

7.1 Introduction
“What determines whether a given piece of DNA along the chromosome is functioning, since it's covered with the histones? You can inherit something beyond the DNA sequence. That's where the real excitement of genetics is now.”
-James Watson, 2003
The term “epigenetics” was coined by Conrad Waddington in 1942 in a discussion of the mechanisms of cell differentiation. Waddington defined epigenetics as “the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being” (Waddington, 1968). The specific epigenetic mechanisms that regulate genetic programming were not discovered until decades after Waddington first coined the term (Griffith and Mahler, 1969; Riggs, 1975). These mechanisms are now known to include histone tail post-translational modifications, DNA methylation, ATP-dependent chromatin remodeling, and non-coding RNA pathways (Tollervey and Lunyak, 2012). With these discoveries, Waddington's original definition of “epigenetics” has changed and evolved to the currently accepted view that “epigenetics (epi- being a Greek prefix for “on top of”) refers to “the study of heritable changes in genes that are not the result of changes in the DNA sequence” (Riggs et al., 1996).
Dr. James Watson won the Nobel Prize for his seminal role in discovering the structure of the DNA double helix structure in 1953, but fifty years later he acknowledged that DNA is not the sole regulator of gene inheritance and expression. Instead, epigenetic changes that occur “above” the DNA may be just as or more important than genetics in terms of their effects on development and disease state. Retinoic acid (RA), a vitamin A derivative that functions as the active metabolite in cellular signaling, induces cell differentiation in stem cells and some cancer cells. Along with the more well known effects of RA signaling on cell lineage specification through transcriptional activation of retinoic acid receptor (RAR)-regulated genes, recent studies are demonstrating that RA also mediates cell differentiation via rapid, profound effects on the epigenome. This observation is opening up a new area of fundamental research into transcriptional regulationas well as pointing the way to new clinical applications of RA. The use of RA in combination with drugs that modify the epigenome is showing promise in the treatment and/or prevention of several types of cancer. This type of combination therapy is increasingly relevant, as many types of cancer exhibit aberrant levels of or mutations in epigenetic regulatory proteins.
7.2 History: Epigenetic regulation is achieved by a number of different mechanisms
Waddington nicely illustrated the idea of genotype-to-phenotype changes along cell development pathways by his drawing of an “epigenetic landscape” (Waddington, 1957) (Fig. 7.1, adapted by Hochedlinger and Plath). A more recent adaptation of this drawing updates the model to include a summary of recent epigenetic research. In this model specific epigenetic modifications are acquired as progenitor cells, depicted as marbles (Fig. 7.1), differentiate and commit to a specific cell fate, conceptualized in the figure as marbles rolling down into one of several valleys. This idea has been substantiated by experimental findings where it has been demonstrated that commitment of cells into specific differentiation pathways is associated with progressive epigenetic modifications (Hochedlinger and Plath, 2009).
Figure 7.1. The Epigenetic Landscape.
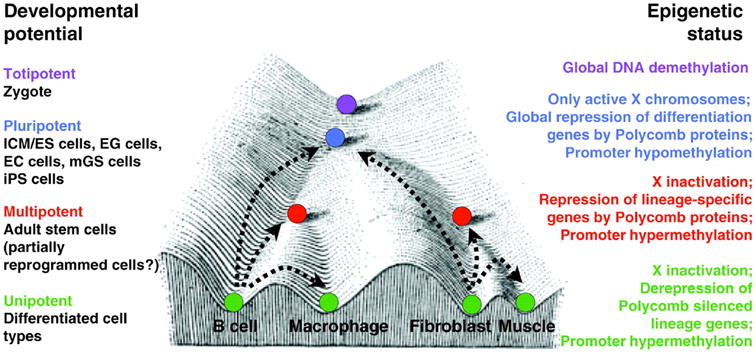
Conrad Waddington's Epigenetic Landscape illustrates the causal mechanisms by which the genotype brings about phenotypic effects. The progressive cellular differentiation (represented by purple, blue, red, and green) into specific cell types (illustrated by the different valleys) is associated with distinct epigenetic modifications. Polycomb repression and promoter hypermethylation are key mechanisms in the regulation of cellular differentiation. The corresponding epigenetic status of the various differentiation states is depicted on the right. (Hochedlinger and Plath, 2009).
Histone protein tail modifications and transcriptional regulation
Cellular chromatin is composed of DNA-wrapped nucleosomes packed into regions of either compacted or loose nucleosomal structure, referred to as hetero- and eu- chromatin, respectively. In general, genes residing in heterochromatic regions are silenced, whereas genes located in euchromatic regions are actively transcribed (Fig. 7.2).
Figure 7.2. The Nucleosome and the Histone Tails.
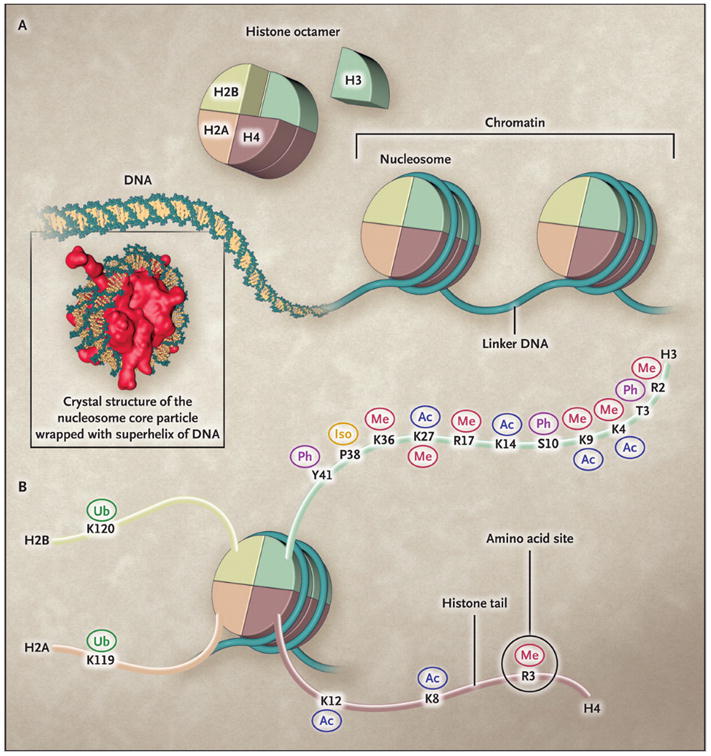
The basic functional unit of chromatin is the nucleosome (Panel A), which is composed of a histone octamer around which DNA is wrapped. Octamers are separated by linker DNA. The histone octamer is assembled from a histone H3:H4 tetramer and two H2A:H2B dimers. The histone tails of all four core histones are subject to a variety of post-translational modifications (Panel B). These include methylation (Me), acetylation (Ac), phosphorylation (Ph), ubiquitylation (Ub), and proline isomerization (Iso), all of which occur at the site of a specific amino acid, such as K4 and K9 on the histone H3 tail. The same histone amino acid may be subject to different post-translational modifications, which may facilitate different biologic outcomes. (Dawson et al., 2012).
The nucleosome is a histone octamer composed of two of each of the core histones H2A, H2B, H3, and H4, and one auxiliary H1 linker histone (Dawson et al., 2012). The histone proteins are each composed of a globular domain with an extended, positively charged N-terminal tail that interacts with the phosphodiester backbone of the DNA. Histone proteins were discovered in 1884, but it was not until 1963 that histone tails were shown to be post-transcriptionally modified (Phillips, 1963). Subsequently, the effects of these histone modifications on gene regulation began to be elucidated (Allfrey et al., 1964). Importantly, the histones, in particular the lysine/arginine rich tails, were shown to be targets for extensive post-transcriptional modifications (Fig. 7.2). Recently, Yuan et al (Yuan et al., 2012) have shown that RA-mediated transcriptional activation of the Cytochrome P450 26a1 gene is associated with a loosening of the chromatin structure, which is required for transcriptional activation.
Histone protein tail regulation is highly complex, and numerous post-translational modificationscan regulate different aspects of gene transcription;these include phosphorylation, sumoylation, ubiquitination, ribosylation, neddylation, ADP-ribosylation, citrullination, and others (Tan et al., 2011; Yang, 2005). The enzymes that regulate these modifications can be divided into three groups of epigenetic regulators: “writers”, “readers”, and “erasers”.
