

Abstract
Acquired resistance of cancer cells to anti-cancer drugs or ionizing radiation (IR) is one of the major obstacles in cancer treatment. Pancreatic cancer is an exceptional aggressive cancer, and acquired drug resistance in this cancer is common. Reactive oxygen species (ROS) play an essential role in cell apoptosis, which is a key mechanism by which radio- or chemo-therapy induces cell killing. Mitochondria are the major source of ROS in cells. Thus, alterations in the expression of mitochondrial proteins involved in ROS production or scavenging may be closely linked to the resistance of cancer cells to radio- or chemo-therapy. In the present study, we generated a stable cell line by exposing pancreatic cancer cells to increasing concentrations of ROS-inducing, anti-cancer compound 2-methoxyestradiol (2-ME) over a three month period. The resulting cell line showed strong resistance to 2-ME and contained an elevated level of ROS. We then used a comparative proteomics method to profile the differential expression of mitochondrial proteins between the parental and the resistant cells. One protein identified to be upregulated in the resistant cells was manganese superoxide dismutase (SOD2), a mitochondrial protein that converts superoxide radicals to hydrogen peroxides. Silencing of SOD2 re-sensitized the resistant cells to 2-ME, and overexpression of SOD2 led the parental cells to 2-ME resistance. In addition, the 2-ME-resistant cells also demonstrated resistance to IR. Our results suggest that upregulation of SOD2 expression is an important mechanism by which pancreatic cancer cells acquire resistance to ROS-inducing, anti-cancer drugs, and potentially also to IR.
Keywords: Acquired drug resistance, pancreatic cancer, ROS, SOD2, 2-methoxyestradiol
Introduction
Pancreatic cancer is an exceptionally aggressive cancer, with the 5-year survival rate of ~2% and median survival after diagnosis of less than 6 months (1). Intrinsic and acquired resistance of most pancreatic cancer patients to radio- and chemo-therapy is one of the major challenges in pancreatic cancer treatment. Currently less than 10% of pancreatic cancer patients benefit from radiation therapy, and 20% of patients from chemotherapy (2, 3). Due to lack or poor therapeutic responses of pancreatic cancer to therapies, the long-term survival rate for patients with organ-confined pancreatic cancer is only 20%, much lower than those for many other major types of cancers (4). Thus, understanding the mechanisms underlying the resistance of pancreatic cancer to radio- and chemo-therapy, and developing novel treatment strategies directed against this devastating disease are greatly needed.
Reactive oxygen species (ROS), such as superoxide (O2•−), hydrogen peroxide (H2O2) and hydroxyl radical (HO•) are the reactive chemical molecules that are constantly produced and eliminated in all living cells in a balanced manner. A loss of the balance between production and elimination would have severe consequences for cells. A moderate increase of cellular ROS accumulation stimulates cell growth and proliferation and promotes the development of cancers (5). However, acute and excessive ROS accumulation triggers apoptotic pathways and leads to cell death (6). Due to oncogenic signaling, increased metabolic activity, mitochondrial malfunction and the hypoxia growth environment of cancer cells, cancer cells usually contain elevated levels of ROS compared with normal cells (7, 8). This unique difference between cancer cells and healthy cells may provide opportunities to develop novel therapeutic strategies to selectively kill cancer cells by inducing cellular ROS burst (6, 7, 9). Ionizing radiation (IR) and many anti-cancer drugs are known to kill cancer cells largely through ROS-mediated apoptosis (10). Thus, identification of the proteins/enzymes that play a critical role in ROS metabolism in relation to cancer drug-resistance may provide valuable information on designing novel anti-cancer drugs to overcome chemo- and radio-resistance in cancer.
Mitochondria are the organelles where most cellular ROS are generated and play a central role in ROS-mediated apoptosis (11). Approximately 90% of the oxygen consumed within a eukaryote is used in mitochondrial respiration, and about 1–2% of the overall oxygen consumption is used for producing ROS (12). ROS formation in mitochondria is mainly due to electron leakage naturally occurring in complexes I and III of the respiratory chain located in the mitochondrial membrane (12). Incomplete oxygen reduction within the mitochondrial respiration chain leads to the formation of the first molecule of ROS, superoxide radicals (O2•−). The superoxide radicals are then dismutased to produce hydrogen peroxides (H2O2) by mitochondrial manganese superoxide dismutase (SOD2), and the produced H2O2 is further converted into water through glutathione peroxidase (GPx) and other molecules (12). The relatively long-lived H2O2 can pass mitochondrial membrane by diffusion into the cytosol and nucleus to activate a range of signaling pathways, including MAPK and PI3K/AKT pathways (6, 13).
2-methoxyestradiol (2-ME) is a natural physiological metabolite of estrogen, 17β-estradiol. A variety of cancer cells, including pancreatic cancer cells, were shown to be sensitive to 2-ME (14–16). 2-ME specifically affects cancer cells with no effects on normal cells (17), and its anti-cancer effects are largely linked to its ability to trigger mitochondrion-dependent apoptosis through inducing accumulation of cellular ROS (15, 18). In order to explore the molecular mechanism underlying the acquired resistance of pancreatic cancer cells to ROS-inducing anti-cancer drugs, we generated a stable cell line by exposing pancreatic cancer cells to increasing concentrations of 2-ME over a period of 3 months. The resulting cell line showed strong resistance to 2-ME and contained an elevated level of ROS. We then used a two dimensional gel electrophoresis (2-DE) based proteomics method to identify the mitochondrial proteins that were differentially expressed between the parental and the resistant cells. One protein identified was SOD2. Functional analysis revealed that SOD2 played a critical role in protecting the 2-ME resistant cells from the excessive oxidative stress, thus conferring resistance to ROS-induced apoptosis to the cells.
