

Abstract
Purpose
To determine the safety and efficacy of gefitinib, an EGFR tyrosine kinase inhibitor, in combination with radiation for newly diagnosed glioblastoma (GBM) patients.
Methods and Materials
Between 3/21/2002 and 5/03/2004 RTOG 0211 enrolled 31 and 147 GBM patients in the phase I and II arms respectively. Treatment consisted of daily oral gefinitnib started at the time of conventional cranial radiotherapy (RT) and continued post RT for 18 months or until progression. Tissue microarrays from 68 cases were analyzed for EGFR expression.
Results
The maximum tolerated dose (MTD) of gefitinib was determined to be 500 mg in patients on non enzyme-inducing anticonvulsant drugs (non-EIAEDs). All patients in the phase II component were treated at a gefitinib dose of 500mg; patients receiving EIADSs could be escalated to 750mg. The most common side-effects of gefitinib in combination with radiation were dermatologic and gastrointestinal. Median survival was 11.5 months for patients treated per protocol. There was no overall survival benefit for patients treated with gefitinib + RT when compared to a historical cohort of patients treated with RT alone, matched by RTOG RPA class distribution. Younger age was significantly associated with better outcome. Per protocol stratification, EGFR expression was not found to be of prognostic value for gefitinib + RT treated patients.
Conclusions
The addition of gefitinib to RT is well tolerated. Median survival of RTOG 0211 patients treated with radiation therapy with concurrent and adjuvant gefitinib was similar to a historical control cohort treated with radiation alone.
Introduction
Glioblastoma remains among the most aggressive of all human malignancies, with median survival times just over one year [1-3]. As radiation therapy (RT) remains one of the primary therapeutic modalities for these tumors and has been found to significantly increase survival compared to surgical resection alone [4, 5], there has been much interest in identifying mechanisms of radiation resistance to enhance radiation efficacy [6-10]. One putative resistance mechanism involves epidermal growth factor receptor (EGFR) signaling. It is well-known that EGFR gene amplification is a common event in GBMs and many GBMs express a mutant variant of EGFR called EGFRvIII, which lacks the extracellular binding domain and is constitutively active [7, 11-14]. EGFR tyrosine kinase inhibitors have been demonstrated to enhance sensitivity to radiation in pre-clinical models [15-17]. Targeting EGFR with the antibody cetuximab in postoperative head and neck cancer patients on RTOG 0234 produced clinical evidence of radiosensitization [18]. In humans, patients with tumors harboring a specific and rare molecular profile appear to benefit from EGFR TKIs, whereas most patients do not [11, 19-21]. Therefore, identifying molecular profiles associated with EGFR TKI sensitivity is a useful goal.
In this context, the Radiation Therapy Oncology Group (RTOG) initiated a single arm Phase I/II study, RTOG 0211, to examine the safety and efficacy of gefitinib, an EGFR tyrosine kinase inhibitor, in combination with radiation (without planned concomitant or adjuvant temozolomide) for newly-diagnosed GBM patients, with integrated tissue collection and correlative endpoints.
To enable improved quantification of expression levels of EGFR a molecular microscopy-based approach using AQUA® (HistoRx, New Haven, CT) was undertaken in lieu of traditional immunohistochemistry.
Methods and Materials
Selection Criteria
Eligibility criteria were: 18 years or older, Zubrod Performance Scale 0-1, histopathologically confirmed newly diagnosed unifocal supratentorial GBM, estimated survival of at least 8 weeks, no prior chemotherapy or RT to the head or neck area (except T1 glottic tumors), no active inflammatory disorders, no major medical/psychiatric illnesses, no malignancy (within three years) except non-melanomatous skin cancer or carcinoma in situ of cervix or bladder, no pregnancy or lactation. Radiotherapy must have been initiated within five weeks after surgery, with gefitinib initiated one week prior to radiotherapy.
