakin IG, Singh UB, Somoskovi A, Skuce RA, van Soolingen D, Streicher EM, Suffys PN, Tortoli E, Tracevska T, Vincent V, Victor TC, Warren RM, Yap SF, Zaman K, Portaels F, Rastogi N, Sola C. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. doi: 10.1186/1471-2180-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ramage HR, Connolly LE, Cox JS. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009;5:e1000767. doi: 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.He L, Fan X, Xie J. Comparative genomic structures of Mycobacterium CRISPR-Cas. J Cell Biochem. 2012;113:2464–2473. doi: 10.1002/jcb.24121. [DOI] [PubMed] [Google Scholar]
- 130.Horan KL, Freeman R, Weigel K, Semret M, Pfaller S, Covert TC, van Soolingen D, Leao SC, Behr MA, Cangelosi GA. Isolation of the genome sequence strain Mycobacterium avium 104 from multiple patients over a 17-year period. J Clin Microbiol. 2006;44:783–789. doi: 10.1128/JCM.44.3.783-789.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Barsom EK, Hatfull GF. Characterization of Mycobacterium smegmatis gene that confers resistance to phages L5 and D29 when overexpressed. Mol Microbiol. 1996;21:159–170. doi: 10.1046/j.1365-2958.1996.6291342.x. [DOI] [PubMed] [Google Scholar]
- 132.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc Natl Acad Sci USA. 1999;96:1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Young R. Bacteriophage lysis: mechanism and regulation. Microbiol Rev. 1992;56:430–481. doi: 10.1128/mr.56.3.430-481.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Berry J, Summer EJ, Struck DK, Young R. The final step in the phage infection cycle: the Rz and Rz1 lysis proteins link the inner and outer membranes. Mol Microbiol. 2008;70:341–351. doi: 10.1111/j.1365-2958.2008.06408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gil F, Catalao MJ, Moniz-Pereira J, Leandro P, McNeil M, Pimentel M. The lytic cassette of mycobacteriophage Ms6 encodes an enzyme with lipolytic activity. Microbiology. 2008;154:1364–1371. doi: 10.1099/mic.0.2007/014621-0. [DOI] [PubMed] [Google Scholar]
- 136.Gil F, Grzegorzewicz AE, Catalao MJ, Vital J, McNeil MR, Pimentel M. Mycobacteriophage Ms6 LysB specifically targets the outer membrane of Mycobacterium smegmatis. Microbiology. 2010;156:1497–1504. doi: 10.1099/mic.0.032821-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Payne K, Sun Q, Sacchettini J, Hatfull GF. Mycobacteriophage Lysin B is a novel mycolylarabinogalactan esterase. Mol Microbiol. 2009;73:367–381. doi: 10.1111/j.1365-2958.2009.06775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Payne KM, Hatfull GF. Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One. 2012;7:e34052. doi: 10.1371/journal.pone.0034052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Catalao MJ, Gil F, Moniz-Pereira J, Pimentel M. The mycobacteriophage Ms6 encodes a chaperone-like protein involved in the endolysin delivery to the peptidoglycan. Mol Microbiol. 2010;77:672–686. doi: 10.1111/j.1365-2958.2010.07239.x. [DOI] [PubMed] [Google Scholar]
