

Abstract
Forward genetic screens using chemical mutagens have been successful in defining the function of thousands of genes in eukaryotic model organisms. The main drawback of this strategy is the time-consuming identification of the molecular lesions causative of the phenotypes of interest. With whole-genome sequencing (WGS), it is now possible to sequence hundreds of strains, but determining which mutations are causative among thousands of polymorphisms remains challenging. We have sequenced 394 mutant strains, generated in a chemical mutagenesis screen, for essential genes on the Drosophila X chromosome and describe strategies to reduce the number of candidate mutations from an average of ∼3500 to 35 single-nucleotide variants per chromosome. By combining WGS with a rough mapping method based on large duplications, we were able to map 274 (∼70%) mutations. We show that these mutations are causative, using small 80-kb duplications that rescue lethality. Hence, our findings demonstrate that combining rough mapping with WGS dramatically expands the toolkit necessary for assigning function to genes.
Systematically defining the function of genes remains one of the most challenging endeavors in biological sciences. Several large forward and reverse genetic efforts have been initiated in mice to address this issue (Justice et al. 1999; Clark et al. 2004; Bradley et al. 2012; White et al. 2013). However, Caenorhabditis elegans and Drosophila melanogaster are still the most coveted systems to perform systematic functional annotation of genes required for development, nervous system function, organogenesis, metabolism, etc. (Venken et al. 2011). To this end, three main approaches are typically used: RNA interference (RNAi), transposon hopping, and chemical mutagenesis. Each of these methods has advantages as well as drawbacks (Mohr et al. 2010; Venken et al. 2011). Chemical mutagens like EMS (ethyl methanesulfonate) have the major advantage of being unbiased and often permit the generation of allelic series. However, mapping the causative mutations using traditional techniques is tedious and time consuming (Venken et al. 2011).
The limitations of chemical mutagenesis, however, can be partially overcome by using low concentrations of mutagen to reduce the mutagenic load, thereby reducing the generation of second site mutations that can modify the phenotype of interest and confound mapping efforts. Moreover, if a method can be developed to efficiently map hundreds of mutations in a relatively short time, a major hurdle would be overcome. Currently, mutations are mapped in Drosophila using duplications (Cook et al. 2010; Venken et al. 2010), deficiencies (Parks et al. 2004; Cook et al. 2012), recombination mapping based on visible markers, single-nucleotide variations (SNVs) (Berger et al. 2001; Hoskins et al. 2001), and/or P-elements (Zhai et al. 2003). The process typically takes several months, depending on the availability of genetic tools, and the methods are not easily scalable to large sets. Hence, a majority of mutations, generated in prior forward genetic EMS-mutagenesis screens, remain unassigned to a gene, even though cursory phenotypic studies have been carried out. Thus, high-throughput strategies, facilitating identification of the causative mutations, are highly desirable.
With the advent of whole-genome sequencing (WGS) (Sarin et al. 2008, 2010; Blumenstiel et al. 2009) and the reduction in cost of sequencing an entire genome (less than $500 per Drosophila genome at 30× coverage) (Hobert 2010), it is now possible to sequence an entire collection of mutant strains. In principle, comparing mutant and wild-type genome sequences should allow for the unambiguous identification of phenotype-causing mutations. However, natural sequence variation between chromosomes from different strains makes it challenging to determine causative mutations. For example, sequencing of 120 wild-type flies revealed one SNV per 25 nucleotides (Mackay et al. 2012). This corresponds to nearly 1 million polymorphisms for the X chromosome, which is 22.4 Mb and contains 2194 genes (http://www.ncbi.nlm.nih.gov/mapview/stats/BuildStats.cgi?taxid=7227&build=9&ver=4&chrm=X). Moreover, even flies that share similar genetic backgrounds exhibit numerous SNVs (Blumenstiel et al. 2009; Keightley et al. 2009). Hence, WGS does not provide a direct solution to the problem of mapping causative mutations.
Thus far, several proof-of-principle studies, each applying different approaches to successfully map a chemically induced mutation using WGS, have been documented in the literature (Sarin et al. 2008, 2010; Blumenstiel et al. 2009; Zhang et al. 2009; Earley and Jones 2011; Fairfield et al. 2011; Andrews et al. 2012; Bull et al. 2013). In general, a subset of SNVs is first removed based on assay-specific criteria upon which some form of mapping is performed to reduce the number of candidate mutations. For instance, Leshchiner et al. (2012) designed an algorithm that identifies SNVs in regions of homozygosity when one combines meiotic mapping with WGS (Leshchiner et al. 2012). Here, every mutant strain is allowed to recombine for several generations with a wild-type strain of different genetic background, upon which a number of individual progeny are pooled and sequenced. This method has allowed the successful mutation identification of a handful of mutants in flies, worms, zebrafish, and mice, but the number of complementation groups that were mapped per report is limited. It thus remains unclear how scalable this approach is or what its success rate is when one attempts to apply WGS to identify their mutant of interest (Doitsidou et al. 2010; Earley and Jones 2011; Andrews et al. 2012; Leshchiner et al. 2012; Bull et al. 2013; Henke et al. 2013). The drawback of combining meiotic mapping and WGS is that (1) recombination mapping requires several generations of back-crossing and is less straightforward when recessive lethal mutations are being mapped, and (2) in order to sequence multiple animals per genotype, animals are typically pooled and sequenced on one lane of the Illumina sequencer at a low coverage (4×–5×), which fails to identify many SNVs that are present in the genome. In addition, it was recently found that, apart from slightly reducing the mutational load, outcrossing mutant strains to wild-type strains also introduces a significant number of variants and may hence complicate mutation identification (Sarin et al. 2010).
Alternatively, independent variants that are found in the same gene can lead to gene identification when the mutants that are part of the same complementation group and exhibit similar phenotypes are sequenced (Sarin et al. 2008; Gonzalez et al. 2012). However, none of the strategies used thus far have been scaled effectively to map numerous causative mutations, and it remains to be determined what the optimal filters are, what fraction of mutations can be identified, and what fraction of multiple versus single alleles can be mapped effectively using the current technologies. Finally, one needs to demonstrate without a doubt that a mutation is causal among hundreds of mutations.
Here, we describe our large-scale effort to map 394 EMS-induced mutations. We performed WGS on mutant lines that were generated in a forward genetic screen for essential genes on the X chromosome (Yamamoto et al. 2014) and have developed a set of filters to reduce the number of SNVs to a manageable level. By combining WGS with a rough mapping strategy (to ∼1.4 Mb), we were able to map 274 (70%) of the mutations. The mutations were shown to be causative by rescuing the lethality with small, molecularly defined P[acman] duplications (Venken et al. 2009, 2010). In summary, we show that WGS can be successfully applied to map EMS-induced mutations on a large scale, permitting forward genetic screens to annotate the function of numerous genes at a much greater pace than currently available methodologies.
Results
30× sequence coverage identifies the majority of SNVs
We generated a collection of EMS-induced lethal mutations on the Drosophila X chromosome, using low EMS concentrations (7.5–10 mM), and screened for numerous phenotypes in mosaic animals to systematically assign phenotypes. Details of the phenotypic analysis of the screen are described elsewhere (Yamamoto et al. 2014). We performed WGS to map the causative mutations in mutants that displayed interesting phenotypes. About 30 virgin females were used for genomic DNA extraction to prevent contamination with sperm DNA, and flies were starved for 4–6 h to clear yeast DNA. We sequenced our isogenized y w FRT19A (FRT19Aiso) at 48× coverage with Illumina HiSeq 2000 (Bentley et al. 2008) to determine the optimal coverage to detect the majority of SNVs in a Drosophila strain when compared to the reference strain in FlyBase (y; cn bw sp) (Adams et al. 2000; St. Pierre et al. 2013). An SNV was considered heterozygous if it was read at least three times and could be detected in ≥10% of the reads (Sarin et al. 2010). If an SNV was detected in ≥90% of the reads, it was considered a homozygous SNV (Challis et al. 2012).