Histone modifications are generated by “writer” enzymes, which include families of lysine/arginine methyltransferases (KMTs/PRMTs), histone lysine acetyltransferases (KATs), and serine/threonine kinases. Methylation of histone tails, includingtrimethylation of histone 3 lysine 9 (H3K9me3) and 27 (H3K27me3), is generally associated with gene repression, whereas the acetylation of the same residues (H3K9and H3K27) correlates strongly with gene activation (Guillemette et al., 2011; Verdone et al., 2006). There are also instances in which methylation of certain residues is associated with gene activation, such as methylation of H3K4 and H3K36 (Schneider et al., 2004; Voigt et al., 2012). Acetylationby KAT proteins, such as KAT3A(p300), KAT3B (CBP), PCAF, or KAT13B (pCIP), promotes the activation of gene expressionby neutralizing the positive charges of histone tails (Verdone et al., 2006), leading to loosening of the negatively charged chromatin and subsequent binding of DNA binding factors that promote gene transcription.
The actions of “writer” enzymes are countered by a group of enzymes known as “erasers”. “Erasers” are responsible for the removal of specific histone modifications. This group includes lysine/arginine demethylases (KDMs/PRDMs), histone deacetylases (HDACs), and serine/threonine phosphatases (summarized in Table 7.1). Typically, these “erasers” counteract the actions of “writer” enzymes, having direct effects on gene transcription. For example, “writer” enzymes of the KAT family mediate the deposition of acetylation marks onto lysine residues of histone tails, thereby neutralizing the attraction of positively charged histones with negatively charged DNA. This allows for the unraveling of DNA, thereby allowing general transcriptional machinery and other proteins to bind that mediate gene activation. Conversely, “eraser” proteins of the HDAC family remove the acetylation mark. This allows the DNA to again wrap around histones, preventing the bindings of general transcription machinery, leading to gene repression.
Table 7.1. Groups of Epigenetic Modifiers and their functions.
“Writers” are a group of enzymes that mediate the addition of epigenetic modifications (marks). “Erasers” are proteins with enzymatic activity that mediate the removal of these marks. “Readers” are proteins, generally with no enzymatic activity, that recognize and bind to posttranslational modifications to mediate downstream effects.
| - | Writer | Eraser | Reader |
|---|---|---|---|
|
| |||
| Histone Marks | - | - | - |
|
| |||
| Lysine methylation | KMT | KDM | CBX proteins |
| Arginine methylation | PRMT | 14-3-3 –proteins | |
| Lysine acetylation | KAT | HDAC | |
| Serine/Threonine phosphorylation | S/T Kinase | Phosphatase | |
|
| |||
| DNA methylation | - | - | - |
|
| |||
| CpG (5meC) | DNMT | DNA demethylase | MeCPMBD1-4 |
Key: Lysine Methyltransferase (KMT); Protein Arginine Methyltransferase (PRMT); Lysine Acetyltransferase (KAT);Serine/Threonine Kinases (S/T Kinase); Lysine Demethylase (KDM); Histone Deacetylase (HDAC); DNA Methyltransferase (DNMT); Methyl-CpG-binding domain proteins (MeCP); Chromobox Homolog (CBX)
“Reader” proteins specifically bind to post-translationally modified chromatin, and recognize these specific histone modifications to alter chromatin structure and dynamics. Often, “reader” proteins are part of larger protein complexes that contain “reader” and/or “eraser” proteins. Without “reader” proteins, posttranslational modifications would not be recognized, and the protein complex or specific “writers” or “erasers” would not be recruited. Alternatively, “writer” or “eraser” proteins themselves can also serve as “readers” proteins. For example, KAT proteins possess a bromodomain that recognizes and binds acetylated lysine residues on histone tails. This allows for KAT proteins to further mediate acetylation at these specific DNA regions. Because of this, these “reader” proteinsalso mediate changes in transcription or DNA replication (Cohen et al., 2011).
The distinction between “writers”, “readers”, and “erasers” is complicated by the fact that protein complexes that add marks(“writer” complexes) are frequently composed of several subunits with different enzymatic properties. For example, the polycomb repressive complex 2 (PRC2) is comprised of at least four subunits which include the Suz12 (zinc finger), Eed, Ezh2 (SET domain with histone methyltransferase activity) and RbAp48 (histone binding domain) proteins. Importantly, the Ezh2 protein has enzymatic activity and can add methyl groups specifically to the H3K27 resulting in trimethylation of this histone residue. This posttranslational modification is deposited onto histone tails at lysine 27by the PRC2 complex – a “writer”, but is recognized by the polycomb repressive complex 1 (PRC1) – a “reader” (Min et al., 2003). However, PRC1 mediates the deposition of ubiquitin, another histone modification, onto histone 2A lysine 119. PRC1 can in other words read the H3K27me3 and write the H2A.K119Ub, and can thus be considered both a “reader” and a “writer” enzyme (Stock et al., 2007). Furthermore, additional modifications of the histone tail (e.g. H3S28ph) proximal to the site of the initial modification (H3K27me3) add another layer complexity. In this example the recognition of H3K27me3 by the INHAT “reader” protein is prevented by phosphorylation of Serine 28 (H3S28ph) (Kim et al., 2012). Thisillustrates how modification of nearby residues can interfere with the recognition of specific histone marks by “reader” proteins. The effect of combinatorial histone modifications is commonly referred to as the histone code, a termcoined by Charles D. Allis in 2001 (Jenuwein and Allis, 2001). As exemplified above by the context dependent recognition of H3K27me3, the emerging view is that the recognition by “reader” proteins is not dictated only by specific histone modifications, but rather by an interplay between different histone modifications
DNA methylation and gene silencing
In contrast to histone modifications, which are relatively transient in nature, DNA methylation provides a more persistent, long-term gene silencing. DNA methylation occurs when a methyl group is deposited on the cytosine of a phosphodiester-bonded cytosine-guanine dinucleotide (CpG) sequence. DNA methylation, e.g. the formation of 5-methylcytosine (5mC), was first proposed as a mechanism for changing gene expression in 1975 (Holliday and Pugh, 1975; Riggs, 1975). CpG sequences are typically concentrated in large clusters called CpG islands, predominantly located at or near gene promoters, but CpG islands are also found in intergenic regions. A family of DNA methyltransferase (DNMT) enzymes transfer methyl groups to DNA and this engenders stable, long term gene silencing (Coskun et al., 2012). DNA methylation is introduced by the recruitment of DNMT3a and 3b by sequence specific repressors that silence gene transcription (Fuks et al., 2001). Newly replicated DNA is transiently hemi-methylated untilDNMT1uses the methylated parent strand to direct deposition of corresponding methylation on the daughter strand, thus maintaining the overall pattern of DNA methylation (Kulis and Esteller, 2010). In the context of DNA methylation, the DNMTs function as “writers”, whereas methyl-CpG-binding domain proteins (MECP), which recognize methylated CpGs, function as “readers” (Sasai and Defossez, 2009).
Recently, researchers have determined that DNA methylation is reversible (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009), which suggests that DNA methylation is a dynamic process rather than a one-way mechanism of gene silencing, as was previously thought to be the case.
Selected groups of epigenetic regulators are listed in Table 7.1, where families of “writers”, “erasers”, and “readers” are listed for each type of epigenetic modification (individual rows). In Table 7.2 are listed a number of commonly investigated epigenetic modifications (individual rows), and their effects on transcription.
Table 7.2. Selected Histone Modifications and Their Enzymatic Regulators.
Specific histone marks involved in transcriptional regulation, and the enzymes that modify these marks. Activating marks are modifications that generally favor transcription (Activation), whereas repressive marks are modifications that favor transcriptional silencing (Repression). Examples are given of specific histone modifications (Histone marks), and of the specific enzymes depositing (“writers”) and removing (“erasers”) these marks. This is a not a comprehensive list, but rather a list of the most well understood regulators of epigenetic changes.
| - | Histone mark | Writer | Eraser |
|---|---|---|---|
| Repression | H3K27me3 | EZH2, NSD3 | KDM6A/B (JMJD3) |
| H3K9me3 | SETDB1/2 SUV39H1/2 |
Lysine specific demethylase 4A/B/C/D | |
| Activation | H3K4me1 | SETD7 | KDM1A KDM5B |
| H3K4me2 | NSD3 | KDM5A/D KDM1A KDM5B |
|
| H3K4me3 | MLL MLL3/4 PRDM9 SETD1A/B SET AND MYND domain-containing protein 3 |
Lysine specific demethylase 4A/B/C/D KDM5B |
|
| H3K36me3 | SETD2 NSD2 |
Lysine specific demethylase 4A Lysine specific demethylase NO66 |
|
| H3K14Ac | PCAF MYST3 |
HDAC3 | |
| H3K9Ac | PCAF KAT13B (pCIP) KAT6A (Moz) |
SIRT1 SIRT6 |
|
| H3K27Ac | KAT3A/B (P300/CBP) | - | |
| H3S28Ph | MAPKKK-MLT MSK1/2 STK5 |
- |
Key: enhancer of zeste homolog 2 (EZH2); Nuclear SET domain-containing protein (NSD3);lysine (K)-specific demethylase (KDM); SET domain, bifurcated(SETDB); methyltransferase variant (SUV39H12); SET domain (SETD); mixed-lineage leukemia (MLL); Positive Regulatory Domain (PRDM); p300/CBP associated factor (PCAF); histone deacetylase (HDAC); Lysine acetyltransferase (KAT); MAP-kinase-kinase-kinase; mitogen- and stress-activated protein kinase (MSK); Aurora kinase (STK).