- 140.Catalao MJ, Gil F, Moniz-Pereira J, Pimentel M. The endolysin-binding domain encompasses the N-terminal region of the mycobacteriophage Ms6 Gp1 chaperone. J Bacteriol. 2011;193:5002–5006. doi: 10.1128/JB.00380-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Berry J, Rajaure M, Pang T, Young R. The spanin complex is essential for lambda lysis. J Bacteriol. 2012;194:5667–5674. doi: 10.1128/JB.01245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Catalao MJ, Gil F, Moniz-Pereira J, Pimentel M. Functional analysis of the holin-like proteins of mycobacteriophage Ms6. J Bacteriol. 2011;193:2793–2803. doi: 10.1128/JB.01519-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stella EJ, Franceschelli JJ, Tasselli SE, Morbidoni HR. Analysis of novel mycobacteriophages indicates the existence of different strategies for phage inheritance in mycobacteria. PLoS One. 2013;8:e56384. doi: 10.1371/journal.pone.0056384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lee MH, Pascopella L, Jacobs WR, Jr, Hatfull GF. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc Natl Acad Sci USA. 1991;88:3111–3115. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology. 2007;153:2711–2723. doi: 10.1099/mic.0.2007/008904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Freitas-Vieira A, Anes E, Moniz-Pereira J. The site-specific recombination locus of mycobacteriophage Ms6 determines DNA integration at the tRNA(Ala) gene of Mycobacterium spp. Microbiology. 1998;144:3397–3406. doi: 10.1099/00221287-144-12-3397. [DOI] [PubMed] [Google Scholar]
- 147.Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF. Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol Microbiol. 2003;50:463–473. doi: 10.1046/j.1365-2958.2003.03723.x. [DOI] [PubMed] [Google Scholar]
- 148.Smith MC, Brown WR, McEwan AR, Rowley PA. Site-specific recombination by phiC31 integrase and other large serine recombinases. Biochem Soc Trans. 2010;38:388–394. doi: 10.1042/BST0380388. [DOI] [PubMed] [Google Scholar]
- 149.Ojha A, Anand M, Bhatt A, Kremer L, Jacobs WR, Jr, Hatfull GF. GroEL1: a dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell. 2005;123:861–873. doi: 10.1016/j.cell.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 150.Ojha AK, Trivelli X, Guerardel Y, Kremer L, Hatfull GF. Enzymatic hydrolysis of trehalose dimycolate releases free mycolic acids during mycobacterial growth in biofilms. J Biol Chem. 2010;285:17380–17389. doi: 10.1074/jbc.M110.112813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Peña CE, Lee MH, Pedulla ML, Hatfull GF. Characterization of the mycobacteriophage L5 attachment site, attP. J Mol Biol. 1997;266:76–92. doi: 10.1006/jmbi.1996.0774. [DOI] [PubMed] [Google Scholar]
- 152.Peña CE, Stoner JE, Hatfull GF. Positions of strand exchange in mycobacteriophage L5 integration and characterization of the attB site. J Bacteriol. 1996;178:5533–5536. doi: 10.1128/jb.178.18.5533-5536.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lewis JA, Hatfull GF. Control of directionality in integrase-mediated recombination: examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res. 2001;29:2205–2216. doi: 10.1093/nar/29.11.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lewis JA, Hatfull GF. Identification and characterization of mycobacteriophage L5 excisionase. Mol Microbiol. 2000;35:350–360. doi: 10.1046/j.1365-2958.2000.01695.x. [DOI] [PubMed] [Google Scholar]
- 155.Lewis JA, Hatfull GF. Control of directionality in L5 integrase-mediated site-specific recombination. J Mol Biol. 2003;326:805–821. doi: 10.1016/s0022-2836(02)01475-4. [DOI] [PubMed] [Google Scholar]
- 156.Pedulla ML, Lee MH, Lever DC, Hatfull GF. A novel host factor for integration of mycobacteriophage L5. Proc Natl Acad Sci USA. 1996;93:15411–15416. doi: 10.1073/pnas.93.26.15411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pena CEA, Kahlenberg JM, Hatfull GF. Protein-DNA complexes in mycobacteriophage L5 integrative recombination. J Bacteriol. 1999;181:454–461. doi: 10.1128/jb.181.2.454-461.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ghosh P, Kim AI, Hatfull GF. The orientation of mycobacteriophage Bxb1 integration is solely dependent on the central dinucleotide of attP and attB. Mol Cell. 2003;12:1101–1111. doi: 10.1016/s1097-2765(03)00444-1. [DOI] [PubMed] [Google Scholar]