To identify the sequencing depth required to identify the majority of SNVs, we randomly down-sampled FRT19Aiso reads to simulate the number of homozygous SNVs that would be identified at specific sequencing depths (Fig. 1A). Beyond a sequencing depth of 30×, the number of identified SNVs did not effectively increase with increased coverage (Fig. 1A). As shown in Figure 1B, at an average of 30× coverage, ∼95% of the X chromosome is sequenced at least 10 times, which represents the depth required to reliably call heterozygous SNVs. This percentage does not include genomic regions containing highly repetitive DNA, as it is very difficult to properly align Illumina’s short ∼100-bp reads to the reference genome when these contain a highly repetitive sequence. For the Drosophila X chromosome, these repetitive sequences encompass 10.3% of the chromosome, and the majority falls into intergenic regions (Smit et al. 1996). About 3.3% of exons are embedded in this repetitive DNA. Hence, we expect to be able to call the SNVs with high confidence for ∼92% of the exons of the X chromosome.
Figure 1.

A sequencing depth of 30× permits identification of 95% of SNVs. (A) Graph displaying the number of identified SNVs at different sequencing depths for the isogenized FRT19A X chromosome (FRT19Aiso). A 30× coverage allows identification of ∼95% of the SNVs identified at 50× coverage. (B) Percentage of the X chromosome that is covered 1×–5× (black), 5×–10× (dark gray), 10×–20× (gray), or ≥20× (light gray) at various average sequencing depths. An average sequencing depth of 30× allows reliable heterozygous SNV-calling (requiring 10 or more reads) of 95% of the X chromosome. (C) Description of SNVs identified in the X chromosome of FRT19Aiso sequenced at 48× when compared to the reference sequence (y; cn bw sp) (Adams et al. 2000).
Isogenized chromosomes facilitate mutation identification
As shown in Figure 1, A and C, at a sequencing depth of 48× we identified 59,414 SNVs in FRT19Aiso, or 2.7 SNVs per kb, when compared to the reference X chromosome in FlyBase (Adams et al. 2000; St. Pierre et al. 2013). Among these SNVs, 1844 are nonsynonymous and 16 are nonsense mutations (Fig. 1C). However, these SNVs are most likely benign polymorphisms, as the newly isogenized FRT19Aiso strain was extensively phenotyped prior to mutagenesis and was selected for its robust health and fertility in homozygous flies (Yamamoto et al. 2014). Given the high genetic diversity between these two strains, extensive filtering is required to identify potential causative mutations in mutant lines.
Identifying candidate mutations requires multiple layers of filtering
For mutations that cause lethality, collecting sufficient DNA to perform WGS from hemizygous mutant animals before they die is tedious. Therefore, we initially crossed different mutant chromosomes [y w (*) FRT19A] to FRT19Aiso and performed WGS on heterozygous females carrying one mutant y w (*) FRT19A and one nonmutagenized y w FRT19Aiso X chromosome to identify the causative mutations. In a pilot experiment of four y w (*) FRT19A/y w FRT19iso, we observed an average of 50,167 SNVs on the X chromosome when compared to the reference genome (Fig. 2B). The majority of these SNVs correspond to variants detected in FRT19Aiso and are therefore not causative of the mutant phenotype. Removing the SNVs that overlap between the heterozygous mutant/FRT19Aiso and the homozygous FRT19Aiso chromosome (Fig. 2A, ΔIso) resulted in an average of 3596 candidate SNVs (Fig. 2B). Removing the SNVs that map to intergenic regions or that correspond to synonymous changes reduced the number of SNVs to 177 (functional in Fig. 2A). Finally, we filtered the remaining SNVs against benign variants that had been identified as homozygous SNVs in wild-type flies that were sequenced for the Drosophila Genetic Reference Panel (ΔDGRP) (Fig. 2A; Mackay et al. 2012). The latter filter reduced the SNVs from 177 to 87 SNVs per heterozygous mutant chromosome (Fig. 2B).
Figure 2.

Filtering process to identify candidate genes in heterozygous mutants. (A) Flowchart of filters applied to identify candidate mutations in heterozygous mutants [y w (*) FRT19A/y w FRT19Aiso]. All identified SNVs (brown) were first filtered against SNVs identified in the isogenized FRT19A X chromosome (ΔIso, orange). Subsequently, only SNVs that affect the coding sequence or splice sites were retained (functional, green). Next, the remaining SNVs were filtered against a database, containing polymorphisms found in a homozygous state in a collection of 205 viable, wild-type strains from the Drosophila Genetic Reference Panel (ΔDGRP, blue). (B, left) Impact of filters, introduced in A, on the total number of SNVs identified on the X chromosome. (Right) In a 1-Mb interval, the number of remaining candidate mutations is ∼4.
Determining which of the remaining 87 SNVs is causative can depend either on failure to complement preexisting mutations or on duplication mapping. On the X chromosome, deficiency mapping is not feasible for essential genes as males only carry a single X chromosome. Hence, we first performed duplication mapping to identify a duplication that can rescue lethality. We obtained a set of 21 partially overlapping duplications that together span 93.5% of the X chromosome (Supplemental Fig. 1A). The duplication set consists of 1- to 2-Mb duplications inserted on the Y chromosome or an autosome or that are free-floating chromosome fragments (Fig. 3A). Many of the selected duplications were only mapped cytologically (Lindsley and Zimm 1992). We therefore performed array comparative genomic hybridization (array CGH) to determine the molecular coverage of most of these lines and were able to identify the coverage of 16 large duplications (Supplemental Fig. 1B,C; Erickson and Spana 2006; Cook et al. 2010). All mutants, generated in the screen, were crossed to one-third of the duplication set, covering ∼50% of the X chromosome in the first round. In subsequent rounds, mutations that failed to be rescued by any duplication in the previous round were crossed to the next set. This allowed us to map the lethality of 72% of mutations to a genomic region of ∼1.4 Mb. Second site lethal mutations account for ∼9.5% of the failures (data not shown), and incomplete coverage of the duplication set accounts for another 7%. The remaining 11% may be due to an enrichment of lethal mutations in the areas that are not covered by duplications. In addition, large duplications create unhealthy male flies that do not always generate sufficient offspring to determine rescue (Supplemental Fig. 1A).
Figure 3.

Mapping and sequencing strategy. General strategy to map lethal mutations on the X chromosome. (A) Duplication (Dp) mapping: For every mutant, lethality was mapped to an ∼1.4-Mb region by Dp mapping. (B) Complementation (Compl) testing: Mutations that map to the same duplication were intercrossed to identify Compl groups. (C) Sequencing: Whole-genome sequencing (WGS) was performed on a total of 394 transheterozygous mutations (mut 1/mut 3) whose lethalities map to a different duplication. The 394 mutations correspond to 258 single alleles and 68 complementation groups with two alleles. (D) Validation: We used 80-kb P[acman] duplications to rescue the lethality and confirm the mapping.