Other epigenetic regulators of gene expression
ATP-dependent remodeling of chromatin structure and long intergenic non-coding RNAs (lincs) are other major epigenetic regulators of gene expression, but to date, little is known about their roles, if any, in RA regulated gene transcription. Here, we will focus on what is known about RA involvement in histone modifications and DNA methylation.
7.3 Development of the Field: Retinoids and Rars mediate histone modifications
RA functions as the ligand for retinoic acid receptors (RARs), and can regulate several developmentally important genes, including the Hox (homeobox) gene clusters (Kashyap et al., 2011; Kashyap et al., 2013; Langston et al., 1997).At these gene clusters as well as at other RA regulated genes, heterodimers of RARγ and retinoid X receptor α (RXRα) recognize and bind to RA responsive DNA elements (RAREs), then inducingepigenetic changes and transcriptional induction in response to RA (Fig. 7.3)(Gillespie and Gudas, 2007a, b). The transcriptional induction by RA is associated with increased levels of the co-activator proteins KAT3A (p300), KAT13B (pCIP), and of RNA polymerase II at target RAREs. Conversely, co-repressor proteins such as SUZ12, a key protein component of PRC2, are associated with specific RAREs, but dissociate in response to RA (Figs. 7.3 and 7.4) (Kashyap et al., 2011; Laursen et al., 2013). Furthermore, the re-association of SUZ12 with RAREs upon RA removal (Gillespie and Gudas, 2007a, b) exemplifies the highly reversible nature of cofactor association. Extensive changes in histone marks can be observed in response to RA, as illustrated by a heat-map showing RA-associated changes in H3K27me3, H3K4me3, and H3ac levels at the Hoxa cluster (Fig. 7.3). Importantly, for the Hoxa cluster the levels of activating marks (H3K4me3 and H3ac) increase, whereas the levels of repressive marks (H3K27me3) decrease in response to RA (Fig. 7.3) (Kashyap et al., 2011). This is not the case for all RA inducible genes; for Nr2F1 the levels of both activating (H3K4me3) and repressive (H3K27me3) marks show an initial increase in response to RA (Fig. 7.4). However, the H3K27me3 levels then start to decline, thereby increasing the extent of the induction (Laursen et al., 2013). The simultaneous presence of active H3K4me3 marks and repressive H3K27me3 marksis referred to as a bivalent domain, and this chromatin structure can often be found at promoters of RA inducible genes (Kashyap et al., 2011; Laursen et al., 2013; Mark et al., 2006; Pan et al., 2007). The bivalent chromatin structure signifies that these genes are in a poised state in which changes in the H3K4me3/H3K27me3 ratio are associated with transcriptional induction (presence of RA; H3K4me3 ↑, H3K27me3 ↓) or silencing (absence or removal of RA; H3K27me3 ↑, H3K4me3 ↓), as illustrated in Fig. 7.4 (Bernstein et al., 2006; Kashyap et al., 2011; Kashyap et al., 2013; Langston et al., 1997; Laursen et al., 2013).
Figure 7.3. Epigenetic changes induced along the Hoxa cluster in response to RA.
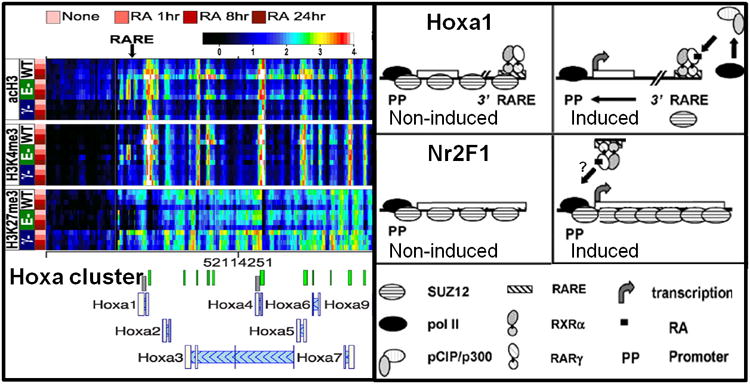
The epigenetic changes of the RA responsive Hoxa gene cluster are shown, with the locations of the Hoxa1 proximal promoter (PP) and RA responsive element (RARE) indicated by arrows. The levels of acH3, H3K4me3, and H3K27me3 determined by ChIP-chip are presented as heatmaps, with rows representing individual timepoints for each genotype, and columns indicating specific genomic regions. The genotypes of thestem cell lines are as follows: WT, RARE-KO (E-), and RARγ-KO (γ-). The cells were cultured in RA for 1, 8, and 24 h, as indicated. The color scale representing log2-transformed ChIP enrichment is indicated at the top of the figure. Note the reduced levels of H3ac and H3K4me3 at Hoxa1 PP and RARE in the RARγ-KO cell line
Models for RA mediated transcription of RA target genes Hoxa1 and Nr2F1. In the absence of RA, RARγ-RXRαheterodimers associated with Hoxa1 RAREs presumably associate with co-repressors, thereby generating a SUZ12-rich environment which represses transcription. Binding of the RA ligand causes a conformational change in the RARγ-RXRα heterodimer bound to the Hoxa1 RARE. This results in the recruitment of pCIP/p300, which generates an euchromatic environment, presumably by acetylating the histone tails. This allows pol II to initiate transcription of Hoxa1. The Nr2F1 promoter region (PP) is bound by SUZ12 in the absence of RA. Upon exposure to RA the increase in activating marks is initially counteracted by a concomitant increase in SUZ12, which attenuates the transcription of Nr2F1. Eventually, the SUZ12 levels decline, allowing the increased transcriptional activation of Nr2F1. (modified from Kashyap et al., 2011 and Gillespie and Gudas, 2007b).
Figure 7.4. Epigenetic Signatures Associated with RA and RAR regulated transcription.
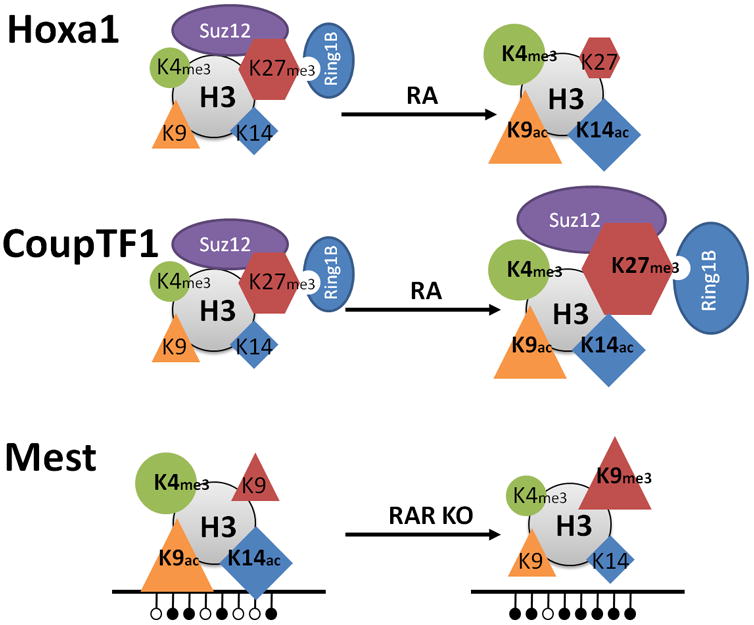
Hoxa1 represents a groupof direct target genes induced by RA (upper panel). The induction is characterized by dissociation of PRCs (ovals) and depletion of the H3K27me3 repressive mark (hexagons), and by increased levels of transcriptionally permissive marks, H3K4me3 (circle), H3K9ac (triangle), and H3K14ac (diamond). CoupTF1 representsa group of target genes with delayed transcriptional induction by RA (middle panel). The induction is characterized by an initial increase of PRCs (ovals) and of the H3K27me3 repressive mark (hexagons), concurrent with increased levels of transcriptionally permissive marks; H3K4me3, H3K9ac, and H3K14ac. The imprinted gene Mest is transcribed in the presence of RARα, but is silenced by DNA methylation upon knockout of RARα (lower panel). The transcriptional silencing of Mest is associated with increased DNA methylation, increased levels of the H3K9me3 repressive mark, and with decreased levels of transcriptionally permissive marks; H3K4me3, H3K9ac, and H3K14ac. Note that for Hoxa1 and CoupTF1 the transcriptionally active state is shown to the right, whereas for Mest the transcriptionally active state is shown to the left, Published in parts (CoupTF1) in Nucleic Acid Research (modified from Laursen, et al., 2013).