Materials and Methods
Cell culture and generation of stable cells with acquired resistance to 2-ME
The pancreatic cancer cells that we used to generate stable cells were MIA PaCa-2, which is an undifferentiated, primary pancreatic cancer cell line sensitive to 2-ME (19, 20). The MIA PaCa-2 cells were routinely maintained in D-MEM supplemented with 10% FBS and 1% penicillin and streptomycin. 2-ME-resistant stable cells were generated by continuously exposing the MIA PaCa-2 cells to stepwise increasing 2-ME concentrations in the range of 0.5 μM to 2.5 μM over a period of 3 months. After the cells adapted to one 2-ME concentration, typically requiring 2 – 3 weeks, exposure to 2-ME was increased by 0.5 μM. The process was repeated until 2-ME concentration reached 2.5 μM. Beyond 2.5 μM, no stable cells were successfully recovered.
ROS measurement
Cellular ROS concentrations were measured with a flow cytometer (BD Biosciences, San Jose, CA) after incubating cells with a fluorescence probe DCFH-DA (Invitrogen, Carlsbad, CA) as described by Gottlieb et al. (21). The parental and the resistant cells were cultured with 2.5 μM 2-ME for one week, and then harvested, washed with cold phosphate-buffered saline (PBS), and incubated with 10 μM DCFH-DA for 30 min. Propidium iodide was added to each sample immediately prior to flow cytometry analysis to differentiate dead and live cells. Fluorescence was analyzed only in the live cells. The cells were also incubated with propidium iodide alone, and the mean fluorescence of live cells of the single-stained cells was subtracted from the live cells of the double-stained cells. For polyethylene glycol (PEG)-catalase or PEG-SOD treatments (22), the parental and the resistant cells were pretreated with control media or media containing 100 units/ml PEG-catalase or 100 units/ml PEG-SOD (Sigma, St. Louis, MO) for 24 h, followed by an incubation with 2.5 μM 2-ME in the media for an additional 16 h. The cells were then washed and incubated with 5 μM CM-H2DCFDA (Invitrogen) for 20 min at 37 °C. After this, the cells were harvested, washed twice with cold PBS, and analyzed by flow cytometry as described above.
Cell viability assay and clonogenic survival assay
Cells were cultured with the indicated concentrations of 2-ME for 48 h, and then used for a viability assay, which was performed with a CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, Madison, WI). Luminescence was measured with a SpectraMax Gemini XS fluorescence microplate reader (Molecular Devices, Sunnyvale, CA). The clonogenic survival assay was performed according to Franken et al. (23). Briefly, the parental and the resistant cells were plated in the 60 mm dishes in D-MEM and cultured for 6 h. 2-ME was then added to each plate resulting in a final 2-ME concentration of 0, 0.25, 0.5, 1.0, or 1.5 μM in the culture medium (or the cells were exposed to different doses of λ-ray). The cells were cultured for 21 days with a medium change every 4 days. Colonies with > 50 cells were counted, and surviving fractions were normalized with plating efficiencies. Triplicate plates were used for each 2-ME concentration or irradiation dose.
Isolation of mitochondria
Mitochondria were purified from cells using differential centrifugation and non-linear sucrose ultracentrifugation (Supplementary Fig. S1) (24, 25).
2-DE
Three hundred micrograms (300 μg) of mitochondrial protein dissolved in a rehydration buffer were loaded onto a 17 cm ReadyStrip IPG strip (pH 3–10), which was in turn kept at room temperature overnight. Other procedures were performed as described previously (26, 27).
Liquid chromatography – tandem mass spectrometry (LC-MS/MS) analysis and database search
The LC-MS/MS analysis was carried out using a LTQ-XL mass spectrometer (Thermo, San Jose, CA) in the Proteomic Facility at the University of Arkansas for Medical Sciences (Little Rock, AR). Proteins were in-gel digested with trypsin (Promega) overnight at 37 °C, and the resulting peptides were dissolved in 20 μl 0.1% formic acid for LC-MS/MS analysis. Mascot (V2.2; Matrix Science, Boston, MA) was used to search against the International Protein Index (IPI) human protein database (V3.68) using LC-MS/MS data as described (26, 27). The parameters for database searching were as follows: (i) 2.0-Da mass error tolerance for MS and 0.65 Da for MS/MS, (ii) a maximum of one missed cleavage, and (iii) variable modifications: acetylation at peptide N terminus, phosphorylation on tyrosine/serine/threonine and oxidation on methionine.
Overexpression and small hairpin RNA (shRNA) silencing of SOD2
For overexpression, the coding sequence of human SOD2 was cloned into the Hind III and Xho I sites of plasmid pcDNA3.1, and the resulting construct was transfected into the parental cells with calcium phosphate method. For silencing of SOD2, shSOD2 sequence (20) was annealed and cloned into the Sal I and Xba I sites of a safety-modified retroviral vector pSuppressor Retro (Imgenex, San Diego, CA). Co-tranfection of the retroviral construct with a packaging vector pCL-10A1 into 293T cells and the subsequent infection of the resistant cells with the resulting viral particles were performed according to the manufacturer’s instructions (Imgenex). A negative control plasmid harboring a scrambled sequence that does not show significant homology to human gene sequences from the same company was transfected and infected in the same way as described above. For both overexpression and shRNA silencing, stable cells were selected with 1 mg/ml G418, and SOD2 overexpressing clones or the clones with knockdown of SOD2 were screened using Western blotting with anti-SOD2 antibodies.
Western blotting
Western blotting was performed as described previously (26, 27). Antibodies were purchased from the following sources: anti-SOD2, peroxiredoxin III (Prx III) and actin from Santa Cruz Biotech (Santa Cruz, CA); and anti-PARP from BD Pharmingen (San Diego, CA).