Patient Treatment
RTOG 0211 was a Phase I/II study combining gefitinib with RT. RT was delivered using 3D-conformal radiation (60Gy in 30 fractions of 2Gy each). An initial target representing the T2/Axial FLAIR volume plus a tailored 2 cm margin was treated to 46 Gy in 23 fractions of 2 Gy each, followed by a 14 Gy boost (in 7 fractions of 2 Gy each) to the contrast-enhancing tumor plus 2.5 cm margin. To determine the maximum tolerated dose (MTD) in phase I, dose was escalated from 250mg QD to 750mg QD in 250mg increments in patients on enzyme-inducing drugs (EIAEDs, group I) and from 250mg to 500mg QD in patients not on EIAED (group II). Post-radiotherapy maintenance was gefitinib alone for 18 months or until disease progression or intolerable toxicity. No patient received concomitant or adjuvant temozolomide as the trial was designed and launched prior to the approval of temozolomide for newly diagnosed GBM.
Immunohistochemistry
Tissue microarrays [22] were deparaffinized and rehydrated using xylenes and ethanol rinses. Antigen retrieval was carried out in Tris-EDTA pH 9.0 using a LabVision PT Module (Labvision, Fremont, CA) programmed to heat, without boiling, for 25 minutes at 102 C. Slide staining was performed on a LabVision 720 Autostainer (Labvision, Fremont, CA) using Peroxidazed blocking reagent (Biocare Medical, Concord, CA) and Background Sniper (Biocare Medical). Mouse monoclonal EGFR (DAKO-M3563, Clone-H11, final conc. 5.9 ug/ml) primary antibody was incubated for one hour at room temperature (triplicate sections). Primary antibodies were included with rabbit anti-GFAP (Dako, Z0334, 1:200). Subsequently slides were rinsed and incubated with a cocktail of mouse EnvisionPlus (Dako) and Alexa555 conjugated anti-rabbit (Invitrogen, A21428, 1:200). Target signal was amplified using the Cy5 tyramide amplification system (Perkin Elmer, SAT705A, 1:50 dilution in amplification buffer) and slides were mounted with Prolong anti-fade with DAPI (Invitrogen, P36931). AQUA (HistoRx, New Haven, CT) scores [23] were calculated as previously described [24, 25].
Statistical Methodology
Dose limiting toxicities (DLT) for phase I were: any grade 3 or 4 nonhematologic toxicity excluding grade 3 nausea/vomiting, fatigue, and skin toxicity – unless there is evidence of erythema multiforme, or toxicity requiring i.v. dehydration, hospitalization, or an interruption of greater than a total of 7 days during RT. If none of the first three patients for a dose, or one of the first three and none of the last three, experiences a DLT, then the next dose will be opened. The highest dose achieved will be considered the MTD.
The phase II study was designed to test whether the addition of gefitinib to RT prolonged survival. Using the Dixon-Simon method of calculating sample size for the comparison of survival against a historical control, a sample size of 140 was calculated (80% probability of detecting a 50% improvement in median survival time at a significance level of 0.05 (one-sided) in patients with high EGFR AQUA scores).
The Kaplan-Meier method was used to estimate overall survival (OS) and progression-free survival (PFS), and the log-rank test was used to compare the different treatment arms. An event for OS was death due to any cause, for PFS the first reported occurrence of progression or death.
Statistical analyses were done using SAS version 9 and R [26]. AQUA scores were log-base-2 transformed. Age was grouped into ten year increments.
Results
Phase I Results
The Phase I component consisted of 31 patients (Supplementary Material 1): 18 patients in Group I (on EIAEDs) and 13 patients in Group II (on non-EIAEDs). The MTD of gefitinib was determined to be 500 mg in Group II patients and 750 mg in Group I patients. The most common side-effects of gefitinib in combination with radiation were dermatologic, gastrointestinal, and fatigue (Table 1 and Supplementary Material 2). In the non-EIAED group at dose level one (250mg) there were two dermatologic (rashes), one hepatic (SGPT elevation), and one metabolic/laboratory (hypokalemia) grade 3 toxicities and no grade 4 toxicities. There were six patients enrolled at dose level 1. On dose level 2 (500mg) there were six patients enrolled in the non-EIAED group and there was one grade 3 skin toxicity, one grade 3 cardiovascular event (DVT), one grade 3 (SGOT/SGPT) and one grade 4 hepatic (SGPT) events. Dose level 2 therefore had more than one dose limiting toxicity and met specifications for maximum tolerated dose. Per protocol for patients not on EIAED gefitinib was dose escalated from 250mg to 500 mg QD and for patients on EIAED gefitinib was dose escalated from 250 mg to 750 mg QD in 250 mg increments. The MTD for patients on EIAED was determined to be 750 mg QD and for patients not on EIAED was determined to be 500 mg QD.