Now the question is; what regulates the levels of histone marks? Since histone marks are actually covalent modifications of histones, the levels need to be regulated by enzymes located in proximity to the histone. This brings us back to the “writers”, “readers”, and “erasers” mentioned in the beginning of the chapter. The levels of the active H3K4me3 mark are regulated by lysine methyl transferases (KMT) “writers” and lysine demethylases (KDM) “erasers”. Specifically, MLL proteins, which are KMT “writers” of the trithorax family, trimethylate H3K4 (Pasini et al., 2008), and KDM5 proteins, which are H3K4me3-specific “erasers,” can subsequently convert the H3K4me3 mark into H3K4me2 (Iwase et al., 2007; Kouzarides, 2007; Pasini et al., 2008). Once H3K4me3 has been converted to H3K4me2, the KDM1A/B “eraser” proteins can remove the remaining methyl marks, thus returning H3K4 to the unmethylated state (Ciccone et al., 2009). Pharmacological inhibition of KDM1A (LSD1) reactivates the RA differentiation pathway in leukemia cells (Schenk et al., 2012), indicating that enzymatic conversion of H3K4 to the unmethylated state by KDM1A plays a key role in antagonizing RA signaling. Curiously, H3K4me3 and H3K27me3 can be found on the same nucleosome, composed of eight histones, but not on the same histone tail (Voigt et al., 2012). Thus, each of the two histone 3 components of the nucleosome can be differentially modified. This specification of a histone as simultaneously activating and repressive is the core of the bivalent domains.
Conversely, the levels of the repressive H3K27me3 mark are regulated by Polycomb group proteins, which are H3K27me1/2 specific KMT “writers” (Cao et al., 2002; Kuzmichev et al., 2002), and the H3K27me2/3-specific KDM “erasers” KDM6A/B (Agger et al., 2007; Lee et al., 2007). The antagonistic effects of H3K4me3 and H3K27me3 are supported by the observation that RA-induced transcription leads to a concomitant decrease in H3K27me3 levels as well as an increase in H3K4me3 levels along the Hoxa cluster (Fig. 7.4). In this respect, it is interesting that the MLL2 complex contains KDM6A, an H3K27 demethylase (Agger et al., 2007). Consequently, the MLL complex combines methyltransferase activity targeting H3K4 with demethylase activity targeting the opposing H3K27me3 mark. A similar “push-pull” effect is observed with the PRC2 complex which contains EZH2, an H3K27 methyltransferase, and KDM5B, an H3K4 demethylase (Zhou et al., 2009). The depletion of H3K27me3 through knockdown of the EZH2 methyltransferase failed to induce Hoxa1 expression (Lee et al., 2007). This suggests that without increased H3K4 methylation the loss of H3K27 methylation is insufficient to induce transcription of Hoxa1. Consequently, the combined actions of H3K4 methyltransferases and H3K27 demethylases may be required for gene transcriptional activation of at least some genes. Also, the transcriptional activation of Hoxa1 precedes the removal of the H3K27me3 mark by many hours, showing that the removal of the H3K27me3 mark is not required for Hoxa1 transcriptional activation by RA (Kashyap et al., 2011).
A different scenario of the “push-pull” effect is observed when RA induces the CoupTF1 (Nr2F1) gene. In this case, activating H3K4me3 and repressive H3K27me3 marks are simultaneously recruited to the CoupTF1 promoter (Fig. 7.4), initiating a repressed or dampened induction characteristic of several late RA target genes (Laursen et al., 2013). While the functional depletion of PRC2 did not enhance RA induction of Hoxa5 and Hoxa1 (early genes), the depletion potently enhanced RA mediated induction of CoupTF1 and CoupTF2 (late genes) (Laursen et al., 2013).This finding is important since it provides a mechanistic rationale for distinguishing between early and late targets of RA induction. It has been shown that PRC2 can sense chromatin density, and thereby distinguish active chromatin (marked by H3K4me3 and H3K36me2/3) from inactive chromatin, on which PRC2 will target H3K27 for methylation. This helps to explain how PRC2 maintains target genes in an inactive, compacted chromatin state for long periods (Yuan et al., 2012). Taken together, these data further point to the presence of a combined “push-pull” effect, wherein the effects of specific KMTs are supported by the effects of specific KDMs, which together place and remove specific lysine methylation marks in a coordinated manner.
RA induced transcription of the Hox genes increases not only histone H3K4 methylation, but also histone H3K9 and H3K14 acetylation (Kashyap et al., 2011) (Fig. 7.3 and 7.4). In addition, H3K27 can be modified by either acetylation or methylation, with opposite effects on the chromatin environment, and thus on the transcriptional activity. Acetylation (Creyghton et al., 2010; Rada-Iglesias et al., 2011) and methylation of H3K27 are mutually exclusive marks positioned by KAT3A/B (CBP/p300) and PRC2 (EZH2), respectively (Pasini et al., 2010; Tie et al., 2012). H3K27 thus provides an example in which the enzymatic activities of KATs/KMTs and HDACs/KDMs converge in regulating gene activity. However, H3K27 is not the only target of acetylation; H3K9 and H3K14 are acetylated concurrently with RA induced transcriptional activation (Figs. 7.3 and 7.4)(Kashyap et al., 2011; Kashyap et al., 2013). The RA-dependent recruitment of the acetyltransferases KAT3B (p300) and KAT13B (NCoA3, Actr, pCIP, Src3) to the RAREs of Hoxa1 (Fig. 7.3) and Cyp26a1 in F9 teratocarcinoma stem cells and ES cells suggests that these KATs also play key roles in RA-induced transcription (Gillespie and Gudas, 2007a, b; Kashyap and Gudas, 2010). Finally, KAT6A (Moz) is involved in H3K9 acetylation of the Hox gene loci, yet RA can activate the Hox loci independently of KAT6A (Voss et al., 2009). The plethora of coregulators involved in RA induced transcription allows for fine-tuning of a highly gene specific response (Fig. 7.3).
7.4 Current State of the Field: DNA demethylation is involved in the RA transcriptional response
Passive DNA demethylation takes place when maintenance methylation is inhibited during DNA replication, while active DNA demethylation requires specific enzymes and can occur without DNA replication (Zhu, 2009). Activation induced cytidinedeaminase (AICDA, AID) is an active, reprogramming DNA demethylase expressed in embryonic stem cells and other cell types (Morgan et al., 2004). A second, more recently discovered DNA demethylase family, Tet 1, 2 and 3, removes DNA methylation through oxidative demethylation, a mechanism also employed by JmjC proteins to demethylate histones (Ito et al., 2010; Tahiliani et al., 2009; Tsukada et al., 2006). Tet1 mediated hydroxylation of 5mC to 5-hydroxylmethylcytosine (5hmU) is enhanced by AICDA, which generates 5hmUas a step towards the demethylation of 5mC. This requires thymine DNA glycosylase (TDG), a base excision repair enzyme, which excises the 5hmU (Guo et al., 2011). Through the active prevention of DNA methylation, TDG maintains bivalent chromatin domains in embryonic stem cells (Cortázar et al., 2011). Considering that several RA primary target genes reside in bivalent domains, it is worth noting that Um et al. (Um et al., 1998) identified interactions between TDG and the RARs/RXRs which may link RA to active demethylation of DNA. TDG forms a complex with AICDA and GADD45a, and is required for the recruitment of the coactivator protein KAT3B (p300) to the promoters of RA-inducible genes (Cortellino et al., 2011). Thus, a loss of TDG activity could result in a decrease in RAR/RA-associated gene transcription and a resultant block in cell differentiation, which would be consistent with the observed increase in DNA methylation of the Mest promoter region in response to knockout of RARα (Laursen et al., 2012). This indicates that RARα (and possibly other RARs) plays a direct role in maintaining gene expression by keeping specific promoters in a hypomethylated state, and conversely, underscores the fact that reduced expression of RARα can have adverse consequences, such as leukemogenesis (Glasow et al., 2008). A reduction in RARα signaling also impairs the survival of tumor reactive CD8 (+) T-cells within the tumor microenvironment (Guo et al., 2012). Whether this is related to RARα 's ability to control the methylation state of certain genes has not yet been elucidated.