Measurements of SOD2 and GPx activities
The parental or resistant cells (~1.8 × 107) were washed with ice cold PBS twice and harvested with a rubber policeman. The cells were lysed in 3 packed cell pellet volumes of 0.05 M phosphate buffer, pH 7.8 by three cycles of sonication (Branson Digital Sonifier 450; Danbury, CT) on ice with a microtip at 15% power (10 × 0.5 sec pulses in each cycle). After centrifugation at 10,000 g for 15 min at 4 °C, protein concentration in the supernatant was determined by a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). SOD2 activity was measured using a SOD Assay Kit-WST (Dojindo Molecular Technologies, Rockville, MD). The measurements were performed according to manufacturer’s instructions with the following specifications: 1) The cell extract was incubated with 5 mM potassium cyanide for 40 min at room temperature to inactivate Cu/Zn-SOD activities; 2) 25 – 40 μg protein was used in each reaction; 3) the standard curve was generated using serial dilutions of SOD from bovine erythrocytes, and one unit of SOD is defined as the amount of enzyme that inhibits the rate of cytochrome c reduction by 50% at pH 7.8 and 25 °C (Sigma). Enzymatic activity was expressed as units per mg of protein.
GPx activity was measured by monitoring the oxidation of NADPH with a UV spectrophotometer at 340 nm (28). The reaction mixture contained 50 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.25 mM NADPH, 2.1 mM reduced glutathione, 0.5 unit glutathione reductase (Sigma), and 0.5 – 1 mg cell extract prepared as described above. The reaction was initiated by adding 300 μM t-butyl hydroperoxide, and the final volume of the reaction mixture was 1 ml. The enzyme-catalyzed oxidation of NADPH was monitored for 5 min at 25 °C.
Statistical Analysis
p values were calculated using a One-way ANOVA (PSI-PLOT, Pearl River, NY).
Results
Generation of stable cell line with acquired resistance to ROS-inducing compound 2-ME
In order to eliminate the complications caused by genetic background variations, we generated a pancreatic cancer isogenic stable cell line that differs in sensitivity to ROS-inducing compound 2-ME. By exposing the pancreatic cancer MIA PaCa-2 cells to increasing concentrations of 2-ME (0.5 – 2.5 μM) over a three month period, we generated an isogenic cell line with acquired resistance to 2-ME, designated as MIA PaCa-2R. As shown in Fig. 1A, the resistant MIA PaCa-2R cells showed strong resistance to 2-ME-mediated cell killing compared with the parental MIA PaCa-2 cells. We also used Western blotting to detect the cleavage of PARP to monitor cell apoptosis. As shown in Fig. 1B, the MIA PaCa-2R cells were more resistant to the 2-ME mediated apoptosis than the parental cells. Apoptosis is not the only form of cell death in most tumors, and clonogenic survival assay is widely regarded as the “gold standard” cellular sensitivity assay due to its ability to measure long-term cell survival and all modes of cell death. We performed clonogenic survival assay on the parental and the resistant cells. As shown in Fig. 1C, the MIA PaCa-2R cells demonstrated markedly enhanced clonogenic survival over the parental cells after the cells were exposed to increasing concentrations of 2-ME.
Figure 1.

Different 2-ME sensitivities between the parental MIA PaCa-2 and the resistant MIA PaCa-2R cells. A, MIA PaCa-2 and MIA PaCa-2R cells were cultured with the indicated concentrations of 2-ME for 72 h, and cell viabilities were examined with the CellTiter-Glo Luminescent Cell Viability Assay. As shown, the MIA PaCa-2R cells were more resistant to 2-ME than the MIA PaCa-2 cells. B, the parental and the resistant cells were cultured with the indicated concentrations of 2-ME for 48 h, and proteins from each population were analyzed by Western blotting. While 2.5 μM 2-ME induced a significant amount of the MIA PaCa-2 to apopotosis, the MIA PaCa-2R cells showed no apoptosis under the same condition. β-actin was used as a loading control. Equal amounts of total protein were loaded in each lane (40 μg/lane). C, clonogenic survival assay. MIA PaCa-2 and MIA PaCa-2R cells were cultured with the presence of indicated concentrations of 2-ME for 21 days. Colonies with > 50 cells were counted. As shown, the MIA PaCa-2R cells were more 2-ME-resistant than the parental cells.
The resistant cells contain a higher level of cellular ROS than the parental cells
In order to examine whether ROS were involved in the resistance of the MIA PaCa-2R cells to 2-ME-mediated cell killing and apoptosis, we measured cellular ROS levels with flow cytometry using a fluorescent probe DCFH-DA (21). In this analysis, once the cell-permeable fluorescent probe DCFH-DA is in the cells, it is cleaved by cellular esterase into its nonfluorescent and non-membrane-permeable form, DCFH, and thus is trapped in the cells. The DCFH is then oxidized by cellular oxidants into its fluorescent form, DCF. After incubating cells with DCFH-DA, it was demonstrated that the fluorescence intensity in the MIA PaCa-2R cells was almost 3-fold of that in the MIA PaCa-2 cells (Fig. 2A). To make sure the DCF fluorescence intensity represented the ROS levels in cells, we incubated cells with cell-permeable PEG-catalase or PEG-SOD to examine whether they could inhibit the fluorescence signal intensity. The results demonstrated that pretreatments of both the parental and resistant cells with PEG-catalase or PEG-SOD significantly inhibited the DCF fluorescent signals (Figs. 2B and 2C; for uncharacterized reasons, PEG-catalase pretreatments resulted in more pronounced decrease in fluorescence signal intensity in the parental cells than in the resistant cells), suggesting that DCF fluorescence signal intensities we measured represented the ROS levels in the cells. In short, results in this section suggest that cellular ROS levels were significantly different between the parental and resistant cells, and cellular ROS may be closely related to the acquired resistance of the MIA PaCa-2R cells to 2-ME.
Figure 2.

The resistant MIA PaCa-2R cells contain a high level of ROS. A, MIA PaCa-2 and MIA PaCa-2R cells were cultured with 2.5 μM 2-ME for one week, and then harvested for ROS measurement with flow cytometry using a fluorescent probe DCFH-DA (see Materials and Methods). Ten thousand (10,000) cells were evaluated and data were expressed as fold increase of ROS generation over that in the parental cells. B, pretreatments of the MIA PaCa-2 cells with PEG-catalase or PEG-SOD inhibit DCF fluorescent signal intensities (thus ROS levels). Data were expressed as fold decrease of ROS generation over the untreated control. C, same as B except that the MIA PaCa-2R cells were tested. Values are the means ± S.E. of three separate sample preparations. ***, **, and * denote statistical significance of p < 0.001, p < 0.01, and p < 0.05, respectively.