Table 1. Group 2 (non-EIACD). Iressa and Acute RT Toxicity. Reported as Definitely, Probably, or Possibly Related to Treatment.
The most common side-effects of gefitinib in combination with radiation were dermatologic, gastrointestinal, and fatigue.
| 250 mg (n=6) Grade | 500 mg (n=136) Grade | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 5 | |
| Allergy/Immunology | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 |
| Auditory/Hearing | 0 | 0 | 0 | 0 | 3 | 5 | 0 | 0 | 0 |
| Blood/Bone marrow | 4 | 1 | 0 | 0 | 48 | 17 | 7 | 1 | 0 |
| Cardiovascular (arrhythmia) | 0 | 0 | 0 | 0 | 2 | 3 | 0 | 0 | 0 |
| Cardiovascular (general) | 1 | 0 | 0 | 0 | 8 | 9 | 10 | 2 | 0 |
| Coagulation | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| Constitutional symptoms | 3 | 1 | 0 | 0 | 49 | 48 | 7 | 5 | 0 |
| Dermatology/Skin | 2 | 0 | 2 | 0 | 40 | 62 | 17 | 2 | 0 |
| Gastrointestinal | 0 | 3 | 0 | 0 | 40 | 48 | 17 | 4 | 1 |
| Hemorrhage | 0 | 0 | 0 | 0 | 7 | 3 | 2 | 2 | 0 |
| Hepatic | 1 | 3 | 1 | 0 | 17 | 19 | 23 | 5 | 0 |
| Infection/Febrile Neutropenia | 0 | 0 | 0 | 0 | 6 | 3 | 5 | 1 | 0 |
| Metabolic/Laboratory | 2 | 0 | 1 | 0 | 19 | 7 | 9 | 1 | 0 |
| Musculoskeletal | 0 | 0 | 0 | 0 | 1 | 3 | 3 | 0 | 0 |
| Neurology | 0 | 0 | 0 | 1 | 11 | 13 | 12 | 5 | 0 |
| Ocular/Visual | 0 | 1 | 0 | 0 | 6 | 7 | 0 | 0 | 0 |
| Pain | 0 | 0 | 0 | 0 | 19 | 16 | 1 | 0 | 0 |
| Pulmonary | 0 | 0 | 0 | 0 | 4 | 4 | 6 | 2 | 0 |
| Renal/Genitourinary | 0 | 1 | 0 | 0 | 19 | 2 | 2 | 1 | 0 |
|
| |||||||||
| Worst Non Hematological | 1 (17%) | 2 (33%) | 2 (33%) | 1 (17%) | 7 (5%) | 41 (30%) | 63 (46%) | 23 (17%) | 1 (1%) |
|
| |||||||||
| Worst Overall | 1 (17%) | 2 (33%) | 2 (33%) | 1 (17%) | 6 (4%) | 41 (30%) | 63 (46%) | 24 (18%) | 1 (1%) |
Phase II Results
The Phase II component (Table 2) consisted exclusively of group II patients treated by RT+gefitinib at 500 mg QD. There were 147 patients enrolled in the phase II component and 136 of the combined phase I/II cohort not receiving EIAED and receiving 500mg gefitinib were eligible for analysis. 119 patients were identified to have been treated per protocol or with acceptable deviation. The progression free survival at 6 months was 40 % (Fig. 1). Median progression free survival was 4.9 months. The median OS for patients treated on RTOG 0211 per protocol or with acceptable deviation was 11.5 months versus 11.0 months for historical controls treated by RT alone (HR (0211 v. historical control) = 1.14; 95%CI: 0.94 – 1.37; p (one-sided) = 0.91), Table 3, Fig. 2). Median OS for all eligible patients was 11.1 months.
Table 2. Pretreatment characteristics by 0211 and historical data set (7401, 7918, 8302, 9006, 9411).
