

Abstract
Thiosemicarbazones (TSCs) are an interesting class of ligands that show a diverse range of biological activity, including anti-fungal, anti-viral and anti-cancer effects. Our previous studies have demonstrated the potent in vivo anti-tumor activity of novel TSCs and their ability to overcome resistance to clinically used chemotherapeutics. In the current study, 35 novel TSCs of 6 different classes were designed using a combination of retro-fragments that appear in other TSCs. Additionally, di-substitution at the terminal N4 atom, which was previously identified to be critical for potent anti-cancer activity, was preserved through the incorporation of an N4-based piperazine or morpholine ring. The anti-proliferative activity of the novel TSCs were examined in a variety of cancer and normal cell-types. In particular, compounds 1d and 3c demonstrated the greatest promise as anti-cancer agents with potent and selective anti-proliferative activity. Structure-activity relationship studies revealed that the chelators that utilized “soft” donor atoms, such as nitrogen and sulfur, resulted in potent anti-cancer activity. Indeed, the N,N,S donor atom set was crucial for the formation of redox active iron complexes that were able to mediate the oxidation of ascorbate. This further highlights the important role of reactive oxygen species generation in mediating potent anti-cancer activity. Significantly, this study identified the potent and selective anti-cancer activity of 1d and 3c that warrants further examination.
Introduction
Iron is an essential element that is necessary for a number of cellular processes, such as cellular proliferation [1], [2], [3]. In fact, the iron-containing enzyme, ribonucleotide redutase, is involved in the rate-limiting step of DNA synthesis and is responsible for the conversion of ribonucleotides to their deoxyribonucleotide counterparts [4], [5]. Importantly, alterations in the iron metabolism of cancer cells relative to their normal counterparts have highlighted the potential of iron chelation therapy to act as a novel treatment avenue. Cancer cells demonstrate a higher requirement for iron than normal cells and this is emphasized by the increased expression of the transferrin receptor 1 (TfR1), that takes up iron from the iron transport protein, transferrin (Tf), on the cell surface [6], [7], [8]. Additionally, the expression of iron-dependent enzyme, ribonucleotide reductase, is markedly higher in tumor cells than normal cells [9]. These factors render tumor cells more sensitive to iron chelation.
Although iron chelators (e.g., desferrioxamine; DFO) have been clinically utilized for the treatment of iron overload disease [1], [3], novel thiosemicarbazone (TSC) chelators have been widely investigated as potential anti-cancer agents [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21]. Although the molecular mechanisms involved in the activity of TSCs have not been completely elucidated, a number of modes of action have been reported [3], [15], [21], [22], [23]. This includes: ( 1 ) the inhibition of cellular iron uptake from Tf [10], [13], [18]; ( 2 ) the mobilization of iron from cells [10], [13], [18]; ( 3 ) the inhibition of the ribonucleotide reductase activity [24], [25]; ( 4 ) the up-regulation of the metastasis suppressor protein, N-myc downstream regulated gene 1 [26], [27], [28], [29]; and ( 5 ) the formation redox active metal complexes that produce reactive oxygen species (ROS) [10], [15], [21], [23], [30]. This latter mechanism is significant, especially as studies have demonstrated the important role of ROS generation in increasing the selective activity of chelators against tumor cells [10], [15], [21].
The TSC, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine; Fig. 1), has been examined in >20 Phase I and II clinical trials as a novel cancer chemotherapeutic [11], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41]. Although clinical trials using Triapine have generally demonstrated limited anti-tumor activity [36], [37], [38], [40], other studies have shown positive results in locally advanced cervical and vaginal cancers when co-administered with cisplatin and radiochemotherapy [33], [34]. Notable side effects observed upon Triapine administration include methemoglobin formation and hypoxia [35], [39], [41] and these problems have necessitated the development of other more active and selective TSCs with potent anti-cancer activity.
Figure 1. Chemical structures of the chelators, Triapine, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT), di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC), 2-benzoylpyridine 4,4-dimethyl-3-thiosemicarbazone (Bp44mT) and quinoline thiosemicarbazone (QT).
Several classes of TSCs have been developed as potential anti-proliferative agents (Fig. 1), a number of which show marked and selective anti-tumor activity both in vitro [10], [15], [21], [42] and in vivo [12], [21], [26], [42]. For example, a five day treatment with di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT; Fig. 1) at 0.4 mg/kg in mice reduced the growth of a murine M109 lung carcinoma to 47% of the control [21]. Additionally, Dp44mT showed potent and selective anti-tumor activity in vitro and in vivo against a range of human tumor xenografts [42] and was able to form redox active metal complexes that generate ROS [15], [21], [30]. Although this TSC showed great potential, it demonstrated cardiac fibrosis at high, non-optimal doses [42]. Thus, further investigations into Dp44mT analogs were necessary and have resulted in the development of di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC; Fig. 1) [12], [26]. DpC has demonstrated selective in vitro and in vivo anti-tumor activity by both the intravenous [12], [26] and oral routes [12] and is currently being further evaluated for entrance into clinical trials. Recently, other TSCs have also been shown to have a novel application as photodynamic therapy enhancers [43].
We have previously examined a variety of quinolone-based TSCs that demonstrate in vitro anti-proliferative activity [16]. In the current investigation, we synthesized 35 novel TSCs of 6 classes that were designed by the combination of active fragments present in previously reported analogs [16], [44]. Earlier studies indicated that di-substitution at the terminal (N4) nitrogen is crucial for effective anti-cancer activity [12], [21]. In the present study, we have preserved di-substitution at the N4 atom through the construction of an N4-based piperazine or morpholine ring, a fragment that is present in several active TSCs [45], [46]. The anti-proliferative activity and selectivity of these novel TSCs was examined in vitro against human cancer cell-types and normal human dermal fibroblast (NHDF) cells. Those series that demonstrated the ability to form redox active iron complexes and mediate the oxidation of ascorbate showed potent anti-proliferative activity, highlighting the importance of ROS generation in their anti-cancer activity.
Materials and Methods
The reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA), ACROS Organics (Geel, Belgium) or Princeton Chemicals Ltd (Luton, Bedfordshire, UK). Silica gel 60 (0.040-0.063 mm; Merck, Darmstadt, Germany) was used for column chromatography. Thin layer chromatography (TLC) experiments were performed on alumina-backed silica gel 40 F254 plates (Merck). The plates were illuminated under UV (254 nm) and evaluated in iodine vapor. The melting points were determined on an Optimelt MPA100 instrument (Stanford Research Systems, Sunnyvale, CA, USA) and are uncorrected. High resolution-mass spectrometry (HRMS) analysis was performed for all new compounds on a Finnigan MAT95 spectrometer (Thermo Fisher Scientific, Bremen, Germany) or on a Mariner ESI-TOF spectrometer (Applied Biosystems, Thermo Fisher Scientific).
All 1H- and 13C-NMR spectra were recorded on a Bruker AM-400 spectrometer (399.95 MHz for 1H; 99.99 MHz for 13C; BrukerBioSpin Corp., Coventry, UK). Chemical shifts are reported in ppm against the internal standard, Si(CH3)4. Easily exchangeable signals were omitted when diffuse. Syntheses were performed on a CEM-DISCOVERY microwave reactor (CEM Corporation, Matthews, NC, USA) with temperature and pressure control.
Log P calc values were calculated using ChemDraw 12 (Perkin-Elmer, Waltham, MA, USA) by performing Crippen's fragmentation [47], Viswanadhan's fragmentation [48] and Broto's method [49] and then calculating the average log P calc.
Synthesis of Thiosemicarbazide Precursors (a–f)
General method I
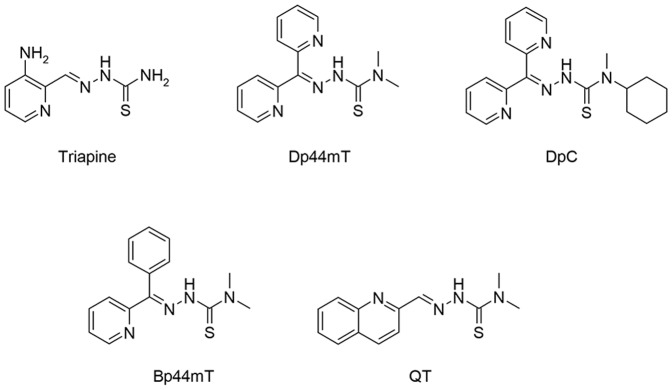
N-alkyl or N-aryl piperazine (6 mmol) was added to a solution of 1,1′-thiocarbonyldiimidazole (6 mmol; 1.068 g) in 25 mL of dichloromethane and the reaction mixture was stirred for 24 h at room temperature. The organic solvent was separated and extracted 3 times using water, then dried over anhydrous magnesium sulfate, filtered and concentrated to provide the crude piperazine derivatives. These crude products were used in the next step without further purification. These intermediates were added to a solution of hydrazine hydrate in 25 mL of ethanol at room temperature. The reaction mixture was refluxed for 3 h and cooled to obtain a white precipitate which was collected via filtration as the final product (Fig. 2). All obtained thiosemicarbazides were crystallized from methanol. See File S1 for structural characterization details and the crystal structure (Fig. S1 in File S1) and data (Table S1 in File S1) of 4-ethylpiperazine-1-carbothiohydrazide (a).
Figure 2. Synthetic route to yield thiosemicarbazides, a–e.