Isolation of mitochondria from cells
For performing comparative proteomic analysis using purified mitochondria as starting materials, we used differential centrifugation and non-linear sucrose ultracentrifugation to purify mitochondria from the MIA PaCa-2 and MIA PaCa-2R cells (24, 25). Western blot analysis with antibodies against marker proteins demonstrated that the purified mitochondria were essentially free of cytosolic and nuclear contaminations but contained endoplasmic reticulum proteins (Supplementary Fig. S1), which are usually difficult to remove using differential centrifugation (24).
SOD2 is upregulated in the resistant MIA PaCa-2R cells
We then used 2-DE to profile protein expression between the MIA PaCa-2 cells and MIA PaCa-2R cells using the purified mitochondria as starting materials. Of the > 650 protein spots resolved, multiple protein spots showed at least a 50% difference in the normalized volume between the MIA PaCa-2 cells and MIA PaCa-2R cells. LC-MS/MS analysis and database search revealed that one protein that showed approximately a 2-fold increase in the normalized volume in the MIA PaCa-2R gel over the counterpart in parental cell gel was SOD2 (Fig. 3A). SOD2 is the mitochondrial enzyme that converts superoxide (O2•−) into H2O2, which is further eliminated by GPx and other molecules (12). Increases of SOD2 expression have been detected in primary cancer tissues of different types of cancers (14, 29, 30). Due to the critical roles of SOD2 in ROS metabolism in normal and cancer cells, we selected this enzyme for further analysis. First, we examined the expression of SOD2 by Western blotting with anti-SOD2 antibodies and confirmed that the expression of SOD2 in the MIA PaCa-2R cells was indeed upregulated (Fig. 3B). To examine whether the increased expression would lead to an enhanced SOD2 enzymatic activity in the resistant cells, we measured the catalytic activity of SOD2. Consistent with the expression data, SOD2 enzymatic activity in the MIA PaCa-2R cells was also approximately 2-fold of that in the parental cells (Fig. 3C).
Figure 3.

SOD2 is upregulated in the resistant MIA PaCa-2R cells. A, portions of the 2-DE gels demonstrating the expression levels of SOD2 in the MIA PaCa-2 cells and the MIA PaCa-2R cells. B, Western blot validation of SOD2 expression in the MIA PaCa-2 cells and the MIA PaCa-2R cells. β-actin was used as a loading control. Equal amounts of total protein were loaded in each lane (40 μg/lane). C, SOD2 enzymatic activities in the MIA PaCa-2 cells and the MIA PaCa-2R cells. Values are the means ± S.E. of three separate sample preparations. **denotes statistical significance of p < 0.01.
Ectopic overexpression of SOD2 decreases the sensitivity of MIA PaCa-2 cells to 2-ME
To examine whether the upregulated SOD2 protected the MIA PaCa-2R cells from 2-ME-mediated cell killing and apoptosis (Fig. 1), we generated a cell line that stably overexpresses SOD2 in MIA PaCa-2 cells (Fig. 4A). Enzymatic activity measurements confirmed that cells overexpressing SOD2 contained significantly (p < 0.001) higher SOD2 activity than the control cells (Fig. 4B). Western blot analysis of PARP cleavage showed that overexpression of SOD2 led MIA PaCa-2 cells to become resistant to 2-ME-mediated apoptosis (Fig. 4C). Consistent with this, cell viability assay demonstrated that overexpression of SOD2 caused the parental cells to be significantly less sensitive to the 2-ME-mediated cell killing as compared with the control cells (Fig. 4D; compare “SOD2over” with control; p < 0.001). However, overexpression of SOD2 alone did not promote the 2-ME resistance of the MIA PaCa-2 cells to the level of the resistance of the MIA PaCa-2R cells (Fig. 4D; compare “SOD2over” with MIA PaCa-2R), suggesting that other factors also contributed to the resistance of MIA PaCa-2R cells to 2-ME. We also performed clonogenic survival assay to compare long-term cell survival between the “SOD2over” cells and the parental cells. The results demonstrated that overexpression of SOD2 enhanced the long-term clonogenic survival of the “SOD2over” cells under different concentrations of 2-ME compared with the parental cells (Fig. 4E). Taken together, results in this section suggest that the upregulated SOD2 in the MIA PaCa-2R cells plays a critical role in conferring the resistance of the MIA PaCa-2R cells to the 2-ME-mediated cell killing and apoptosis.
Figure 4.

Overexpression of SOD2 in MIA PaCa-2 cells enhances resistance to 2-ME-mediated cell killing and apoptosis. A, Western blot analysis of SOD2 expression in vector-transformed cells (control) and the stable cells that overexpresses SOD2 (SOD2over). B, enzymatic activities of SOD2 in the parental and the SOD2over cells. C, the control and the SOD2over cells were cultured with indicated concentrations of 2-ME for 48 h, and then harvested for analysis of PARP cleavage by Western blotting. In both A and C, equal amounts of total protein were loaded in each lane (40 μg/lane), and β-actin was used as a loading control. D, the control, SOD2over, and MIA PaCa-2R cells were cultured with 2.5 μM 2-ME for 48 h and then harvested for viability analysis. E, clonogenic survival of the parental cells and the SOD2over cells. The parental and the SOD2over cells were cultured with presence of 2-ME at the indicated concentrations for 21 days, and the colonies with > 50 cells were counted for calculation of cell survival. Values in B, D and E are the means ± S.E. of three separate sample preparations. ***denotes statistical significance of p < 0.001.