There appear to be no differences in overall survival, neither in the group of all patients nor in patient groups stratified by.RPA class.
| RTOG 0211 | Historical control | |||
|---|---|---|---|---|
| n | % | n | % | |
| Age | ||||
| <50 | 30 | 22 | 487 | 33 |
| >= 50 | 106 | 78 | 970 | 67 |
| Zubrod | ||||
| 0 | 57 | 42 | 629 | 43 |
| 1 | 79 | 58 | 684 | 47 |
| 2,3 | 0 | 0 | 143 | 10 |
| unknown/missing | 0 | 0 | 1 | <1 |
| Surgery | ||||
| biopsy | 31 | 22 | 241 | 17 |
| part.resect | 68 | 51 | 878 | 60 |
| tot.resect | 36 | 26 | 328 | 23 |
| other | 1 | 1 | 8 | 1 |
| unknown/missing | 0 | 0 | 2 | <1 |
| Neurologic Function | ||||
| none/minor | 107 | 79 | 794 | 54 |
| moderate | 27 | 20 | 582 | 40 |
| hospital | 0 | 0 | 77 | 5 |
| unknown/missing | 2 | 1 | 4 | <1 |
| RPA Class | ||||
| III | 12 | 9 | 250 | 17 |
| IV | 83 | 61 | 652 | 45 |
| V | 41 | 30 | 555 | 38 |
Figure 1. Progression-Free Survival.
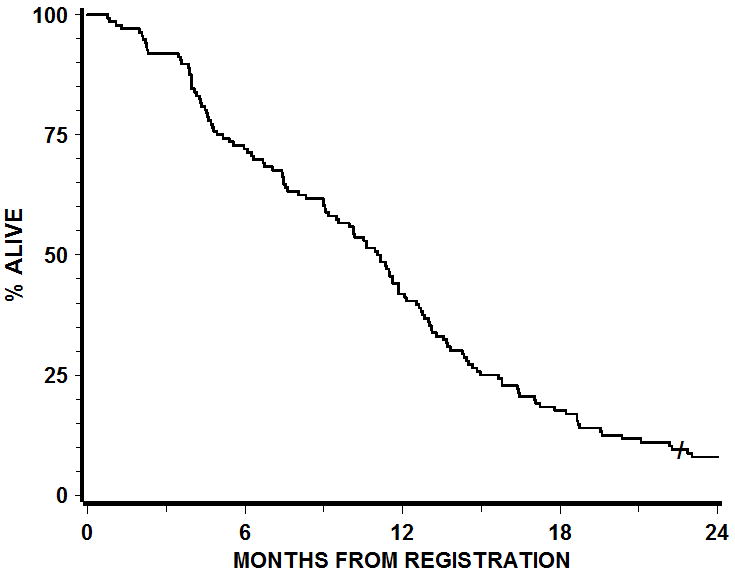
119 patients were identified to have been treated per protocol or with acceptable deviation. The progression free survival at 6 months was 40 %.
Table 3. Overall Survival by 0211 (per protocol/acceptable) vs. historical control.
The median OS for patients treated on RTOG 0211 per protocol or with acceptable deviation was 11.5 months versus 11.0 months for historical controls treated by RT alone (HR (0211 v. historical control) = 1.14; 95%CI: 0.94 – 1.37; p (one-sided) = 0.91). Median OS for all eligible patients was 11.1 months.
| Months | 0211 Per protocol/Acceptable | Historical control | ||
|---|---|---|---|---|
| Survival | # at Risk | Survival | # at Risk | |
| 0 | 100% | 119 | 100% | 1457 |
| 3 | 97% | 115 | 90% | 1315 |
| 6 | 78% | 93 | 77% | 1115 |
| 9 | 65% | 77 | 61% | 890 |
| 12 | 45% | 54 | 44% | 640 |
| 15 | 26% | 31 | 32% | 458 |
| 18 | 19% | 23 | 23% | 328 |
| 24 | 9% | 10 | 13% | 188 |
|
| ||||
| Median Survival Time (MST) | 11.5 months | 11.0 months | ||
| Dead/Total | 117/119 | 1405/1457 | ||
| Hazard ratio (λ0211/λhistorical) (95% CI) | 1.14 (0.94 to 1.37) | |||
| p-value(one-sided log rank) | 0.91 | |||
Figure 2. Overall survival.
