

Abstract
Respiratory infections are well-known triggers of chronic respiratory diseases. Recently, culture-independent tools have indicated that lower airway microbiota may contribute to pathophysiologic processes associated with asthma and chronic obstructive pulmonary disease (COPD). However, the relationship between upper airway microbiota and chronic respiratory diseases remains unclear. This study was undertaken to define differences of microbiota in the oropharynx of asthma and COPD patients relative to those in healthy individuals. To account for the qualitative and quantitative diversity of the 16S rRNA gene in the oropharynx, the microbiomes of 18 asthma patients, 17 COPD patients, and 12 normal individuals were assessed using a high-throughput next-generation sequencing analysis. In the 259,572 total sequence reads, α and β diversity measurements and a generalized linear model revealed that the oropharynx microbiota are diverse, but no significant differences were observed between asthma and COPD patients. Pseudomonas spp. of Proteobacteria and Lactobacillus spp. of Firmicutes were highly abundant in asthma and COPD. By contrast, Streptococcus, Veillonella, Prevotella, and Neisseria of Bacteroidetes dominated in the healthy oropharynx. These findings are consistent with previous studies conducted in the lower airways and suggest that oropharyngeal airway microbiota are important for understanding the relationships between the various parts of the respiratory tract with regard to bacterial colonization and comprehensive assessment of asthma and COPD.
Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are common chronic inflammatory diseases of the airways, and their prevalence and mortality rates continue to increase, establishing them as leading causes of disability worldwide [1]. The World Health Organization (WHO) estimates that 235 million people are annually affected by asthma and approximately 250,000 people die per year worldwide [2]. COPD also affects 329 million people worldwide, resulting in approximately 3 million deaths annually [3], [4].
Clinical studies of chronic respiratory diseases have been focused on their association with common respiratory viruses. Respiratory viral infections are common and usually self-limiting illnesses in healthy adults but are a major cause of exacerbations in patients with asthma and COPD [5]. One of the triggers for development of asthma and COPD is impaired lung function, caused due to bacterial infections. In asthma, Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis are clinically relevant contributors to asthma exacerbations, particularly when sinusitis is present [1], [2]. Atypical bacteria such as Chlamydophila pneumoniae and Mycoplasma pneumoniae may also be associated with asthma [6]–[8]. The role that bacterial pathogens play in acute exacerbations of COPD is controversial. Pseudomonas aeruginosa, Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae are known to be associated with a significantly increased risk of exacerbation of COPD. However, the prevalence of bacterial pathogens in the lower airways was found to be the same during acute exacerbations and stable disease; thus, changes in bacterial load are unlikely to be an important mechanism underlying exacerbation of COPD [9]–[11].
Human microbiota is diverse across individuals and different sites in the body. Oral microbiota are known to be involved in upper and lower respiratory infections, which can also develop into atopic airway diseases such as allergic rhinitis and bronchial asthma [12]. The oropharynx is constantly exposed to both inhaled and ingested microbes, including those cleared by mucociliary mechanisms from the upper and lower respiratory tracts and those contained in saliva [13]. The clinical relevance of the microbiota of the oropharyngeal wall is unknown. However, it is known that the diversity of the oral microbiota can be affected by antibiotics, probiotics, diet, and gut microbiota [14].
Oral microbiota can trigger and strike as a critical component in conditions like Bronchial asthma and COPD [15], particularly considering that the normal healthy lung is not a sterile organ as previously assumed [16]. To date, the relationship between airway microbiota and chronic respiratory diseases has been investigated only in lower airway samples such as sputum and bronchoalveolar lavage (BAL) samples [17], [18], [19]. However, the microbial inhabitants of the upper respiratory tract in asthma and COPD patients has been insufficiently characterized. The induction or suppression of systemic immune tolerance to challenge antigens can be attributed to the link between oral tolerance and airway tolerance. Although strong evidence exists to implicate bacterial infections in the course and pathogenesis of airway diseases, systematic studies of organisms in the airways are lacking [12]. So it becomes crucial to define the characteristics of the oropharyngeal microbiota, unfolding the pathogenic roles of these microorganisms. This in turn can lead to identification of potential targets and the discovery of novel therapies for the prevention and treatment of oral complications [20].
Traditionally identification of bacteria in the oral cavity has been done by culturing methods. Analysis of microbiota through culture-dependent methodologies is both expensive and time-consuming in comparison with many of the available culture-independent methods. It is evident to assess minor or non-dominant populations, culture-independent strategies are important for analysis of microbial ecology. Previous studies demonstrate culture-independent approaches have revealed more diverse microbiota compared to culture-dependent microbiota, of which nearly 40–60% bacterial taxa in the mouth were uncultured or validly described [21].
Recent high throughput methods like next-generation sequencing have revolutionized the way diversity of microbial communities in varied environments, including the human body is analyzed [22]. In this study, we aimed to comprehensively characterize the bacterial communities of the oropharyngeal airway in asthma and COPD patients relative to healthy individuals using a high-throughput 16S rRNA gene-pyrosequencing analysis.
Methods
Study subjects
All of the samples were collected from individuals with newly diagnosed asthma and COPD in accordance with the ethical guidelines mandated by the commission a protocol (number #12-0015) that was approved by the University’s Human Subjects Institutional Review Board (IRB) of Chung-Ang University College of Medicine, Seoul, Korea. Human subjects remain anonymous and the informed consent was written. All participants gave written informed consent for participation in the study. The current diagnosis of asthma was based on a history of wheezing, shortness of breath, and cough, which were variable in severity and frequency [23], [24]. COPD patients were further classified into four distinct stages according to Global Initiative on Obstructive Lung Disease (GOLD) guidelines [25]. Patients with bronchial asthma had mild intermittent disease and were treated as being naive. For the oropharyngeal swabs, 3M quick swabs (3M Microbiology Products; St. Paul, MN, USA) were inserted into the oral cavity until resistance was met at the oropharynx. The swabs were then rotated 180 degrees and withdrawn while taking care not to contaminate the swabs with oral bacteria. After swabbing, the swabs were transported immediately to the lab for identification and subsequent microbiomic analysis. Each swab sample was aseptically placed in a microtube and centrifuged at 14,000 rpm for 10 min, and the supernatant was removed. To detect possible contamination, negative controls were prepared and subjected to the same procedures.