General method II
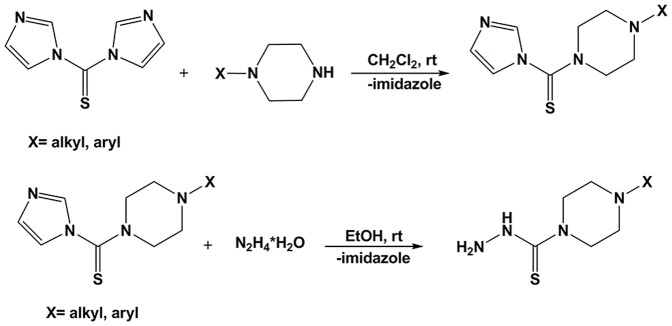
Carbon disulfide (CS2) (0.2 mol, 12.06 mL) was added dropwise over 15 min to N-methylcyclohexylamine (0.2 mol, 26.3 mL) in NaOH solution (0.8 M, 250 mL). The reaction mixture was intensively stirred for 20 min and then sodium chloroacetate (0.2 mol) was added to the solution and allowed to stir for 15 h. The reaction mixture was neutralized with concentrated HCl (20 mL) to give the carboxymethylthiocarbamate intermediate as a white precipitate. The obtained carboxymethylthiocarbamate intermediate (0.08 mol) was then reacted with hydrazine hydrate (0.08 mol) in water (10 mL). This reaction mixture was gently refluxed for 2 h to give white crystals of the thiosemicarbazide (Fig. 3). The final product was crystalized from methanol-water (1/1, v/v).
Figure 3. Synthetic route to yield the thiosemicarbazide, f.
Preparation of TSC Derivatives
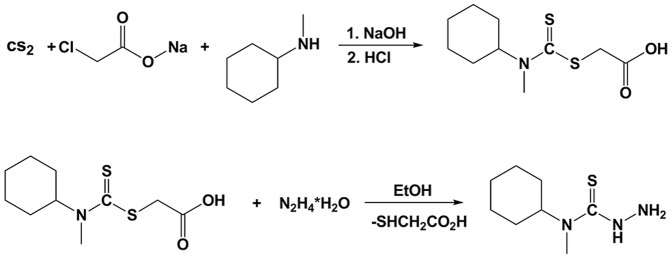
The heteroaromatic TSC analogs were synthesized by reacting the respective heteroaromatic ketone or carbaldehyde and thiosemicarbazide under microwave irradiation. Equimolar quantities of the appropriate thiosemicarbazide (0.5 mmol) and carbonyl compound (0.5 mmol) were dissolved in 4 mL of EtOH with the addition of 0.1 mL of acetic acid as catalyst (Fig. 4). The resulting mixture was heated in a microwave reactor at 83°C/30 min (max. microwave power 50 W). After cooling, the precipitated solid was filtered and washed with ether. The final product was purified using crystallization (from ethanol or methanol) or column chromatography. See File S1 for structural characterization details, the crystal structure (Fig. S2 in File S1) and data (Table S1 in File S1) of Z-N′-(di(pyridin-2-yl)methylene)-4-(pyridin-2-yl)piperazine-1-carbothiohydrazide (1d) and isosbestic curves (Fig. S3 in File S1).
Figure 4. Synthetic route to yield the final thiosemicarbazones.
HPLC Purity Data
The final purity of the TSCs was determined using the following general conditions: Gynkotek HPLC System (Gynkotek); Pump T580; Autosampler GINA50 (Gynkotek); Detector DAAD UVD340U; Column: Hilic Kinetex 100 Å (Phenomenex, Torrance, CA, USA); Flow: 0.5 mL/min (0–1 min), 0.5–1.2 mL/min (1–3 min), 1.2 mL/min (3–7 min), 1.2–0.5 mL/min (7–8 min), 90% CH2Cl2, 10% CH3OH; UV at 250 nm; Software: Chromeleon (Thermo Scientific, Waltham, MA, USA). See File S1 for HPLC purity data (Table S2 in File S1).
Cell Culture
The human cancer cell types (neuroepithelioma (SK-N-MC), colon cancer (HCT116 with wild-type p53 (p53+/+)), Burkitt's lymphoma (Raji) and cervical carcinoma (HeLa)) cells and normal human dermal fibroblasts (NHDF) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). HCT116 null p53 (p53−/−) cells were obtained from the Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Poland.
The SK-N-MC cells were cultured in minimum essential medium (MEM; Life Technologies, Grand Island, New York, USA) containing 10% (v/v) fetal bovine serum (FBS; Life Technologies), 1 mM sodium pyruvate (Life Technologies), 1% (v/v) non-essential amino acids (Life Technologies), 2 mM L-glutamine (Life Technologies), 100 U/mL penicillin (Life Technologies), 100 µg/mL streptomycin (Life Technologies) and 0.28 µg/mL fungizone (Bristol Myers Squibb Pharmaceuticals, Montreal, Canada). The HCT116 and NHDF cells were grown in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 12% (v/v) heat-inactivated FBS (HCT116; Life Technologies) or 15% (v/v) FBS (NHDF; Life Technologies), 100 µg/mL of gentamicin (Polfa Warszawa S.A., Warsaw, Poland), 100 µg/mL of streptomycin (Polfa) and 100 U/mL of penicillin (Polfa). The Raji and HeLa cells were cultured in Roswell Park Memorial Institute medium (RPMI-1640; Sigma-Aldrich) with the addition of 10% (v/v) heat-inactivated FBS (Life Technologies) and the supplements described for SK-N-MC cells above. All cell lines were cultured under standard conditions at 37°C in a humidified atmosphere at 5% CO2 and were subcultured every 3–4 days as required.
Proliferation Assay
The cells were seeded in 96-well plates (SK-N-MC: 1.5×104 cells/well; HeLa: 5.0×103 cells/well; Raji: 3.0×103 cells/well; HCT116: 3.5×103 cells/well; NHDF: 3.0×103 cells/well) 24 h prior to the addition of the novel compounds. The assays were performed using a 72 h (SK-N-MC, Raji, HeLa) or 96 h (HCT 116, NHDF) incubation period with the agents at 37°C. These seeding and growth conditions were utilized so that the cells did not come to confluence during the incubation periods. Additionally, DFO and Dp44mT were included as positive controls in all experiments as their activity is well characterized [21], [42], [50].
Chelator stock solutions were prepared in DMSO and diluted in media so that the final [DMSO] <0.05%. The results were expressed as a percentage of the control and the resulting IC50 values were calculated using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). The IC50 was defined as the concentration necessary to reduce the absorbance to 50% of the untreated control. Each individual compound was tested in triplicate in a single experiment, with each experiment being repeated three times. After incubation of HCT 116 and NHDF cells with the tested compounds, 20 µL of the CellTiter 96 Aqueous One Solution - MTS (Promega, Madison, WI, USA) solution was added to each well (with 100 µL of DMEM without phenol red) and incubated for 1 h/37°C. The optical density of the samples was analyzed at 490 nm.
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) was used to evaluate the anti-proliferative effects of the chelators in SK-N-MC and Raji cells. Following incubation with the investigated compounds, 10 µL of MTT (5 mg/mL in PBS; Sigma-Aldrich) was added to each well. After a 2 h (SK-N-MC) or 4 h (Raji) incubation, the plates were centrifuged, and the cells were lysed with 100 µL of 10% SDS-50% isobutanol in 0.01 M HCl (SK-N-MC) or DMSO (Raji). The absorbance was measured at 570 nm. The anti-proliferative effects of these novel agents on HeLa cells were estimated using crystal violet staining (0.5% crystal violet solution for 10 min). Finally, the wells were rinsed with water and the cells were lysed with 2% SDS. The optical density of the samples was analyzed at 595 nm.
Labeling of Transferrin with 59Fe
The iron transport protein, Tf (Sigma-Aldrich), was labeled with 59Fe (PerkinElmer Life and Analytical Sciences, Boston, MA) to form 59Fe2-Tf using standard methods [6], [7]. Unbound 59Fe was removed by passage through a Sephadex G25 column and was followed by exhaustive dialysis [6], [7].
Effect of the Chelators on Mobilizing Cellular 59Fe
To examine the ability of the novel chelators to mobilize 59Fe from SK-N-MC cells, 59Fe efflux experiments were performed using established techniques [50], [51]. The monolayer of SK-N-MC cells was prelabeled for 3 h at 37°C in MEM containing 59Fe2-Tf (0.75 µM). Cells were then washed four times with ice-cold PBS and incubated for a further 3 h at 37°C with medium alone (the control) or medium containing the chelator (25 µM). After this incubation, the overlying medium that contained the released 59Fe was separated from the cells using a Pasteur pipette. Radioactivity was measured in both the cells and supernatant using a γ-scintillation counter (Wallac Wizard 3, Turku, Finland). In these studies, the novel ligands were compared to the previously characterized chelators, DFO and Dp44mT, as their ability to mobilize cellular 59Fe has been extensively examined in these cells [10], [12], [17], [18], [50].
Effect of Chelators at Preventing Cellular 59Fe Uptake
The ability of the chelators to prevent the cellular uptake of 59Fe from 59Fe2-Tf was examined using standard methods [10], [14], [17], [18], [50]. A monolayer of SK-N-MC cells was incubated with medium containing 59Fe2-Tf (0.75 µM) and the chelator (25 µM) for 3 h at 37°C. The medium was then removed and the cells were washed four times with ice-cold PBS. The cells were then incubated for 30 min at 4°C with the general protease, Pronase (1 mg/mL; Sigma-Aldrich), to remove membrane-bound 59Fe. The cells were removed using a plastic spatula and centrifuged at 14,000 rpm/1 min to separate internalized from membrane-bound 59Fe. The cell pellet was resuspended in 1 mL of PBS and the internalized 59Fe was measured on a γ-scintillation counter. Internalized 59Fe uptake was calculated as a percentage of the control (medium alone). Again, the novel ligands were compared to DFO and Dp44mT as their ability to inhibit cellular 59Fe uptake has been extensively characterized using SK-N-MC cells [10], [14], [17], [18], [50].