- 159.Ghosh P, Pannunzio NR, Hatfull GF. Synapsis in phage Bxb1 integration: selection mechanism for the correct pair of recombination sites. J Mol Biol. 2005;349:331–348. doi: 10.1016/j.jmb.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 160.Ghosh P, Bibb LA, Hatfull GF. Two-step site selection for serine-integrase-mediated excision: DNA-directed integrase conformation and central dinucleotide proofreading. Proc Natl Acad Sci USA. 2008;105:3238–3243. doi: 10.1073/pnas.0711649105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ghosh P, Wasil LR, Hatfull GF. Control of phage Bxb1 excision by a novel recombination directionality factor. PLoS Biol. 2006;4:e186. doi: 10.1371/journal.pbio.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Savinov A, Pan J, Ghosh P, Hatfull GF. The Bxb1 gp47 recombination directionality factor is required not only for prophage excision, but also for phage DNA replication. Gene. 2012;495:42–48. doi: 10.1016/j.gene.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nesbit CE, Levin ME, Donnelly-Wu MK, Hatfull GF. Transcriptional regulation of repressor synthesis in mycobacteriophage L5. Mol Microbiol. 1995;17:1045–1056. doi: 10.1111/j.1365-2958.1995.mmi_17061045.x. [DOI] [PubMed] [Google Scholar]
- 164.Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell. 2013;49:237–248. doi: 10.1016/j.molcel.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sarkis GJ, Jacobs WR, Jr, Hatfull GF. L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol Microbiol. 1995;15:1055–1067. doi: 10.1111/j.1365-2958.1995.tb02281.x. [DOI] [PubMed] [Google Scholar]
- 166.Garcia M, Pimentel M, Moniz-Pereira J. Expression of mycobacteriophage Ms6 lysis genes is driven by two sigma(70)-like promoters and is dependent on a transcription termination signal present in the leader RNA. J Bacteriol. 2002;184:3034–3043. doi: 10.1128/JB.184.11.3034-3043.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rybniker J, Nowag A, van Gumpel E, Nissen N, Robinson N, Plum G, Hartmann P. Insights into the function of the WhiB-like protein of mycobacteriophage TM4: a transcriptional inhibitor of WhiB2. Mol Microbiol. 2010;77:642–657. doi: 10.1111/j.1365-2958.2010.07235.x. [DOI] [PubMed] [Google Scholar]
- 168.Ptashne M, Jeffrey A, Johnson AD, Maurer R, Meyer BJ, Pabo CO, Roberts TM, Sauer RT. How the lambda repressor and cro work. Cell. 1980;19:1–11. doi: 10.1016/0092-8674(80)90383-9. [DOI] [PubMed] [Google Scholar]
- 169.Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF. Transcrip-tional silencing by the mycobacteriophage L5 repressor. EMBO J. 1997;16:5914–5921. doi: 10.1093/emboj/16.19.5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bandhu A, Ganguly T, Chanda PK, Das M, Jana B, Chakrabarti G, Sau S. Antagonistic effects Na+ and Mg2+ on the structure, function, and stability of mycobacteriophage L1 repressor. BMB Rep. 2009;42:293–298. doi: 10.5483/bmbrep.2009.42.5.293. [DOI] [PubMed] [Google Scholar]
- 171.Bandhu A, Ganguly T, Jana B, Mondal R, Sau S. Regions and residues of an asymmetric operator DNA interacting with the monomeric repressor of temperate mycobacteriophage L1. Biochemistry. 2010;49:4235–4243. doi: 10.1021/bi9020956. [DOI] [PubMed] [Google Scholar]