Suppression of SOD2 expression moderately re-sensitizes MIA PaCa-2R cells to 2-ME
To ensure that SOD2 is indeed involved in the acquired resistance to 2-ME, we selectively suppressed SOD2 expression in MIA PaCa-2R cells with retrovirus-mediated shRNA (Fig. 5A). SOD2 enzymatic activity assay confirmed that the stable cells with SOD2 being silenced contained significantly lower activity of SOD2 compared with the resistant cells (Fig. 5B). Western blot analysis demonstrated that silencing of the SOD2 expression re-sensitized the MIA PaCA-2R cells to 2-ME-mediated apoptosis (Fig. 5C). In agreement with this, suppression of SOD2 in the MIA PaCa-2R cells led to a moderate increase in sensitivity to 2-ME-mediated cell killing (Fig. 5D; compare “shSOD2” with MIA PaCa-2R; p = 0.0712). However, the suppression of SOD2 expression did not completely reverse the 2-ME resistant phenotype of the MIA PaCa-2R cells (Fig. 5D; compare “shSOD2” with MIA PaCa-2). These results were consistent with the overexpression data (Fig. 4), and both sets of results imply that SOD2 plays an important role in the acquired resistance of pancreatic cancer cells to 2-ME but is not the sole factor responsible for the acquired resistance.
Figure 5.

Suppression of SOD2 expression by shRNA moderately re-sensitizes MIA PaCa-2R cells to 2-ME-mediated apoptosis and cell killing. A, SOD2 expression in the MIA PaCa-2R cells and the cells with SOD2 knockdown by shRNA (shSOD2). B, enzymatic activities of SOD2 in the resistant and the shSOD2 cells. C, MIA PaCa-2R and the shSOD2 cells were cultured with 2.5 μM 2-ME for 48 h and then harvested for Western blot analysis with anti-PARP antibody. In both A and C, equal amounts of total protein were loaded in each lane (40 μg/lane), and β-actin was used as a loading control. D, the indicated cells were cultured with 2.5 μM 2-ME for 48 h, and then harvested for cell viability assay. Values in B and D are the means ± S.E. of three separate sample preparations. **denotes statistical significance of p < 0.01.
MIA PaCa-2R cells also show a tendency to have IR resistance
In addition to mediating drug-induced apoptosis and cell killing, ROS are also major components involved in IR-induced apoptosis and cell killing (10). To test whether the sensitivity of pancreatic cells to 2-ME is linked to their sensitivity to IR, we first treated the MIA PaCa-2 and MIA PaCA-2R cells with 5 Gy of λ-radiation and measured cell apoptosis and viability. The results demonstrated the 2-ME-resistant MIA PaCa-2R cells were also more resistant to IR-induced apoptosis (Fig. 6A) and showed a moderately higher cell viability after the exposure (Fig. 6B; p = 0.0975). We then performed a clonogenic survival assay to compare the long-term survival of the two types of cells after exposing the cells to different doses of IR. As shown in Fig. 6C, the resistant MIA PaCa-2R cells demonstrated significantly (p < 0.01) enhanced long-term clonogenic survival over the parental cells after the cells were exposed to IR of > 3 Gy. These results suggest that the acquired 2-ME resistance of pancreatic cancer cells is not restricted to ROS-inducing compound 2-ME, but also linked to radio-resistance of the cells. The observed radio-resistance of the MIA PaCa-2R cells might be related to the increased expression of SOD2 in the resistant cells, as overexpression of SOD2 has been shown to result in IR resistance in different types of cells (31).
Figure 6.

The 2-ME resistant MIA PaCa-2R cells are also moderately resistant to γ-ray-mediated apoptosis and cell killing. A and B, the MIA PaCa-2 cells and the MIA PaCa-2R cells were treated with 5 Gy of γ-ray, followed by a recovery culture at 37 °C for 48 h. The cells were then harvested for Western blotting with anti-PARP antibodies (panel A) and cell viability assay (panel B). In Western blotting, equal amounts of total protein were loaded in each lane (40 μg/lane), and β-actin was used as a loading control. C, clonogenic survival of the parental and the resistant cells. The parental and the resistant cells were treated with the indicated doses of γ-ray and then cultured for 21 days. The colonies with > 50 cells were counted for calculation of cell survival. Values in B and C are the means ± S.E. of three separate sample preparations. The p value in panel B was 0.0975, and the p value at each dose > 3 Gy in panel C was < 0.01.
The expression or activity of important proteins involved in detoxifying H2O2 in mitochondria is not enhanced
H2O2, the product of SOD2, is also toxic to cells. Multiple factors are known to be involved in converting H2O2 into water in mammalian cells, including catalase, GPx and peroxiredoxin (32). Catalase, a major H2O2 detoxifying enzyme in cells, is mainly located in peroxisomes (33) and has not been found in the mitochondria of most tissues except for heart tissue (34, 35). Even in heart mitochondria, the contribution of catalase to H2O2 detoxification is negligible (36). In mitochondria, GPx and a mitochondrion-specific peroxiredoxin, Prx III, appear to play a major role in removing H2O2 (12, 36, 37). In order to examine whether these enzymes are involved in the 2-ME-resistance of the MIA PaCa-2R cells, we compared the expression of Prx III and the activity of GPx between the parental cells and the resistant cells. The expression of Prx III was similar between the parental and the resistant cells (Fig. 7A). In contrast to the changes in SOD2 expression and activity (Fig. 1), the catalytic activity of GPx in the resistant cells was significantly (p < 0.05) lower than that in the parental cells (Fig. 7B). A decrease in GPx catalytic activity could result in increased sensitivity of cells to ROS (instead of enhanced resistance to ROS). These results suggest that Prx III and GPx may not positively contribute to the increased 2-ME-resistance of the MIA PaCa-2R cells.
Figure 7.