Ascorbate Oxidation Assay
The ability of the iron complexes of the novel ligands to mediate the oxidation of the physiological substrate, ascorbate, was examined using established methods [10], [18], [22], [52], [53]. Ascorbic acid (100 µM) was prepared immediately prior to each experiment and was incubated in the presence of FeIII (10 µM; as FeCl3), the chelator (1–60 µM) and a 50-fold molar excess of citrate (500 µM). The absorbance at 265 nm was measured after 10 and 40 min and the difference in absorbance at these time points was calculated. The results of these experiments were expressed as iron-binding equivalents (IBE) due to the different denticity of the chelators examined. The chelators, Dp44mT, DFO and ethylenediaminetetraacetic acid (EDTA) were used as controls as the ability of their iron complexes to oxidize ascorbate has been extensively characterized [10], [15], [52], [53].
Statistical Analysis
Data was expressed as mean ±S.D. of at least 3 experiments. Statistical analyses were performed using Prism v6 (GraphPad Software, Inc.) implementing a one-way ANOVA with Bonferroni's post-hoc test.
Results and Discussion
Drug Design
Molecular properties such as molecular weight (MW) and calculated octanol-water partition coefficient (log P calc) are key factors in the successful development of drug candidates [54], [55]. In general, the average MW and log P calc of drugs that are launched on the market are 300–450 Da and 2–4, respectively [56], [57]. Considering this, the clinically trialed TSC, Triapine (MW: 195 Da; log P calc: 0.761), represents an interesting lead molecule with a considerable reserve in MW and log P calc for modification.
Identifying functional fragments for drug design is a complex problem that involves different approaches, including those with an experimental and theoretical basis. The latter consist of a variety of methods among which are those identifying advantageous sub-structures, scaffolds and/or linkers on the basis of previously reported compounds. Alternatively, the fragmentation of organic molecules into smaller moieties is an important method in retrosynthetic analysis and has inspired various pseudo-retrosynthetic approaches [58]. This has identified fragments that may be useful for drug design. For example, the di-2-pyridyl [12], [15], [21], [59], quinolinyl [60], piperazinyl [60], [61], [62], morpholinyl [63] and quinoxalinyl [64] motifs are common fragments in other anti-cancer agents, which have been incorporated into the design of the novel TSCs reported herein.
In the current study, the main rationale underlying our approach was that the newly synthesized TSCs should preserve the potent anti-cancer activity of their TSC precursors, while retaining appropriate MW and log P calc values to show substantial promise as drug candidates for pharmaceutical development. We used a combination of retro-fragments that appear in other TSC precursors [15], [16], [21], [44], [59] and other anti-cancer agents [60], [61], [62], [63], [64]. Additionally, di-substitution at the terminal N4 atom was preserved through the construction of an N4-based piperazine or morpholine ring (Fig. 5), a moiety that is observed in several potent TSCs [45], [46].
Figure 5. Chemical structures of the thiosemicarbazone chelators of series 1–6.

Synthesis of Novel Ligands
The thiosemicarbazide precursors, a–f (Fig. 5), were synthesized from commercially available reagents in a two-step process that gave moderate to high yields (47–95%). The treatment of bis(imidazole)thioketone with the appropriate piperazine, followed by the reaction with hydrazine hydrate gave the N-substituted piperazine-based thiosemicarbazides in high yield (File S1). The final thiosemicarbazone series, 1–6 (Fig. 5), were synthesized in moderate to high yield (58–96%) by Schiff-base condensation of the appropriate ketone/aldehyde with the prepared thiosemicarbazides. The purity of the thiosemicarbazones was confirmed by HPLC and was >95% (see Table S2 in File S1).
Anti-Proliferative Activity of the Novel Thiosemicarbazones Against a Variety of Cancer Cell-Types
The ability of the novel thiosemicarbazone series 1–6 to inhibit the cellular proliferation of tumor cells was examined in a variety of cancer cell-types (Table 1), including human p53 wild-type and null colon cancer (HCT116 p53+/+ and HCT116 p53−/−, respectively), Burkitt's lymphoma (Raji), human cervical carcinoma (HeLa) and neuroepithelioma (SK-N-MC) cancer cells. The ability of these novel agents to selectivity target cancer cells was assessed by examining their effects on the cellular proliferation of a mortal cell-type, namely normal human dermal fibroblast (NHDF) cells. The anti-proliferative activity of series 1–6 is presented in Table 1 and is also represented as color maps (see Fig. S4 in File S1). The effects of these novel ligands were then compared to the following chelators that were used as positive controls: ( 1 ) DFO, that is clinically used for the treatment of iron overload disease [1], [3] and ( 2 ) Dp44mT, a chelator with potent anti-proliferative activity in vitro and in vivo [15], [21].
Table 1. Anti-proliferative activity (IC50 values) of the novel thiosemicarbazones in comparison to DFO and Dp44mT in several tumor cell-types and normal human dermal fibroblast (NHDF) cells.
| IC50 (µM) | ||||||
| Chelator | HCT116 p53+/+ | HCT116 p53−/− | Raji | HeLa | SK-N-MC | NHDF |
| DFO | >25 | >25 | 4.74±1.80 | 14.34±0.96 | 15.06±5.2 | >25 |
| Dp44mT | 0.002±0.001 | 0.005±0.002 | 0.007±0.001 | 0.04±0.01 | 0.012±0.001 | 15.38±5.06 |
| 1a | 0.020±0.002 | 0.040±0.003 | 0.08±0.02 | 0.48±0.31 | 0.18±0.09 | 0.017±0.004 |
| 1b | 0.0008±0.0001 | 0.0008±0.0001 | 0.0003±0.00003 | 0.57±0.16 | 0.05±0.01 | 0.0017±0.0007 |
| 1c | 0.014±0.005 | 0.014±0.006 | 0.003±0.004 | 2.21±0.47 | 1.21±0.35 | 0.016±0.006 |
| 1d | 0.002±0.008 | 0.009±0.001 | 0.0013±0.0001 | 0.03±0.02 | 0.02±0.01 | 0.16±0.04 |
| 1e | 0.038±0.003 | 0.031±0.003 | 0.04±0.02 | 0.72±0.07 | 0.07±0.05 | 0.014±0.003 |
| 2a | 1.44±0.69 | 3.55±1.23 | 0.06±0.02 | 0.43±0.16 | 2.54±0.86 | 9.62±1.80 |
| 2b | 0.042±0.003 | 0.09±0.04 | 0.04±0.01 | 0.20±0.07 | 0.33±0.02 | 0.033±0.007 |
| 2c | 1.49±0.02 | 1.56±1.12 | 0.06±0.01 | 1.75±0.33 | 1.40±0.69 | 3.27±0.88 |
| 2d | 0.39±0.15 | 0.27±0.16 | 0.05±0.02 | 5.14±2.87 | 0.52±0.16 | 0.032±0.008 |
| 2e | 0.088±0.044 | 0.48±0.15 | 0.29±0.11 | 1.56±0.44 | 0.48±0.10 | 9.44±3.84 |
| 2f | 0.026±0.001 | 0.035±0.003 | 0.81±0.10 | 0.94±0.45 | 0.82±0.19 | 0.015±0.005 |
| 3a | 9.25±1.42 | 8.83±0.63 | 0.20±0.02 | 3.41±1.46 | 0.82±0.61 | 15.8±4.6 |
| 3b | 4.18±1.46 | 4.67±2.04 | 0.19±0.02 | 8.45±2.09 | >6.25 | 8.88±1.07 |
| 3c | 0.004±0.002 | 0.017±0.006 | 0.02±0.01 | 0.18±0.07 | >6.25 | 10.6±3.5 |
| 3d | 8.41±1.78 | 8.46±0.25 | 0.12±0.04 | 6.59±0.13 | >6.25 | 10.2±2.4 |
| 3e | 0.05±0.03 | 5.01±2.55 | 0.04±0.02 | 2.39±0.02 | >6.25 | 12.1±3.6 |
| 3f | 3.11±1.92 | 4.35±1.35 | 0.01±0.04 | 2.57±0.46 | >6.25 | 5.36±1.99 |
| 4a | 20.2±0.3 | 20.3±0.9 | 3.35±1.11 | 11.3±1.6 | 5.09±0.47 | 12.1±4.2 |
| 4b | 7.45±0.66 | 8.88±2.53 | 2.23±1.20 | 4.38±0.95 | 4.46±0.78 | 10.1±1.2 |
| 4c | 20.1±0.3 | 19.8±4.9 | 3.53±1.87 | 10.2±1.2 | 4.97±0.95 | >25 |
| 4d | 9.15±1.19 | 6.84±5.48 | 1.55±0.40 | 2.73±0.55 | 4.54±1.60 | 10.1±2.7 |
| 4e | 10.1±0.7 | 9.44±1.35 | 4.55±2.52 | 3.49±0.04 | 4.17±1.44 | 9.56±1.44 |
| 4f | 19.0±0.5 | 18.9±3.6 | 0.46±0.23 | 13.3±1.0 | >6.25 | 9.18±3.68 |
| 5a | 6.15±1.28 | 8.72±1.64 | 0.11±0.02 | 1.12±0.03 | ND | 2.73±1.77 |
| 5b | 3.92±0.74 | 2.24±1.47 | 0.50±0.05 | 6.05±2.73 | 3.44±1.12 | 9.61±0.90 |
| 5c | 2.03±1.24 | 5.84±1.65 | 0.24±0.04 | 0.86±0.09 | 2.87±1.25 | 11.6±1.8 |
| 5d | 9.4±0.8 | 9.41±1.04 | 0.49±0.09 | 1.78±0.77 | 2.33±0.09 | 7.13±0.82 |
| 5e | 5.73±0.75 | 8.49±3.05 | 0.27±0.01 | 3.77±1.71 | 1.98±0.68 | 15.8±5.1 |
| 5f | 1.24±0.45 | 1.02±0.54 | 0.72±0.29 | 4.09±2.07 | 2.04±0.34 | 0.50±0.21 |
| 6a | >25 | >25 | 10.2±6.6 | 22.1±5.4 | >6.25 | >25 |
| 6b | 6.9±2.3 | 8.86±1.46 | 1.96±0.85 | 5.80±0.48 | 5.13±0.51 | 13.5±4.8 |
| 6c | 15.4±1.7 | 16.4±3.1 | 3.79±0.08 | 18.2±1.3 | >6.25 | >25 |
| 6d | 9.7±2.5 | 10.6±1.2 | 2.60±0.33 | 16.4±4.5 | 5.42±0.64 | 18.4±1.9 |
| 6e | 10.9±0.7 | 10.7±1.0 | 9.83±2.05 | 19.2±2.5 | >6.25 | >25 |
| 6f | >25 | >25 | 0.69±0.51 | 10.1±3.6 | >6.25 | >25 |
Results are mean ±SD (3 experiments).
