

Abstract
In recent decades, comparative chromosomal banding, chromosome painting, and gene-order studies have shown strong conservation of gross chromosome structure and gene order in mammals. However, findings from the human genome sequence suggest an unprecedented degree of recent (<35 million years ago) segmental duplication. This dynamism of segmental duplications has important implications in disease and evolution. Here we present a chromosome-wide view of the structure and evolution of the most highly homologous duplications (⩾1 kb and ⩾90%) on chromosome 22. Overall, 10.8% (3.7/33.8 Mb) of chromosome 22 is duplicated, with an average sequence identity of 95.4%. To organize the duplications into tractable units, intron-exon structure and well-defined duplication boundaries were used to define 78 duplicated modules (minimally shared evolutionary segments) with 157 copies on chromosome 22. Analysis of these modules provides evidence for the creation or modification of 11 novel transcripts. Comparative FISH analyses of human, chimpanzee, gorilla, orangutan, and macaque reveal qualitative and quantitative differences in the distribution of these duplications—consistent with their recent origin. Several duplications appear to be human specific, including a ∼400-kb duplication (99.4%–99.8% sequence identity) that transposed from chromosome 14 to the most proximal pericentromeric region of chromosome 22. Experimental and in silico data further support a pericentromeric gradient of duplications where the most recent duplications transpose adjacent to the centromere. Taken together, these data suggest that segmental duplications have been an ongoing process of primate genome evolution, contributing to recent gene innovation and the dynamic transformation of genome architecture within and among closely related species.
Introduction
Our current understanding of overall mammalian genome organization and evolution is derived mainly from cytogenetic banding and painting studies, as well as gene-order mapping (Yunis and Prakash 1982; Nadeau and Sankoff 1998; O'Brien et al. 1999; Murphy et al. 2001). These data suggest strong conservation of genome architecture, with few noticeable rearrangements—one per 10 million years, on average (Wienberg et al. 1997; Murphy et al. 2001). In fact, it has been stated that the human genome has evolved from a common primate ancestor (∼60–70 million years ago) in as few as seven translocation steps. These conclusions are based largely on comparative FISH analysis using whole-chromosomal paint probes (Muller et al. 1999; O'Brien et al. 1999). Although examples like the highly rearranged karyotype of gibbons illustrate that certain genomes have undergone extensive rearrangement (Jauch et al. 1992), on the whole, strong conservation is the norm. Even human comparisons to distantly related mammals demonstrate strong conservation, with the estimated number of rearrangements varying from 17 in felines (Wienberg et al. 1997) to 180 in mice (Nadeau and Taylor 1984; O'Brien et al. 1999). Such a static view of genome architecture recently has been challenged, as a result of the identification of highly homologous segmental duplications within the human genome (IHGSC 2001) (see sidebar) and their potential role in genomic rearrangement (Stankiewicz et al. 2001).
Duplicitous Duplications and Human Genetics.
The central task of human genetics is the correlation of human phenotype and human genotype. Much of this effort depends on our ability to track unique DNA by association or linkage with phenotype. The revelation that a significant fraction (∼5%) of our genome is composed of recent segmental duplications has a serious impact on the work of human geneticists. Segmental duplications may be hundreds of kb in size, may share a high degree of sequence identity (>99%), may harbor genes, and, unlike other classes of repetitive sequence, cannot be distinguished as such a priori. In essence, these properties have made a portion of our genome intractable, by the standard molecular techniques applied within our field. The inability to develop STSs, for example, in regions of the genome that are completely devoid of “unique” sequence information, translates into an inability to track the inheritance pattern of these regions through human pedigrees. The development of human SNP maps is similarly hampered, leading to misleadingly high density of SNPs over duplicated regions. Duplicated segments pose serious problems for the assembly of the human genome. Human cytogeneticists, who now depend on this assembly to select probes to interrogate human chromosomal rearrangements, are often confounded by ambiguous results when multiple signals are encountered. Finally, it has become increasingly apparent that the segmental duplications themselves provide the molecular basis for many human genetic disorders, including complex genetic-disease traits. To be sure, there are solutions to the problems posed by the duplicated sequence. First and foremost, however, it is essential that such highly paralogous regions be identified, their locations refined, and their sequence correctly assembled into the human reference genome. Understanding the biology and evolution of these regions is critical for a complete understanding of the genetic basis of human disease.
Segmental duplications consist of the duplicative transposition of genomic DNA, ranging in size from one to hundreds of kilobases (for reviews, see Ji et al. 2000; Shaffer and Lupski 2000; Eichler 2001). This scale is much smaller than the detection limits of 5 Mb (for “chromosomal painting” experiments) and several Mb (for most studies of gene order). Segmental duplications appear to be “normal DNA” and may be composed of genic sequence with introns and exons, as well as common repeats, such as Alus and L1 elements. In general, the duplications are highly homologous (sharing an average of 95.5% sequence identity). This has been taken as evidence that many duplications have arisen recently during the evolution of our species (<35 million years ago). To date, they have no distinguishing sequence features that facilitate a priori detection, and the mechanism underlying their movement remains unknown (Eichler 2001). Duplications may occur in tandem, but they are usually interspersed, occurring both within (intrachromosomal duplications) and between (interchromosomal duplications) homologous chromosomes. Anecdotal observations have implied that interchromosomal duplications are biased in favor of pericentromeric (Tomlinson et al. 1994; Eichler et al. 1996, 1997; Regnier et al. 1997; Zimonjic et al. 1997) and subtelomeric regions (Trask et al. 1998b; Wong et al. 1999). Many pericentromeric regions that have been studied in detail appear to be composed of duplicated stretches of mosaic or juxtaposed sequences originating from different regions of the genome (Jackson et al. 1999; Loftus et al. 1999; Ruault et al. 1999; Guy et al. 2000; Horvath et al. 2000a; IHGSC 2001). Recent analysis of the entire genome suggests that interchromosomal duplications are at least 4.5-fold enriched in pericentromeric regions and at least 2.7-fold enriched in subtelomeric regions (Bailey et al. 2001).