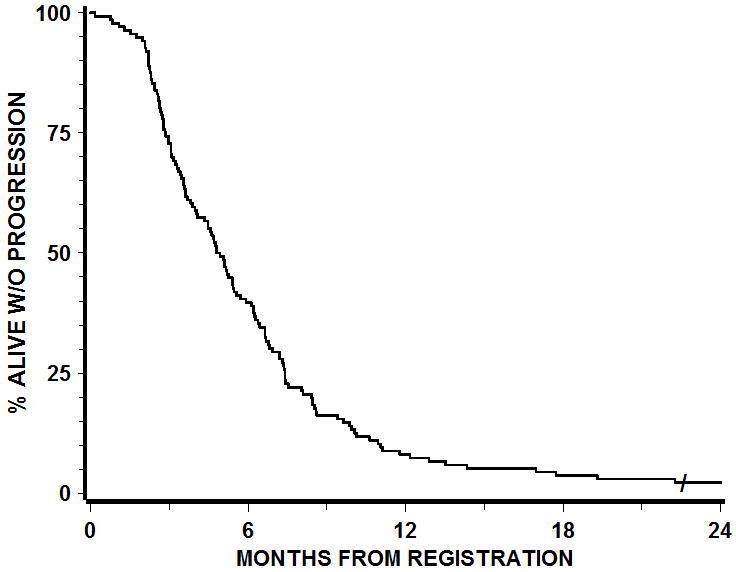
The median OS for patients treated on RTOG 0211 per protocol or with acceptable deviation was 11.5 months versus 11.0 months for historical controls treated by RT alone (HR (0211 v. historical control) = 1.14; 95%CI: 0.94 – 1.37; p (one-sided) = 0.91).
Toxicities observed during the phase II portion of the study are summarized in Table 1 (N=136). The incidence of grade 1 / 2 and 3 / 4 rash was 75 and 13% respectively. Grade 3/4 cardiovascular complications related to thromboembolic disease (8.8%), (which is an expected incidence for this patient population). Grade 3 /4 liver function tests abnormalities were observed in 21% of patients. Seventy two percent of the observed Grade 3 / 4 GI toxicity (15%) were due to diarrhea; grade 1 /2 diarrhea was ~51%.
Correlative results
Stratified by the RTOG recursive partitioning analysis (RPA) criteria, there did not appear to be a clinical group of patients who benefited from the addition of gefitinib to RT (Supplementary Material 3-5). Only younger age was significantly associated with improved clinical outcome in RTOG 0211-treated patients [HR (50+ v. <50) = 1.86 (95%CI: 1.22 – 2.83; p = 0.0037]. Therefore correlative analysis of EGFR expression was adjusted for age. Tissues were obtained from 68 out of 136 eligible patients in the Phase II component of RTOG 0211. There were no significant differences in pretreatment demographics or patient outcomes between patients with or without tissue submission.
EGFR over expression was not found to be of prognostic significance for patients treated with radiation therapy with concurrent and adjuvant gefitinib (HR 0.99). Additional correlative analysis will be reported separately.
Discussion
The biological significance of EGFR signaling in GBMs has galvanized much interest in investigating EGFR tyrosine kinase inhibitors by themselves or in combination with radiotherapy or chemotherapy. In the recurrent setting, it has become clear that only a relatively modest subset of GBM patients demonstrate an objective response to EGFR TKIs. RTOG 0211 was a single arm phase I/II study which demonstrated safety but no efficacy of gefitinib therapy when combined with radiation therapy. Per protocol analysis of EGFR expression did not identify a patient subset that benefited from radiation therapy with concurrent and adjuvant gefitinib. This is consistent with results from N0177, a NCCTG phase I/II trial of concurrent and adjuvant erlotinib and temozolomide and radiation therapy for newly diagnosed GBM. There was no survival benefit on N0177 when compared to RT/TMZ treated patients and EGFR gene amplification was not of prognostic value [27].