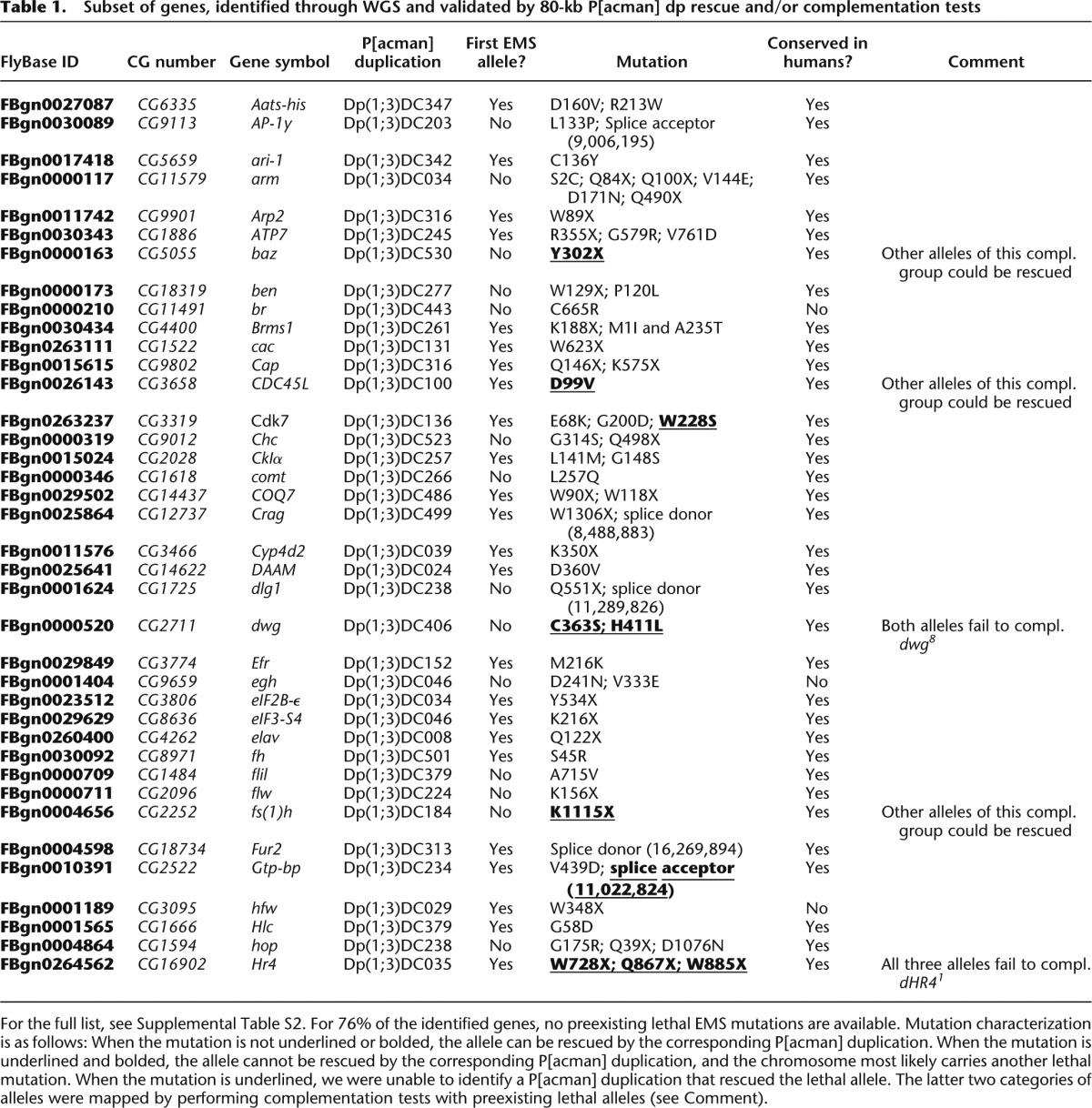
Since the lethality-causing mutation of interest should be located within the region that is covered by the rescuing duplication, we were able to reduce the number of SNVs to on average four to five per mutant (Fig. 2B). We next rescued the lethality with P[acman] duplications, prioritizing based on the severity of the mutation. The P[acman] X chromosome duplication kit consists of a library of partially overlapping 80-kb constructs that span the entire X chromosome (Venken et al. 2010). We were able to rescue lethality and hence confirm the molecular identity of the genetic aberrations underlying the four sequenced heterozygous mutant/iso lines (CG17766 [R1294X], CG6189 [Q860X], CG11579 [Q84X], and CG3794 [V124E]) (Table 1; Supplemental Table 2). No other mutations were identified in the coding regions contained in the ∼80 kb covered by the P[acman] clones. Hence, by combining rough mapping with WGS, we were able to identify the genetic lesion that underlies a mutant phenotype.
Table 1.
Subset of genes, identified through WGS and validated by 80-kb P[acman] dp rescue and/or complementation tests
Mapping two mutations at once
As with most EMS screens, we expected to isolate from one to numerous alleles per gene. To reduce the number of lines that had to be sequenced, we determined which alleles fail to complement each other when they were rescued by the same duplication and belonged to the same phenotypic class (Fig. 3B). Thus far, this allowed us to establish 109 complementation groups consisting of multiple alleles (5.1 alleles per gene) and 935 mutant strains that either contain single alleles or have not been assigned to a complementation group yet. To reduce the cost, we performed WGS on females carrying two complementing mutations mapping to different duplications, thereby halving the sequencing costs (Fig. 3C). We generated and sequenced 197 transheterozygous lines, including 258 single alleles and 136 alleles (68 × 2) of genes represented by multiple alleles.
Additional filters greatly facilitate mutation identification
Data analysis of the first 20 sequenced transheterozygous mutants revealed that the same SNVs could be found in multiple strains, suggesting that these alleles are present in a substantial fraction of the genomes. These alleles should have been eliminated in the isogenization process, but they have likely appeared in the generations after the isogenization of the chromosome. Regardless of their origin, these variations are unlikely to be causative of lethality. We therefore built a database to exclude these background-specific SNVs (ΔXscreen filter) (Fig. 4A). This database can be built and modified, depending on the number of lines that are sequenced. We tested the ΔXscreen database based on recurring SNVs found in either the first six, 12, 24, 48, or 96 transheterozygous mutant flies (Fig. 4B). Applying this filter to the sequence files for all 352 chromosomes that were sequenced led to a very significant decrease in SNVs when the data of the first 12 transheterozygous genomes were included (Fig. 4B). We therefore decided to build our ΔXscreen database based on the sequences acquired with the first 12 genomes.
Figure 4.

Filtering strategy to identify candidate genes in transheterozygous (mut 1/mut 2) mutants. (A) The same filters were applied as in Figure 2, and additional filters were added to remove SNVs identified repeatedly in multiple sequenced genomes (ΔXscreen [red]). A final filter was added to exclude genes that appear to be difficult to sequence (technical [purple]). (B) Building a background-specific filter (∆Xscreen). The largest drop in SNVs is seen when the ΔXscreen filter is built based on recurring SNVs found in 12 transheterozygous mutant genomes. (C) Building a technique-specific filter (technical). Approximately 95 genes appear difficult to sequence or analyze, since SNVs in these genes are called in nearly every sequenced genome. Hence, these genes were excluded from analysis (see Supplemental Table 1). (D) Distribution of the number of SNVs per chromosome that were identified in all analyzed sequence files. On average, 15 to 25 SNVs were identified for the two X chromosomes sequenced in the same reaction. (E) Distribution of the number of identified candidate mutations in an ∼1.4-Mb region to which lethality was mapped by duplication mapping. On average, one to two candidate mutations were found per duplication. (F) Mapping efficiency. For complementation groups consisting of multiple alleles, the causative mutation could be identified in 85% of the sequenced lines, as they could be rescued by an 80-kb P[acman] construct. For single alleles, the mutation could be validated in 62% of the sequenced lines. (G) Characteristics of the identified mutations.