As expected from our previous studies [10], [14], [17], [18], [50], the control chelator, DFO, demonstrated poor anti-proliferative effects in all cancer and normal cell-types, with IC50 values ranging between 4.74 to >25 µM (Table 1). In contrast, Dp44mT showed selective and potent anti-cancer activity with IC50 values of 0.002–0.04 µM, but was markedly less effective in mortal NHDF cells (IC50: 15.38 µM; Table 1).
Considering the development of new therapeutics against cancer, it is notable that a lack of expression of the tumor suppressor protein, p53, in some tumors aids the progression of neoplastic cells and promotes resistance against chemotherapeutics [65], [66]. Hence, the use of agents that are active against both wild-type and null p53 tumors are vital, particularly considering the high prevalence of p53 mutations in advanced cancers [42], [67]. Thus, we initially examined the anti-proliferative activity of the novel TSC series 1–6 against human HCT116 p53+/+ and HCT116 p53−/− colon cancer cells (Table 1). Significantly, the anti-proliferative effects of most of the novel TSCs showed the same general pattern of activity against HCT116 p53+/+ and HCT116 p53−/− cells, irrespective of p53 status (Table 1). One notable exception in the activity between these two cell-types was observed for compound 3e, which showed a 100-fold decrease in anti-proliferative efficacy when comparing HCT116 p53+/+ (IC50: 0.05 µM) and HCT116 p53−/− cells (IC50: 5 µM; Table 1). However, overall, the anti-proliferative activity of the TSCs was generally independent of p53 status (Table 1), as shown for other compounds in this class [16], [42]. Hence, this is an important property of the TSCs that could explain their marked activity that has been observed with related agents across a variety of cancer cell-types [42].
Of all the series of TSCs synthesized in this study, those analogs derived from di-2-pyridyl (1a–e) demonstrated the most potent anti-proliferative activity in HCT116 p53+/+ and p53−/− cells, resulting in IC50 values ranging from 0.0008–0.04 µM (Table 1). While the analogs derived from quinolin-2-yl (2a–f) and 8-hydroxyquinolin-2-yl (3a–f) demonstrated moderate anti-proliferative effects (IC50: 0.026–3.55 and 0.004–9.25 µM, respectively). In contrast, those chelators derived from the 7-hydroxyquinolin-8-yl (4a–f), quinoxalin-2-yl (5a–f) and salicylic (6a–f) moieties showed poor anti-cancer activity (IC50: 1.02−>25 µM) in HCT116 p53+/+ and p53−/− cells (Table 1).
A similar general trend in the anti-proliferative activity of the novel TSCs to those observed in HCT116 p53+/+ and p53−/− cells was also observed in Raji, HeLa and SK-N-MC cells (Table 1). Indeed, those chelators containing the di-2-pyridyl moiety (1a–e) were the most potent anti-cancer (IC50: 0.0003–2.21 µM) analogs examined. Similarly to the HCT116 data, compounds from series 2 generally displayed moderate anti-proliferative activity (IC50: 0.04–5.14 µM) in Raji, HeLa and SK-N-MC cells, while series 3, 4, 5 and 6 generally showed moderate to poor anti-proliferative effects (Table 1).
Only a weak to moderate correlation (R 2 = 0.01–0.6) was evident between the anti-cancer activity of series 1–6 and their log P calc values, suggesting that other factors besides their ability to transverse the cellular membrane by passive diffusion were critical in their anti-proliferative effects.
Anti-Proliferative Activity of the Novel Thiosemicarbazones Against Normal, Mortal Cells
Importantly, for an agent to be useful as an anti-cancer drug, it must show preferential anti-proliferative activity against tumor cells over normal, mortal cell-types. Hence, the selectivity of the novel TSCs was examined in mortal cells, namely NHDF cells. Of all the analogs examined, 1b, 1d, 2b, 2f and 3c showed the greatest anti-proliferative activity in the majority of cancer cell-types (Table 1). Thus, to examine the selectivity of the 5 best TSCs identified above, namely 1b, 1d, 2b, 2f and 3c, a “therapeutic index” was calculated by dividing the NHDF cell IC50 by the IC50 of the neoplastic HCT116 p53+/+ or HCT116 p53−/− cell-types (Table 2). Notably, the selectivity index of Dp44mT comparing NHDFs to HCT116 p53+/+ or HCT116 p53−/− cell-types was marked, being 7690 and 3076, respectively.
Table 2. The selectivity of Dp44mT relative to the 5 most potent anti-cancer TSCs, namely 1b, 1d, 2b, 2f and 3c, was examined by calculating their “therapeutic indices”.
| Therapeutic index | ||
| Chelator | NHDF vs. HCT116 p53+/+ | NHDF vs. HCT116 p53−/− |
| Dp44mT | 7690 | 3076 |
| 1b | 2 | 2 |
| 1d | 80 | 18 |
| 2b | 0.8 | 0.4 |
| 2f | 0.6 | 0.4 |
| 3c | 2650 | 624 |
The latter was calculated by dividing the NHDF cell IC50 by the IC50 of the neoplastic HCT116 p53+/+ or HCT116 p53−/− cell-types.
Of the 5 most potent anti-cancer TSCs described herein (Table 1), the greatest therapeutic indices were identified for 1d and 3c and were 18–2650 (Table 2). This high selectivity against the cancer cells was due to the fact that both 1d and 3c showed potent anti-cancer activity (IC50: 0.002–0.017 µM) in HCT116 cells, but their cytotoxicity was greatly reduced in NHDF cells (IC50: 0.16–10.6 µM). In contrast, analogs 1b, 2b and 2f demonstrated poor selectivity with IC50 values of 0.0008–0.09 µM in HCT116 cells and 0.0017–0.03 µM in mortal NHDF cells (Table 1). In fact, compounds 2b and 2f generally showed greater anti-proliferative effects in mortal NHDF cells relative to HCT116 cells, resulting in very low therapeutic indices of 0.4–0.8 (Table 2). Compound 1b demonstrated limited selectivity and resulted in a therapeutic index of 2 in mortal NHDF cells relative to HCT116 cells. Hence, of all the analogs examined, compounds 1d and 3c demonstrated the greatest promise as anti-cancer agents, having both potent and selective anti-proliferative effects.
Ability of Novel Thiosemicarbazones to Mobilize Cellular 59Fe
As the ability of chelators to bind cellular iron can play a role in their anti-proliferative effects [50], we examined the ability of these novel ligands (25 µM) to mobilize cellular 59Fe from prelabeled SK-N-MC cells (Fig. 6). The chelator-mediated release of intracellular 59Fe was compared to the positive controls, DFO and Dp44mT (Fig. 6), as their ability to mobilize 59Fe has been extensively characterized in these cells [10], [17], [18], [52], [53]. As previously observed [10], [17], [18], [52], [53], the control medium alone resulted in the release of very little 59Fe, namely 4±1% of cellular 59Fe (Fig. 6). The chelators, DFO and Dp44mT, significantly (p<0.001) increased the mobilization of cellular 59Fe to 11±2 and 35±2%, respectively, relative to the control medium (Fig. 6).
Figure 6. Effect of (A) series 1, (B) series 2, (C) series 3, (D) series 4, (E) series 5 and (F) series 6 chelators, relative to the controls DFO and Dp44mT, on 59Fe mobilization from prelabeled SK-N-MC cells.

Cells were incubated for 3 h/37°C with 59Fe-transferrin (0.75 µM), washed 4 times with ice-cold PBS and then reincubated for 3 h/37°C in the presence or absence of the chelators (25 µM). Release of 59Fe was then assessed using a γ-scintillation counter. Results are mean ±SD (3 experiments).
Compounds of series 1, 3, 4 and 6 generally displayed high efficacy at mobilizing cellular 59Fe (Fig. 6). In particular, the ligands, 1c, 1e, 3b–3f, 4a, 4c, and 6b–6f all demonstrated comparable 59Fe mobilization efficacy to that of Dp44mT, resulting in the efflux of 31–39% of cellular 59Fe. Apart from 1b, all ligands of series 1, 3, 4 and 6 were significantly (p<0.001) more effective than DFO in mediating the release of cellular 59Fe. Interestingly, ligands of series 1 and 4 demonstrated a similar pattern in terms of their ability to mobilize cellular 59Fe (Fig. 6A,D). In both series 1 and 4, the ligands containing the more hydrophilic fragments, a, c and e, showed increased efficacy at mobilizing cellular 59Fe relative to those containing the more lipophilic fragments b, d and f (Fig. 6A,D). Of all the ligands examined, chelators of series 3 and 6 demonstrated the greatest efficacy as 59Fe mobilization agents, mediating the release of 25–39% of cellular 59Fe (Fig. 6C,F).
In contrast, the series based on quinolin-2-yl (2) and quinoxalin-2-yl (5) generally demonstrated poor ability to mobilize cellular 59Fe and resulted in the release of 5–28% of cellular 59Fe (Fig. 6B,E). All ligands of series 2 and 5 were significantly (p<0.001) less effective than Dp44mT in mediating the release of cellular 59Fe. In fact, ligand 2a demonstrated comparable 59Fe mobilization efficacy to that of the control medium (Fig. 6B), while 2b, 2d, 2f and 5f showed comparable release of 59Fe relative to DFO (Fig. 6B,E).