DNA extraction from oropharyngeal swab samples
The first step in the analysis was extraction of DNA from the bacterial pellet that was obtained from oropharyngeal swab samples, followed by polymerase chain reaction (PCR) analysis. Samples were extracted individually by using the cetyltrimethylammonium bromide method [26]. Purified DNA was dissolved in sterile water with 40 µg/mL RNase A and quantified using an Infinite 200 NanoQuant microplate reader (Tecan; Männedorf, Switzerland) at a wavelength of 260 nm.
GS-FLX 454 pyrosequencing by using 16S rRNA gene amplification
PCR amplification of the 16S rRNA gene was performed following established procedures [27]. The V1–V3 region of the 16S rRNA gene was amplified with primers 8F (5′-CTGCTGCCTYCCGTA-3′) and 530R (5′-GTATTACCGCGGCTGCTG-3′) with the MID sequence. PCR was performed in final reaction mixtures of 25 µL containing 5–25 ng of genomic DNA, each primer at 0.4 mM, 0.2 mM dNTPs (Takara Bio; Shiga, Japan), 1.5 mM MgCl2 (Invitrogen; Carlsbad, CA, USA), 2.0 U of Hot Start Taq polymerase (Takara), and 1.0 µL of reaction buffer (Invitrogen). PCR amplification was performed on a GeneAmp PCR system 9700 thermocycler (Applied Biosystems; Foster City, CA, USA) under the following conditions: initial denaturation for 5 min at 94°C, followed by 30 cycles of 30 s at 94°C, 30 s at 55°C, and 1 min at 72°C, with a final extension for 10 min at 72°C and cooling to 4°C. The PCR products were resolved on a 1.2% Seakem LE agarose gel (FMC Bioproducts; Rockland, ME, USA) and visualized after ethidium bromide staining. 454 sequencing was carried out on the pooled PCR products of equal amounts. Amplicon pyrosequencing was performed using a 454/Roche GS-FLX Titanium instrument (Roche Molecular Systems; Branchburg, NJ, USA). After sequencing, the sequence file (.fna) and quality file (.qual) were recovered by MID groups. Quality control of the post-processing sequences was performed to eliminate sequences with ambiguous base calls, homopolymers >6, quality scores <Q25, read lengths <250 bp, and low-quality sequence ends and those without a 100% match to barcodes. Random sampling was used to analyze the diversity of the low reads of the sequence and the high reads of the sequence set. Tests of the sequence analysis methods are described in PRINSEQ [28]. The raw data were submitted to the National Center for Biotechnology Information-Sequence Read Archive (NCBI-SRA; http://www.ncbi.nlm.nih.gov/sra) with accession number SRX316595.
16S rRNA gene sequence analysis
The nucleotide sequence homology of the amplified 16S rRNA gene V1–V3 regions was determined using the nucleotide BLAST (BLASTn) program, which is available at the SILVA website (http://www.arb-silva.de/, Release 115) and the BLAST webpage of the NCBI. Accurate identification of organisms through comparative analysis of 16S rRNA gene sequences is strongly dependent on the quality of the database that is used. The curated Ribosomal Database Project (RDP; http://rdp.cme.msu.edu, Release10, update 32) provides bacterial and archaeal small subunit rRNA gene sequences in an aligned and annotated format, and has achieved major improvements in the detection of sequence anomalies [29]. Among all of the online tools provided by the RDP website, the RDP pyrosequencing pipeline includes information of Shannon-Wiener, Chao1, and ACE and operational taxonomic units (OTUs) through the rarefaction analysis tool. A phylogenetic tree was built using the neighbor-joining method in Clustal_X [30] with 1,000 bootstrap replicates. Fast UniFrac was used to evaluate the relatedness of samples [31], [32] (http://unifrac.colorado.edu/static/welcome.html). Weighted and normalized PCoA and Jackknife Environment Clusters were used to evaluate similarity among samples, where each sample represented an environment. Hierarchical cluster analysis was performed using complete linkage and UniFrac distances as a measure of similarity [33]. Weighted and normalized Principal Coordinate Analysis (PCoA) was performed to evaluate similarity among the samples, where each sample represented an environment. R software (version 2.15.2) with gplots and the RColorBrewer package was used for all other analytical processes.
Statistical analysis
Factor analysis was applied to track the important variables from an observed set of variables which are influencing the final response. Principal components were identified with over 70% of the cumulative variance. The component matrix of the chosen principal components was calculated, and variables whose factor loadings lower than 0.6 were deleted. A generalized linear model (GLM) was constructed using the final set of chosen variables. The following equation (a) represents the GLM:
| (a) |
where t is the total number of occurrences,  , and
, and 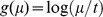 in the pooled group of samples from asthmatic patients, COPD patients, and healthy controls.
in the pooled group of samples from asthmatic patients, COPD patients, and healthy controls.
The parameter beta was tested at a 5% significance level by using (a).
Results
Study subjects
The 47 characteristics of the subjects are summarized in Table 1. For the current microbiota study, subjects were segregated into three groups: 18 patients with asthma (10 males, mean age 53.4±17.1 years; 8 females, mean age 55.0±13.0 years). All patients had a history of wheezing, cough, and dyspnea, with predicted post-bronchodilator FEV1/FVC 75.4±6.0, FEV1% 108.1±25.6, and FVC% 105.4±23.7. COPD subjects (15 males, mean age 68.9±7 years; 22 females, mean age 74.5±0.7 years) received a diagnosis of stage I (n = 6), stage II (n = 10), stage III (n = 1), or stage IV (n = 0) based on Global Initiative on Obstructive Lung Disease (GOLD) guidelines [25], with predicted post-bronchodilator FEV1/FVC 60.3±13.3, FEV1% 78.2±26.0, and FVC% 105.4±23.7. Healthy control subjects (six males, mean age 50.2±22.6 years; six females, mean age 59.3±17.7 years) exhibited no evidence of underlying respiratory disease.
