

Abstract
The application of designer nucleases allows the induction of DNA double-strand breaks (DSBs) at user-defined genomic loci. Due to imperfect DNA repair mechanisms, DSBs can lead to alterations in the genomic architecture, such as the disruption of the reading frame of a critical exon. This can be exploited to generate somatic knockout cell lines. While high genome editing activities can be achieved in various cellular systems, obtaining cell clones that contain all-allelic frameshift mutations at the target locus of interest remains a laborious task. To this end, we have developed an easy-to-follow deep sequencing workflow and the evaluation tool OutKnocker (www.OutKnocker.org), which allows convenient, reliable, and cost-effective identification of knockout cell lines.
Advances in targeted genome editing technologies have opened new avenues for addressing challenging questions in the field of life sciences. The recent introduction of designer nucleases such as ZFNs (Carroll 2011), TALENs (Miller et al. 2011), or CRISPR/Cas systems (Jinek et al. 2012; Cong et al. 2013; Mali et al. 2013) allows for highly efficient, flexible, and specific induction of DNA double-strand breaks (DSB) in eukaryotic genomes. DSBs trigger two distinct repair pathways that can be exploited to specifically modify gene architecture (Carroll 2011). While the process of homologous recombination (HR) accurately repairs DSBs using the sister chromatid as a template, nonhomologous end-joining (NHEJ) repair is an error-prone end-joining mismatch repair pathway that frequently leads to genetic alterations (Lieber 2010; Chiruvella et al. 2013). Providing a donor construct with appropriate homology arms as a template, the pathway of DSB-triggered HR can be used to site-specifically introduce heterologous genetic material into cells (Carroll 2011). For example, it is possible to generate gene knockouts in somatic cell lines by introducing marker cassettes with premature stop codons. However, this strategy is time consuming and laborious and therefore not optimal for high-throughput approaches. The DSB-induced NHEJ repair pathway, on the other hand, leads to insertions or deletions (indels) (Lieber 2010) that can result in frameshift mutations and thus loss-of-function phenotypes if located within early coding exons.
While in HR-based genome editing approaches marker genes can be introduced to select for the desired genotype starting from a polyclonal cell culture, frameshift mutations induced by NHEJ are difficult to select for unless the editing event provides a survival benefit. To this end, single-cell cloning and subsequent sequencing of the genetic locus is required to obtain cells with the desired gene disruption. Sanger sequencing is most commonly used to identify modified alleles. However, in addition to being costly, this method requires a locus-specific PCR to be subcloned in order to sequence single alleles, and thus is not practical for large-scale projects. Moreover, the ploidy of the genome may vary between cell lines and even between loci, which may require the sequencing of a considerable number of PCR subclones to reliably identify cell clones with all-allelic frameshift mutations. Small benchtop deep sequencing machines can achieve a far greater throughput. Theoretically, even low sequencing capacities are sufficient to analyze hundreds of clones in parallel, without the need to subclone PCR products. However, analysis of deep sequencing data remains challenging and no streamlined workflow has been described that would allow full exploitation of deep sequencing capacities in gene disruption projects.
Here we describe OutKnocker, a web-based application that facilitates the analysis of deep sequencing data to identify knockout cells obtained from designer nuclease-mediated genome editing. We aimed at developing an evaluation tool to genotype single-cell clones at a confined genomic region for indel mutations, as they are typically induced by designer nuclease targeting. As such, we established an algorithm that focuses on identifying a single indel event per sequencing read around a predefined target site, while ignoring SNPs or point mutations originated during sequencing. Optionally, our software also allows the detection of specific point mutations introduced by targeted mutagenesis. To fully exploit sequencing capacities, OutKnocker was designed to analyze data of sequencing runs that have been multiplexed to evaluate the same or different genomic target regions in parallel, while only requiring a limited number of unidirectional sequencing reads. OutKnocker is operated from a web browser making it conveniently accessible to any user.