Apart from these recurring SNVs, a last filter was implemented. We observed that some genes contain multiple SNVs in the first set of 20 sequenced genomes that could not be found in the reference genome or FRT19Aiso (Fig. 4C). For example, SNVs in CG32580 were found in 91% of the sequenced lines and carried, on average, 22 SNVs per X chromosome. Hence, SNVs that map to these genes are very unlikely to be causative of the phenotypes, and the corresponding genes were excluded from our analysis as they lead to an elevated number of false positives. In total, this filter excludes ∼5% of the X chromosome genes (technical in Fig. 4A,C; Supplemental Table 1).
Adding these two filters to the previous sets yields an average of 29 SNVs per mutant X chromosome (Fig. 4D). These SNVs are from now on referred to as candidate mutations. Hence, if a mutation can be mapped to an ∼1.4-Mb region (the average size of the duplications) as described above, only one to two candidate mutations should remain on average, which is indeed what we observed (Fig. 4E).
Sequencing of two independently generated alleles per complementation group
For two alleles of a complementation group, we compared only the SNVs that fall into the region to which lethality was mapped (∼1.4 Mb). For most complementation groups, this led to the identification of a single gene for which both sequenced alleles carried a different candidate mutation. Subsequently, these candidate mutations were shown to be causal for lethality, as mutant flies carrying a P[acman] duplication covering the gene are viable, and no other coding SNVs are present in the DNA covered by the P[acman] construct (Fig. 3D). We were able to identify and confirm a lethal mutation in 115/136 (85%) of the sequenced pairs (Fig. 4F). This percentage includes strains that carry second site hits (∼10%). A strain was labeled as carrying a second site hit if it carried a mutation in the same gene as the other sequenced allele of the complementation group and failed to complement this allele, yet could not be rescued by a P[acman] duplication (see the underlined mutations in Table 1 and Supplemental Table 2). As discussed below, this success rate can be further improved with better sequencing technology and data analysis.
Sequencing single alleles
We sequenced 258 mutations in genes that carry single alleles. Although the average number of SNVs per mutant is 29, duplication mapping permitted us to reduce the number of SNVs to an average of 2.44 candidate mutations per mutant, ranging from zero to nine (Fig. 4E). For ∼50% of the mutations, only one of the candidates was a nonsense mutation, and P[acman] duplications were used to rescue the lethality of these mutations first. We were able to rescue lethality and confirm the molecular identity for 159/258 of the single alleles, an overall efficiency of 62% (Fig. 4F; Table 1; Supplemental Table 2).
In total, we identified and validated 274 mutations in 148 genes. Of these, 20 were randomly selected and all were verified by Sanger sequencing (data not shown). Fifty-five percent of all validated mutations were transitions (A↔G and C↔T changes) (Fig. 4G). Eight percent of the identified mutations impair splicing, and 44% are nonsense mutations. Interestingly, 81 (55%) of the identified genes are uncharacterized, and for 111 (76%) of the genes, the described mutations represent the first lethal EMS alleles that are publicly available (Table 1; Supplemental Table 2). Hence, the mutations that were generated through the X chromosome screen and were mapped through WGS provide a substantial expansion of the toolkit necessary for assigning function to genes.
Discussion
Here, we report the first large-scale mapping of chemically induced lethal mutations in a higher eukaryote. We show that WGS can be applied on a large scale in Drosophila to identify ∼70% of the mutations, provided that SNVs are extensively filtered and rough mapping is performed. Upon filtering out those SNVs that are in noncoding regions and those that lead to synonymous changes, the number of SNVs is typically reduced by about 20-fold. By removing SNVs that were found as homozygous SNVs in wild-type flies, we further reduced the SNVs by about twofold. Removing SNVs that occur frequently reduces the SNVs by about fivefold. Hence, by combining WGS with a rough mapping approach, we typically identified one to three candidates. Combining these strategies with rescue experiments using the P[acman] BAC transgenic collection allowed us to identify 274 out of 394 EMS-induced mutations. We also provide compelling evidence that the mutations are causative, as we rescue the lethality associated with the mutations with relatively small, molecularly defined duplications. Currently, a collection of strains carrying P[acman] duplications that together span the entire chromosome is only available for the X chromosome (Venken et al. 2010). However, the generation of strains for the second and third chromosome is in progress (R Chen and G Mardon, pers. comm.). It is important to emphasize that even low doses of EMS induce, on average, 3500 SNVs per mutant chromosome (about one SNV per 35 kb) (Fig. 2B) emphasizing the need of rescuing the phenotypes of EMS-induced mutations.
Enhancing mapping efficiency
As described above, when multiple alleles are sequenced from a complementation group, we were able to successfully map 85% of the sequenced lines. Why only 85% of the mutations? First, the Illumina sequencing technology has several shortcomings. Due to the relatively short sequence reads (∼100 bp), highly repetitive genomic regions are difficult to align to the genome. Such misaligned regions would lead to the calling of numerous false-positive SNVs and are therefore generally excluded from analysis. As determined by RepeatMasker, a program designed to detect and filter out highly repetitive regions, 3.3% of the coding region of the Drosophila X chromosome is excluded from analysis (Smit et al. 1996). Second, we found that a subset of the X chromosome genes (5%) contains multiple SNVs in many WGS reactions. The 100 excluded genes (Fig. 4C; Supplemental Table 1) are not significantly bigger than the average gene on the X chromosome (data not shown), yet SNVs are observed too frequently to be causative. These identified variants are therefore unlikely to represent true SNVs. Rather, we surmise that these genes represent a challenge for the Illumina sequencing technology. Indeed, similar findings were recently documented when human exome sequences were analyzed, and the investigators generated similar lists of genes and chromosome regions that should be excluded from analysis (Fuentes Fajardo et al. 2012). These SNVs could be the result of misalignments in low complexity regions or of duplicated genes, paralogs, or pseudogenes. However, the underlying reason remains unclear.
Third, at 30× sequencing depth, we are unable to call SNVs for 4.7% of the X chromosome (Fig. 1B). Fourth, all functional predictions are based on genes annotated in FlyBase version 5.12 of the Drosophila genome, and newly annotated or unannotated genes were not screened (St. Pierre et al. 2013). Fifth, EMS induces small insertions and deletions (indels) in ∼2.4% of the mutant chromosomes (Cooper et al. 2008). Indeed, when indels were analyzed in 20/120 chromosomes for which we failed to identify a causative mutation, two causative indels were identified (CG8949 [16965894DelC] and CG7280 [18741343InTG]). We therefore estimate that 1%–3% of the total number of mutations are indels. Finally, we did not analyze mutations that affect transcriptional regulation. Based on the above data, the fraction of regulatory mutations among the lethal, EMS-induced mutation fractions appears to be very small. For the sequenced alleles that are part of a complementation group, we reached an efficiency of 85%. If we add the genes that were not covered at 30× coverage (4.7%), the excluded genes (3.3% + 5.0%), and the causative indels (1%–3%), the fraction of lethality-causing regulatory mutations that is induced by EMS is very low.