Limited and somewhat controversial previous data have suggested that patients that respond to EGFR RTK inhibition harbor the EGFRvIII gene and express wild-type PTEN implying that the proliferation drive was predominantly through the EGFR pathway, and hence shutting it off could be therapeutically useful [11]. At the time of protocol development this data was not available, and therefore no stratification based on EGFRvIII was undertaken. For that reason only EGFR expression was prespecified as a stratification variable. Outside of the protocol specified analysis we will report additional correlative analysis including EGFRvIII and PTEN status separately. The recent discovery that oncogenic FGFR-TACC fusions are present in a small percentage of GBM patients is very interesting [28] and hopefully will result in development of an appropriate therapy for this patient subpopulation. At this time is remains to be determined if this or other molecular alteration can predict response to EGFR-TKi such as gefitinib.
Also, at the time of protocol development the standard of care for newly diagnosed glioblastoma patients did not include temozolomide.
Supplementary Material
Acknowledgments
NIH/NCI awards R01CA108633 (To AC and PM); RC2CA148190 (To AC and WC); the Brain Tumor Funders Collaborative Group (To AC and MM); and German Research Foundation DFG LA2510/1-1 (to AC and TL). “This publication (journal, article, etc.) was supported by RTOG grant U10 CA21661, and CCOP grant U10 CA37422 from the National Cancer Institute. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute.
The authors wish to acknowledge the valuable contributions of Robert Pinard, Agnes Ang, Natasha Rabinowitz, Maya Panjikaran, Leo Goodrich, Liqun Xu, and Don Waldron of HistoRX in data analysis and preparation.
Authors
Arnab Chakravarti, MD, Department of Radiation Oncology, Arthur G. James Comprehensive Cancer Center and Richard L. Solove Research Institute, The Ohio State University Medical School, 300 W. 10th Avenue, Columbus, Ohio 43210, Arnab.chakravarti@osumc.edu, Phone: 614-293-0672, Fax: 614-293-0573
Meihua Wang, PhD, Radiation Therapy Oncology Group/ACR, 1818 Market Street, Suite 1600, Philadelphia, PA 19013, mwang@acr-arrs.org, Phone: 215-574-3207, Fax: 215-928-0153
H. Ian Robins, MD, PhD, Department of Medicine Human Oncology & Neurology, Paul P Carbone Comprehensive Cancer Center, University of Wisconsin, K4/534 CSC, 600 Highland Avenue, Madison WI 53792, hirobins@wisc.edu, Phone office: 608-263-1416, Administrative Phone: 608-265-0861, Fax: 608-265-8133
Tim Lautenschlaeger, MD, Department of Radiation Oncology, Arthur G. James Comprehensive Cancer Center and Richard L. Solove Research Institute, The Ohio State University Medical School, Wiseman Hall 385 G, 410 W. 12th Ave., Columbus, OH 43210, tim.lautenschlaeger@osumc.edu, Phone: 6140247-6432, Fax: 614-292-5275
Walter Curran, MD, FACR, Winship Cancer Institute, Emory University, 1365-C Clifton Road, NE, Atlanta, GA 30322, curran@radonc.emory.org, Phone: 404-778-5323, Fax: 404-778-5152
David G. Brachman, MD, Department of Radiation Oncology, Arizona Oncology Services Foundation, 350 W. Thomas Rd, Phoenix, AZ 85013, dbrachman@azoncology.com, Phone: 602 406 3170, Fax: 602 263 7816
Christopher J. Schultz, MD, Department of Radiation Oncology, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI 53226, cshcultz@radonc.mcw.edu, Phone: 414-805-4480
Ali K. Choucair, MD, Norton Healthcare System, Neuroscience Institute, 210 E Gray Str, Suite 1102, Louisville, KY 40202, ali.choucair@nortonhealthcare.org, Phone: 502-629-5510, Fax: 502-629-5512
Marisa Dolled-Filhart, PhD, HistoRx Inc., 35 Northeast Industrial Road, Branford, Connecticut 06405, Marisa.Dolled-Filhart@historx.com
Jason Christiansen, PhD, Genoptix Medical Laboratory, 2110 Rutherford Rd, Carlsbad, CA 92008, jchristiansen@genoptix.com, Phone: 760-516-5484, Fax: 760-516-6201