Table 1. Characteristics of 47 subjects in the asthma, COPD, and healthy control groups.
| Asthma patients | COPD patients | Healthy controls | |
| No. of subjects | 18 | 17 | 12 |
| Gender | M (10), F (8) | M (15), F (2) | M (6), F (6) |
| Average age (range) | 53.4±17.1/55.0±13.0 | 68.9±7.2/74.5±0.7 | - |
| FEV1% | 108.1±25.6 | 78.2±26.0 | - |
| FVC % | 105.4±23.7 | 90.3±23.6 | - |
| FEV1/FVC | 75.4±6.0 | 60.3±13.3 | - |
| Gold stage | - | I 6 | - |
| - | II 10 | - | |
| - | III 1 | - | |
| - | IV 0 | - | |
| Wheezing | 18 | - | - |
| Cough | 18 | - | - |
| Dyspnea | 18 | - | - |
FEV1, forced expiratory volume in 1 s; FVC, functional vital capacity.
Date for FEV1% and FVC % are expressed as mean 6 SD.
Sequence reads of the 16S rRNA gene
The diversity of microbiota in the oropharyngeal swab sample was characterized by sequencing the V1–V3 region of the 16S rRNA gene, which was amplified from genomic DNA samples of 18 asthmatic patients, 17 COPD patients, and 12 healthy controls. In total, 259,572 bacterial sequence reads were identified for three group samples with average read lengths of 273–476 bp (mean ± standard deviation, 398.9±41.9) (Table 2). The 191,616 sequence reads obtained after trimming of low-quality sequences were then analyzed (mean ± standard deviation, control: 3214±1136; asthma: 4140±3723; and COPD: 4619±5710).
Table 2. Characteristics of the study participants and sequencing results.
| No. | Samplename | Sex | Age | Trimmedsequencesreads | OTUs | Avg. readlength | Chao1 | Shannonindex |
| 1 | Normal 1 | Male | 32 | 3768 | 410 | 431.4 | 503.0 | 4.8 |
| 2 | Normal 2 | Male | 74 | 3254 | 159 | 397.6 | 229.7 | 3.1 |
| 3 | Normal 3 | Male | 71 | 1189 | 132 | 273.6 | 177.2 | 3.2 |
| 4 | Normal 4 | Male | 67 | 3610 | 195 | 423.6 | 270.7 | 3.2 |
| 5 | Normal 5 | Male | 30 | 634 | 77 | 313.3 | 113.3 | 3.2 |
| 6 | Normal 7 | Male | 27 | 3608 | 280 | 437.7 | 384.1 | 4.2 |
| 7 | Normal 8 | Female | 79 | 3868 | 203 | 403.9 | 245.4 | 3.3 |
| 8 | Normal 10 | Female | 61 | 3857 | 232 | 418.5 | 303.0 | 3.9 |
| 9 | Normal 11 | Female | 53 | 3803 | 327 | 398.7 | 477.1 | 3.9 |
| 10 | Normal 14 | Female | 71 | 4286 | 242 | 406.5 | 292.7 | 3.8 |
| 11 | Normal 15 | Female | 64 | 3832 | 81 | 410.4 | 87.2 | 2.0 |
| 12 | Normal 16 | Female | 28 | 2864 | 144 | 416.4 | 198.1 | 3.6 |
| Normal average | 3214±1136 | 207±99 | 394±49 | 3.5±0.7 | ||||
| 13 | Asthma 17 | Male | 55 | 3897 | 32 | 417.4 | 37.0 | 1.1 |
| 14 | Asthma 18 | Male | 55 | 3251 | 165 | 407.0 | 224.9 | 3.6 |
| 15 | Asthma 19 | Male | 58 | 3598 | 150 | 421.8 | 187.6 | 2.5 |
| 16 | Asthma 20 | Male | 57 | 3115 | 29 | 476.7 | 32.5 | 0.3 |
| 17 | Asthma 21 | Male | 28 | 8046 | 214 | 419.8 | 254.5 | 3.3 |
| 18 | Asthma 22 | Male | 23 | 1509 | 105 | 390.6 | 165.0 | 3.0 |
| 19 | Asthma 23 | Male | 69 | 2897 | 120 | 421.2 | 151.5 | 3.1 |
| 20 | Asthma 24 | Male | 62 | 2075 | 155 | 402.8 | 313.0 | 2.8 |
| 21 | Asthma 25 | Male | 74 | 2341 | 169 | 378.3 | 218.9 | 3.2 |
| 22 | Asthma 26 | Female | 74 | 4063 | 78 | 433.3 | 88.1 | 1.3 |
| 23 | Asthma 27 | Female | 51 | 1160 | 126 | 341.7 | 171.0 | 2.7 |
| 24 | Asthma 28 | Female | 70 | 1924 | 41 | 451.8 | 54.0 | 1.4 |
| 25 | Asthma 29 | Female | 51 | 4070 | 72 | 415.3 | 106.4 | 0.8 |
| 26 | Asthma 30 | Female | 47 | 3821 | 211 | 425.9 | 311.6 | 3.8 |
| 27 | Asthma 31 | Female | 30 | 890 | 29 | 346.6 | 45.5 | 1.8 |
| 28 | Asthma 32 | Female | 55 | 2693 | 131 | 431.1 | 170.7 | 3.2 |
| 29 | Asthma 83 | Female | 55 | 16657 | 189 | 420.1 | 213.8 | 2.5 |
| 30 | Asthma 84 | Female | 62 | 8509 | 286 | 378.6 | 371.2 | 3.2 |
| Asthma average | 4140±3723 | 128±73 | 410±89 | 2.4±1 | ||||
| 31 | COPD 85 | Male | 77 | 2914 | 190 | 411.9 | 245.1 | 3.7 |
| 32 | COPD 86 | Male | 69 | 10568 | 297 | 340.8 | 372.0 | 3.8 |
| 33 | COPD 87 | Male | 73 | 4214 | 157 | 390.2 | 218.9 | 3.0 |
| 34 | COPD 88 | Male | 62 | 2563 | 75 | 420.1 | 110.8 | 1.5 |
| 35 | COPD 89 | Female | 74 | 2998 | 124 | 391.7 | 159.3 | 3.2 |
| 36 | COPD 90 | Male | 66 | 471 | 46 | 333.9 | 61.1 | 2.6 |