Results
OutKnocker deep sequencing analysis tool
The graphic user interface of OutKnocker retrieves the genomic reference locus and the nuclease target site from the user (Supplemental Fig. 1). The user enters the reference locus so that its 5′ end matches the 5′ end of the amplicon of the genotyping PCR that is positioned ∼100 nucleotides (nt) upstream of the nuclease target site (Fig. 1A). Raw sequencing data reads are loaded in FASTQ format, with up to 96 individual sequencing files analyzed in parallel. Upon execution, OutKnocker then identifies sequencing reads that are relevant to the reference locus by aligning the first 50 bases to the reference sequence (>75% identities, no gaps allowed). This simple and rapid alignment method is possible given the fact that deep sequencing reads start at a defined base position. Next, the algorithm extends the alignment of the 50-nt seed in the sequencing direction to locate a possible indel (Fig. 1B). An indel position is called at the first mismatch position of a 10-nt word that does not match the reference sequence (<60% identities, no gaps allowed). Upon identification of an indel position, a local alignment 10 nt downstream from the first mismatch position is performed using a 20-nt word size (>75% identities, no gaps allowed). When successful, the alignment offset is considered as the indel length and a two-dimensional mutation counting matrix is incremented based on the indel position and length (Fig. 1B). In cases where the 3′ end of the nuclease target site is reached without calling an indel event and without discarding the read due to one of the aforementioned criteria, the read is counted to match the reference sequence. To increase the stringency of the mismatch calls, the user can nominate a phred score threshold. Optionally, single-nucleotide exchanges introduced by targeted mutagenesis approaches using donor oligonucleotides can also be analyzed (Supplemental Note 1) (Chen et al. 2011; Bedell et al. 2012; Yang et al. 2013).
Figure 1.
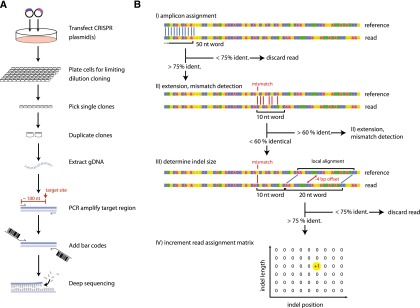
The workflow to generate and identify knockout cell clones using OutKnocker. (A) Schematic view of the developed workflow to obtain gene-targeted cell clones for subsequent deep sequencing analysis. Forty-eight hours after transfection of a designer nuclease, cells are seeded under limiting dilution conditions and cultured for 2 wk. Grown single-cell clones are picked and duplicated. One duplicate is lysed to perform a locus-specific PCR and a subsequent second PCR is performed to attach barcodes and sequencing adapters. Obtained PCR products are then pooled and subjected to deep sequencing. (B) Schematic view of the alignment and indel calling algorithm used by OutKnocker (see text for details).
When all reads have been processed, uploaded sequencing files (corresponding to the individual barcodes) are displayed as individual pie charts, whereas uniquely identified indel events that exceed a user-defined threshold are displayed as pieces within a pie chart (Supplemental Fig. 2). The size of each pie chart corresponds to the number of reads evaluated, while the size of its pieces correlates with the relative frequency of the unique mutations observed. Moreover, the color of the pieces indicates the impact an indel mutation has on a putative reading frame: Red colors indicate indels with a size of 3n + 1 or 3n + 2 bases; blue colors indicate indel events with a size of 3n bases; and gray represents sequencing reads that are found devoid of indel events. Consequently, when evaluating the impact of a genome-editing event in a protein-coding region, functional knockout clones are visible as pie charts that are entirely red. By clicking on an individual pie chart, a list of uniquely identified indel events is displayed. The respective sequence is obtained by averaging the individual base calls of all raw sequence reads assigned to this particular indel event (alignment view). Deletions are indicated as gaps, and inserted bases are depicted below the position of the respective insertion.