Two alleles per complementation group facilitate mutation identification
We obtained a different mapping efficiency for complementation groups consisting of single (62%) or multiple (85%) alleles. The reasons for this discrepancy are twofold. First, comparing two sequence files and probing for variants that affect the same gene is straightforward and has a higher chance of success. Second, when multiple alleles are available, it is possible to determine whether a second site lethal mutation is present on the chromosome. Indeed, we were unable to rescue ∼10% of the mutations that are part of a complementation group using a P[acman] construct, although the lethality associated with another allele of the same complementation group can be rescued (Table 1; Supplemental Table 2). We therefore surmise that ∼10% of the single alleles contain a second site lethal mutation. As the EMS dosage determines the number of induced mutations, it is important to treat the animals with the lowest possible dose, as this will facilitate mapping (i.e., reduce the number of candidate SNVs) in addition to reducing the mutagenic load. Obviously, this requires that the screening assay is simple as the number of animals that need to be screened is inversely proportional to the dose of EMS.
Mutation identification with WGS: What is feasible?
Numerous mutagenesis screens have been performed since the introduction of Drosophila as a model organism (St. Johnston 2002). These screens have been very successful at describing gene function, yet the majority of mutations are unassigned to a gene. In the absence of any mapping or preexisting complementation test, it will be highly unlikely that one can identify the causative mutation. However, when two alleles of a single complementation group are available and our set of filters is applied, the number of genes containing an SNV on both X chromosomes is 1.7 ± 0.8 SNVs (Supplemental Fig. 2A), permitting rapid gene identification without the need of rough mapping. Unfortunately, for screens that were performed in the past, the isogenized strain may no longer be available, and several filters that we used will not be available when only two or fewer alleles are sequenced. In addition, one may be interested in mapping only a single complementation group rather than the hundreds that were sequenced for this project. Is it possible to use WGS to identify the underlying genetic aberrations in these mutant strains? To address this question, we simulated the mapping for 40 different complementation groups using filters that do not require any preexisting knowledge (Supplemental Fig. 2B). We first eliminated all the SNVs shared by the two chromosomes, the background-specific SNVs (background) (Supplemental Fig. 2B). Second, we applied the functional filter to retain only the SNVs that alter the protein coding sequence or that affect splicing. Third, we filtered out genes that contained three or more mutations in the same gene, which corresponded mostly to genes excluded in the technical filter described above. For the last filter, we calculated the genome mappability score (GMS), which represents the probability that a read can be aligned properly at a given position (Lee and Schatz 2012), as genes that have a low mappability score are more likely to be misaligned. For genes on the X chromosome, the average GMS is 96 ± 13 (a score of 100 represents a perfect chance of aligning to the correct position in the genome), and we excluded genes with a mappability score below 85, the technical filter. Applying this set of filters yielded an average of 3.5 ± 1.5 SNVs per X chromosome that affect the same gene in both alleles of a complementation group (Supplemental Fig. 2), including the gene we identified previously. It should therefore be feasible to map mutations from preexisting mutant collections using WGS as long as two alleles or more of a complementation group are available.
Extrapolation of our data suggests that if a behavioral screen is performed, WGS should be able to identify the genes of interest if the following conditions are met: First, the screen is performed on an isogenized chromosome; second, the screened phenotype needs to be robust such that it can be unambiguously mapped to large deficiencies or duplications by complementation tests; and third, two or more alleles have been identified for a given complementation group. If these conditions are met, one can generate and apply filters (Supplemental Fig. 2B, right) that will lead to the identification of an average of 3.5 candidate mutations. The mutants can then be crossed to flies containing an 80-kb P[acman] construct that spans the variant of interest. To allow this on the second and third chromosome, a collection of strains carrying 80-kb P[acman] constructs is generated (R Chen and G Mardon, pers. comm.). As some behavioral phenotypes are extremely sensitive to alterations in the genetic background, mapping the lesion with deficiencies may be difficult or impossible. We estimate that for this type of mutants, the number of remaining candidate mutations upon filtering will be about 20-fold higher than estimated in Supplemental Figure 2B. In summary, mapping will only be possible if the phenotypes are not easily subject to genetic variation and if multiple alleles are available.
To facilitate data analysis of WGS, we have established a web-based interactive tool where sequence files can be uploaded and filtered according to our protocol (http://www.iipl.fudan.edu.cn/FlyVar). Here, individual filters can be selected, allowing one to, for instance, solely filter out the SNVs that were found as a homozygous variant in the DGRP collection. In addition, we assembled a file (Bellen-FRT19Aiso-variants.vcf) containing all SNVs that were identified in our isogenized FRT19A strain upon filtering with the DGRP filter (Supplemental File 1). Finally, we generated a file (Bellen-EMS-mutations.vcf) that contains all variants, identified in the sequenced mutants, remaining upon filtering against the DGRP variants (Supplemental File 2). Both files can be downloaded from http://www.iipl.fudan.edu.cn/FlyVar/sourcefordownloading.jsp.
In conclusion, given the high number of SNVs between two Drosophila chromosomes, it is imperative to initiate a screen with an isogenized chromosome, since more than 50,000 SNVs can be filtered out (Fig. 2B). Next, a low dose of mutagen will facilitate the mapping process. It reduces the generation of second site hits that increase the total number of SNVs that potentially modify the phenotype of interest and confound mapping efforts. Hence, an isogenized strain that is treated with a low dose of EMS will greatly facilitate the mapping process. Upon screening for a phenotype of interest, the remaining mutants should be mapped to ∼1 Mb. For the X chromosome, this is most easily achieved with duplication mapping as sets of duplications are available (Cook et al. 2010; Venken et al. 2010). For autosomes, we recommend P-element mapping, as it is arguably the fastest, cheapest, and least labor-intensive technique to map to an ∼1-Mb interval in our laboratory (Zhai et al. 2003). Alternatively, SNP mapping (Berger et al. 2001; Hoskins et al. 2001) or deficiency mapping (Parks et al. 2004; Cook et al. 2012) can be used. Finally, the information obtained from WGS with the appropriate filters described here can be combined with the rough mapping data (Supplemental Fig. 3), resulting in the identification of one to five candidate mutations. This number is then reduced to a single gene by complementation tests with small deficiencies on the autosomes or P[acman] duplications on the X chromosome. This should allow for the relatively fast identification of the majority of mutations generated by forward mutagenesis screens and should significantly alleviate the biggest burden of this type of screen.
Methods
EMS mutagenesis
In short, mutagenesis was performed on 6-d-old, isogenized y w FRT19A iso males that were starved for 6–12 h by feeding them a sucrose solution containing a low concentration (7.5–10 mM) of EMS for 15 h. After recovery from mutagenesis, these males were mated en masse with Df(1)JA27/FM7c Kr > GFP virgin females for 3 d. In the F1 generation, y w mut* FRT19A/FM7c Kr>GFP (mut* indicates the EMS-induced mutation) virgins were collected and 33,887 females were crossed with FM7c Kr>GFP males to establish balanced stocks; 5859 lines carried lethal mutations, and the remaining stocks were discarded.