Importantly, a positive correlation (R2 = 0.911) between log P calc and 59Fe mobilization efficacy was observed for ligands of series 1 (Fig. 7), suggesting that their lipophilic/hydrophilic balance played a role in their ability to permeate the cellular membrane to reach intracellular 59Fe. Interestingly, the more hydrophobic ligands of series 1 were less effective than their more hydrophilic counterparts, suggesting that the more hydrophobic ligands or their resultant 59Fe complexes may be sequestered in cellular membranes, preventing 59Fe release. In contrast, a poor to moderate correlation (R2 = 0.38–0.67) between log P calc and 59Fe mobilization efficacy was evident for series 2–6. A weak correlation (R2 = 0.37) was observed between the anti-proliferative activity of these novel ligands and their ability to mobilize cellular 59Fe in SK-N-MC cells, indicating that other factors besides their ability to induce 59Fe mobilization were responsible for their anti-cancer effects.
Figure 7. Relationship between the cellular iron released (% total) and lipophilicity (logP calc) of series 1 chelators using SK-N-MC neuroepithelioma cells.
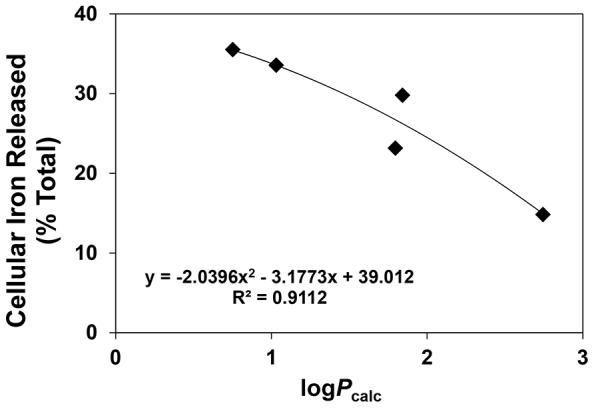
Lines were fitted in using Microsoft Excel 2010 (Microsoft, Redmond, WA).
Ability of Novel Thiosemicarbazones to Inhibit the Cellular Uptake of 59Fe from 59Fe2-Tf
The anti-proliferative activity and iron chelation efficacy of a ligand is dependent not only on its ability to mobilize cellular iron, but also on its ability to prevent the cellular uptake of iron from Fe2-Tf [50]. Thus, the ability of the novel ligands (25 µM) to inhibit the cellular uptake of 59Fe from 59Fe2-Tf was assessed in SK-N-MC cells (Fig. 8). As utilized in the 59Fe efflux experiments, the chelators, DFO and Dp44mT, were included as positive controls as their ability to inhibit 59Fe from 59Fe2-Tf has been extensively assessed [10], [14], [17], [18], [50], [52], [53]. The results were expressed as a percentage of the 59Fe uptake observed by cells incubated with 59Fe2-Tf in control medium (i.e., in the absence of chelator; Fig. 8).
Figure 8. Effect of (A) series 1, (B) series 2, (C) series 3, (D) series 4, (E) series 5 and (F) series 6 chelators, relative to the controls DFO and Dp44mT, on 59Fe uptake from 59Fe-transferrin by SK-N-MC cells.

Cells were incubated for 3 h/37°C with 59Fe-transferrin (0.75 µM) in the presence or absence of the chelators (25 µM). At the end of this incubation, cells were washed 4 times with ice-cold PBS. Internalization of 59Fe was assessed by incubation for 30 min/4°C with the protease, Pronase (1 mg/mL). Cellular 59Fe was then assessed using a γ-scintillation counter. Results are mean ±SD (3 experiments).
As shown previously [10], [52], DFO was able to significantly (p<0.001) inhibit 59Fe uptake from 59Fe2-Tf to 83% of that found for the control (Fig. 8), while Dp44mT significantly (p<0.001) and markedly prevented the uptake of 59Fe, reducing it to 6% of the control (Fig. 8). In general, those analogs that displayed high efficacy at mobilizing cellular 59Fe (Fig. 6) also potently inhibited the cellular uptake of 59Fe from 59Fe2-Tf (Fig. 8), with a linear correlation (R2 = 0.81) evident between these 2 factors (Fig. 9). Generally, the ligands of series 1, 3, 4 and 6 potently inhibited the uptake of 59Fe from 59Fe2-Tf and were significantly (p<0.001) more effective than DFO (Fig. 8A,C,D,F). In fact, the ligands, 1c, 3f, 5c, 6c and 6f demonstrated comparable inhibition of cellular 59Fe uptake to that of Dp44mT, resulting in 3.6–11.5% of cellular 59Fe uptake relative to the control (Fig. 8). Interestingly, those ligands containing the more hydrophilic fragment, c, were generally the most effective chelators of each series (Fig. 8).
Figure 9. Relationship between the internalized iron uptake (% control) and cellular iron released (% total) of series 1–6 using SK-N-MC neuroepithelioma cells.
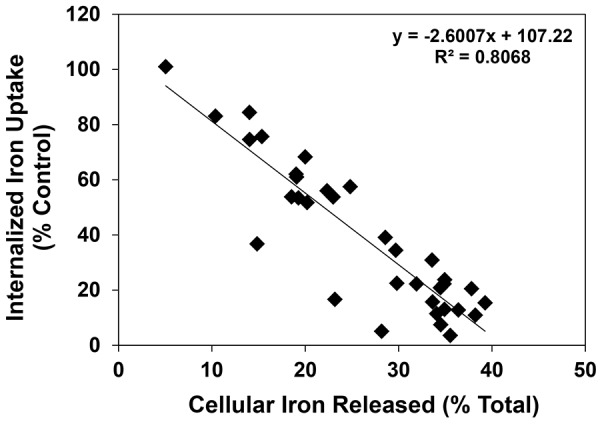
Lines were fitted in using Microsoft Excel 2010 (Microsoft, Redmond, WA).
Similarly to the 59Fe efflux experiments, those ligands based on the quinolin-2-yl (2) and quinoxalin-2-yl (5) moieties generally showed poor ability to prevent the uptake of 59Fe from 59Fe2-Tf (Fig. 8B,E). In fact, compound 2a showed comparable ability to inhibit 59Fe uptake to the control medium, while compounds 2b, 2d, 2f and 5f demonstrated comparable inhibition of 59Fe uptake to that of DFO (Fig. 8B,E). All analogs of series 2 and 5, except 5c, were significantly (p<0.001) less effective than Dp44mT at inhibiting cellular 59Fe uptake.
No strong correlation (R2 = 0.20–0.21) was observed between the ability of the ligands to prevent cellular 59Fe uptake and either log P calc or anti-proliferative activity in SK-N-MC cells, suggesting other factors besides inhibition of 59Fe uptake played a role in their anti-cancer activity.
Ability of the Iron Complexes of Novel Thiosemicarbazones to Mediate the Oxidation of Ascorbate
The ability of the iron complexes of the novel TSCs to catalyze the oxidation of the physiological substrate, ascorbate, was important to assess as redox cycling may play a critical role in their anti-proliferative activity [10], [15], [18], [52], [53]. Thus, the oxidation of ascorbate mediated by the iron complexes of series 1–6 were examined in comparison to the iron complexes of the control compounds, DFO, EDTA and Dp44mT, as the ability of the latter to oxidize ascorbate is well characterized [10], [15], [52], [53], [68]. The results were expressed as a percentage of the control (no chelator) at iron-binding equivalents (IBEs) of 0.1 (excess “free” iron), 1 (iron-chelator complexes with a fully filled coordination sphere), or 3 (excess free chelator), due to the different denticity of the chelators examined (Fig. 10).
Figure 10. Effect of the iron complexes of (A) series 1, (B) series 2, (C) series 3, (D) series 4, (E) series 5 and (F) series 6 chelators, relative to DFO, Dp44mT and EDTA, on ascorbate oxidation.

Chelators at iron-binding equivalent (IBE) ratios of 0.1, 1, and 3 were incubated in the presence of FeIII (10 µM) and ascorbate (100 µM). The UV-Vis absorbance at 265 nm was recorded after 10 and 40 min, and the difference between the time points was calculated. Results are mean ±SD (3 experiments).
As previously observed, the redox-inactive iron complex of DFO resulted in very limited ascorbate oxidation (Fig. 10) [15], [68], with the ligand acting in a protective manner, significantly (p<0.05) inhibiting ascorbate oxidation at IBEs of 1 and 3 to 47% and 23% of the control, respectively (Fig. 10). In contrast, the iron complex of the positive control, EDTA, significantly (p<0.001) increased ascorbate oxidation at IBEs of 0.1, 1 and 3 to 208%, 449% and 936%, respectively, in comparison to the control (Fig. 10). This is in agreement with our previous studies that showed its ability to mediate ascorbate oxidation [10], [15], [52], [53]. Additionally, the Dp44mT-iron complex also mediated ascorbate oxidation, significantly (p<0.01–0.001) increasing it to 129%, 203% and 196% at IBEs of 0.1, 1 and 3, respectively, relative to the control (Fig. 10), as demonstrated before [10], [15], [52], [53].
The iron complexes of series 1 and 2 generally enhanced ascorbate oxidation, with compounds 1b, 1d, 1e, 2a and 2e significantly (p<0.05–0.001) increasing ascorbate oxidation to 200–265% of the control at an IBE of 3 (Fig. 10A,B). In fact, all iron chelator complexes of series 1 and 2, except for 2f, showed comparable levels of ascorbate oxidation to the Dp44mT-iron complex at an IBE of 3 (Fig 10A,B). These data suggest that the ligands of series 1 and 2 form iron complexes that are redox active.