- 172.Ganguly T, Bandhu A, Chattoraj P, Chanda PK, Das M, Mandal NC, Sau S. Repressor of temperate mycobacteriophage L1 harbors a stable C-terminal domain and binds to different asymmetric operator DNAs with variable affinity. Virol J. 2007;4:64. doi: 10.1186/1743-422X-4-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ganguly T, Chanda PK, Bandhu A, Chattoraj P, Das M, Sau S. Effects of physical, ionic, and structural factors on the binding of repressor of mycobacteriophage L1 to its cognate operator DNA. Protein Pept Lett. 2006;13:793–798. doi: 10.2174/092986606777841262. [DOI] [PubMed] [Google Scholar]
- 174.Ganguly T, Chattoraj P, Das M, Chanda PK, Mandal NC, Lee CY, Sau S. A point mutation at the C-terminal half of the repressor of temperate mycobacteriophage L1 affects its binding to the operator DNA. J Biochem Mol Biol. 2004;37:709–714. doi: 10.5483/bmbrep.2004.37.6.709. [DOI] [PubMed] [Google Scholar]
- 175.Jain S, Hatfull GF. Transcriptional regulation and immunity in mycobacteriophage Bxb1. Mol Microbiol. 2000;38:971–985. doi: 10.1046/j.1365-2958.2000.02184.x. [DOI] [PubMed] [Google Scholar]
- 176.Sau S, Chattoraj P, Ganguly T, Lee CY, Mandal NC. Cloning and sequencing analysis of the repressor gene of temperate mycobacteriophage L1. J Biochem Mol Biol. 2004;37:254–259. doi: 10.5483/bmbrep.2004.37.2.254. [DOI] [PubMed] [Google Scholar]
- 177.Chaudhuri B, Sau S, Datta HJ, Mandal NC. Isolation, characterization, and mapping of temperature-sensitive mutations in the genes essential for lysogenic and lytic growth of the mycobacteriophage L1. Virology. 1993;194:166–172. doi: 10.1006/viro.1993.1246. [DOI] [PubMed] [Google Scholar]
- 178.Bandhu A, Ganguly T, Jana B, Chakravarty A, Biswas A, Sau S. Biochemical characterization of L1 repressor mutants with altered operator DNA binding activity. Bacteriophage. 2012;2:79–88. doi: 10.4161/bact.21157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Petrovski S, Seviour RJ, Tillett D. Genome sequence and characterization of a Rhodococcus equi phage REQ1. Virus Genes. 2013 doi: 10.1007/s11262-013-0887-1. [Epu. ahead of print] [DOI] [PubMed] [Google Scholar]
- 180.Broussard GW, Hatfull GF. Evolution of genetic switch complexity. Bacteriophage. 2013;3:e24186. doi: 10.4161/bact.24186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Marinelli LJ, Hatfull GF, Piuri M. Recombineering: a powerful tool for modification of bacteriophage genomes. Bacteriophage. 2012;2:5–14. doi: 10.4161/bact.18778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Marinelli LJ, Piuri M, Swigonova Z, Balachandran A, Oldfield LM, van Kessel JC, Hatfull GF. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One. 2008;3:e3957. doi: 10.1371/journal.pone.0003957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.van Kessel JC, Marinelli LJ, Hatfull GF. Recombineering mycobacteria and their phages. Nat Rev Microbiol. 2008;6:851–857. doi: 10.1038/nrmicro2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- 185.van Kessel JC, Hatfull GF. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol Microbiol. 2008;67:1094–1107. doi: 10.1111/j.1365-2958.2008.06109.x. [DOI] [PubMed] [Google Scholar]
- 186.van Kessel JC, Hatfull GF. Mycobacterial recombineering. Methods Mol Biol. 2008;435:203–215. doi: 10.1007/978-1-59745-232-8_15. [DOI] [PubMed] [Google Scholar]