Validation of the OutKnocker tool
To validate this analysis tool, we targeted the toll-like receptor 2 (TLR2) and unc-93 homolog B1 (C. elegans) (UNC93B1) genes in the human monocyte-like cell line THP-1 (Fig. 2A; Supplemental Fig. 3A). TLR2 is involved in the recognition of bacterial cell wall components (Aliprantis et al. 1999), whereas UNC93B1 is an ER-resident trafficking molecule that is essential for the function of endosomal TLRs (Tabeta et al. 2006). To this end, CRISPR target sites were chosen to disrupt the reading frame of the respective genes in close proximity to their start codons. THP-1 cells were electroporated with a construct containing CMV-mCherry-Cas9 and an U6-gRNA cassette, and cells expressing high levels of mCherry-Cas9 were sorted and subsequently plated under limiting dilution conditions. After 2 wk, single-cell clones were selected and duplicated into new culture vessels, and one replicate was used for PCR genotyping. In a first PCR, the targeted genomic locus was amplified, and in a second PCR, barcodes and adapter sequences for Illumina deep sequencing were added. All PCR amplicons were pooled and subjected to deep sequencing (Fig. 1A). OutKnocker was then used to analyze the sequencing data for each target region individually. In total, we analyzed 96 clones for TLR2 (Fig. 2B) and 33 clones for UNC93B1 (Supplemental Fig. 3B) genome editing. A total of 52 out of 96 clones for TLR2 and eight out of 33 clones for UNC93B displayed at least one edited allele. For the majority of clones, two distinct edited regions with roughly equal frequency could be identified, which can be interpreted as two edited alleles. However, we also detected clones that contain only one specific mutation (Fig. 2B, e.g., clones 13, 27, 36), which could be due to the fact that either all alleles bear the same indel mutation or that the clone lost one allele, which cannot be distinguished by sequencing. Data displaying more than two distinct edited sequences (Fig. 2B, clone 9) were most likely derived from two or more clones growing in a single well despite limiting dilution. At the same time, this could also be due to prolonged Cas9-mediated genome-editing activity during clonal expansion of the cells. In total, 30 of 96 (TLR2) and two of 33 clones (UNC93B1) carried frameshift mutations in all uniquely identified genomic regions (pie charts all in red color), thus representing functional knockout clones.
Figure 2.

Application of the OutKnocker analysis tool to generate TLR2 knockout THP-1 cells. (A) The genomic locus of the human TLR2 gene is depicted. Small, black square represents noncoding exons, whereas the large, gray square represents coding exons. The red arrow highlights the target site of the CRISPR that is magnified below. (B) Shown is the analysis performed by OutKnocker of 96 THP-1 clones that were treated with a CRISPR targeting human TLR2. Every pie chart represents a clone, whereas the size of each chart corresponds to the number of reads that were analyzed to evaluate the clone (see legend in the top right). Colors of the individual pie areas indicate in-frame mutations (blue), out-of-frame mutations (red), or no indel calls (gray) (see legend in the bottom right). (C) The identified indel mutations of two knockout clones (5 and 40) are depicted. Orange letters highlight the PAM sequence and red letters indicate the CRISPR target site. (D) Two knockout clones (clones 5 and 40) as well as two unmodified clones (clones 6 and 8) were stimulated with increasing amounts of either the TLR4 ligand LPS (serial dilutions 1:10 from 1 µg/mL to 0.1 ng/mL) or with the TLR2 ligand Pam3CSK4 (serial dilutions 1:10 from 1 µg/mL to 0.1 ng/mL). A representative result out of two independent experiments is depicted as mean value + SEM of biological duplicates.
To confirm these genotyping results, selected clones were expanded and stimulated with TLR ligands, measuring TNF production as a readout for TLR-induced signaling. THP-1 cells were either treated with the synthetic triacylated lipopeptide Pam3CSK4, a classical TLR2 stimulus, or with the TLR4 agonist lipopolysaccharide (LPS) as a control. As expected, clones recovered with TLR2 frameshift mutations (clones 5 and 40) showed no response to Pam3CSK4, whereas unmodified clones (clones 6 and 8) secreted TNF in a dose-dependent manner (Fig. 2C,D). As a control, all clones showed a dose-dependent response to TLR4 stimulation. Analogous data were obtained for UNC93B1 (Supplemental Fig. 3C,D). In addition to the target regions shown here (TLR2 and UNC93B1), we have generated knockout cell lines for more than 100 independent genes to date using this approach (Ablasser et al. 2013, 2014; Zhu et al. 2014; data not shown). Of note, in the case of previously characterized target genes, the phenotype of these knockout cell lines always reflected the expected outcome.