Mark Gustavson, PhD, HistoRx Inc., 35 Northeast Industrial Road, Branford, Connecticut 06405, Mark.Gustavson@historx.com, Phone: 203-498-7500
Annette Molinaro, MA, PhD, University of California, San Francisco, Department of Neurological Surgery, 505 Parnassus Ave. Rm. M779, San Francisco, CA 94143-0112, MolinaroA@neurosurg.ucsf.edu, Phone: 415-353-7500, Fax 415-353-2889
Paul Mischel, MD, Ludwig Institute for Cancer Research, University of California, San Diego, CMM-East, Room 2021, 9500 Gilman Drive, La Jolla, CA 92039, pmischel@ucsd.edu, Lab phone: 858-534-6079, Office phone: 858-534-6080
Adam P. Dicker, MD, PhD, Radiation Oncology Department, Kimmel Cancer Center at Thomas Jefferson University, 111 South 11th Street, Room G-301, Bodine Center, Philadelphia, PA 19107, adam.dicker@mail.tju.edu, Phone: 215-955-6700, Fax: 215- 503-0013
Markus Bredel, MD, PhD, Department of Neurosurgery, Neurocenter, and Comprehensive Cancer Center, University of Freiburg, Germany, Department of Radiation Oncology, UAB Comprehensive Cancer Center, 303 E Superior Street, Lurie Rm. 6-111, Chicago, IL 60611-3015, m-bredel@northwestern.edu, Phone: 312.503.1727, Fax: 312.503.5607
Minesh Mehta, MD, Radiation Oncology Department, Northwestern University Feinberg School of Medicine, NMH/Arkes Family Pavilion Suite 1820, 676 N Saint Clair, Chicago IL 60611, mineshpmehta@gmail.com, Phone: 312-926-2520, Clinical Fax: 312-926-6374
Conflict of Interest Statements
Arnab Chakravarti has no conflict to declare.
Meihua Wang has no conflict of interest to declare.
H. Ian Robins has served as a consultant to Genentech, Abbott, and Novocure.
Tim Lautenschlaeger has not conflict of interest to declare.
Walter Curran has no conflict of interest to declare.
David Brachman has no conflict of interest to declare.
Christopher Schultz has no conflict of interest to declare.
Ali Chouchair has no conflict of interest to declare.
Marisa Dolled-Filhart has no conflict to declare.
Jason Christiansen has no conflict to declare.
Mark Gustafson has no conflict of interest to declare.
Annette Molinaro has no conflict of interest to declare.
Paul Mischel has no conflict of interest to declare.
Adam Dicker has no conflict of interest to declare.
Markus Bredel has no conflict of interest to declare
Minesh Mehta has prior or current consulting and/or speaking relationships with Abbott, Adnexus, Bristol-Meyers, Elekta, Genentech, Merck, Novartis, Novocure, Schering Plough, and Tomotherapy; served on the DSMB for Apogenix; is on the Medical Advisory Board for Colby, and Stemina; is on the Board of Directors of Pharmacyclics; and has stock options in Colby, Pharmacyclics and Stemina, and previously in Tomotherapy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buckner JC, Ballman KV, Michalak JC, et al. Phase III trial of carmustine and cisplatin compared with carmustine alone and standard radiation therapy or accelerated radiation therapy in patients with glioblastoma multiforme: North Central Cancer Treatment Group 93-72-52 and Southwest Oncology Group 9503 Trials. J Clin Oncol. 2006;24(24):3871–9. doi: 10.1200/JCO.2005.04.6979. [DOI] [PubMed] [Google Scholar]
- 2.Pichlmeier U, Bink A, Schackert G, et al. Resection and survival in glioblastoma multiforme: an RTOG recursive partitioning analysis of ALA study patients. Neuro Oncol. 2008;10(6):1025–34. doi: 10.1215/15228517-2008-052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Keime-Guibert F, Chinot O, Taillandier L, et al. Radiotherapy for glioblastoma in the elderly. N Engl J Med. 2007;356(15):1527–35. doi: 10.1056/NEJMoa065901. [DOI] [PubMed] [Google Scholar]
- 5.Walker MD, Alexander E, Jr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49(3):333–43. doi: 10.3171/jns.1978.49.3.0333. [DOI] [PubMed] [Google Scholar]