During gametogenesis, maternal or paternal genomes can be modified so that one parental allele is expressed, whereas the other is transcriptionally silenced. This genomic imprinting typically occurs through DNA methylation of CpG islands (Prickett and Oakey, 2012). An exciting, recent, findingsuggests that RARα, independently of RA, maintains the DNA methylation status of specific imprinted genes (Laursen et al., 2012). This was highlighted by the identification of several aberrantly expressed, imprinted, genes in RARα-/- F9 stem cells (Laursen et al., 2012). Under normal conditions RARα associates with the promoter region of the paternally expressed gene, Mest;upon RARα knockout, resulting in the absence of RARα, the levels of H3K9me3 and the DNA methylation of the Mest promoter region significantly increase (Laursen et al., 2012) (Fig. 7.4). Several of the changes in gene expression associated with the RARα knockout are similar to those observed during the differentiation of stem-like progenitors to hypertrophic chondrocytes in the developing growth plate (Constância et al., 2005). This similarity between the in vivo and in vitro data supports the idea that in vivo imprinting may be regulated by RARα, and highlights the important roles of specific RARs in regulating epigenetic changes during development. Further exploration of this topic is expected to deepen our understanding of genomic imprinting and to expand the realm of RAR regulated transcription beyond the well-known ligand-induced regulation of gene activity.
7.5 Relevance: RA Regulated epigenetic changes in carcinogenesis
Retinoid signaling is often disrupted during carcinogenesis, suggesting that restoration of retinoid signaling may be a viable option for cancer prevention and/or treatment (Mongan and Gudas, 2005; Tang and Gudas, 2011). Synthetic retinoids modify the levels of the various RARs during breast carcinogenesis (Bosch et al., 2012), and RA inhibits the growth of human osteosarcoma by promoting cell differentiation (Yang et al., 2012). In a glioma animal model, RA also promoted the differentiation of cancer stem cells (Campos et al., 2010). As a result, retinoids are currently being tested and/or used for treatment of many different cancers, including breast, ovarian, renal, head and neck, melanoma, leukemias, and prostate cancers. However, epigenetic changes, such as histone modifications and DNA methylation, and subsequent changes in gene expression are also thought to play major roles in cancer initiation and progression. Therefore, in line with the aforementioned “push-pull” model, combination cancer therapies that include retinoids together with epigenetic therapeutic agents are believed to be moreeffectivein treatingdifferentcancers.
Histone deacetylase inhibitors, such as suberoylanilidehydroxamic acid (SAHA) have been extensively studied as potential cancer therapies, and are currently being used to treat multiple cancers, including cutaneous T-cell lymphoma and non-small cell lung cancer (Bantscheff et al., 2011). Chemoproteomics profiling of HDAC inhibitors revealed selective targeting of histone deacetylase (HDAC) complexes as promising cancer therapies (Bantscheff et al., 2011).
It is believed that treatment with HDAC inhibitors together with retinoid therapies may be an even more effective treatment regimen for certain cancers. When combined with HDAC inhibitors such as TSA and valproic acid, RA can re-induce RARβ expression in kidney (Touma et al., 2005) or breast (Mongan and Gudas, 2005) cancers, and inhibit cell proliferation in many types of cancers (Cimino et al., 2006; Feng et al., 2012; Kato et al., 2007; Pili et al., 2012; Qi and Ratnam, 2006; Savickiene et al., 2012; Tavares et al., 2008; Trus et al., 2005; Wang et al., 2005). Furthermore, RA synergizes with valproic acid to promote the degradation of the PML-RARα oncoprotein, destroying the leukemia initiating cells in vivo (Leiva et al., 2012). Recently a phase I trial using valproic acid and liposomal RA for patients with solid tumors yielded positive results, suggesting that this therapy maybe used for various solid tumors (David et al., 2010).
Another promising treatment approach is the co-administration of retinoids with DNA methyltransferase inhibitors. Mice treated with a combination of RA and the DNA methyltransferase inhibitor 5-Aza-2′-deoxycytidine (5-Aza) exhibit a decreased incidence of oral cancer after carcinogen treatment (Tang et al., 2009), and valproic acid, 5-Aza, and RA promoted growth arrest and cell differentiation of cultured human head and neck squamous cell carcinoma (HNSCC) cells (Gan et al., 2012). Additionally, a phase II clinical trial for patients with acute myeloid leukemia combining 5-Azawith RA was just completed with promising results (Lübbert et al., 2012).
Finally, other studies have examined the potential efficacy of treatments with RA, HDAC inhibitors, and DNA methyltransferases together. RA treatment in the presence of both valproic acid and 5-Aza promotes the re-expression of RARβ and inhibits cell growth in breast cancer cell lines (Mongan and Gudas, 2005). Additionally, promyelocytic leukemia cells exhibit cell growth inhibition and increased granulocyte differentiation after treatment with all three drugs (Savickiene et al., 2012). Overall, these studies indicate that combinations of retinoids and epigenetic modulating drugs are promising treatment options for multiple types of cancer, in part because of their actions in promoting cell differentiation and the inhibition of cell proliferation. Depicted in Fig. 7.5 are various epigenetic machinery inhibitors that are being intensely studied as possible cancer treatments (Rodríguez-Paredes and Esteller, 2011), and thesecould potentially be even more effective in combination with RA.
Figure. 7.5. Current drugs targeting various epigenetic machinery.
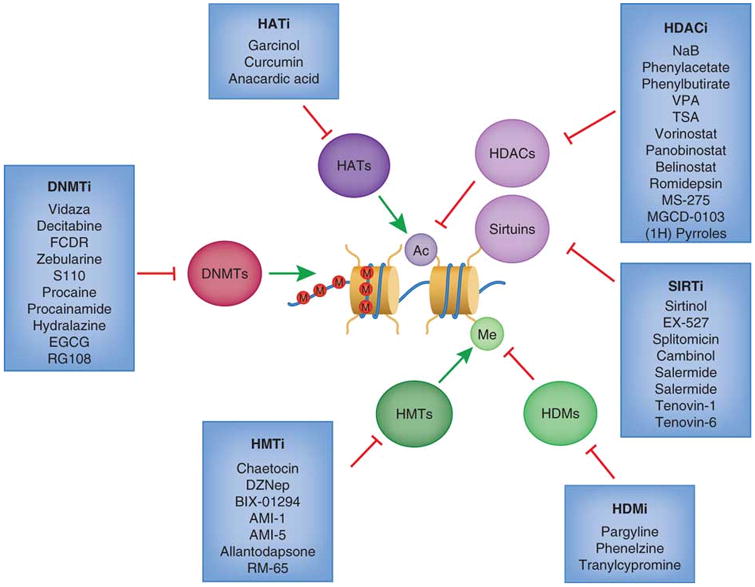
Histone acetyltransferase inhibitors (HATi) may be effective treatments since HAT proteins are sometimes considered to be tumor promoters. Histone deacetylase inhibitors (HDACi) such as vorinostat (SAHA) and romidepsin have been FDA approved for treatments of certain hematological cancers. SIRT inhibitors (SIRTi) specifically target the SIRT family of histone deacetylases. Histone demethyltransferase inhibitors (HDMi) and histone methylasetransferase (HMTi) have importantly become more selective towards specific marks, since methylation can act as both a repressive and an activating mark. DNA methyltransferase inhibitors (DNMTi) may serve as promising drugs for re-sensitizing certain cancer cells to chemotherapy, and two (vidaza and decitabine) are FDA approved for cancer treatment. (Rodríguez-Paredes et al., 2011)
7.6 The Future: RA action and epigenetics, cell differentiation and cancer
Further studies are needed to determine the roles and specificities of various KATs and KDMs with respect to RA transcriptional activation and to develop a better understanding of how RARα (and possibly other RARs) plays a direct role in maintaining gene expression by keeping specific promoters in a hypomethylated state. Many different epigenetic changes must take place for stem cells to differentiate properly, and when these changes do not proceed normally, increased tumorigenesis can result. Aberrant expression of the polycomb protein EZH2, a core component of PRC2, has been found in human breast, prostate, bladder, and colon cancers, and this overexpression is correlated with a poor prognosis (Mills, 2010; Raman et al., 2005). Overexpression of EZH2 in hematopoietic stem cells (HSCs) eliminates the exhaustion of the long-term repopulation potential of these stem cells during multiple, sequential transplantations (Kamminga et al., 2006). EZH2 also enhances leukemogenesis by enhancing the differentiation block in acute myeloid leukemia (Neff et al., 2012; Tanaka et al., 2012). Thus, these epigenetic modifications by EZH2 have profound consequences in terms of reducing the ability of HSCs to differentiate and enhancing tumorigenesis. Likewise, in prostate cancer EZH2 can block differentiation by affecting transcriptional regulation by the androgen receptor (Crea et al., 2011). The recent development of EZH2 inhibitors for treatment of lymphomas shows the power of manipulating epigenetic modifications for cancer treatment (Béguelin et al., 2013).