Array CGH
Array CGH to determine the molecular coverage of large cytologically mapped duplications was performed as previously described (Erickson and Spana 2006). In brief, male flies that carry X duplications were crossed with virgin females carrying a wild-type X chromosome. The male progeny carrying the X chromosome duplication was selected based on the markers present on the duplication, and genomic DNA was extracted using the PureLink Genomic DNA mini kit (Invitrogen). Labeling, hybridization, and detection were performed at the Duke University Microarray Facility using operon array-ready 70mer oligo arrays. Array CGH data for Dp(5678) were kindly provided by Drs. Eric Spana (Duke University) and Kevin Cook (Indiana University). Data for Dp5459 were kindly provided by Dr. Ela Serpe (NICHD). We did not perform array CGH for Dp(761), Dp5594, Dp948, Dp929, and Dp5273 due to technical reasons.
Duplication rescue and rough mapping using large duplications
Virgin females from mutant lines were crossed to males carrying different X chromosome duplications. Progenies were scored to determine whether the duplication rescued the lethality of the mutation. The duplication mapping was performed in three rounds.
Round 1: Df(1)svr, Nspl-1 ras2 fw1/Dp(1;Y)y267g19.1/C(1)DX, y1 f1 (Dp901), Df(1)64c18, g1 sd1/Dp(1;2;Y)w+/C(1)DX, y1 w1 f1 (Dp936), Df(1)JC70/Dp(1;Y)dx + 5, y+/C(1)M5 (Dp5279), Dp(1;Y)619, y+ BS/w1 oc9/C(1)DX, y1 f1 (Dp5678), y1 nejQ7 v1 f1/Dp(1;Y)FF1, y+/C(1)DX, y1 w1 f1 (Dp5292), Df(1)v-N48, f*/Dp(1;Y)y + v + #3/C(1)DX, y1 f1 (Dp3560), Dp(1;Y)BSC1, y+/w67c23 P{lacW}SmrG0060/C(1)RA, y1 (Dp5596), Dp(1;Y)W73, y31d B1, f+, BS/C(1)DX, y1 f1/y1 bazEH171 (Dp1537), Dp1538, Df(1)R20, y1/C(1)DX, y1 w1 f1/Dp(1;Y)y + mal + (Dp3033)
Round 2: Dp(1;f)R, y+/y1 dor8 (Dp761), Df(1)dhd81, w1118/C(1)DX, y1 f1; Dp(1;2)4FRDup/+ (Dp5594), Df(1)ct-J4, In(1)dl-49, f1/C(1)DX, y1 w1 f1; Dp(1;3)sn13a1/+ (Dp948), winscy/Dp(1;Y)8-28-8A/C(1)DX, y1 w1 f1 (Dp8-28-8A) (gift from Dr. Kevin Cook, Indiana University), Df(1)v-L15, y1/C(1)DX, y1 w1 f1; Dp(1;2)v+75d/+ (Dp929), C(1;Y)6, y1 w* P{white-un4}BE1305 mew023/C(1)RM, y1 pn1 v1; Dp(1;f)y+ (Dp5459), w* l(1)dd4xr16/ FM7a/Dp(1;Y)y + g + (Dp26276), Df(1)19, f1/C(1)DX, y1 w1 f1; Dp(1;4)r + l (Dp5273)
Round 3: Dp(1;Y)BSC231, y+ P{3′.RS5 + 3.3′}BSC27, BS/Df(1)ED7265, w1118 P{3′.RS5 + 3.3′}ED7265/C(1)RA, In(1)scJ1, In(1)sc8, l(1)1Ac1, scJ1 sc8 (Dp33250), Dp(1;Y)BSC223, y+ P{3′.RS5 + 3.3′}BSC16, BS/Df(1)ED7344, w1118 P{3′.RS5 + 3.3′}ED7344/ C(1)RA, In(1)scJ1, In(1)sc8, l(1)1Ac1, scJ1 sc8 (Dp33244), Dp(1;Y)BSC129, y+ P{3′.RS5 + 3.3′}BSC22, BS/Df(1)ED7441, w1118 P{3′.RS5 + 3.3′}ED7441/C(1)RA, In(1)scJ1, In(1)sc8, l(1)1Ac1, scJ1 sc8 (Dp30450)
Rescued males were crossed to a stock that carries a compound X chromosome (C(1)DX) or to the original mutant stock to establish stocks that stably produce rescued male flies. For Dp5459, this was not possible due to technical reasons.
DNA preparation for Illumina sequencing
Twenty to 50 flies were collected, starved for 4–6 h, and frozen at −80°C. Subsequently, flies were homogenized in Buffer G2 (20 mM EDTA, 100 mM NaCl, 1% Triton X-100, 500 mM guanidine-HCl, 10 mM Tris at pH 7.9). DNase-free RNase A was added (20 μg/mL), and lysates were incubated for 30 min at 37°C. Samples were subsequently subjected to proteinase K treatment (0.8 mg/mL) for 2 h at 50°C. Lysates were spun at 14,000 rpm for 20 min before loading to pre-equilibrated Qiagen G-20 columns. Next, a standard DNA purification protocol was followed to obtain DNA.
DNA libraries for sequencing were generated according to Illumina’s sample preparation protocol for genomic DNA. Briefly, 1 μg of genomic DNA was sheared into 300- to 500-bp fragments. DNA fragments were end-repaired using polynucleotide kinase and Klenow. The 5′ ends of the DNA fragments were phosphorylated and a single adenine base was added to the 3′ ends using Klenow exonuclease. Illumina Y-shaped index adaptors were ligated to the repaired ends, then the DNA fragments were PCR-amplified for eight cycles and fragments of 200–500 bp were isolated by bead purification. The libraries were quantified using the PicoGreen fluorescence assay and their size distributions determined by the Agilent 2100 Bioanalyzer. Libraries were sequenced on the Illumina HiSeq 2000 as 100-bp paired-end reads (or 50-bp single-end reads for a small number of samples), following the manufacturer’s protocols.
Illumina data analysis and variant detection
Repetitive sequences, present in the D. melanogaster reference genome (dm3), were first masked using the RepeatMasker software (Smit et al. 1996). Sequence reads were then aligned to this masked reference genome (dm3) using Burrows-Wheeler Aligner software (BWA version 0.5.4) (Li and Durbin 2009) and calibrated with the Genome Analysis Toolkit (GATK version 1.0.3299) (McKenna et al. 2010). Variants (SNVs and small indels) were called using the Atlas2 variant analysis software (Challis et al. 2012). At least three reads were required to support variant calling (Sarin et al. 2010). In addition to variant calls, the application collects coverage information to estimate the likely genotype of each variant site. For heterozygous sites, the cutoff was set at 0.1 of the allele fraction, whereas it was set at 0.9 for homozygous sites.
Filtering
All called variants that map to the X chromosome were subjected to several rounds of filtering to remove noncausal polymorphisms. First, homozygous and heterozygous SNVs that had also been detected in FRT19Aiso were removed. Next, SNVs that map to coding regions were extracted with an in-house perl script that also allows identification of variants that affect either splicing or the amino acid sequence (FlyBase Release 5.12 genome annotation downloaded from UCSC http://genome.ucsc.edu/cgi-bin/hgTrackUi?g=flyBaseGene&db=dm3) (St. Pierre et al. 2013). Only these SNVs were retained. The remaining SNVs were compared to a database that we built based on data from the D. melanogaster Genetic Reference Panel (Mackay et al. 2012). SNVs that were detected at least once in a homozygous state in this data set were considered as not essential for viability and were included in our database. The next filter is based on a database consisting of variants that recur in the sequenced mutant strains. Variants that appear at least once in a homozygous state or at least four times in a heterozygous state were included in this database. SNVs of all sequenced transheterozygous mutants were filtered against this database, which was based on the first 12 sequenced genomes. The final filter excludes SNVs that map to genes that appear to be difficult to sequence with the current Illumina sequencing technology. The average number of SNVs per gene was calculated based on all remaining variants detected in the 307 sequenced genomes. The average number of SNVs per gene, per genome was calculated. Based on the 95% confidence interval, the top 5% outliers were incorporated in the final filter.