In contrast to series 1 and 2, the iron complexes of series 3, 4, 5 and 6 generally did not enhance ascorbate oxidation, with the majority acting in a protective manner (Fig. 10C,D,E,F). Although the iron complexes of 3a and 5e mediated significantly (p<0.01–0.001) increased levels of ascorbate oxidation, the majority of the iron complexes of series 3, 4, 5 and 6 resulted in comparable ability to catalyze ascorbate oxidation relative to the control at an IBE of 3. In fact, the iron complexes of 5d, 5f, 6a, 6d and 6f significantly (p<0.05) decreased ascorbate oxidation relative to the control at an IBE of 3. Additionally, all iron complexes of series 3, 4, 5 and 6, apart from 3a, 4a, 4e and 5e, mediated comparable ascorbate oxidation to that of DFO at an IBE of 3, resulting in 4–111% of ascorbate oxidation relative to the control (Fig. 10C,D,E,F). Thus, the majority of iron complexes of series 3, 4, 5 and 6 acted in a protective manner, suggesting the formation of redox inactive iron complexes.
Structure-Activity Relationships Linking Anti-Proliferative Activity, Ascorbate Oxidation and Donor Atom Identity
In the current study, series 1 analogs based on the di-2-pyridyl moiety (Fig. 5) generally demonstrated the greatest anti-proliferative activity of all the novel ligands examined (Table 1). Series 1 ligands, that utilize the N,N,S donor atom set, showed high iron chelation efficacy (Figs. 6A,8A) and their iron complexes generally resulted in the increased oxidation of ascorbate (Fig. 10A). These data suggest that the potent anti-proliferative activity of series 1 stemmed from their ability to effectively chelate cellular iron and result in the formation of redox active iron complexes that mediate oxidative damage. This is in agreement with our previous studies on other di-2-pyridyl-based thiosemicarbazones, such as Dp44mT and DpC [12], [15], and di-2-pyridyl-based thiohydrazones [52], which also utilize the N,N,S donor atom set and demonstrate potent anti-cancer activity via the formation of redox active complexes. The use of “soft” donor atoms, such as nitrogen and sulfur, play a critical role in facilitating reversible FeIII/II redox cycling reactions that are important for potent anti-cancer effects [12], [15], [52].
The ligands of series 2, based on the quinolin-2-yl moiety (Fig. 5), also utilized the N,N,S donor atom set and their resultant iron complexes generally increased levels of ascorbate oxidation (Fig. 10B). This suggested that, similarly to series 1, ligands of series 2 may form redox active iron complexes. However, in contrast to series 1, series 2 compounds demonstrated poor iron chelation efficacy, which may be a contributing factor in the moderate anti-proliferative activity observed for series 2 relative to series 1 (Table 1). This is consistent with our previous studies, in which the presence of the quinolin-2-yl moiety in 2-quinolinecarboxaldehyde isonicotinoyl hydrazones [59] and other quinoline-based thiosemicarbazones [16] were found to have poor iron mobilization efficacy and anti-proliferative effects in SK-N-MC cells. Our current data support these previous findings, suggesting that the quinolin-2-yl moiety confers poor anti-proliferative and iron chelation efficacy when the quinoline nitrogen acts as a donor atom [16], [59].
Series 3 analogs, that differ from series 2 by the addition of a hydroxy group at position 8 of the quinoline ring (Fig. 5), generally demonstrated moderate to poor anti-proliferative activity (Table 1). In contrast to series 2 that showed poor iron mobilization efficacy and the ability to mediate ascorbate oxidation, series 3 ligands displayed high iron chelation efficacy (Figs. 6C,8C). However, their iron complexes generally did not enhance the oxidation of ascorbate (Fig. 10C). In fact, the majority of series 3 iron complexes acted in a protective manner and inhibited ascorbate oxidation (Fig. 10C). It is probable that series 3 ligands can bind iron either in a tridentate manner, using the N,N,S donor atom set similarly to series 2, or in a bidentate system similar to that of the structurally-related chelator, clioquinol [69] that utilizes the quinoline nitrogen and 8-hydroxy oxygen as donor atoms [70]. Due to the contrasting activity of series 3 relative to series 2 with regards to anti-proliferative effects (Table 1), iron chelation efficacy (Figs. 6&8) and ascorbate oxidation (Fig. 10), this suggests that the series 3 analogs may act as bidentate ligands that utilize the 8-hydroxyl oxygen and quinoline nitrogen as donor atoms. Significantly, the use of the “hard” oxygen donor atom in series 3 may result in the formation of redox-inactive iron complexes that cannot oxidize ascorbate and consequently show poor anti-proliferative activity [1], [52].
Those ligands based on the 7-hydroxyquinolin-8-yl (series 4) moiety bind iron in an analogous manner to that of 2-hydroxynaphthaldehyde thiosemicarbazone [71] or 2-hydroxy-1-naphthaldehyde thiobenzoyl hydrazone (H2NTBH; [52]) and utilize the O,N,S donor atom set [16]. Similarly, those chelators of series 6, based on the salicylic moiety, also bind iron via the O,N,S donor atom set in a similar manner to salicylaldehyde thiobenzoyl hydrazone (H2STBH; [52]). Although the series 4 and 6 analogs demonstrated high iron mobilization efficacy (Figs. 6&8), these ligands showed poor anti-proliferative effects (Table 1) and their iron complexes did not mediate or prevented the oxidation of ascorbate (Fig. 10). This is in agreement with our previous studies in which the O,N,S-thiohydrazones, H2STBH or H2NTBH, mediated high iron chelation efficacy, but showed poor anti-proliferative activity and the inability of their iron complexes to mediate the oxidation of ascorbate [52]. Importantly, these results suggest that iron complexes of series 4 and 6 ligands cannot mediate the formation of ROS, which greatly reduces their anti-proliferative effects and this relates to the inclusion of the hard oxygen donor (Fig. 5).
The analogs based on quinoxalin-2-yl (5a–f) utilize the N,N,S donor atoms and differ from series 2 by an additional non-coordinating nitrogen located at position 4 of the quinoline moiety (Fig. 5). This structural modification resulted in a large decrease in the anti-proliferative activity of series 5 relative to series 2 (Table 1), although both series showed poor iron mobilization efficacy (Figs. 6&8). Importantly, the iron complexes of series 5 acted in a protective manner and inhibited the oxidation of ascorbate, while iron complexes of series 2 were able to promote ascorbate oxidation (Fig. 10). This further highlights the critical role of the formation of redox active iron complexes in the anti-proliferative activity of novel thiosemicarbazones [10], [15], [21], [52]. Significantly, our previous studies on methyl pyrazinylketone isonicotinoyl hydrazone (MPIH) analogs demonstrated that the incorporation of a second, non-coordinating, electron-withdrawing nitrogen in the aromatic ring played a major role in the formation of redox-inactive iron complexes that prevented the formation of ROS [53], [72]. Similarly to series 5, the iron complexes of the MPIH series prevented the oxidation of ascorbate [53]. Thus, the replacement of the quinoline moiety of series 2 with the quinoxaline group of series 5 resulted in decreased anti-proliferative activity due to the formation of redox-inactive iron complexes.
Conclusions
In the current study, several novel classes of TSCs were designed that retained the appropriate MW and log P calc values to show promise as drug candidates for pharmaceutical development. A combination of retro-fragments that appear in other TSC precursors were utilized and di-substitution at the terminal N4 atom was preserved through the incorporation of an N4-based piperazine or morpholine ring. The selectivity and anti-cancer activity of the novel TSCs were examined in a variety of cancer cell-types. In particular, of all the compounds examined, 1d and 3c demonstrated the greatest promise as anti-cancer agents with both potent and selective anti-proliferative activity (Tables 1 and 2).
Structure-activity relationship studies revealed that the combination of the donor atoms used, rather than the identity of fragments a–f, played a crucial role in their anti-cancer activity. Indeed, the chelators that utilized “soft” donor atoms, such as nitrogen and sulfur, formed redox active iron complexes capable of mediating ascorbate oxidation. This further highlights the important role of ROS generation in mediating potent anti-cancer effects. Significantly, this study identified potent and selective anti-cancer chelators that warrant further in vivo examination.
Supporting Information
This file reports the chemical characterization of thiosemicarbazides and thiosemicarbazones, X-ray data for selected thiosemicarbazones and thiosemicarbazides, HPLC purity data, isosbestic curves and anti-proliferative activity color maps. Figure S1, The crystal structure of 4-ethylpiperazine-1-carbothiohydrazide (a). Figure S2, The crystal structure of Z-N′-(di(pyridin-2-yl)methylene)-4-(pyridin-2-yl)piperazine-1-carbothiohydrazide (1d). Figure S3, The absorbance spectrum of 2f and its Fe3+ complexes prepared in situ to obtain 1∶1, 2∶1, 4∶1, 5∶1, and 10∶1 ligand:Fe ratios. Figure S4, Color maps of the anti-proliferative activity of series 1–6 in several tumor cell-types and normal human dermal fibroblast (NHDF) cells. Red represents the thiosemicarbazones with the greatest anti-proliferative activity (IC50: <1 µM), yellow represents the thiosemicarbazones with moderate activity (IC50: 1–6.25 µM) and grey represents those analogs with poor anti-proliferative effects (IC50>6.25 µM). Table S1, Crystal data of a and 1d. Table S2, HPLC purity data for all chelators of series 1–6.
(PDF)
Acknowledgments
We acknowledge Drs. Zaklina Kovacevic, Patric J. Jansson, Angelica M. Merlot and Vera Richardson for their feedback prior to submission.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information file.