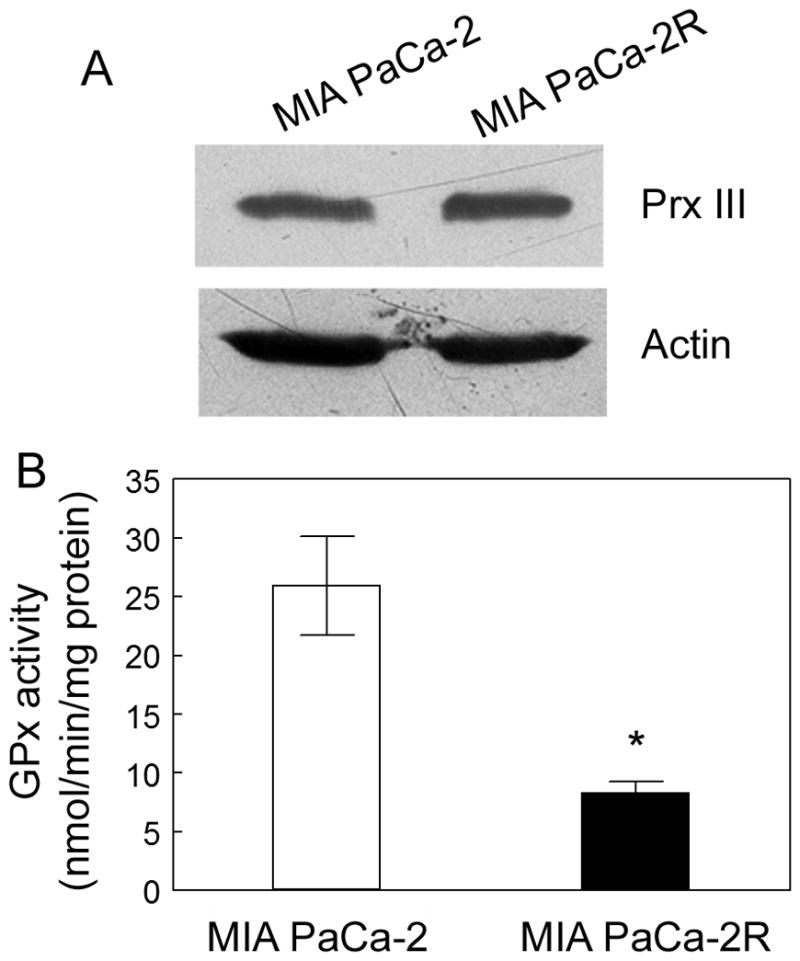
The expression or activity of important proteins involved in detoxifying H2O2 in the mitochondria. A, the expression of Prx III in the parental and the resistant cells. Equal amounts of total protein were loaded in each lane (40 μg/lane), and β-actin was used as a loading control. B, the activity of GPx in the parental and the resistant cells. Values are the means ± S.E. of three separate sample preparations. *denotes statistical significance of p < 0.05.
Discussion
Through exposure of pancreatic cancer MIA PaCa-2 cells to increasing concentrations of ROS-inducing, anti-cancer compound 2-ME over a three month period, we generated a cell line, MIA PaCa-2R, that was strongly resistant to 2-ME (Fig. 1). The ROS level in the MIA PaCa-2R cells was almost 3-fold of that in the parental cells (Fig. 2). Comparative proteomic analysis revealed that the expression of SOD2, a key antioxidant enzyme in mitochondria, was upregulated (Fig. 3). Functional analyses of SOD2 suggest that SOD2 plays a critical role in the acquired resistance of the MIA PaCa-2R cells to 2-ME (Figs. 4 and 5), and potentially also to IR (Fig. 6).
Compared with other major types of cancers, one unique feature of pancreatic cancer is that approximately 90% of pancreatic cancers contain Kras mutations (38), the highest in all cancers. Kras is involved in the G-protein signal transduction pathway, modulating cellular proliferation and differentiation. Mutations of Kras result in constitutive activation of the MAPK and PI3K/AKT signal transduction pathways and consequently, unregulated proliferation and impaired differentiation (39), which lead to increased generation of ROS (7). The elevated levels of ROS in turn can further activate MAPK and PI3K/AKT pathways (6, 13), potentially forming a positive-feedback loop. The activating mutations of Kras usually occur at a very early stage of pancreatic cancer development, suggesting that Kras oncogenes play a key role in the pancreatic cancer pathogenesis (38). Tumor cells harboring activated Ras mutation are usually resistant to chemo- and radio-therapy (40). In line with this notion, transformation of human ovarian epithelial cells with mutated Ras increases the threshold of ROS tolerance (hence the resistance to the oxidative agent-mediated cell killing and apoptosis) by upregulating the overall antioxidant capacity of cells (41). Furthermore, a recent study demonstrated that the IR resistance of cancer stem cells was associated with low levels of ROS resulting from enhanced ROS defense in the cells (42). All these lines of evidence suggest that ROS may play a critical role in the exceptional intrinsic and acquired resistance of pancreatic cancer cells to IR and ROS-inducing chemo-drugs. SOD2 plays a vital role in ROS metabolism in that it converts highly toxic superoxide (O2•−) into less toxic hydrogen peroxides (H2O2) in the mitochondria, a site that is vulnerable to ROS attack and important to cell apoptosis. SOD2 knockout mice die within 10 days after birth (43). Consistent with its essential functions, enforced expression of SOD2 suppresses the malignant phenotypes of several types of human cancer cells including pancreatic cancer, suggesting that SOD2 may function as a tumor suppressor (19, 44, 45). However, in the present study we found that SOD2 expression was upregulated instead of being downregulated in the resistant MIA PaCa-2R cells (Fig. 3). This contradiction suggests that SOD2 must function as a tumor suppressor different from most other conventional tumor suppressors, which are usually inhibited or lost in malignant cancer cells (39). Since the resistant MIA PaCa-2R cells contain high levels of ROS and are thus under excessive oxidative stress, the cells must have evolved a molecular mechanism to defend against the excessive stress for survival (i.e., to evade the ROS-induced apoptosis). Upregulation of SOD2 expression must be one of those molecular mechanisms evolved in the MIA PaCa-2R cells. From this perspective, it is logical to speculate that the increased expression of SOD2 in the MIA PaCa-2R cells is an adaptive response of the cells to the excessive oxidative cellular environment, which can be reflected by the higher levels of ROS in the cells (Fig. 2). Since IR and many anti-cancer drugs kill cancer cells largely through inducing ROS (6, 9, 10), upregulation of SOD2 is likely to be a common molecular mechanism underlying acquired radio- or chemo-resistance in cancer cells. This may be particularly true for those types of cancers that harbor a mutated Ras.