| 37 | COPD 91 | Male | 73 | 24809 | 363 | 434.1 | 452.5 | 2.0 |
| 38 | COPD 92 | Female | 75 | 2505 | 83 | 376.8 | 108.1 | 2.7 |
| 39 | COPD 93 | Male | 57 | 3485 | 46 | 436.8 | 51.6 | 1.4 |
| 40 | COPD 94 | Male | 68 | 3942 | 115 | 463.3 | 128.8 | 2.1 |
| 41 | COPD 95 | Male | 76 | 2506 | 186 | 322.5 | 261.9 | 3.8 |
| 42 | COPD 96 | Male | 77 | 4384 | 281 | 387.5 | 354.8 | 4.0 |
| 43 | COPD 97 | Male | 75 | 1044 | 201 | 326.1 | 274.6 | 4.3 |
| 44 | COPD 99 | Male | 66 | 2116 | 97 | 397.9 | 122.8 | 2.6 |
| 45 | COPD 100 | Male | 72 | 6678 | 280 | 393.5 | 352.0 | 3.8 |
| 46 | COPD 101 | Male | 53 | 1481 | 21 | 346.2 | 23.0 | 1.1 |
| 47 | COPD 102 | Male | 69 | 1849 | 108 | 346.0 | 147.5 | 3.0 |
| COPD average | 4619±5710 | 157±100 | 384±83 | 2.9±1 | ||||
| Total reads | 191,616 | 159±94 | ||||||
The Chao1 estimator of total species richness sequences in healthy controls and asthmatic and COPD patients is described in Table 2. Ecological diversity was also estimated by using the Shannon–Wiener index, which takes into account relative abundance. This index is widely applied in ecological studies as a measurement of biodiversity, as it accounts for both the number of different taxa and their relative abundance. For this purpose, the 16S rRNA fragments were identified from the normalized pyrosequencing datasets and classified using the RDP classifier. Only 16S rRNA sequences of genus level with at least 80% confidence was used for Shannon-Wiener index. The resulting Shannon-Wiener index value demonstrated that the normal group was more diverse than the COPD and asthmatic groups because the sequence data obtained from the 454/Roche GS FLX Titanium sequencer provided a high estimate of biodiversity.
Species richness, diversity, and rarefaction calculations were used to analyze whether the diversity of the oropharyngeal microbial community was sufficiently covered by the sequence data. Species coverage, richness, evenness, and diversity were estimated for the combined data set for each of the bacterial sequences from the oropharyngeal samples. In addition, OTUs (5% difference) were calculated for the healthy control group and patients with asthma and COPD. Rarefaction analyses with RDP analysis curves at the OTUs reached saturation for the subjects with lower microbial diversity when the sequences of each sample type and the total number of OTUs were estimated. The diversity ranged from 29 to 410 OTUs with 95% similarity in each of the 47 samples (mean ± standard deviation, control: 207±99; asthma: 128±73; and COPD: 157±100) (Table 2). The calculation of rarefaction curves was used for all the samples, which showed that additional sampling would not have provided any additional OTUs (Figure S1) with very few exceptions. The samples from the asthma, COPD, and healthy control groups contained a total of 190 different bacterial genera representing 12 different bacterial phyla (Table S1).
Phylum identification of bacterial communities
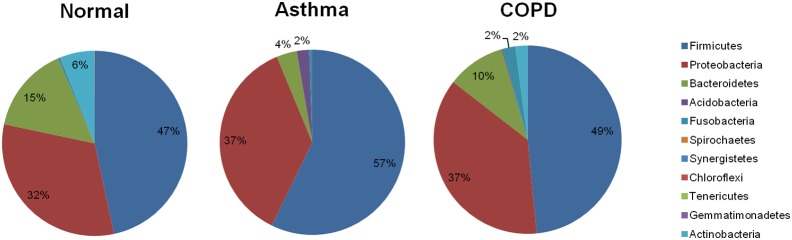
Seven bacterial phyla were detected, but most sequences were assigned to three phyla: Firmicutes, Proteobacteria, and Bacteroidetes. In 17 of 18 asthmatic patients, the majority of bacteria belonged to Firmicutes (59.8%), at a proportion higher than that in the healthy controls (48.4%). In addition, in 17 of 17 COPD patients, 61.6% of the bacterial phyla belonged to Firmicutes. Therefore, an increase in the prevalence of Firmicutes from 59.8% in the asthma sample to 61.6% in the COPD sample was observed. In the 12 healthy control samples, Proteobacteria and Bacteroidetes composed 29.02% and 16.20% of the bacteria community, respectively. In asthmatic and COPD patients, Proteobacteria accounted for 35.61% and 28.04% of the bacteria community, respectively. However, the percentage of Bacteroidetes was drastically decreased in the asthma (2.62%) and COPD (7.17%) groups (Table 3).
Table 3. Abundance table of normal, asthma and COPD.