Funding Statement
The authors appreciated the financial support of TWING and DoktoRIS PhD scholarships, NCN (DEC-2011/01/N/NZ4/01166, 2013/09/B/NZ7/00423 and N405/068440), and the National Centre for Research and Development, Warsaw (ORGANOMET No: PBS2/A5/40/2014 grants). This work was also supported by a Project Grant from the National Health and Medical Research Council (NHMRC) Australia to DRR [Grant 632778]; and DSK [Grant 1048972]; a NHMRC Senior Principal Research Fellowship to DRR [Grant 571123]; and a Helen and Robert Ellis Fellowship to DSK from the Sydney Medical School Foundation of The University of Sydney. EP and TS also appreciate the support of a Czech Science Foundation Grant [13-15008S]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kalinowski DS, Richardson DR (2005) The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev 57: 547–583. [DOI] [PubMed] [Google Scholar]
- 2. Lieu PT, Heiskala M, Peterson PA, Yang Y (2001) The roles of iron in health and disease. Mol Aspects Med 22: 1–87. [DOI] [PubMed] [Google Scholar]
- 3. Merlot AM, Kalinowski DS, Richardson DR (2013) Novel chelators for cancer treatment: where are we now? Antioxid Redox Signal 18: 973–1006. [DOI] [PubMed] [Google Scholar]
- 4. Kolberg M, Strand KR, Graff P, Andersson KK (2004) Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta 1699: 1–34. [DOI] [PubMed] [Google Scholar]
- 5. Thelander L, Reichard P (1979) Reduction of ribonucleotides. Annu Rev Biochem 48: 133–158. [DOI] [PubMed] [Google Scholar]
- 6. Richardson D, Baker E (1992) Two mechanisms of iron uptake from transferrin by melanoma cells. The effect of desferrioxamine and ferric ammonium citrate. J Biol Chem 267: 13972–13979. [PubMed] [Google Scholar]
- 7. Richardson DR, Baker E (1990) The uptake of iron and transferrin by the human malignant melanoma cell. Biochim Biophys Acta 1053: 1–12. [DOI] [PubMed] [Google Scholar]
- 8. Trinder D, Zak O, Aisen P (1996) Transferrin receptor-independent uptake of differic transferrin by human hepatoma cells with antisense inhibition of receptor expression. Hepatology 23: 1512–1520. [DOI] [PubMed] [Google Scholar]
- 9. Elford HL, Freese M, Passamani E, Morris HP (1970) Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J Biol Chem 245: 5228–5233. [PubMed] [Google Scholar]
- 10. Kalinowski DS, Yu Y, Sharpe PC, Islam M, Liao YT, et al. (2007) Design, synthesis, and characterization of novel iron chelators: structure-activity relationships of the 2-benzoylpyridine thiosemicarbazone series and their 3-nitrobenzoyl analogues as potent antitumor agents. J Med Chem 50: 3716–3729. [DOI] [PubMed] [Google Scholar]
- 11. Kunos C, Radivoyevitch T, Abdul-Karim FW, Fanning J, Abulafia O, et al. (2012) Ribonucleotide reductase inhibition restores platinum-sensitivity in platinum-resistant ovarian cancer: a Gynecologic Oncology Group Study. J Transl Med 10: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lovejoy DB, Sharp DM, Seebacher N, Obeidy P, Prichard T, et al. (2012) Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J Med Chem 55: 7230–7244. [DOI] [PubMed] [Google Scholar]
- 13. Lukmantara AY, Kalinowski DS, Kumar N, Richardson DR (2013) Synthesis and biological evaluation of substituted 2-benzoylpyridine thiosemicarbazones: novel structure-activity relationships underpinning their anti-proliferative and chelation efficacy. Bioorg Med Chem Lett 23: 967–974. [DOI] [PubMed] [Google Scholar]
- 14. Richardson DR, Kalinowski DS, Richardson V, Sharpe PC, Lovejoy DB, et al. (2009) 2-Acetylpyridine thiosemicarbazones are potent iron chelators and antiproliferative agents: redox activity, iron complexation and characterization of their antitumor activity. J Med Chem 52: 1459–1470. [DOI] [PubMed] [Google Scholar]
- 15. Richardson DR, Sharpe PC, Lovejoy DB, Senaratne D, Kalinowski DS, et al. (2006) Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J Med Chem 49: 6510–6521. [DOI] [PubMed] [Google Scholar]
- 16. Serda M, Kalinowski DS, Mrozek-Wilczkiewicz A, Musiol R, Szurko A, et al. (2012) Synthesis and characterization of quinoline-based thiosemicarbazones and correlation of cellular iron-binding efficacy to anti-tumor efficacy. Bioorg Med Chem Lett 22: 5527–5531. [DOI] [PubMed] [Google Scholar]
- 17. Stefani C, Jansson PJ, Gutierrez E, Bernhardt PV, Richardson DR, et al. (2013) Alkyl substituted 2′-benzoylpyridine thiosemicarbazone chelators with potent and selective anti-neoplastic activity: novel ligands that limit methemoglobin formation. J Med Chem 56: 357–370. [DOI] [PubMed] [Google Scholar]
- 18. Stefani C, Punnia-Moorthy G, Lovejoy DB, Jansson PJ, Kalinowski DS, et al. (2011) Halogenated 2′-benzoylpyridine thiosemicarbazone (XBpT) chelators with potent and selective anti-neoplastic activity: relationship to intracellular redox activity. J Med Chem 54: 6936–6948. [DOI] [PubMed] [Google Scholar]
- 19. Yu Y, Kalinowski DS, Kovacevic Z, Siafakas AR, Jansson PJ, et al. (2009) Thiosemicarbazones from the old to new: iron chelators that are more than just ribonucleotide reductase inhibitors. J Med Chem 52: 5271–5294. [DOI] [PubMed] [Google Scholar]
- 20. Yu Y, Suryo Rahmanto Y, Richardson DR (2012) Bp44mT: an orally active iron chelator of the thiosemicarbazone class with potent anti-tumour efficacy. Br J Pharmacol 165: 148–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuan J, Lovejoy DB, Richardson DR (2004) Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood 104: 1450–1458. [DOI] [PubMed] [Google Scholar]
- 22. Chaston TB, Lovejoy DB, Watts RN, Richardson DR (2003) Examination of the antiproliferative activity of iron chelators: multiple cellular targets and the different mechanism of action of triapine compared with desferrioxamine and the potent pyridoxal isonicotinoyl hydrazone analogue 311. Clin Cancer Res 9: 402–414. [PubMed] [Google Scholar]
- 23. Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P, et al. (2011) Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res 71: 5871–5880. [DOI] [PubMed] [Google Scholar]
- 24. Shao J, Zhou B, Di Bilio AJ, Zhu L, Wang T, et al. (2006) A Ferrous-Triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol Cancer Ther 5: 586–592. [DOI] [PubMed] [Google Scholar]
- 25. Zhu L, Zhou B, Chen X, Jiang H, Shao J, et al. (2009) Inhibitory mechanisms of heterocyclic carboxaldehyde thiosemicabazones for two forms of human ribonucleotide reductase. Biochem Pharmacol 78: 1178–1185. [DOI] [PubMed] [Google Scholar]
- 26. Kovacevic Z, Chikhani S, Lovejoy DB, Richardson DR (2011) Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1: a new strategy for the treatment of pancreatic cancer. Mol Pharmacol 80: 598–609. [DOI] [PubMed] [Google Scholar]
- 27. Kovacevic Z, Chikhani S, Lui GY, Sivagurunathan S, Richardson DR (2013) The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid Redox Signal 18: 874–887. [DOI] [PubMed] [Google Scholar]
- 28. Kovacevic Z, Sivagurunathan S, Mangs H, Chikhani S, Zhang D, et al. (2011) The metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1), upregulates p21 via p53-independent mechanisms. Carcinogenesis 32: 732–740. [DOI] [PubMed] [Google Scholar]
- 29. Le NT, Richardson DR (2004) Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood 104: 2967–2975. [DOI] [PubMed] [Google Scholar]
- 30. Bernhardt PV, Sharpe PC, Islam M, Lovejoy DB, Kalinowski DS, et al. (2009) Iron chelators of the dipyridylketone thiosemicarbazone class: precomplexation and transmetalation effects on anticancer activity. J Med Chem 52: 407–415. [DOI] [PubMed] [Google Scholar]
- 31. Feun L, Modiano M, Lee K, Mao J, Marini A, et al. (2002) Phase I and pharmacokinetic study of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) using a single intravenous dose schedule. Cancer Chemother Pharmacol 50: 223–229. [DOI] [PubMed] [Google Scholar]
- 32. Karp JE, Giles FJ, Gojo I, Morris L, Greer J, et al. (2008) A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk Res 32: 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kunos CA, Radivoyevitch T, Waggoner S, Debernardo R, Zanotti K, et al. (2013) Radiochemotherapy plus 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in advanced-stage cervical and vaginal cancers. Gynecol Oncol 130: 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kunos CA, Waggoner S, von Gruenigen V, Eldermire E, Pink J, et al. (2010) Phase I trial of pelvic radiation, weekly cisplatin, and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) for locally advanced cervical cancer. Clin Cancer Res 16: 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma B, Goh BC, Tan EH, Lam KC, Soo R, et al. (2008) A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Invest New Drugs 26: 169–173. [DOI] [PubMed] [Google Scholar]
- 36. Mackenzie MJ, Saltman D, Hirte H, Low J, Johnson C, et al. (2007) A Phase II study of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) and gemcitabine in advanced pancreatic carcinoma. A trial of the Princess Margaret hospital Phase II consortium. Invest New Drugs 25: 553–558. [DOI] [PubMed] [Google Scholar]
- 37. Nutting CM, van Herpen CM, Miah AB, Bhide SA, Machiels JP, et al. (2009) Phase II study of 3-AP Triapine in patients with recurrent or metastatic head and neck squamous cell carcinoma. Ann Oncol 20: 1275–1279. [DOI] [PubMed] [Google Scholar]