DZNep, an S-adenosylhomocysteine (SAH) hydrolase inhibitor, can eradicate tumor initiating cells in hepatocellular carcinoma cells and induce apoptosis in acute myeloid leukemia (Chiba et al., 2012; Fiskus et al., 2009; Zhou et al., 2011). DZNep can also inhibit tumorigenicity and progression in prostate cancer (Crea et al., 2011). The inhibition of SAH hydrolase causes an increase in SAH, resulting in inhibition of S-adenosyl-L-methionine dependent methyltransferases such as EZH2. We recently showed that human colon cancer cells, when exposed to RA, DZNep, or to a genetic knockdown of the PRC2 core protein SUZ12, exhibited enhanced PTEN mediated apoptosis, whereas the survival of embryonic stem cells was unaffected (Benoit et al., 2013b). The apoptotic effects of RA, DZNep, or SUZ12 depletion were further enhanced by combination with the TRAIL death receptor (Benoit et al., 2013a). The synergy between TRAIL and RA was confirmed by another report in which the authors demonstrate that treatment with retinyl acetate (another vitamin A metabolite) in combination with TRAIL not only induced apoptosis specifically in intestinal polyps, but also inhibited tumor growth and prolonged survival in a murine model of human colon cancer (Zhang et al., 2010). These results suggest that one mechanism by which RA enhances TRAIL associated apoptosis is via removing PRC2 complexes from various genes involved in differentiation and/or apoptosis (Fig. 7.6).
Figure. 7.6. Representation of RA and DZNep effects on apoptosis regulation in human colon cancer cells.
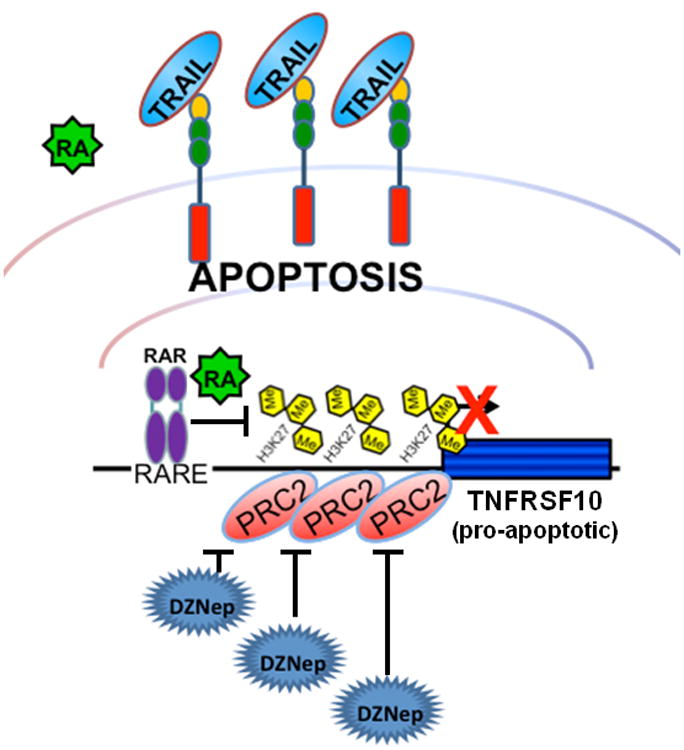
Retinoic acid treatment promotes TRAIL-related apoptosis in RARβ/RARγ–positive HT29 cells, but not in SW480 cells which express only low levels of RARβ/RARγ. The functional depletion of PRC2 by either inhibition with DZNep or by knockdown of SUZ12 increases TRAIL-mediated apoptosis in both HT29 and SW480 cell lines. In this scenario the PRC2-mediated repression is alleviated, thereby activating TNFRSF10 even in the absence of RARβ/RARγ.
Research in this field will be enhanced by the recent development of more specific EZH2 inhibitors (Knutson et al., 2012; McCabe et al., 2012), and by the evaluation of new drug combinations that more efficiently target specific epigenetic regulators. The fact that so many different types of cancer exhibit altered epigenetic profiles and/or mutations in proteins that modify the epigenome indicates that this will be a fruitful area of research that will provide major benefits to cancer patients in terms of new combination therapies.
The APC gene product, whichcontrols intestinal cell fate, can, togetherwith RA, downregulatedemethylase components; this promotes DNA methylation of key target genes and increases the commitment of progenitors to differentiation (Rai et al., 2010). We recently showed that, when combined with the TNF-related apoptosis-inducing ligand (TRAIL), RA exhibited enhanced apoptotic effects on human colorectal cancer cells (Benoit et al., 2013a). This additive effect of RA could be substituted by either inhibition or functional depletion of the PRC2 complex by 3-Deazaneplanocin A (DZNep) or knockdown of SUZ12, respectively (Benoit et al., 2013a), which suggests that the observed synergy with TRAIL is a result the RA induced dissociation of SUZ12 (described in section 7.2).
Acknowledgments
We would like to thank Weill Cornell and the NIH (NCI R01-CA043796 to LJG, NIDCR R01-DE010389 to LJG), and (NIAAA F32-AA021045 to AU) for support for this chapter. We also thank members of the Gudas lab for suggestions and comments, and in particular, Dr. Yannick Benoit for critically reading this chapter. We thank Tamara Weissman for editing this chapter.
Abbreviations
- SAH
S-adenosylhomocysteine
- SAHA
suberoylanilidehydroxamic acid
- KMT/PRMT
lysine/arginine methyltransferase
- KDM/PRDM
lysine/arginine demethylase
- ES
embryonic stem cells
- iPS
induced pluripotent stem cells
- HAT
histone acetyltransferase proteins
- HDAC
histone deacetylase
- CpG
phosphodiester-bonded cytosine-guanine dinucleotide
- MECP
methyl-CpG-binding domain proteins
- DNMT
DNA methyltransferase
- RAR
retinoic acid
- RA
all-trans retinoic acid
- Hox
homeobox
- ChIP
chromatin immunoprecipitation
- RXR
retinoid X receptor
- RARE
retinoic acid responsive DNA element
- PRC
Polycomb repressive complex
- 5hmU
5-hydroxymethyluracil
- 5-Aza
5-Aza-2′-deoxycytidine
- DZNep
3-deazaneplanocin A
- HSCs
hematopoietic stem cells
- TRAIL
TNF-related apoptosis-inducing ligand
- WT
Wild type
Footnotes
Standardized Gene Names/Nomenclature: KDM1: LSD1/2
KDM4A: JMJD2A
KDM5A: Jarid1A/B/C/D
KDM6: JMJD3/UTX/(UTY)
KAT3A/B: CBP/p300
KAT6A: MOZ
KAT6B: MORF
MT2A: MLL1
KMT2B/C: MLL2/3
Contributor Information
Alison Urvalek, Email: alu2004@med.cornell.edu, Complete Address, Weill Cornell Medical College, Department of Pharmacology, 1300 York Avenue, New York, NY 10065 USA, Phone: 212-746-6262, Fax: 212-746-8835.
Kristian Bruun Laursen, Email: krl2004@med.cornell.edu, Complete Address, Weill Cornell Medical College, Department of Pharmacology, 1300 York Avenue, New York, NY 10065 USA, Phone:212-746-6262, Fax: 212-746-8835.
Lorraine J. Gudas, Email: ljgudas@med.cornell.edu, Complete Address, Weill Cornell Medical College, Department of Pharmacology, 1300 York Avenue, New York, NY 10065 USA, Phone: 212-746-6250, Fax: 212-746-8858.