| Classification | Normal (n = 12) | Asthma (n = 18) | COPD (n = 17) | |||||||
| Totalreads | % | Occurred | Totalreads | % | Occurred | Totalreads | % | Occurred | ||
| Phylum | Firmicutes | 18067 | 48.37 | 12 | 48172 | 59.76 | 17 | 51317 | 61.57 | 17 |
| Proteobacteria | 10840 | 29.02 | 12 | 28702 | 35.61 | 17 | 23376 | 28.04 | 16 | |
| Bacteroidetes | 6052 | 16.2 | 12 | 2109 | 2.62 | 17 | 5974 | 7.17 | 16 | |
| Actinobacteria | 2190 | 5.86 | 12 | 1306 | 1.62 | 16 | 1409 | 1.69 | 15 | |
| Fusobacteria | 167 | 0.45 | 1 | 308 | 0.38 | 14 | 1189 | 1.43 | 14 | |
| Cyanobacteria | 16 | 0.45 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Spirochaetes | 9 | 0.02 | 4 | 2 | 0 | 2 | 2 | 0 | 2 | |
| Tenericutes | 7 | 0.02 | 3 | 1 | 0 | 1 | 2 | 0 | 2 | |
| Acidobacteria | 1 | 0 | 1 | 0 | 0 | 0 | 82 | 0.1 | 1 | |
| Chloroflexi | 0 | 0 | 0 | 4 | 0 | 3 | 3 | 0 | 2 | |
| Genus | Streptococcus | 12749 | 33.05 | 12 | 21117 | 28.34 | 17 | 18467 | 23.52 | 17 |
| Neisseria | 8368 | 21.69 | 12 | 4661 | 6.26 | 13 | 4417 | 5.62 | 15 | |
| Prevotella | 5178 | 13.42 | 12 | 1852 | 2.49 | 15 | 3307 | 4.21 | 16 | |
| Veillonella | 3090 | 8.01 | 12 | 2598 | 3.49 | 15 | 3125 | 3.98 | 15 | |
| Leptotrichia | 2397 | 6.21 | 12 | 151 | 0.20 | 14 | 1076 | 1.37 | 14 | |
| Actinomyces | 1492 | 3.87 | 11 | 551 | 0.74 | 14 | 522 | 0.66 | 11 | |
| Fusobacterium | 696 | 1.80 | 10 | 153 | 0.21 | 10 | 99 | 0.13 | 10 | |
| Porphyromonas | 566 | 1.47 | 9 | 106 | 0.14 | 8 | 106 | 0.13 | 8 | |
| Halomonas | 437 | 1.13 | 10 | 840 | 1.13 | 13 | 359 | 0.46 | 12 | |
| Gemella | 340 | 0.88 | 10 | 249 | 0.33 | 13 | 331 | 0.42 | 13 | |
| Acinetobacter | 109 | 0.28 | 7 | 5 | 0.01 | 3 | 4242 | 5.40 | 7 | |
| Lactobacillus | 26 | 0.07 | 6 | 22109 | 29.67 | 14 | 27093 | 34.50 | 15 | |
| Pseudomonas | 25 | 0.06 | 3 | 11775 | 15.80 | 10 | 7955 | 10.13 | 8 | |
| Enterobacter | 7 | 0.02 | 2 | 983 | 1.32 | 9 | 237 | 0.30 | 7 | |
| Stenotrophomonas | 4 | 0.01 | 2 | 2190 | 2.94 | 8 | 253 | 0.32 | 6 | |
| Citrobacter | 0 | 0.00 | 0.00 | 1319 | 1.77 | 3 | 10.00 | 0.01 | 3 | |
| Leuconostoc | 0 | 0.00 | 0.00 | 901 | 1.21 | 4 | 1090 | 1.39 | 6 | |
The relative abundance of the main phyla identified in the normal control group and in the asthmatic and COPD patients was analyzed using an R script with gplots and the RColorBrewer package (Figure 1). The Firmicutes and Proteobacteria phyla were highly abundant in the asthma and COPD groups. The Bacteroidetes and Actinobacteria phyla had low abundance in the COPD group but were highly abundant in the healthy control group. These results indicate that the increase in microbial communities in the asthma and COPD groups was associated with an abundance of Proteobacteria and Firmicutes along with a reduction in the number of Bacteroidetes.
Figure 1. Oropharynx microbial diversity of phyla in the asthma, chronic obstructive pulmonary disease (COPD), and healthy control groups.
Genus identification of bacterial communities
To diversity of the microbial communities present in the oropharyngeal swab samples, was determined from BLASTn analysis, where the assembled sequences showed closest relationship with their neighbouring genera. Although the three data sets showed broad taxonomic similarity, there was notable variation in each sample at the genus level. In total, 190 different bacterial genera were identified in this study: 105, 131, and 133 different genera were present in the healthy control, asthma, and COPD groups, respectively (Table 3). Most sequences in the microbiome library from the healthy control group were Streptococcus spp. (33.05%), Neisseria spp. (21.69%), Prevotella spp. (13.42%), and Veillonella spp. (8.01%). Most sequences in the bronchial asthma library were identified as Lactobacillus spp. (29.67%), Streptococcus spp. (28.34%), Pseudomonas spp. (15.80%), Neisseria spp. (6.26%), Prevotella spp. (2.49%), and Veillonella spp. (3.46%). Intriguingly, Stenotrophomonas spp. (2.94%) was present in the asthmatic group (eight of 18 patients), but largely absent from the healthy control group (0.01%) and COPD group (0.32%). Most sequences in the COPD library were identified as Lactobacillus spp. (34.50%), Streptococcus spp. (23.52%), Pseudomonas spp. (10.13%), Neisseria spp. (5.26%), Prevotella spp. (4.21%), and Veillonella spp. (3.98%).
In the healthy control group, Neisseria spp. (Proteobacteria) and Prevotella spp. (Bacteroidetes) constituted 21.69% and 13.42% of the sequences, respectively, and thus comprised more than 40% of the bacterial community in the healthy control group compared to just over 10% in the asthmatic and COPD patients. When compared to the healthy controls, Lactobacillus spp. (29.67% and 34.50%) and Pseudomonas spp. (15.80% and 10.13%) were much more prevalent in patients with asthma or COPD, respectively. Lactobacillus spp. was present in 14 of 18 asthmatic patients and 15 of 17 patients with COPD, and Pseudomonas spp. was present in 10 of 18 asthmatic patients and eight of 17 patients with COPD. This was in contrast to the low frequencies of Lactobacillus spp. (0.07%) and Pseudomonas spp. (0.06%) in the healthy control population, i.e., six and four, respectively, of a total of 12 individuals. The proportion of Pseudomonas spp. decreased from 15.80% in the asthma sample to 10.13% in the COPD sample, and that of Lactobacillus increased from 29.67% in the asthma sample to 34.50% in the COPD sample. Among the phylum Bacteroidetes, populations of Prevotella spp. that were observed in the healthy control group were replaced with Pseudomonas spp. of Proteobacteria in the asthma and COPD groups (Table 3). It should be noted that this analysis was coverage-limited.
Identification by statistical analysis
We found that the three phyla and six genera in the normal controls and patients with asthma and COPD that were identified by sequencing and BLASTn analysis were significantly different in terms of frequency among samples, as indicated by the GLM statistical analysis (Table 4). In the statistical analysis that estimated the number of bacterial genera in each sample, if the value was less than 0, the genus was considered to occur at a greater frequency in the healthy sample. If the value was greater than 0, the genus was considered to occur at a greater frequency in the patient samples. Pseudomonas spp. (5.471) and Lactobacillus spp. (6.0006) were found with greater frequencies in the asthmatic and COPD patients. Streptococcus spp., Neisseria spp., Veillonella spp., and Prevotella spp. were found with greater frequencies in the healthy controls (p<0.0001). Jackknife clustering and PCoA were performed using Fast UniFrac [34].