- 6.Murat A, Migliavacca E, Gorlia T, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26(18):3015–24. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee B, McEllin B, Camacho CV, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69(10):4252–9. doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker FG, II, Simmons ML, Chang SM, et al. EGFR overexpression and radiation response in glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2001;51(2):410–8. doi: 10.1016/s0360-3016(01)01609-1. [DOI] [PubMed] [Google Scholar]
- 9.Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002;62(1):200–7. [PubMed] [Google Scholar]
- 10.Chakravarti A, Chakladar A, Delaney MA, et al. The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a RAS-dependent manner. Cancer Res. 2002;62(15):4307–15. [PubMed] [Google Scholar]
- 11.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 12.Yadav AK, Renfrow JJ, Scholtens DM, et al. Monosomy of chromosome 10 associated with dysregulation of epidermal growth factor signaling in glioblastomas. Jama. 2009;302(3):276–89. doi: 10.1001/jama.2009.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu KV, Zhu S, Cvrljevic A, et al. Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009;69(17):6889–98. doi: 10.1158/0008-5472.CAN-09-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonyak MA, Moscatello DK, Wong AJ. Constitutive activation of c-Jun N-terminal kinase by a mutant epidermal growth factor receptor. J Biol Chem. 1998;273(5):2817–22. doi: 10.1074/jbc.273.5.2817. [DOI] [PubMed] [Google Scholar]
- 15.Eller JL, Longo SL, Kyle MM, et al. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery. 2005;56(1):155–62. doi: 10.1227/01.neu.0000145865.25689.55. discussion 162. [DOI] [PubMed] [Google Scholar]
- 16.Sarkaria JN, Carlson BL, Schroeder MA, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12(7 Pt 1):2264–71. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 17.Stea B, Falsey R, Kislin K, et al. Time and dose-dependent radiosensitization of the glioblastoma multiforme U251 cells by the EGF receptor tyrosine kinase inhibitor ZD1839 (‘Iressa’) Cancer Lett. 2003;202(1):43–51. doi: 10.1016/j.canlet.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Harari PM, Harris J, Kies MS. Phase II Randomized Trial of Surgery Followed by Chemoradiation Plus Cetuximab for High-Risk Squamous Cell Carcinoma of the Head and Neck (RTOG 0234) Int J Radiat Oncol Biol Phys. 2007;(69):S13. [Google Scholar]
- 19.Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22(1):133–42. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 20.Pfeffer MR, Levitt ML, Aderka D. Gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22(13):2755–6. doi: 10.1200/JCO.2004.99.299. [DOI] [PubMed] [Google Scholar]
- 21.van den Bent MJ, Brandes AA, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27(8):1268–74. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rimm DL, Camp RL, Charette LA, et al. Tissue microarray: a new technology for amplification of tissue resources. Cancer J. 2001;7(1):24–31. [PubMed] [Google Scholar]
- 23.Gustavson MD, Bourke-Martin B, Reilly D, et al. Standardization of HER2 immunohistochemistry in breast cancer by automated quantitative analysis. Arch Pathol Lab Med. 2009;133(9):1413–9. doi: 10.5858/133.9.1413. [DOI] [PubMed] [Google Scholar]
- 24.Dolled-Filhart M, McCabe A, Giltnane J, et al. Quantitative in situ analysis of beta-catenin expression in breast cancer shows decreased expression is associated with poor outcome. Cancer Res. 2006;66(10):5487–94. doi: 10.1158/0008-5472.CAN-06-0100. [DOI] [PubMed] [Google Scholar]
- 25.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med. 2002;8(11):1323–7. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 26.Ihaka R, Gentleman R. A language for data analysis and graphics. Journal of Computational and Graphical Statistics. 1996;(5):299–314. [Google Scholar]
- 27.Brown PD, Krishnan S, Sarkaria JN, et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol. 2008;26(34):5603–9. doi: 10.1200/JCO.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh D, Minhow Chan J, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):1231–5. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.