It has been well established that 2-ME induces cellular ROS accumulation (15, 18), and ROS triggers the expression of SOD2 (29, 46). Concomitantly, ROS have also been shown to activate transcription factor NF-kB, which in turn control the expression of SOD2 (47, 48). Thus, the increased expression of SOD2 in the MIA PaCa-2R could be a result of NF-kB activation induced by the markedly elevated ROS levels in the resistant cells (Fig. 2). In addition to NF-kB, the promoter of SOD2 also contains binding site(s) for transcription factor AP-1 (49). Furthermore, mutations or methylations of the SOD2 promoter sequence were also reported to affect the expression of SOD2 in cancer cells (20, 50). These results suggest that multiple factors, including NF-kB, AP-1, promoter mutations and methylations, may function independently or in combination to regulate the expression of SOD2 in different tissues or under different environments. Noticeably, SOD2 expression in primary pancreatic cancer cells (including MIA PaCa-2) is repressed (19, 20). Further investigations on how the SOD2 expression is de-repressed during the development of acquired drug-resistance may provide valuable information on designing novel therapeutic strategies to re-sensitize drug-resistant pancreatic cancer cells to radio- and chemo-therapy.
2-ME has been shown to trigger O2•− generation in cells via the mitochondrial electron transport chain (18). Interestingly, in the present study we found that the enzymatic activity of GPx, a key enzyme in detoxifying H2O2 in the mitochondria (12, 36), in the resistant cells was significantly (p < 0.05) decreased compared with the parental cells (Fig. 7B). The decreased GPx could potentially contribute to the increased ROS levels in the MIA PaCa-2R cells (Fig. 2). Future studies on examining the relationship between the reduced activity of GPx and the increased ROS level in the MIA PaCa-2R cells may provide new information on how cellular ROS is built up when cancer cells are exposed to ROS-inducing compounds such as 2-ME.
Supplementary Material
Acknowledgments
Financial support: Y. Du: Two grants from the NIH, and one grant from the Arkansas Biosciences Institute.
Grant support: This work was supported by NIH grants 1R03CA135549-01A1 and 1R15GM087671-01, and a grant from the Arkansas Biosciences Institute (Y. Du).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Mangray S, King TC. Molecular pathobiology of pancreatic adenocarcinoma. Front Biosci. 1998;3:D1148–60. doi: 10.2741/a351. [DOI] [PubMed] [Google Scholar]
- 2.Jessup JM, Steele G, Jr, Mayer RJ, Posner M, Busse P, Cady B, et al. Neoadjuvant therapy for unresectable pancreatic adenocarcinoma. Arch Surg. 1993;128:559–64. doi: 10.1001/archsurg.1993.01420170093014. [DOI] [PubMed] [Google Scholar]
- 3.White R, Lee C, Anscher M, Gottfried M, Wolff R, Keogan M, et al. Preoperative chemoradiation for patients with locally advanced adenocarcinoma of the pancreas. Ann Surg Oncol. 1999;6:38–45. doi: 10.1007/s10434-999-0038-z. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Clegg LX, Ward E, Ries LA, Wu X, Jamison PM, et al. Annual report to the nation on the status of cancer, 1975–2001, with a special feature regarding survival. Cancer. 2004;101:3–27. doi: 10.1002/cncr.20288. [DOI] [PubMed] [Google Scholar]
- 5.Nicotera TM, Privalle C, Wang TC, Oshimura M, Barrett JC. Differential proliferative responses of Syrian hamster embryo fibroblasts to paraquat-generated superoxide radicals depending on tumor suppressor gene function. Cancer Res. 1994;54:3884–8. [PubMed] [Google Scholar]
- 6.Fruehauf JP, Meyskens FL., Jr Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13:789–94. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 7.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]
- 9.Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002;3:420–5. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong Q, Lillehei KO. Antioxidant inhibitors for cancer therapy. Med Hypotheses. 1998;51:405–9. doi: 10.1016/s0306-9877(98)90036-6. [DOI] [PubMed] [Google Scholar]
- 11.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 12.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta A, Rosenberger SF, Bowden GT. Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis. 1999;20:2063–73. doi: 10.1093/carcin/20.11.2063. [DOI] [PubMed] [Google Scholar]
- 14.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–19. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 15.Qanungo S, Basu A, Das M, Haldar S. 2-Methoxyestradiol induces mitochondria dependent apoptotic signaling in pancreatic cancer cells. Oncogene. 2002;21:4149–57. doi: 10.1038/sj.onc.1205508. [DOI] [PubMed] [Google Scholar]
- 16.Schumacher G, Kataoka M, Roth JA, Mukhopadhyay T. Potent antitumor activity of 2-methoxyestradiol in human pancreatic cancer cell lines. Clin Cancer Res. 1999;5:493–9. [PubMed] [Google Scholar]
- 17.Seegers JC, Lottering ML, Grobler CJ, van Papendorp DH, Habbersett RC, Shou Y, et al. The mammalian metabolite, 2-methoxyestradiol, affects P53 levels and apoptosis induction in transformed cells but not in normal cells. J Steroid Biochem Mol Biol. 1997;62:253–67. doi: 10.1016/s0960-0760(97)00043-5. [DOI] [PubMed] [Google Scholar]
- 18.Chauhan D, Li G, Sattler M, Podar K, Mitsiades C, Mitsiades N, et al. Superoxide-dependent and -independent mitochondrial signaling during apoptosis in multiple myeloma cells. Oncogene. 2003;22:6296–300. doi: 10.1038/sj.onc.1206734. [DOI] [PubMed] [Google Scholar]
- 19.Cullen JJ, Weydert C, Hinkhouse MM, Ritchie J, Domann FE, Spitz D, et al. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res. 2003;63:1297–303. [PubMed] [Google Scholar]