- 187.Pitcher RS, Tonkin LM, Daley JM, Palmbos PL, Green AJ, Velting TL, Brzostek A, Korycka-Machala M, Cresawn S, Dziadek J, Hatfull GF, Wilson TE, Doherty AJ. Mycobacteriophage exploit NHEJ to facilitate genome circularization. Mol Cell. 2006;23:743–748. doi: 10.1016/j.molcel.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 188.Jacobs WR, Jr, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, Kieser T, Sarkis GJ, Hatfull GF, Bloom BR. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science. 1993;260:819–822. doi: 10.1126/science.8484123. [DOI] [PubMed] [Google Scholar]
- 189.Jain P, Hartman TE, Eisenberg N, O’Donnell MR, Kriakov J, Govender K, Makume M, Thaler DS, Hatfull GF, Sturm AW, Larsen MH, Moodley P, Jacobs WR., Jr phi(2)GFP10, a high-intensity fluorophage, enables detection and rapid drug susceptibility testing of Mycobacterium tuberculosis directly from sputum samples. J Clin Microbiol. 2012;50:1362–1369. doi: 10.1128/JCM.06192-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Piuri M, Jacobs WR, Jr, Hatfull GF. Fluoromycobacterio-phages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS One. 2009;4:e4870. doi: 10.1371/journal.pone.0004870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 193.Lamichhane G, Zignol M, Blades NJ, Geiman DE, Dougherty A, Grosset J, Broman KW, Bishai WR. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2003;100:7213–7218. doi: 10.1073/pnas.1231432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Murry JP, Sassetti CM, Lane JM, Xie Z, Rubin EJ. Transposon site hybridization in Mycobacterium tuberculosis. Methods Mol Biol. 2008;416:45–59. doi: 10.1007/978-1-59745-321-9_4. [DOI] [PubMed] [Google Scholar]
- 195.Fullner KJ, Hatfull GF. Mycobacteriophage L5 infection of Mycobacterium bovis BCG: implications for phage genetics in the slow-growing mycobacteria. Mol Microbiol. 1997;26:755–766. doi: 10.1046/j.1365-2958.1997.6111984.x. [DOI] [PubMed] [Google Scholar]
- 196.da Silva JL, Piuri M, Broussard G, Marinelli LJ, Bastos GM, Hirata RD, Hatfull GF, Hirata MH. Application of BRED technology to construct recombinant D29 reporter phage expressing EGFP. FEMS Microbiol Lett. 2013;344:166–172. doi: 10.1111/1574-6968.12171. [DOI] [PubMed] [Google Scholar]
- 197.Lee MH, Hatfull GF. Mycobacteriophage L5 integrase-mediated site-specific integration in vitro. J Bacteriol. 1993;175:6836–6841. doi: 10.1128/jb.175.21.6836-6841.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huff J, Czyz A, Landick R, Niederweis M. Taking phage integration to the next level as a genetic tool for mycobacteria. Gene. 2010;468:8–19. doi: 10.1016/j.gene.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pascopella L, Collins FM, Martin JM, Lee MH, Hatfull GF, Stover CK, Bloom BR, Jacobs WR., Jr Use of in vivo complementation in Mycobacterium tuberculosis to identify a genomic fragment associated with virulence. Infect Immun. 1994;62:1313–1319. doi: 10.1128/iai.62.4.1313-1319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 201.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Jr, Bloom BR. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 202.Springer B, Sander P, Sedlacek L, Ellrott K, Bottger EC. Instability and site-specific excision of integration-proficient mycobacterio-phage L5 plasmids: development of stably maintained integrative vectors. Int J Med Microbiol. 2001;290:669–675. doi: 10.1016/S1438-4221(01)80004-7. [DOI] [PubMed] [Google Scholar]
- 203.Martin C, Mazodier P, Mediola MV, Gicquel B, Smokvina T, Thompson CJ, Davies J. Site-specific integration of the Strepto-myces plasmid pSAM2 in Mycobacterium smegmatis. Mol Microbiol. 1991;5:2499–2502. doi: 10.1111/j.1365-2958.1991.tb02095.x. [DOI] [PubMed] [Google Scholar]