Discussion
Here we describe an easy-to-follow workflow to genotype functional knockout or base-specifically mutated cell clones by utilizing a web browser–based evaluation tool for deep sequencing data. The data evaluation algorithm was implemented in javascript, which allows researchers to conveniently run the tool as a platform-independent service with an intuitive graphic user interface and without the requirement for transferring large amounts of data to a web server. The genotyping algorithm was streamlined for frame-disrupting allele identification while ignoring SNPs and sequencing errors typically observed with deep sequencing. This approach allows maximum information yield from limited read numbers. However, we also included the option to identify specific point mutations introduced by targeted mutagenesis. With the setup described here, thousands of cell clones can be multiplexed in a benchtop deep sequencing run (e.g., 15 × 106 250-bp reads), rendering the per-clone sequencing cost below 10 cents.
Methods
THP-1 cell culture
THP-1 cells were cultivated at 37° and 5% CO2 in RPMI supplemented with 10% FCS, 1 mM sodium pyruvate, and 0.1 mg/L Ciprofloxacin. For differentiation, cells were seeded overnight in complete medium containing 100 nM PMA. On the next day cells were washed with PBS, and seeded in flat-bottom 96-well plates at a density of 5 × 105/mL. Once attached, cells were stimulated with LPS (serial dilutions 1:10 from 1 µg/mL to 0.1 ng/mL), Pam3CSK4 (serial dilutions 1:10 from 1 µg/mL to 0.1 ng/mL), or R848 (serial dilutions 1:2 from 10 to 0.04 µg/mL).
CRISPR constructs
We used a plasmid encoding a CMV-mCherry-Cas9 expression cassette and a gRNA under the U6 promoter (Ablasser et al. 2013). The CRISPR target sites used were GACTGTACCCTTAATGGAGTTGG (TLR2) and GCACGTTCTTGAGCACGCCCAGG (UNC93B1).
Primer design
A genomic amplicon of 220- to 270-nt length was chosen with the nuclease target site located in the middle. Primers of 23 nt each with a GC content of ∼50% within the entire primer sequence and a GC content of exactly 50% in the two 3′-terminal primer positions were chosen. The primer sequences were elongated by the following sequences: Fwd: ACACTCTTTCCCTACACGACGctcttccgatct - N23 and Rev: TGACTGGAGTTCAGACGTGTGctcttccgatct - N23.
Electroporation
THP-1 cells were plated at a density of 2 × 105/mL. After 24 h, 2.5 × 106 cells were resuspended in 250 μL Opti-MEM, mixed with 5 µg plasmid DNA in a 4-mm cuvette, and electroporated using an exponential pulse at 250 V and 950 μF utilizing a Gene Pulser electroporating device (Bio-Rad Laboratories). Cells were allowed to recover for 2 d in 6-well plates filled with 4 mL medium per well.
FACS sorting
FACS sorting of 20,000 mCherry-positive cells was performed on a BD FACSAria III (BD Biosciences) sorting device.
Limiting dilution cloning
Cells were plated at a density of 4, 8, or 16 cells per well of nine round-bottom 96-well plates and grown for 2 wk. Then, plates were scanned for absorption at 600 nm. Growing clones were identified using custom software and picked and duplicated by a Biomek FXp (Beckman Coulter) liquid handling system.
Cell lysis
The medium was discarded and the cells were lysed in 30 μL of lysis buffer: 0.2 mg/mL proteinase K, 1 mM CaCl2, 3 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10 mM Tris (pH 7.5). The reactions were incubated for 10 min at 65°C and 15 min at 95°C.