References
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez-Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, Van Aller GS, Kruger RG, Liu Y, McHugh CF, Scott DW, Chung YR, Kelleher N, Shaknovich R, Creasy CL, Gascoyne RD, Wong KK, Cerchietti L, Levine RL, Abdel-Wahab O, Licht JD, Elemento O, Melnick AM. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit YD, Laursen KB, Witherspoon MS, Lipkin SM, Gudas LJ. Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J Cell Physiol. 2013a;228:764–772. doi: 10.1002/jcp.24224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit YD, Witherspoon MS, Laursen KB, Guezguez A, Beauséjour M, Beaulieu JF, Lipkin SM, Gudas LJ. Pharmacological inhibition of polycomb repressive complex-2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res. 2013b doi: 10.1016/j.yexcr.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bosch A, Bertran SP, Lu Y, Garcia A, Jones AM, Dawson MI, Farias EF. Reversal by RARalpha agonist Am580 of c-Myc-induced imbalance in RARalpha/RARgamma expression during MMTV-Myc tumorigenesis. Breast Cancer Res. 2012;14:R121. doi: 10.1186/bcr3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos B, Wan F, Farhadi M, Ernst A, Zeppernick F, Tagscherer KE, Ahmadi R, Lohr J, Dictus C, Gdynia G, Combs SE, Goidts V, Helmke BM, Eckstein V, Roth W, Beckhove P, Lichter P, Unterberg A, Radlwimmer B, Herold-Mende C. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin Cancer Res. 2010;16:2715–2728. doi: 10.1158/1078-0432.CCR-09-1800. [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Chiba T, Suzuki E, Negishi M, Saraya A, Miyagi S, Konuma T, Tanaka S, Tada M, Kanai F, Imazeki F, Iwama A, Yokosuka O. 3-Deazaneplanocin A is a promising therapeutic agent for the eradication of tumor-initiating hepatocellular carcinoma cells. Int J Cancer. 2012;130:2557–2567. doi: 10.1002/ijc.26264. [DOI] [PubMed] [Google Scholar]
- Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461:415–418. doi: 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- Cimino G, Lo-Coco F, Fenu S, Travaglini L, Finolezzi E, Mancini M, Nanni M, Careddu A, Fazi F, Padula F, Fiorini R, Spiriti MA, Petti MC, Venditti A, Amadori S, Mandelli F, Pelicci PG, Nervi C. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer Res. 2006;66:8903–8911. doi: 10.1158/0008-5472.CAN-05-2726. [DOI] [PubMed] [Google Scholar]
- Cohen I, Poręba E, Kamieniarz K, Schneider R. Histone modifiers in cancer: friends or foes? Genes Cancer. 2011;2:631–647. doi: 10.1177/1947601911417176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constância M, Angiolini E, Sandovici I, Smith P, Smith R, Kelsey G, Dean W, Ferguson-Smith A, Sibley CP, Reik W, Fowden A. Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc Natl Acad Sci U S A. 2005;102:19219–19224. doi: 10.1073/pnas.0504468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortázar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, Steinacher R, Jiricny J, Bird A, Schär P. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun V, Tsoa R, Sun YE. Epigenetic regulation of stem cells differentiating along the neural lineage. Curr Opin Neurobiol. 2012;22:762–767. doi: 10.1016/j.conb.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE, Danesi R, Farrar WL. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer. 2011;10:40. doi: 10.1186/1476-4598-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David KA, Mongan NP, Smith C, Gudas LJ, Nanus DM. Phase I trial of ATRA-IV and Depakote in patients with advanced solid tumor malignancies. Cancer Biol Ther. 2010;9:678–684. doi: 10.4161/cbt.9.9.11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–657. doi: 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- Feng D, Cao Z, Li C, Zhang L, Zhou Y, Ma J, Liu R, Zhou H, Zhao W, Wei H, Ling B. Combination of valproic acid and ATRA restores RARβ2 expression and induces differentiation in cervical cancer through the PI3K/Akt pathway. Curr Mol Med. 2012;12:342–354. doi: 10.2174/156652412799218949. [DOI] [PubMed] [Google Scholar]
- Fiskus W, Buckley K, Rao R, Mandawat A, Yang Y, Joshi R, Wang Y, Balusu R, Chen J, Koul S, Joshi A, Upadhyay S, Atadja P, Bhalla KN. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol Ther. 2009;8:939–950. doi: 10.4161/cbt.8.10.8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan CP, Hamid S, Hor SY, Zain RB, Ismail SM, Wan Mustafa WM, Teo SH, Saunders N, Cheong SC. Valproic acid: growth inhibition of head and neck cancer by induction of terminal differentiation and senescence. Head Neck. 2012;34:344–353. doi: 10.1002/hed.21734. [DOI] [PubMed] [Google Scholar]
- Gillespie RF, Gudas LJ. Retinoic acid receptor isotype specificity in F9 teratocarcinoma stem cells results from the differential recruitment of coregulators to retinoic response elements. J Biol Chem. 2007a;282:33421–33434. doi: 10.1074/jbc.M704845200. [DOI] [PubMed] [Google Scholar]
- Gillespie RF, Gudas LJ. Retinoid regulated association of transcriptional co-regulators and the polycomb group protein SUZ12 with the retinoic acid response elements of Hoxa1, RARbeta(2), and Cyp26A1 in F9 embryonal carcinoma cells. J Mol Biol. 2007b;372:298–316. doi: 10.1016/j.jmb.2007.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasow A, Barrett A, Petrie K, Gupta R, Boix-Chornet M, Zhou DC, Grimwade D, Gallagher R, von Lindern M, Waxman S, Enver T, Hildebrandt G, Zelent A. DNA methylation-independent loss of RARA gene expression in acute myeloid leukemia. Blood. 2008;111:2374–2377. doi: 10.1182/blood-2007-05-088344. [DOI] [PubMed] [Google Scholar]
- Griffith JS, Mahler HR. DNA ticketing theory of memory. Nature. 1969;223:580–582. doi: 10.1038/223580a0. [DOI] [PubMed] [Google Scholar]
- Guillemette B, Drogaris P, Lin HH, Armstrong H, Hiragami-Hamada K, Imhof A, Bonneil E, Thibault P, Verreault A, Festenstein RJ. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 2011;7:e1001354. doi: 10.1371/journal.pgen.1001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Pino-Lagos K, Ahonen CA, Bennett KA, Wang J, Napoli JL, Blomhoff R, Sockanathan S, Chandraratna RA, Dmitrovsky E, Turk MJ, Noelle RJ. A Retinoic Acid--Rich Tumor Microenvironment Provides Clonal Survival Cues for Tumor-Specific CD8+ T Cells. Cancer Res. 2012;72:5230–5239. doi: 10.1158/0008-5472.CAN-12-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509–523. doi: 10.1242/dev.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. [PubMed] [Google Scholar]
- Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kamminga LM, Bystrykh LV, de Boer A, Houwer S, Douma J, Weersing E, Dontje B, de Haan G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107:2170–2179. doi: 10.1182/blood-2005-09-3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap V, Gudas LJ. Epigenetic regulatory mechanisms distinguish retinoic acid-mediated transcriptional responses in stem cells and fibroblasts. J Biol Chem. 2010;285:14534–14548. doi: 10.1074/jbc.M110.115345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap V, Gudas LJ, Brenet F, Funk P, Viale A, Scandura JM. Epigenomic reorganization of the clustered Hox genes in embryonic stem cells induced by retinoic acid. J Biol Chem. 2011;286:3250–3260. doi: 10.1074/jbc.M110.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap V, Laursen KB, Brenet F, Viale AJ, Scandura JM, Gudas LJ. RARγ is essential for retinoic acid induced chromatin remodeling and transcriptional activation in embryonic stem cells. J Cell Sci. 2013;126:999–1008. doi: 10.1242/jcs.119701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Salumbides BC, Wang XF, Qian DZ, Williams S, Wei Y, Sanni TB, Atadja P, Pili R. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol Cancer Ther. 2007;6:70–81. doi: 10.1158/1535-7163.MCT-06-0125. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim KB, Son HJ, Chae YC, Oh ST, Kim DW, Pak JH, Seo SB. H3K27 methylation and H3S28 phosphorylation-dependent transcriptional regulation by INHAT subunit SET/TAF-Iβ. FEBS Lett. 2012;586:3159–3165. doi: 10.1016/j.febslet.2012.06.026. [DOI] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston AW, Thompson JR, Gudas LJ. Retinoic acid-responsive enhancers located 3′ of the Hox A and Hox B homeobox gene clusters. Functional analysis. J Biol Chem. 1997;272:2167–2175. doi: 10.1074/jbc.272.4.2167. [DOI] [PubMed] [Google Scholar]
- Laursen KB, Mongan NP, Zhuang Y, Ng MM, Benoit YD, Gudas LJ. Polycomb recruitment attenuates retinoic acid-induced transcription of the bivalent NR2F1 gene. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen KB, Wong PM, Gudas LJ. Epigenetic regulation by RARalpha maintains ligand-independent transcriptional activity. Nucleic Acids Res. 2012;40:102–115. doi: 10.1093/nar/gkr637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- Leiva M, Moretti S, Soilihi H, Pallavicini I, Peres L, Mercurio C, Dal Zuffo R, Minucci S, de Thé H. Valproic acid induces differentiation and transient tumor regression, but spares leukemia-initiating activity in mouse models of APL. Leukemia. 2012;26:1630–1637. doi: 10.1038/leu.2012.39. [DOI] [PubMed] [Google Scholar]