- 20.Hurt EM, Thomas SB, Peng B, Farrar WL. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br J Cancer. 2007;97:1116–23. doi: 10.1038/sj.bjc.6604000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottlieb E, Vander Heiden MG, Thompson CB. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol. 2000;20:5680–9. doi: 10.1128/mcb.20.15.5680-5689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai H, Li Z, Dikalov S, Holland SM, Hwang J, Jo H, et al. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J Biol Chem. 2002;277:48311–7. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- 23.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 24.Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem. 2001;276:11615–23. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- 25.Rezaul K, Wu L, Mayya V, Hwang SI, Han D. A systematic characterization of mitochondrial proteome from human T leukemia cells. Mol Cell Proteomics. 2005;4:169–81. doi: 10.1074/mcp.M400115-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Zhou J, Ruan C, Du Y. Inhibition of Type I Interferon Production via Suppressing IKK-Gamma Expression: A New Strategy for Counteracting Host Antiviral Defense by Influenza A Viruses? J Proteome Res. 2012;11:217–23. doi: 10.1021/pr200894t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Z, Zhou J, Du Y. Estrogen receptor alpha interacts with mitochondrial protein HADHB and affects beta-oxidation activity. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M111.011056. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park SY, Chang I, Kim JY, Kang SW, Park SH, Singh K, et al. Resistance of mitochondrial DNA-depleted cells against cell death: role of mitochondrial superoxide dismutase. J Biol Chem. 2004;279:7512–20. doi: 10.1074/jbc.M307677200. [DOI] [PubMed] [Google Scholar]
- 29.Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem. 2005;280:39485–92. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 30.Malafa M, Margenthaler J, Webb B, Neitzel L, Christophersen M. MnSOD expression is increased in metastatic gastric cancer. J Surg Res. 2000;88:130–4. doi: 10.1006/jsre.1999.5773. [DOI] [PubMed] [Google Scholar]
- 31.Sun J, Chen Y, Li M, Ge Z. Role of antioxidant enzymes on ionizing radiation resistance. Free Radic Biol Med. 1998;24:586–93. doi: 10.1016/s0891-5849(97)00291-8. [DOI] [PubMed] [Google Scholar]
- 32.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–14. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 33.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 34.Phung CD, Ezieme JA, Turrens JF. Hydrogen peroxide metabolism in skeletal muscle mitochondria. Arch Biochem Biophys. 1994;315:479–82. doi: 10.1006/abbi.1994.1528. [DOI] [PubMed] [Google Scholar]
- 35.Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991;266:22028–34. [PubMed] [Google Scholar]
- 36.Antunes F, Han D, Cadenas E. Relative contributions of heart mitochondria glutathione peroxidase and catalase to H(2)O(2) detoxification in in vivo conditions. Free Radic Biol Med. 2002;33:1260–7. doi: 10.1016/s0891-5849(02)01016-x. [DOI] [PubMed] [Google Scholar]
- 37.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–84. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 38.Motojima K, Tsunoda T, Kanematsu T, Nagata Y, Urano T, Shiku H. Distinguishing pancreatic carcinoma from other periampullary carcinomas by analysis of mutations in the Kirsten-ras oncogene. Ann Surg. 1991;214:657–62. doi: 10.1097/00000658-199112000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 40.Brunner TB, Cengel KA, Hahn SM, Wu J, Fraker DL, McKenna WG, et al. Pancreatic cancer cell radiation survival and prenyltransferase inhibition: the role of K-Ras. Cancer Res. 2005;65:8433–41. doi: 10.1158/0008-5472.CAN-05-0158. [DOI] [PubMed] [Google Scholar]
- 41.Young TW, Mei FC, Yang G, Thompson-Lanza JA, Liu J, Cheng X. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res. 2004;64:4577–84. doi: 10.1158/0008-5472.CAN-04-0222. [DOI] [PubMed] [Google Scholar]
- 42.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–81. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 44.Church SL, Grant JW, Ridnour LA, Oberley LW, Swanson PE, Meltzer PS, et al. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc Natl Acad Sci U S A. 1993;90:3113–7. doi: 10.1073/pnas.90.7.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weydert C, Roling B, Liu J, Hinkhouse MM, Ritchie JM, Oberley LW, et al. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol Cancer Ther. 2003;2:361–9. [PubMed] [Google Scholar]
- 46.Chang LY, Kang BH, Slot JW, Vincent R, Crapo JD. Immunocytochemical localization of the sites of superoxide dismutase induction by hyperoxia in rat lungs. Lab Invest. 1995;73:29–39. [PubMed] [Google Scholar]
- 47.Das KC, Lewis-Molock Y, White CW. Thiol modulation of TNF alpha and IL-1 induced MnSOD gene expression and activation of NF-kappa B. Mol Cell Biochem. 1995;148:45–57. doi: 10.1007/BF00929502. [DOI] [PubMed] [Google Scholar]
- 48.Warner BB, Stuart L, Gebb S, Wispe JR. Redox regulation of manganese superoxide dismutase. Am J Physiol. 1996;271:L150–8. doi: 10.1152/ajplung.1996.271.1.L150. [DOI] [PubMed] [Google Scholar]
- 49.Wan XS, Devalaraja MN, St Clair DK. Molecular structure and organization of the human manganese superoxide dismutase gene. DNA Cell Biol. 1994;13:1127–36. doi: 10.1089/dna.1994.13.1127. [DOI] [PubMed] [Google Scholar]
- 50.Xu Y, Krishnan A, Wan XS, Majima H, Yeh CC, Ludewig G, et al. Mutations in the promoter reveal a cause for the reduced expression of the human manganese superoxide dismutase gene in cancer cells. Oncogene. 1999;18:93–102. doi: 10.1038/sj.onc.1202265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.