- 38. Ocean AJ, Christos P, Sparano JA, Matulich D, Kaubish A, et al. (2010) Phase II trial of the ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehydethiosemicarbazone plus gemcitabine in patients with advanced biliary tract cancer. Cancer Chemother Pharmacol 68: 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Odenike OM, Larson RA, Gajria D, Dolan ME, Delaney SM, et al. (2008) Phase I study of the ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde-thiosemicarbazone (3-AP) in combination with high dose cytarabine in patients with advanced myeloid leukemia. Invest New Drugs 26: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wadler S, Makower D, Clairmont C, Lambert P, Fehn K, et al. (2004) Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. J Clin Oncol 22: 1553–1563. [DOI] [PubMed] [Google Scholar]
- 41. Yen Y, Margolin K, Doroshow J, Fishman M, Johnson B, et al. (2004) A phase I trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone in combination with gemcitabine for patients with advanced cancer. Cancer Chemother Pharmacol 54: 331–342. [DOI] [PubMed] [Google Scholar]
- 42. Whitnall M, Howard J, Ponka P, Richardson DR (2006) A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A 103: 14901–14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mrozek-Wilczkiewicz A, Serda M, Musiol R, Malecki G, Szurko A, et al. (2014) Iron chelators in photodynamic therapy revisited: Synergistic effect by novel highly active thiosemicarbazones. ACS Med Chem Lett 5: 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Serda M, Mrozek-Wilczkiewicz A, Jampilek J, Pesko M, Kralova K, et al. (2012) Investigation of the biological properties of (hetero)aromatic thiosemicarbazones. Molecules 17: 13483–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hu WX, Zhou W, Xia CN, Wen X (2006) Synthesis and anticancer activity of thiosemicarbazones. Bioorg Med Chem Lett 16: 2213–2218. [DOI] [PubMed] [Google Scholar]
- 46. Stanojkovic TP, Kovala-Demertzi D, Primikyri A, Garcia-Santos I, Castineiras A, et al. (2010) Zinc(II) complexes of 2-acetyl pyridine 1-(4-fluorophenyl)-piperazinyl thiosemicarbazone: Synthesis, spectroscopic study and crystal structures - potential anticancer drugs. J Inorg Biochem 104: 467–476. [DOI] [PubMed] [Google Scholar]
- 47. Ghose AK, Crippen GM (1987) Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure−activity relationships. 2. Modeling dispersive and hydrophobic interactions. J Chem Inf Comput Sci 27: 21–35. [DOI] [PubMed] [Google Scholar]
- 48. Viswanadhan VN, Ghose AK, Revankar GR, Robins RK (1987) Atomic physiochemical parameters for 3-dimensional-structure directed quantitative structure−activity relationships 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally-occuring nucleoside antibiotics. J Chem Inf Comput Sci 29: 163–172. [Google Scholar]
- 49. Broto P, Moreau G, Vandycke C (1984) Molecular structures: Perception, autocorrelation descriptor and SAR studies. System of atomic contributions for the calculation of the n-octanol/water partition coefficients. Eur J Med Chem Chim Theor 19: 71–78. [Google Scholar]
- 50. Richardson DR, Tran EH, Ponka P (1995) The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood 86: 4295–4306. [PubMed] [Google Scholar]
- 51. Baker E, Richardson D, Gross S, Ponka P (1992) Evaluation of the iron chelation potential of hydrazones of pyridoxal, salicylaldehyde and 2-hydroxy-1-naphthylaldehyde using the hepatocyte in culture. Hepatology 15: 492–501. [DOI] [PubMed] [Google Scholar]
- 52. Kalinowski DS, Sharpe PC, Bernhardt PV, Richardson DR (2007) Design, synthesis, and characterization of new iron chelators with anti-proliferative activity: structure-activity relationships of novel thiohydrazone analogues. J Med Chem 50: 6212–6225. [DOI] [PubMed] [Google Scholar]
- 53. Kalinowski DS, Sharpe PC, Bernhardt PV, Richardson DR (2008) Structure-activity relationships of novel iron chelators for the treatment of iron overload disease: the methyl pyrazinylketone isonicotinoyl hydrazone series. J Med Chem 51: 331–344. [DOI] [PubMed] [Google Scholar]
- 54. Hann MM, Keseru GM (2012) Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discov 11: 355–365. [DOI] [PubMed] [Google Scholar]
- 55. Meanwell NA (2011) Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol 24: 1420–1456. [DOI] [PubMed] [Google Scholar]
- 56. Gleeson MP (2008) Generation of a set of simple, interpretable ADMET rules of thumb. J Med Chem 51: 817–834. [DOI] [PubMed] [Google Scholar]
- 57. Hann MM (2011) Molecular obesity, potency and other addictions in drug discovery. Med Chem Commun 2: 349–355. [Google Scholar]
- 58.Proschak E, Tanrikulu Y, Schneider G (2008) Chapter 7: Fragment-based de novo design of drug-like molecules. In: Varnek A, Tropsha A, editors.Chemoinformatics approaches to virtual screening.Cambridge: Royal Society of Chemistry. pp.217–239. [Google Scholar]
- 59. Becker EM, Lovejoy DB, Greer JM, Watts R, Richardson DR (2003) Identification of the di-pyridyl ketone isonicotinoyl hydrazone (PKIH) analogues as potent iron chelators and anti-tumour agents. Br J Pharmacol 138: 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Y, Ai J, Chen Y, Wang L, Liu G, et al. (2011) Synthesis and c-Met kinase inhibition of 3,5-disubstituted and 3,5,7-trisubstituted quinolines: identification of 3-(4-acetylpiperazin-1-yl)-5-(3-nitrobenzylamino)-7- (trifluoromethyl)quinoline as a novel anticancer agent. J Med Chem 54: 2127–2142. [DOI] [PubMed] [Google Scholar]
- 61. Chetan B, Bunha M, Jagrat M, Sinha BN, Saiko P, et al. (2010) Design, synthesis and anticancer activity of piperazine hydroxamates and their histone deacetylase (HDAC) inhibitory activity. Bioorg Med Chem Lett 20: 3906–3910. [DOI] [PubMed] [Google Scholar]
- 62. Hou X, Ge Z, Wang T, Guo W, Cui J, et al. (2006) Dithiocarbamic acid esters as anticancer agent. Part 1: 4-substituted-piperazine-1-carbodithioic acid 3-cyano-3,3-diphenyl-propyl esters. Bioorg Med Chem Lett 16: 4214–4219. [DOI] [PubMed] [Google Scholar]
- 63. Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, et al. (2009) Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res 69: 6232–6240. [DOI] [PubMed] [Google Scholar]
- 64. Gao H, Yamasaki EF, Chan KK, Shen LL, Snapka RM (2003) DNA sequence specificity for topoisomerase II poisoning by the quinoxaline anticancer drugs XK469 and CQS. Mol Pharmacol 63: 1382–1388. [DOI] [PubMed] [Google Scholar]
- 65.Silva JL, Gallo CV, Costa DC, Rangel LP (2014) Prion-like aggregation of mutant p53 in cancer. Trends Biochem Sci In Press April 25, 2014. [DOI] [PubMed]
- 66. Breen L, Heenan M, Amberger-Murphy V, Clynes M (2007) Investigation of the role of p53 in chemotherapy resistance of lung cancer cell lines. Anticancer Res 27: 1361–1364. [PubMed] [Google Scholar]
- 67. Whibley C, Pharoah PD, Hollstein M (2009) p53 polymorphisms: cancer implications. Nat Rev Cancer 9: 95–107. [DOI] [PubMed] [Google Scholar]
- 68. Mladenka P, Kalinowski DS, Haskova P, Bobrovova Z, Hrdina R, et al. (2009) The novel iron chelator, 2-pyridylcarboxaldehyde 2-thiophenecarboxyl hydrazone, reduces catecholamine-mediated myocardial toxicity. Chem Res Toxicol 22: 208–217. [DOI] [PubMed] [Google Scholar]
- 69. Deraeve C, Pitie M, Meunier B (2006) Influence of chelators and iron ions on the production and degradation of H2O2 by beta-amyloid-copper complexes. J Inorg Biochem 100: 2117–2126. [DOI] [PubMed] [Google Scholar]
- 70. Di Vaira M, Bazzicalupi C, Orioli P, Messori L, Bruni B, et al. (2004) Clioquinol, a drug for Alzheimer's disease specifically interfering with brain metal metabolism: Structural characterization of Its zinc(II) and copper(II) complexes. Inorg Chem 43: 3795–3797. [DOI] [PubMed] [Google Scholar]
- 71. Lovejoy DB, Richardson DR (2002) Novel "hybrid" iron chelators derived from aroylhydrazones and thiosemicarbazones demonstrate selective antiproliferative activity against tumor cells. Blood 100: 666–676. [DOI] [PubMed] [Google Scholar]
- 72. Bernhardt PV, Wilson GJ, Sharpe PC, Kalinowski DS, Richardson DR (2008) Tuning the antiproliferative activity of biologically active iron chelators: characterization of the coordination chemistry and biological efficacy of 2-acetylpyridine and 2-benzoylpyridine hydrazone ligands. J Biol Inorg Chem 13: 107–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file reports the chemical characterization of thiosemicarbazides and thiosemicarbazones, X-ray data for selected thiosemicarbazones and thiosemicarbazides, HPLC purity data, isosbestic curves and anti-proliferative activity color maps. Figure S1, The crystal structure of 4-ethylpiperazine-1-carbothiohydrazide (a). Figure S2, The crystal structure of Z-N′-(di(pyridin-2-yl)methylene)-4-(pyridin-2-yl)piperazine-1-carbothiohydrazide (1d). Figure S3, The absorbance spectrum of 2f and its Fe3+ complexes prepared in situ to obtain 1∶1, 2∶1, 4∶1, 5∶1, and 10∶1 ligand:Fe ratios. Figure S4, Color maps of the anti-proliferative activity of series 1–6 in several tumor cell-types and normal human dermal fibroblast (NHDF) cells. Red represents the thiosemicarbazones with the greatest anti-proliferative activity (IC50: <1 µM), yellow represents the thiosemicarbazones with moderate activity (IC50: 1–6.25 µM) and grey represents those analogs with poor anti-proliferative effects (IC50>6.25 µM). Table S1, Crystal data of a and 1d. Table S2, HPLC purity data for all chelators of series 1–6.
(PDF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information file.