- 204.Murry J, Sassetti CM, Moreira J, Lane J, Rubin EJ. A new site-specific integration system for mycobacteria. Tuberculosis (Edinb) 2005;85:317–323. doi: 10.1016/j.tube.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 205.Saviola B, Bishai WR. Method to integrate multiple plasmids into the mycobacterial chromosome. Nucleic Acids Res. 2004;32:e11. doi: 10.1093/nar/gnh005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Parish T, Lewis J, Stoker NG. Use of the mycobacteriophage L5 excisionase in Mycobacterium tuberculosis to demonstrate gene essentiality. Tuberculosis (Edinb) 2001;81:359–364. doi: 10.1054/tube.2001.0312. [DOI] [PubMed] [Google Scholar]
- 207.Pashley CA, Parish T. Efficient switching of mycobacterio-phage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol Lett. 2003;229:211–215. doi: 10.1016/S0378-1097(03)00823-1. [DOI] [PubMed] [Google Scholar]
- 208.Thorpe HM, Smith MC. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc Natl Acad Sci USA. 1998;95:5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR, Jr, Fidock DA. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 inte-grase. Nat Methods. 2006;3:615–621. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Huang J, Ghosh P, Hatfull GF, Hong Y. Successive and targeted DNA integrations in the Drosophila genome by Bxb1 and phiC31 integrases. Genetics. 2011;189:391–395. doi: 10.1534/genetics.111.129247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Thomson JG, Chan R, Smith J, Thilmony R, Yau YY, Wang Y, Ow DW. The Bxb1 recombination system demonstrates heritable transmission of site-specific excision in Arabidopsis. BMC Biotechnology. 2012;12:9. doi: 10.1186/1472-6750-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Russell JP, Chang DW, Tretiakova A, Padidam M. Phage Bxb1 integrase mediates highly efficient site-specific recombination in mammalian cells. Biotechniques. 2006;40:460–462. 464. doi: 10.2144/000112150. [DOI] [PubMed] [Google Scholar]
- 213.Bonnet J, Subsoontorn P, Endy D. Rewritable digital data storage in live cells via engineered control of recombination directionality. Proc Natl Acad Sci USA. 2012;109:8884–8889. doi: 10.1073/pnas.1202344109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bonnet J, Yin P, Ortiz ME, Subsoontorn P, Endy D. Amplifying genetic logic gates. Science. 2013;340:599–603. doi: 10.1126/science.1232758. [DOI] [PubMed] [Google Scholar]
- 215.Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu Rev Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 216.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Murphy KC. Phage recombinases and their applications. Adv Virus Res. 2012;83:367–414. doi: 10.1016/B978-0-12-394438-2.00008-6. [DOI] [PubMed] [Google Scholar]
- 218.Datta S, Costantino N, Zhou X, Court DL. Identification and analysis of recombineering functions from Gram-negative and Grampositive bacteria and their phages. Proc Natl Acad Sci USA. 2008;105:1626–1631. doi: 10.1073/pnas.0709089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Swingle B, Bao Z, Markel E, Chambers A, Cartinhour S. Recombineering using RecTE from Pseudomonas syringae. Appl Environ Microbiol. 2010;76:4960–4968. doi: 10.1128/AEM.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Jain P, Thaler DS, Maiga M, Timmins GS, Bishai WR, Hatfull GF, Larsen MH, Jacobs WR. Reporter phage and breath tests: emerging phenotypic assays for diagnosing active tuberculosis, antibiotic resistance, and treatment efficacy. J Infect Dis. 2011;204(Suppl 4):S1142–S1150. doi: 10.1093/infdis/jir454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wilson SM, al-Suwaidi Z, McNerney R, Porter J, Drobniewski F. Evaluation of a new rapid bacteriophage-based method for the drug susceptibility testing of Mycobacterium tuberculosis. Nat Med. 1997;3:465–468. doi: 10.1038/nm0497-465. [DOI] [PubMed] [Google Scholar]