Dual PCR barcoding
First-level PCR reactions were performed using 1 μL PCR-compatible lysate as a template for a 6.25-μL Phusion (Thermo Scientific) PCR reaction according to the manufacturer’s protocol (annealing temperature: 60°C; elongation time: 15 sec, 19 cycles). Of this reaction, 1 μL was transferred to a second-level PCR using the same cycling conditions and a combination of barcode primers that is unique for each clone to be analyzed. For all primer sequences see Supplemental Table 1.
Deep sequencing
Crude PCR products were pooled and size-separated using a 1.5% agarose gel run at 100 V. After visualization with ethidium bromide under UV light, DNA bands from 300 bp to 450 bp were cut out and purified using Jena Analytik innuPREP gel extraction kit according to the manufacturer’s protocol. Eluted DNA was precipitated by adding 0.1 volumes of 3 M NaOAc (pH 5.2) and 1.1 volumes of isopropanol. After centrifugation for 15 min at 4°C, the resulting pellets were washed once in 70% EtOH and air-dried. A total of 30 μL water was added, nonsoluble fractions were spun down and removed, and the DNA concentration was quantified using a Nanodrop spectrophotometer system (Thermo Fisher). Deep sequencing was performed according to the manufacturer’s protocol using the MiSeq (Illumina) benchtop sequencing system. Data were obtained in FASTQ format.
Data evaluation
Data evaluation by OutKnocker was performed using an Apple MacBook Pro (2.3 GHz Dual-Core, 4 GB RAM) on Firefox version 27.0.1. OutKnocker also runs on current versions of Safari and Google Chrome.
Data access
The sequence data generated for this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP044260. OutKnocker is available as open source software at http://www.OutKnocker.org.
Supplementary Material
Acknowledgments
This work was supported by a scholarship from the Studienstiftung des Deutschen Volkes for J.L.S.-B. V.H. is a member of the excellence cluster ImmunoSensation and supported by grants from the German Research Foundation (SFB704 and SFB670) and the European Research Council (ERC 2009 StG 243046). We kindly thank E. Endl for excellent FACS sorting. We thank Julia Reinhardt, Stefanie Riesenberg, and Christine DeNardo for critical reading of the manuscript.
Author contributions: J.L.S.-B., T.S., and V.H. developed the methodology, designed the experiments, analyzed the data, and wrote the manuscript. J.L.S.-B. programmed the software and J.L.S.-B. and T.S. performed most of the experiments. T.S.E. established the THP-1 targeting. M.M.G. performed the TLR2 studies, whereas K.P. and E.L. performed the UNC93B1 experiments. V.H. conceived and supervised the study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.176701.114.
References
- Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. 2013. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503: 530–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. 2014. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 192: 5993–5997 [DOI] [PubMed] [Google Scholar]
- Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. 1999. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285: 736–739 [DOI] [PubMed] [Google Scholar]
- Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG 2nd, Tan W, Penheiter SG, Ma AC, Leung AY, et al. 2012. In vivo genome editing using a high-efficiency TALEN system. Nature 491: 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. 2011. Genome engineering with zinc-finger nucleases. Genetics 188: 773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Pruett-Miller SM, Huang Y, Gjoka M, Duda K, Taunton J, Collingwood TN, Frodin M, Davis GD. 2011. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods 8: 753–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiruvella KK, Liang Z, Wilson TE. 2013. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol 5: a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79: 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. 2011. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148 [DOI] [PubMed] [Google Scholar]
- Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, et al. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 7: 156–164 [DOI] [PubMed] [Google Scholar]
- Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Rios X, et al. 2013. Optimization of scarless human stem cell genome editing. Nucleic Acids Res 41: 9049–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang Y, Ghosh A, Cuevas RA, Forero A, Dhar J, Ibsen MS, Schmid-Burgk JL, Schmidt T, Ganapathiraju MK, et al. 2014. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40: 936–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.