Sanger sequencing validation
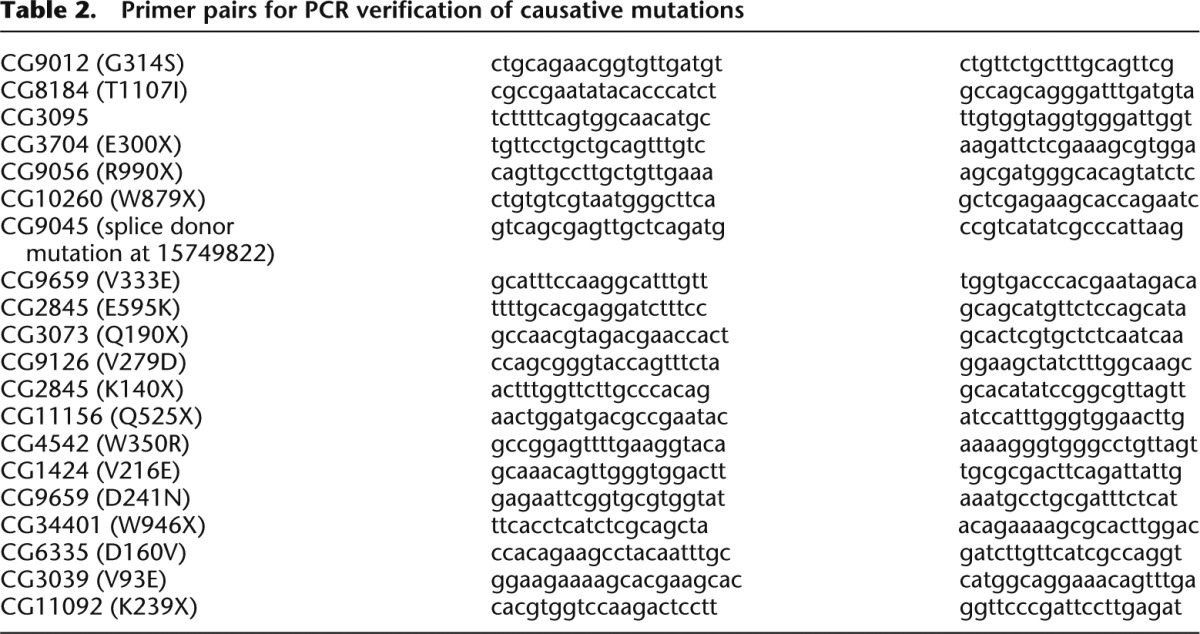
For PCR verification of causative mutations, DNA was isolated from 10 to 15 third instar larvae using the PureLink Genomic DNA mini kit (Invitrogen). PCR reaction conditions were as follows: 1 μL DNA, 1 μL primer F (10 μM), 1 μL primer R (10 μM), 2 μL 10× buffer, 0.16 μL dNTPs (25 mM each), 0.08 μL Qiagen HotStarTaq DNA polymerase (Qiagen), and 14.76 μL milliQ water. PCR cycling conditions in PTC-225 or DNA Engine (MJ Research) were as follows: denaturation for 10 min at 94°C; 35 cycles for 30 sec at 94°C, for 30 sec at 60°C, and for 60 at 72°C; and post-amplification extension for 10 min at 72°C. PCR was performed with mutation-specific primers (see Table 2).
Table 2.
Primer pairs for PCR verification of causative mutations
P[acman] duplication mapping
Balanced mutant females [y w (*) FRT19A/FM7c Kr>GFP] were crossed to a transgenic male, containing an 80-kb P[acman] duplication that covers a single candidate mutation. Progenies were scored to determine whether the duplication rescued the lethality of the mutation. A duplication was considered to cover the causative mutation if viable, unbalanced, hemizygous, mutant males could be detected in the progeny of this cross.
Data access
The sequencing data for this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number PRJNA239441. We have also generated a database that allows web-based query and data filtering of sequence files (http://www.iipl.fudan.edu.cn/FlyVar). Files containing identified variants can be found in the Supplemental Material (Supplemental Files 1, 2) and at http://www.iipl.fudan.edu.cn/FlyVar/sourcefordownloading.jsp. The mutant strains have been deposited at the Bloomington Drosophila Stock Center (http://flystocks.bio.indiana.edu/).
Competing interest statement
Rui Chen and Graeme Mardon own Genetivision, a company that is generating a collection of fly strains containing 80 kb P[acman] duplications to cover the autosomes of Drosophila.
Supplementary Material
Acknowledgments
We thank N. Giagtzoglou, H.A. Dierick, K. Venkatachalam, H. Jafar-Nejad, and T. Li for critical reading of the manuscript. We thank the members of the Bloomington Drosophila Stock Center for maintaining and providing flies. We thank K. Cook, E. Spana, and E. Serpe for contributing to defining the molecular boundaries of duplications. We thank Y.W. Wan and Z. Liu for help with bioinformatical analysis of candidate mutations. We thank Y. Chen, C. Benitez, X. Shi, S. Gibbs, H. Wang, and L. Wang for help with the Drosophila screen, and past and present members of the Bellen laboratory for flies, help, and/or stimulating discussions. We thank S. Richards for discussions concerning the DGRP project. We thank H. Wang, N.N. Xu, and S. Xu for managing the Illumina sequencing experiments. H.J.B. is an investigator with the Howard Hughes Medical Institute. This work was supported by the National Institutes of Health (NIH) through an RC4-grant (1RC4GM096355-01) to H.J.B. and R.C. V.B. was supported by the NIH (5T32-HD055200) and the Edward and Josephine Hudson Scholarship Fund. H.S. was supported by NIH 5R01GM067858 and the Research Education and Career Horizon Institutional Research and Academic Career Development Award Fellowship 5K12GM084897. B.X. was supported by the Houston Laboratory and Population Science Training Program in Gene-Environment Interaction from the Burroughs Wellcome Fund (grant no. 1008200). W.-L.C. was supported by the Taiwan Merit Scholarships Program sponsored by the National Science Council (NSC-095-SAF-I-564-015-TMS). K.J.T.V. is supported by the NIH (1R21HG006726) and the McNair Medical Institute. S.Y. was supported by a fellowship from the Nakajima Foundation and is currently supported by the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital.