Table 4. Statistical analysis of the healthy control, asthma, and chronic obstructive pulmonary disease (COPD) groups.
| Phylum | Genus | Estimate | Standard Error | Chi-squared | p-value | ||||
| Asthma | COPD | Asthma | COPD | Asthma | COPD | Asthma | COPD | ||
| Firmicutes | Streptococcus spp. | –0.244 | –0.341 | 0.0112 | 0.0146 | 470.19 | 541.05 | <0.0001 | <0.0001 |
| Firmicutes | Lactobacillus spp. | 6.006 | 6.203 | 0.1962 | 0.1965 | 936.78 | 996.15 | <0.0001 | <0.0001 |
| Proteobacteria | Pseudomonas spp. | 5.471 | 4.890 | 0.2002 | 0.2008 | 746.37 | 593.05 | <0.0001 | <0.0001 |
| Proteobacteria | Neisseria spp. | –1.177 | –2.069 | 0.0187 | 0.0209 | 3955.36 | 9841.56 | <0.0001 | <0.0001 |
| Firmicutes | Veillonella spp. | –0.893 | –1.191 | 0.0267 | 0.0306 | 1116.00 | 1517.35 | <0.0001 | <0.0001 |
| Bacteroidetes | Prevotella spp. | –1.773 | –1.046 | 0.0271 | 0.0303 | 4284.72 | 1193.36 | <0.0001 | <0.0001 |
The identification of microbiota in the healthy control, asthma, and COPD groups (Figure 2A), proved to be fairly robust with the Jackknife clustering method (75% bootstrap on all nodes but one). This is also supported by Fast UniFrac PCoA plot results (Figure 2B). The dendrogram and PCoA plots depict well, the differences in overall microbial community structure in each group, highlighting the differences between three groups. It also apparent from these figures that the microbiotas in asthma samples were closer to outlying COPD samples. On the contrary in PCoA plots, asthma samples clustered with both sides of normal and COPD samples. There is a distant overlap between asthma and COPD subjects with many outliers including the controls, emphasizing absence of tight clustering between the disease states. A derangement of some of the samples was observed with asthma no 18, no 25, no 30, and COPD no 85 falling in the normal control cluster, while COPD no 88, no 93, no 94 clustered into the asthma group. Normal control sample no 3, no 5, no 15 and asthma sample no 17, no 21, no 22, no 27, no 83, no 84 were found to be a part of the COPD group.
Figure 2. Fast UniFrac analysis of V1–V3 16S sequences of the oropharynx microbial community in the asthma, chronic obstructive pulmonary disease (COPD), and healthy control groups.

(A) Jackknife clustering of the environments in the Fast UniFrac dataset. A phylogenetic tree was generated by the neighbor-joining method as implemented in Clustal W with 1,000 bootstrap replicates. The numbers next to the nodes represent the number of times that a particular node was observed in a random sample from the whole dataset. (B) Principal coordinate analysis (PCoA) scatter plot of microbiota from individuals in the healthy control, asthma, and COPD groups (green: healthy control; blue: asthma; red: COPD).
Discussion
Respiratory diseases include the common cold, influenza, pharyngitis, sinusitis, bronchitis, pneumonia, asthma, and COPD, which account for more than 10% of visits to primary care physicians [35]. Among these, conditions like asthma and COPD are heterogeneous syndromes of chronic airway inflammation with undetermined pathogenesis and high mortality rates [36]. There are many guidelines for the diagnosis and management of asthma and COPD, but to date both diseases have often been misdiagnosed and undertreated [36], [37]. The key to confront this problem is to understand and characterize the microbiome of these disease conditions in comparison to the healthy state. The human microbiome studies can identify the link between healthy and diseased states and have the capacity to reveal disease mechanisms.
So far the studies of the microbial community in asthma and COPD of human airways, have targeted the lower respiratory tracts using samples such as BAL and the sputum, showing controversial results about the role of the microbiota. There are still gaps when compared to previous studies in this aspect, for instance trying to understand the role of the microbial populations in the lower respiratory tract of oropharyngeal airway in conditions like asthma and COPD which is still unclear. So it becomes imperative to get a true representation of the microbiome in these disease states.
In present decade it has become possible to capture the true microbial diversity with modern high throughput culture-independent techniques which aid in comparing the microbiome between the healthy and diseased conditions. These studies can provide new information on the oropharyngeal microbiota in asthma and COPD patients. Previously, several studies investigated the links between airway bacterial concentrations and COPD, but with targeted molecular typing methods aimed at detecting specific bacterial species in the sputum [10], [11]. These studies demonstrated that changes in the bacterial load in the sputum were unlikely to be an important mechanism in the exacerbation of COPD. These results were also supported by a study of the microbiome of healthy smokers and individuals with COPD using 454 pyrosequencing analysis, which showed that relatively low numbers of diverse types of bacteria existed in the lung [19].
In the current study, we gathered comprehensive information concentrating on the whole microbial diversity of the lower respiratory tract as opposed to previous studies with a non-targeted culture independent approach. We have three main conclusions from this study. First in terms of overall diversity, the oropharyngeal airway microbiota are quite diverse but reveal only few differences between asthma and COPD patients. Secondly, Pseudomonas spp. of the phylum Proteobacteria and Lactobacillus spp. of the phylum Firmicutes prove to be the most dominant populations in asthma and COPD patients but however were not detected in the healthy oropharynx. The final conclusion from this study is the dominance of groups like, Streptococcus, Veillonella, Prevotella, and Neisseria, belonging to the phylum Bacteroidetes, in the healthy oropharynx.
Briefly highlighting the methodology, sequences were generated using a high-throughput GS-FLX 454 pyrosequencer and aligned using tools like PRINSEQ, RDP, and Clustal_X. A GLM analysis of the 259,572 total sequence reads reveals a rich microbial environment in the oropharyngeal airway in asthma and COPD individuals. Distance-based statistical UniFrac tests were used for detecting the changes in abundance and rare lineages [33]. Minimal differences were observed between asthma and COPD individuals (Proteobacteria, Firmicutes; p<0.0001). As observed Pseudomonas (estimated asthma: 5.471; COPD: 4.890) and Lactobacillus spp. (estimated asthma: 6.006; COPD: 6.203) were found to be in abundance in asthma and COPD subjects when compared to healthy individuals. This is consistent with the findings of previous diversity studies in the bronchi of asthma and COPD patients, by 16S rRNA gene-based Sanger sequencing and by the 16S rRNA PhyloChip methods in endotracheal aspirates [38], [39].