- 222.Watterson SA, Wilson SM, Yates MD, Drobniewski FA. Comparison of three molecular assays for rapid detection of rifampin resistance in Mycobacterium tuberculosis. J Clin Microbiol. 1998;36:1969–1973. doi: 10.1128/jcm.36.7.1969-1973.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.McNerney R, Kiepiela P, Bishop KS, Nye PM, Stoker NG. Rapid screening of Mycobacterium tuberculosis for susceptibility to rifampicin and streptomycin. Int J Tuberc Lung Dis. 2000;4:69–75. [PubMed] [Google Scholar]
- 224.Pai M, Kalantri S, Pascopella L, Riley LW, Reingold AL. Bacteriophage-based assays for the rapid detection of rifampicin resistance in Mycobacterium tuberculosis: a meta-analysis. J Infect. 2005;51:175–187. doi: 10.1016/j.jinf.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 225.Minion J, Pai M. Bacteriophage assays for rifampicin resistance detection in Mycobacterium tuberculosis: updated meta-analysis. Int J Tuberc Lung Dis. 2010;14:941–951. [PubMed] [Google Scholar]
- 226.Pearson RE, Jurgensen S, Sarkis GJ, Hatfull GF, Jacobs WR., Jr Construction of D29 shuttle phasmids and luciferase reporter phages for detection of mycobacteria. Gene. 1996;183:129–136. doi: 10.1016/s0378-1119(96)00530-6. [DOI] [PubMed] [Google Scholar]
- 227.Riska PF, Su Y, Bardarov S, Freundlich L, Sarkis G, Hatfull G, Carriere C, Kumar V, Chan J, Jacobs WR., Jr Rapid film-based determination of antibiotic susceptibilities of Mycobacterium tuberculosis strains by using a luciferase reporter phage and the Bronx Box. J Clin Microbiol. 1999;37:1144–1149. doi: 10.1128/jcm.37.4.1144-1149.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bardarov S, Jr, Dou H, Eisenach K, Banaiee N, Ya S, Chan J, Jacobs WR, Jr, Riska PF. Detection and drug-susceptibility testing of M. tuberculosis from sputum samples using luciferase reporter phage: comparison with the Mycobacteria Growth Indicator Tube (MGIT) system. Diagn Microbiol Infect Dis. 2003;45:53–61. doi: 10.1016/s0732-8893(02)00478-9. [DOI] [PubMed] [Google Scholar]
- 229.Carriere C, Riska PF, Zimhony O, Kriakov J, Bardarov S, Burns J, Chan J, Jacobs WR., Jr Conditionally replicating luciferase reporter phages: improved sensitivity for rapid detection and assessment of drug susceptibility of Mycobacterium tuberculosis. J Clin Microbiol. 1997;35:3232–3239. doi: 10.1128/jcm.35.12.3232-3239.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Banaiee N, Bobadilla-Del-Valle M, Bardarov S, Jr, Riska PF, Small PM, Ponce-De-Leon A, Jacobs WR, Jr, Hatfull GF, Sifuentes-Osornio J. Luciferase reporter mycobacteriophages for detection, identification, and antibiotic susceptibility testing of Mycobacterium tuberculosis in Mexico. J Clin Microbiol. 2001;39:3883–3888. doi: 10.1128/JCM.39.11.3883-3888.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Banaiee N, Bobadilla-del-Valle M, Riska PF, Bardarov S, Jr, Small PM, Ponce-de-Leon A, Jacobs WR, Jr, Hatfull GF, Sifuentes-Osornio J. Rapid identification and susceptibility testing of Mycobacterium tuberculosis from MGIT cultures with luciferase reporter mycobacterio-phages. J Med Microbiol. 2003;52:557–561. doi: 10.1099/jmm.0.05149-0. [DOI] [PubMed] [Google Scholar]