Author contributions: Drosophila screen mutagenesis, phenotypic screening, and large duplication mapping were performed by N.A.H., V.B., H.S., B.U., K.L.T., K.Z., B.X., W.-L.C., T.B., A.J., G.D., M.J., and S.Y. Whole-genome sequencing, variant calling, bioinformatics, and support: L.J., Y.L., and R.C. Candidate mutations were identified by N.A.H., B.U., and K.L.T. P[acman] duplication rescue experiments were performed by N.A.H., D.B., and K.J.T.V. N.A.H., L.J., V.B., R.C., and H.J.B. designed the study and wrote the manuscript. All authors edited the manuscript and agreed with the submission.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.174615.114.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195 [DOI] [PubMed] [Google Scholar]
- Andrews TD, Whittle B, Field MA, Balakishnan B, Zhang Y, Shao Y, Cho V, Kirk M, Singh M, Xia Y, et al. 2012. Massively parallel sequencing of the mouse exome to accurately identify rare, induced mutations: an immediate source for thousands of new mouse models. Open Bio 2: 120061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Suzuki T, Senti KA, Stubbs J, Schaffner G, Dickson BJ. 2001. Genetic mapping with SNP markers in Drosophila. Nat Genet 29: 475–481 [DOI] [PubMed] [Google Scholar]
- Blumenstiel JP, Noll AC, Griffiths JA, Perera AG, Walton KN, Gilliland WD, Hawley RS, Staehling-Hampton K. 2009. Identification of EMS-induced mutations in Drosophila melanogaster by whole-genome sequencing. Genetics 182: 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley A, Anastassiadis K, Ayadi A, Battey JF, Bell C, Birling M-C, Bottomley J, Brown SD, Bürger A, Bult CJ, et al. 2012. The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome 23: 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull KR, Rimmer AJ, Siggs OM, Miosge LA, Roots CM, Enders A, Bertram EM, Crockford TL, Whittle B, Potter PK, et al. 2013. Unlocking the bottleneck in forward genetics using whole-genome sequencing and identity by descent to isolate causative mutations. PLoS Genet 9: e1003219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, Milosavljevic A, Gibbs RA, Yu F. 2012. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics 13: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AT, Goldowitz D, Takahashi JS, Vitaterna MH, Siepka SM, Peters LL, Frankel WN, Carlson GA, Rossant J, Nadeau JH, et al. 2004. Implementing large-scale ENU mutagenesis screens in North America. Genetica 122: 51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook RK, Deal ME, Deal JA, Garton RD, Brown CA, Ward ME, Andrade RS, Spana EP, Kaufman TC, Cook KR. 2010. A new resource for characterizing X-linked genes in Drosophila melanogaster: systematic coverage and subdivision of the X chromosome with nested, Y-linked duplications. Genetics 186: 1095–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook RK, Christensen SJ, Deal JA, Coburn RA, Deal ME, Gresens JM, Kaufman TC, Cook KR. 2012. The generation of chromosomal deletions to provide extensive coverage and subdivision of the Drosophila melanogaster genome. Genome Biol 13: R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JL, Greene EA, Till BJ, Codomo CA, Wakimoto BT, Henikoff S. 2008. Retention of induced mutations in a Drosophila reverse-genetic resource. Genetics 180: 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsidou M, Poole RJ, Sarin S, Bigelow H, Hobert O. 2010. C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS ONE 5: e15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley EJ, Jones CD. 2011. Next-generation mapping of complex traits with phenotype-based selection and introgression. Genetics 189: 1203–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JN, Spana EP. 2006. Mapping Drosophila genomic aberration breakpoints with comparative genome hybridization on microarrays. Methods Enzymol 410: 377–386 [DOI] [PubMed] [Google Scholar]
- Fairfield H, Gilbert GJ, Barter M, Corrigan RR, Curtain M, Ding Y, D’Ascenzo M, Gerhardt DJ, He C, Huang W, et al. 2011. Mutation discovery in mice by whole exome sequencing. Genome Biol 12: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes Fajardo KV, Adams D, NISC Comparative Sequencing Program, Mason CE, Sincan M, Tifft C, Toro C, Boerkoel CF, Gahl W, Markello T. 2012. Detecting false-positive signals in exome sequencing. Hum Mutat 33: 609–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Van Booven D, Hulme W, Ulloa R, Lebrigio R, Osterloh J, Logan M, Freeman M, Zuchner S. 2012. Whole genome sequencing and a new bioinformatics platform allow for rapid gene identification in D. melanogaster EMS screens. Biology 1: 766–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke K, Bowen ME, Harris MP. 2013. Perspectives for identification of mutations in the zebrafish: making use of next-generation sequencing technologies for forward genetic approaches. Methods 62: 185–196 [DOI] [PubMed] [Google Scholar]
- Hobert O. 2010. The impact of whole genome sequencing on model system genetics: get ready for the ride. Genetics 184: 317–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Phan AC, Naeemuddin M, Mapa FA, Ruddy DA, Ryan JJ, Young LM, Wells T, Kopczynski C, Ellis MC. 2001. Single nucleotide polymorphism markers for genetic mapping in Drosophila melanogaster. Genome Res 11: 1100–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. 1999. Mouse ENU mutagenesis. Hum Mol Genet 8: 1955–1963 [DOI] [PubMed] [Google Scholar]
- Keightley PD, Trivedi U, Thomson M, Oliver F, Kumar S, Blaxter ML. 2009. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res 19: 1195–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Schatz MC. 2012. Genomic dark matter: the reliability of short read mapping illustrated by the genome mappability score. Bioinformatics 28: 2097–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchiner I, Alexa K, Kelsey P, Adzhubei I, Austin-Tse CA, Cooney JD, Anderson H, King MJ, Stottmann RW, Garnaas MK, et al. 2012. Mutation mapping and identification by whole-genome sequencing. Genome Res 22: 1541–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Zimm GG. 1992. The genome of Drosophila melanogaster. Academic Press, San Diego, CA [Google Scholar]
- Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. 2012. The Drosophila melanogaster Genetic Reference Panel. Nature 482: 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S, Bakal C, Perrimon N. 2010. Genomic screening with RNAi: results and challenges. Annu Rev Biochem 79: 37–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, Tan LR, Winter CG, Bogart KP, Deal JE, et al. 2004. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat Genet 36: 288–292 [DOI] [PubMed] [Google Scholar]
- Sarin S, Prabhu S, O’Meara MM, Pe’er I, Hobert O. 2008. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods 5: 865–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, Bertrand V, Bigelow H, Boyanov A, Doitsidou M, Poole RJ, Narula S, Hobert O. 2010. Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing. Genetics 185: 417–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. 1996. RepeatMasker Open 3.0. http://www.repeatmasker.org
- St. Johnston D. 2002. The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet 3: 176–188 [DOI] [PubMed] [Google Scholar]
- St. Pierre SE, Ponting L, Stefancsik R, McQuilton P, FlyBase Consortium . 2013. FlyBase 102: advanced approaches to interrogating FlyBase. Nucleic Acids Res 42: D780–D788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, de Jong PJ, White KP, et al. 2009. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nature 6: 431–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Popodi E, Holtzman SL, Schulze KL, Park S, Carlson JW, Hoskins RA, Bellen HJ, Kaufman TC. 2010. A molecularly defined duplication set for the X chromosome of Drosophila melanogaster. Genetics 186: 1111–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Simpson JH, Bellen HJ. 2011. Genetic manipulation of genes and cells in the nervous system of the fruit fly. Neuron 72: 202–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Gerdin A-K, Karp NA, Ryder E, Buljan M, Bussell JN, Salisbury J, Clare S, Ingham NJ, Podrini C, et al. 2013. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154: 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Jaiswal M, Charng W, Gambin T, Karaca E, Mirzaa G, Wiszniewski W, Sandoval H, Haelterman NA, Xiong B, et al. 2014. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Hiesinger PR, Koh T-W, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, et al. 2003. Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci 100: 10860–10865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Alpert D, Francis R, Chatterjee B, Yu Q, Tansey T, Sabol SL, Cui C, Bai Y, Koriabine M, et al. 2009. Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy. Proc Natl Acad Sci 106: 3219–3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.