Hilty et al. [39] analyzed 5,054 16S rRNA bacterial clones in the bronchi and found that the members of Proteobacteria were in dominance in asthmatic children, whereas Bacteroidetes, particularly Prevotella, were more frequent in controls than in adult or pediatric asthmatic or COPD patients. Sze et al. [40] evaluated the lung tissue microbiomes of patients with severe COPD at the time of lung transplantation and noted increased bacterial diversity of the phylum Firmicutes, attributable to an increase in Lactobacillus species. Using 16S rRNA-based culture-independent profiling methods, Huang et al. [41] reported increased microbiota at exacerbation of COPD primarily belonging to phylum Proteobacteria, including some non-typical COPD pathogens. In another study a combination of high-density 16S ribosomal RNA microarray and parallel clone library-sequencing analysis in asthmatic patients revealed a rich microbial environment in the oropharyngeal airway [42].
In the oropharyngeal airway of asthma and COPD patients, Pseudomonas, Stenotrophomonas, and Lactobacillus were found to be dominant. Whereas the main phyla reported in the bronchoalvelolar lavage fluid of COPD patients are Actinobacteria, Firmicutes, and Proteobacteria, with dominance of Pseudomonas, Haemophilus, or Stenotrophomonas. in the respective phyla [19]. However, Haemophilus was not found in our study of the oropharynx. Pseudomonas and Stenotrophomonas maltophilia are the most common pathogenic bacteria associated with cystic fibrosis [43]–[45]. In COPD, Pseudomonas was also reported to a risk factor for severe acute exacerbation [19] and Lactobacillus is known to be involved in severe COPD and fatal lung abscess [19], [40]. Therefore, further study is necessary to understand the pathogenesis of diseases at microanatomic sites of the respiratory tract.
In our study the healthy controls showed higher frequencies of organisms from Streptococcus, Veillonella, Prevotella, and Neisseria of phylum Firmicutes when compared to asthmatic and COPD patients. Normally, Streptococcus species are considered to be commensal bacteria of the oral cavity; however, S. pneumoniae, S. mitis, and S. oralis are members of the viridians streptococci and are associated with pneumonia and endocarditis and are considered as opportunistic pathogens [46]–[49]. Veillonella is a type of subgingival microflora found at periodontal disease sites and is known to be a periodontopathogenic bacteria [50]. Recently, Prevotella copri was reported to be involved in inflammation [51].
Previously, several studies reported less abundance of Neisseria in smokers [16], [52], [53]. This study also showed that Neisseria was less dominant in asthmatic and COPD patients, implying that Neisseria is an important indicator of the health of the oral and upper respiratory tract. Therefore, the potential relationship between these oropharynx microbiota and chronic respiratory diseases should be evaluated.
Previously exquisite study of the bronchial microbiome obtained from lower respiratory samples avoiding upper airway contamination in the healthy adults revealed overlapping of certain groups of bacteria similar to upper airways in six subjects [16]. Therefore, our results suggest that the distribution and profile of microbiota are consistent between the oropharynx and bronchi in asthma and COPD patients and that the pathogenic role of oropharyngeal commensal bacteria changes with n the progression of chronic respiratory diseases.
Supporting Information
Bacterial diversity in respiratory tract samples. The graph shows rarefaction curves indicating the number of assigned operational taxonomic units in relation to the number of 16S rRNA sequences.
(TIF)
Abundance table of normal, asthma and COPD.
(DOC)
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, which is funded by the Ministry of Education (2013R1A1A2A10012148).
Funding Statement
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, which is funded by the Ministry of Education (2013R1A1A2A10012148). The funders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Janeiro RD (2013) Differentiating chronic obstructive pulmonary disease from asthma in clinical practice. HUPE Journal 12: 57–64. [Google Scholar]
- 2.World Health Organization, Office of Health Communications and Public Relations. (2006) Asthma. Geneva: World Health Organization. 3 p. [Google Scholar]
- 3. Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3: e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization “The 10 leading causes of death in the world, 2000 and 2011”, July 2013. Retrieved 2013 November 29.
- 5. Kurai D, Saraya T, Ishii H, Takizawa H (2013) Virus-induced exacerbations in asthma and COPD. Front Microbiol 4: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnston SL, Martin RJ (2005) Chlamydophila pneumoniae and Mycoplasma pneumoniae: a role in asthma pathogenesis? Am J Respir Crit Care Med 172: 1078–1089. [DOI] [PubMed] [Google Scholar]
- 7. Kraft M, Cassell GH, Pak J, Martin RJ (2002) Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma: effect of clarithromycin. Chest 121: 1782–1788. [DOI] [PubMed] [Google Scholar]
- 8. Kraft M, Adler KB, Ingram JL, Crews AL, Atkinson TP, et al. (2008) Mycoplasma pneumoniae induces airway epithelial cell expression of MUC5AC in asthma. Eur Respir J 31: 43–46. [DOI] [PubMed] [Google Scholar]
- 9. Alamoudi OS (2007) Bacterial infection and risk factors in outpatients with acute exacerbation of chronic obstructive pulmonary disease: a 2-year prospective study. Respirology 12: 283–287. [DOI] [PubMed] [Google Scholar]
- 10. Dang HT, Kim SA, Park HK, Shin JW, Park SG, et al. (2013) Analysis of Oropharyngeal Microbiota between the Patients with Bronchial Asthma and the Non-Asthmatic Persons. J Bacteriol Virol 43: 270–278. [Google Scholar]
- 11. Sethi S, Sethi R, Eschberger K, Lobbins P, Cai X, et al. (2007) Airway bacterial concentrations and exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 176: 356–361. [DOI] [PubMed] [Google Scholar]
- 12. Bourdin A, Gras D, Vachier I, Chanez P (2009) Upper airway x 1: allergic rhinitis and asthma: united disease through epithelial cells. Thorax 64: 999–1004. [DOI] [PubMed] [Google Scholar]
- 13.Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, et al. (2010) Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. MBio 1. [DOI] [PMC free article] [PubMed]
- 14. Gerritsen J, Smidt H, Rijkers GT, de Vos WM (2011) Intestinal microbiota in human health and disease: the impact of probiotics. Genes Nutr 6: 209–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramsey CD, Gold DR, Litonjua AA, Sredl DL, Ryan L, et al. (2007) Respiratory illnesses in early life and asthma and atopy in childhood. J Allergy Clin Immunol 119: 150–156. [DOI] [PubMed] [Google Scholar]