- Lübbert M, Rüter BH, Claus R, Schmoor C, Schmid M, Germing U, Kuendgen A, Rethwisch V, Ganser A, Platzbecker U, Galm O, Brugger W, Heil G, Hackanson B, Deschler B, Döhner K, Hagemeijer A, Wijermans PW, Döhner H. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica. 2012;97:393–401. doi: 10.3324/haematol.2011.048231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol. 2006;46:451–480. doi: 10.1146/annurev.pharmtox.46.120604.141156. [DOI] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer. 2010;10:669–682. doi: 10.1038/nrc2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongan NP, Gudas LJ. Valproic acid, in combination with all-trans retinoic acid and 5-aza-2′-deoxycytidine, restores expression of silenced RARbeta2 in breast cancer cells. Mol Cancer Ther. 2005;4:477–486. doi: 10.1158/1535-7163.MCT-04-0079. [DOI] [PubMed] [Google Scholar]
- Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, Xie H, Orkin SH, Armstrong SA. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A. 2012;109:5028–5033. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Pasini D, Hansen KH, Christensen J, Agger K, Cloos PA, Helin K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008;22:1345–1355. doi: 10.1101/gad.470008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON, Helin K. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010;38:4958–4969. doi: 10.1093/nar/gkq244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DM. The presence of acetyl groups of histones. Biochem J. 1963;87:258–263. doi: 10.1042/bj0870258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, Carducci MA, Rudek MA. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106:77–84. doi: 10.1038/bjc.2011.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickett AR, Oakey RJ. A survey of tissue-specific genomic imprinting in mammals. Mol Genet Genomics. 2012;287:621–630. doi: 10.1007/s00438-012-0708-6. [DOI] [PubMed] [Google Scholar]
- Qi H, Ratnam M. Synergistic induction of folate receptor beta by all-trans retinoic acid and histone deacetylase inhibitors in acute myelogenous leukemia cells: mechanism and utility in enhancing selective growth inhibition by antifolates. Cancer Res. 2006;66:5875–5882. doi: 10.1158/0008-5472.CAN-05-4048. [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Sarkar S, Broadbent TJ, Voas M, Grossmann KF, Nadauld LD, Dehghanizadeh S, Hagos FT, Li Y, Toth RK, Chidester S, Bahr TM, Johnson WE, Sklow B, Burt R, Cairns BR, Jones DA. DNA demethylase activity maintains intestinal cells in an undifferentiated state following loss of APC. Cell. 2010;142:930–942. doi: 10.1016/j.cell.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman JD, Mongan NP, Tickoo SK, Boorjian SA, Scherr DS, Gudas LJ. Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder. Clin Cancer Res. 2005;11:8570–8576. doi: 10.1158/1078-0432.CCR-05-1047. [DOI] [PubMed] [Google Scholar]
- Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- Riggs AD, Matienssen RA, Russo VEA. Epigenetic mechanisms of gene regulation. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1996. [Google Scholar]
- Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- Sasai N, Defossez PA. Many paths to one goal? The proteins that recognize methylated DNA in eukaryotes. Int J Dev Biol. 2009;53:323–334. doi: 10.1387/ijdb.082652ns. [DOI] [PubMed] [Google Scholar]
- Savickiene J, Treigyte G, Jazdauskaite A, Borutinskaite VV, Navakauskiene R. DNA methyltransferase inhibitor RG108 and histone deacetylase inhibitors cooperate to enhance NB4 cell differentiation and E-cadherin re-expression by chromatin remodelling. Cell Biol Int. 2012;36:1067–1078. doi: 10.1042/CBI20110649. [DOI] [PubMed] [Google Scholar]
- Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, Casero RA, Marton L, Woster P, Minden MD, Dugas M, Wang JC, Dick JE, Müller-Tidow C, Petrie K, Zelent A. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Stock JK, Giadrossi S, Casanova M, Brookes E, Vidal M, Koseki H, Brockdorff N, Fisher AG, Pombo A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9:1428–1435. doi: 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, Suzuki Y, Sugano S, Nakaseko C, Yokote K, Koseki H, Iwama A. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107–1117. doi: 10.1182/blood-2011-11-394932. [DOI] [PubMed] [Google Scholar]
- Tang XH, Albert M, Scognamiglio T, Gudas LJ. A DNA methyltransferase inhibitor and all-trans retinoic acid reduce oral cavity carcinogenesis induced by the carcinogen 4-nitroquinoline 1-oxide. Cancer Prev Res (Phila) 2009;2:1100–1110. doi: 10.1158/1940-6207.CAPR-09-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- Tavares TS, Nanus D, Yang XJ, Gudas LJ. Gene microarray analysis of human renal cell carcinoma: the effects of HDAC inhibition and retinoid treatment. Cancer Biol Ther. 2008;7:1607–1618. doi: 10.4161/cbt.7.10.6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Banerjee R, Conrad PA, Scacheri PC, Harte PJ. Histone demethylase UTX and chromatin remodeler BRM bind directly to CBP and modulate acetylation of histone H3 lysine 27. Mol Cell Biol. 2012;32:2323–2334. doi: 10.1128/MCB.06392-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey J, Lunyak VV. Epigenetics: Judge, jury and executioner of stem cell fate. Epigenetics. 2012;7:823–840. doi: 10.4161/epi.21141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma SE, Goldberg JS, Moench P, Guo X, Tickoo SK, Gudas LJ, Nanus DM. Retinoic acid and the histone deacetylase inhibitor trichostatin a inhibit the proliferation of human renal cell carcinoma in a xenograft tumor model. Clin Cancer Res. 2005;11:3558–3566. doi: 10.1158/1078-0432.CCR-04-1155. [DOI] [PubMed] [Google Scholar]
- Trus MR, Yang L, Suarez Saiz F, Bordeleau L, Jurisica I, Minden MD. The histone deacetylase inhibitor valproic acid alters sensitivity towards all trans retinoic acid in acute myeloblastic leukemia cells. Leukemia. 2005;19:1161–1168. doi: 10.1038/sj.leu.2403773. [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Um S, Harbers M, Benecke A, Pierrat B, Losson R, Chambon P. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1998;273:20728–20736. doi: 10.1074/jbc.273.33.20728. [DOI] [PubMed] [Google Scholar]
- Verdone L, Agricola E, Caserta M, Di Mauro E. Histone acetylation in gene regulation. Brief Funct Genomic Proteomic. 2006;5:209–221. doi: 10.1093/bfgp/ell028. [DOI] [PubMed] [Google Scholar]
- Voigt P, Leroy G, Drury WJ, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. Asymmetrically modified nucleosomes. Cell. 2012;151:181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss AK, Collin C, Dixon MP, Thomas T. Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev Cell. 2009;17:674–686. doi: 10.1016/j.devcel.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Waddington CH. The Strategy of Genes. Allen & Unwin; London: 1957. [Google Scholar]
- Waddington CH. Towards a Theoretical Biology. Edinburgh University Press; Edinburgh, Scotland: 1968. [Google Scholar]
- Wang XF, Qian DZ, Ren M, Kato Y, Wei Y, Zhang L, Fansler Z, Clark D, Nakanishi O, Pili R. Epigenetic modulation of retinoic acid receptor beta2 by the histone deacetylase inhibitor MS-275 in human renal cell carcinoma. Clin Cancer Res. 2005;11:3535–3542. doi: 10.1158/1078-0432.CCR-04-1092. [DOI] [PubMed] [Google Scholar]
- Yang QJ, Zhou LY, Mu YQ, Zhou QX, Luo JY, Cheng L, Deng ZL, He TC, Haydon RC, He BC. All-trans retinoic acid inhibits tumor growth of human osteosarcoma by activating Smad signaling-induced osteogenic differentiation. Int J Oncol. 2012;41:153–160. doi: 10.3892/ijo.2012.1426. [DOI] [PubMed] [Google Scholar]
- Yang XJ. Multisite protein modification and intramolecular signaling. Oncogene. 2005;24:1653–1662. doi: 10.1038/sj.onc.1208173. [DOI] [PubMed] [Google Scholar]
- Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, Han Z, Chai J, Zhou XJ, Gao S, Zhu B. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science. 2012;337:971–975. doi: 10.1126/science.1225237. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ren X, Alt E, Bai X, Huang S, Xu Z, Lynch PM, Moyer MP, Wen XF, Wu X. Chemoprevention of colorectal cancer by targeting APC-deficient cells for apoptosis. Nature. 2010;464:1058–1061. doi: 10.1038/nature08871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, Koh TL, Yu Q, Chng WJ. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118:2830–2839. doi: 10.1182/blood-2010-07-294827. [DOI] [PubMed] [Google Scholar]
- Zhou W, Chen H, Zhang L. The PcG protein hPc2 interacts with the N-terminus of histone demethylase JARID1B and acts as a transcriptional co-repressor. BMB Rep. 2009;42:154–159. doi: 10.5483/bmbrep.2009.42.3.154. [DOI] [PubMed] [Google Scholar]
- Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–166. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]