- 232.Banaiee N, January V, Barthus C, Lambrick M, Roditi D, Behr MA, Jacobs WR, Jr, Steyn LM. Evaluation of a semi-automated reporter phage assay for susceptibility testing of Mycobacterium tuberculosis isolates in South Africa. Tuberculosis (Edinb) 2008;88:64–68. doi: 10.1016/j.tube.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rondon L, Piuri M, Jacobs WR, Jr, de Waard J, Hatfull GF, Takiff H. Evaluation of fluoromycobacteriophages for detecting drug resistance in Mycobacterium tuberculosis. J Clin Microbiol. 2011;49:1838–1842. doi: 10.1128/JCM.02476-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hazbon MH, Guarin N, Ferro BE, Rodriguez AL, Labrada LA, Tovar R, Riska PF, Jacobs WR., Jr Photographic and luminometric detection of luciferase reporter phages for drug susceptibility testing of clinical Mycobacterium tuberculosis isolates. J Clin Microbiol. 2003;41:4865–4869. doi: 10.1128/JCM.41.10.4865-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lu B, Fu Z, Xu S. Rapid rifampicin susceptibility test by using recombinant mycobacteriophages. Zhonghua Jie He He Hu Xi Za Zhi. 2000;23:480–484. (In Chinese) [PubMed] [Google Scholar]
- 236.Piuri M, Rondon L, Urdaniz E, Hatfull GF. Generation of affinity-tagged fluoromycobacteriophages by mixed assembly of phage capsids. Appl Environ Microbiol. 2013 doi: 10.1128/AEM.01016-13. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Summers WC. Felix d’Herelle and the Origins of Molecular Biology. Yale University Press; New Haven, CT: 1999. [Google Scholar]
- 238.Chang KC, Yew WW. Management of difficult multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis: update 2012. Respirology. 2013;18:8–21. doi: 10.1111/j.1440-1843.2012.02257.x. [DOI] [PubMed] [Google Scholar]
- 239.Sula L, Sulova J, Stolcpartova M. Therapy of experimental tuberculosis in guinea pigs with mycobacterial phages DS-6A, GR-21 T, My-327. Czech Med. 1981;4:209–214. [PubMed] [Google Scholar]
- 240.Broxmeyer L, Sosnowska D, Miltner E, Chacon O, Wagner D, McGarvey J, Barletta RG, Bermudez LE. Killing of Mycobacterium avium and Mycobacterium tuberculosis by a mycobacteriophage delivered by a nonvirulent mycobacterium: a model for phage therapy of intracellular bacterial pathogens. J Infect Dis. 2002;186:1155–1160. doi: 10.1086/343812. [DOI] [PubMed] [Google Scholar]
- 241.Danelishvili L, Young LS, Bermudez LE. In vivo efficacy of phage therapy for Mycobacterium avium infection as delivered by a nonvirulent mycobacterium. Microb Drug Resist. 2006;12:1–6. doi: 10.1089/mdr.2006.12.1. [DOI] [PubMed] [Google Scholar]
- 242.Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23:1026–1028. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- 243.Pimentel M. Genetics of phage lysis. Microbiol Spectrum. 2014;2(1) doi: 10.1128/microbiolspec.MGM2-0017-2013. MGM2-0017-2013. [DOI] [PubMed] [Google Scholar]
- 244.Smith MC, Hendrix RW, Dedrick R, Mitchell K, Ko CC, Russell D, Bell E, Gregory M, Bibb MJ, Pethick F, Jacobs-Sera D, Herron P, Buttner MJ, Hatfull GF. Evolutionary relationships among actinophages and a putative adaptation for growth in Streptomyces spp. J Bacteriol. 2013;195:4924–4935. doi: 10.1128/JB.00618-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Jacobs WR., Jr . Gene transfer in Mycobacterium tuberculosis: shuttle phasmids to enlightenment. In: Hatfull GF, Jacobs WR Jr, editors. Molecular Genetics of Mycobacteria. 2. ASM Press; Washington, DC: in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lee S, Kriakov J, Vilcheze C, Dai Z, Hatfull GF, Jacobs WR., Jr Bxz1, a new generalized transducing phage for mycobacteria. FEMS Microbiol Lett. 2004;241:271–276. doi: 10.1016/j.femsle.2004.10.032. [DOI] [PubMed] [Google Scholar]