- 16. Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, et al. (2011) Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med 184: 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramsey CD, Gold DR, Litonjua AA, Sredl DL, Ryan L, et al. (2007) Respiratory illnesses in early life and asthma and atopy in childhood. J Allergy Clin Immunol 119: 150–156. [DOI] [PubMed] [Google Scholar]
- 18. Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, et al. (2011) Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 6: e16384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garzoni C, Brugger SD, Qi W, Wasmer S, Cusini A, et al. (2013) Microbial communities in the respiratory tract of patients with interstitial lung disease. Thorax 68: 1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Noverr MC, Huffnagle GB (2005) The ‘microflora hypothesis’ of allergic diseases. Clin Exp Allergy 35: 1511–1520. [DOI] [PubMed] [Google Scholar]
- 21. Jumpstart Consortium Human Microbiome Project Data Generation Working Group (2012) Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One 7: e39315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li K, Bihan M, Yooseph S, Methe BA (2012) Analyses of the microbial diversity across the human microbiome. PLoS One 7: e32118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. British Thoracic Society (2003) British guideline on the management of asthma. Scottish Intercollegiate Guidelines Network. Thorax 58: 1–94. [Google Scholar]
- 24.National Institutes of Health (1997) Guidelines for the diagnosis and management of asthma. National Asthma Education and Prevention Program Expert Panel Report 2. Z Naturforsch C. Washington, DC: U.S. Government Printing Office. 97–4051. [Google Scholar]
- 25. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS (2001) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 163: 1256–1276. [DOI] [PubMed] [Google Scholar]
- 26.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JGS, et al. (1993) Miniprep of bacterial genomic DNA. Current Protocols in Molecular Biology. New York. 2.4. p.
- 27. Woese CR (1987) Bacterial evolution. Microbiol Rev 51: 221–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, et al. (2007) The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 35: D169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lozupone C, Hamady M, Knight R (2006) UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, et al. (2012) Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 28: 2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamady M, Lozupone C, Knight R (2010) Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bates JH, Campbell GD, Barron AL, McCracken GA, Morgan PN, et al. (1992) Microbial etiology of acute pneumonia in hospitalized patients. Chest 101: 1005–1012. [DOI] [PubMed] [Google Scholar]
- 36. Buist AS (2003) Similarities and differences between asthma and chronic obstructive pulmonary disease: treatment and early outcomes. Eur Respir J Suppl 39: 30s–35s. [DOI] [PubMed] [Google Scholar]
- 37. Athanazio R (2012) Airway disease: similarities and differences between asthma, COPD and bronchiectasis. Clinics (Sao Paulo) 67: 1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang YJ, Kim E, Cox MJ, Brodie EL, Brown R, et al. (2010) A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS 14: 9–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, et al. (2010) Disordered microbial communities in asthmatic airways. PLoS One 5: e8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, et al. (2012) The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 185: 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, et al. (2014) Airway Microbiome Dynamics in Exacerbations of Chronic Obstructive Pulmonary Disease. J Clin Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, et al. (2011) Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol 127: 372–381 e371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Vrankrijker AM, Wolfs TF, van der Ent CK (2010) Challenging and emerging pathogens in cystic fibrosis. Paediatr Respir Rev 11: 246–254. [DOI] [PubMed] [Google Scholar]
- 44. Emerson J, McNamara S, Buccat AM, Worrell K, Burns JL (2010) Changes in cystic fibrosis sputum microbiology in the United States between 1995 and 2008. Pediatr Pulmonol 45: 363–370. [DOI] [PubMed] [Google Scholar]
- 45. Millar FA, Simmonds NJ, Hodson ME (2009) Trends in pathogens colonising the respiratory tract of adult patients with cystic fibrosis, 1985–2005. J Cyst Fibros 8: 386–391. [DOI] [PubMed] [Google Scholar]
- 46. Balkundi DR, Murray DL, Patterson MJ, Gera R, Scott-Emuakpor A, et al. (1997) Penicillin-resistant Streptococcus mitis as a cause of septicemia with meningitis in febrile neutropenic children. J Pediatr Hematol Oncol 19: 82–85. [DOI] [PubMed] [Google Scholar]
- 47. Cabellos C, Viladrich PF, Corredoira J, Verdaguer R, Ariza J, et al. (1999) Streptococcal meningitis in adult patients: current epidemiology and clinical spectrum. Clin Infect Dis 28: 1104–1108. [DOI] [PubMed] [Google Scholar]
- 48. Lu HZ, Weng XH, Zhu B, Li H, Yin YK, et al. (2003) Major outbreak of toxic shock-like syndrome caused by Streptococcus mitis . J Clin Microbiol 41: 3051–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lyytikainen O, Rautio M, Carlson P, Anttila VJ, Vuento R, et al. (2004) Nosocomial bloodstream infections due to viridans streptococci in haematological and non-haematological patients: species distribution and antimicrobial resistance. J Antimicrob Chemother 53: 631–634. [DOI] [PubMed] [Google Scholar]
- 50. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr (1998) Microbial complexes in subgingival plaque. J Clin Periodontol 25: 134–144. [DOI] [PubMed] [Google Scholar]
- 51. Hofer U (2014) Microbiome: pro-inflammatory Prevotella? Nat Rev Microbiol 12: 5. [DOI] [PubMed] [Google Scholar]
- 52. Tamada K, Tomonaga S, Hatanaka F, Nakai N, Takao K, et al. (2010) Decreased exploratory activity in a mouse model of 15q duplication syndrome; implications for disturbance of serotonin signaling. PLoS One 5: e15126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, et al. (2013) Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 187: 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial diversity in respiratory tract samples. The graph shows rarefaction curves indicating the number of assigned operational taxonomic units in relation to the number of 16S rRNA sequences.
(TIF)
Abundance table of normal, asthma and COPD.
(DOC)