Duplications of genomic DNA have two main biological consequences. In terms of genetic disease, they underlie the molecular basis of many recurrent chromosomal structural-rearrangement syndromes. During meiosis, highly paralogous sequences can align and undergo homologous recombination, producing rearrangements in the gametes. Examples of genomic disorders mediated by such aberrant recombination include microdeletions—such as Smith-Magenis syndrome (SMS [MIM 182290]), Prader-Willi and Angelman syndromes (PWS [MIM 176270] and AS [MIM 105830], respectively), Neurofibromatosis type 1 (NF1 [MIM 162200]), and velocardiofacial and DiGeorge syndromes (VCFS [MIM 192430] and DGS [MIM 188400], respectively)—and microduplications, such as cat-eye syndrome (CES [MIM 115470]) and Charcot-Marie-Tooth disease type 1A (CMT1A [MIM 118220]) (Chen et al. 1997; Amos-Landgraf et al. 1999; Edelmann et al. 1999a, 1999b; Dorschner et al. 2000; Shaikh et al. 2000). Recent analysis has further suggested that duplicated sequences may also predispose to large-scale mitotic polymorphisms associated with complex genetic traits (Gratacos et al. 2001).
In addition to their mechanistic role in disease, duplications have long been viewed as a major pathway of gene evolution—mainly through whole-genome duplication (Ohno 1968, 1970). Although whole-genome duplication during vertebrate evolution is a widely held hypothesis, the significance of such events, compared to that of smaller segmental duplications, has recently been questioned (Eichler 2001; Hughes et al. 2001; IHGSC 2001; Venter et al. 2001). Certainly, for primate evolution, genomewide duplications have played little role in recent gene evolution, since the last postulated whole-genome duplication event occurred an estimated 430 million years ago (Skrabanek and Wolfe 1998). Since that time, numerous genes and gene families have arisen through segmental duplications such as zinc-finger, olfactory-receptor, certain globin, opsin, coagulation, and fibrinolytic genes. Zinc-finger genes and globins are examples of clustered duplications in which the entire gene has been duplicated in the form of repetitive tandem arrays. Coagulation and fibrinolytic genes are postulated examples of duplicative shuffling, where exons from different genes have been reassembled to create a new gene with a new function (Patthy 1996). To date, there are just a few examples, in the literature of recently evolved genes, where the duplicated intronic and flanking DNA still maintains a high degree of similarity (Teglund et al. 1994; Eichler et al. 1998; Seroussi et al. 1999; Edelmann et al. 2001). One recent example is intrachromosomal duplications on chromosome 16 that appear to be mediating the rapid expansion, through adaptive selection, of a new gene family (morpheus) in the great apes (Johnson et al. 2001). Such “newly” evolving genes may, at least in part, account for underlying phenotypic differences between humans and closely related species such as chimpanzee and gorilla.
The objective of this study was to investigate the structure, organization, and evolution of recent segmental duplications at a chromosome-wide level. Combining resources from the Human Genome Project and in silico data mining with experimental data, we present an evolutionary perspective of the fine-scale remodulation of chromosome-22 architecture through recent duplication processes. In previous analyses, the systematic study of duplications has been hindered by the inability to differentiate allelic overlap from highly similar duplications without resorting to experimental verification. Such determinations are made even more difficult by the error-prone nature of draft sequences and their resulting assembly, where perceived duplications with >98.5% identity are usually missed allelic overlaps (Bailey et al. 2001; IHGSC 2001). This is further complicated by the fact that duplicated sequences are prone to being unassigned or misassigned in the genome assembly (Bailey et al. 2001). As the first “completely sequenced” chromosome, chromosome 22 provides a high-quality sequence and assembly from which to study segmental duplications. Chromosome 22q11 has also long been implicated in genomic disease where segmental duplications, termed “low copy repeats” (LCRs) have been found to underlie several genomic diseases, including der(22), VCFS, and CES (Halford et al. 1993; Edelmann et al. 1999a, 1999b; Shaikh et al. 2000; Footz et al. 2001). Although the disease-causing LCRs have been extensively studied, a global sequence analysis to define and identify segmental duplications across the entire chromosome has never been undertaken. Here, we present a chromosome-wide in silico analysis, combined with a comparative FISH analysis, of both interchromosomal and intrachromosomal segmental duplications on chromosome 22, to detail their sequence properties, organization, and potential role in gene evolution.
Material and Methods
Detection and Quantitation of Segmental Duplications
To detect segmental duplications, we used the method we have described elsewhere (Bailey et al. 2001). Briefly, this method aims to detect large alignments, despite disruption by large deletions and/or insertions. For this study of chromosome 22, we analyzed the January 2001 assembly (oo23) of the October 2000 sequence freeze (see UCSC Human Genome Assembly Web site). This genome assembly contains the finished published assembly of chromosome 22 (Dunham et al. 1999), with a few additions to close several gaps. We have adhered to the genome-assembly coordinates, which add a proximal 13 Mb of ambiguous nucleotides to represent the unknown sequence of the p arm and centromere. The basic methodology included the identification of common repeat elements (by RepeatMasker), extraction of the repeats, and global BLAST comparisons of the putatively unique DNA. BLAST results (⩾250 bp and ⩾85% identity) were retained to increase our sensitivity. Repeats were reincorporated into the sequence, and the alignment ends were trimmed to better define the duplication boundaries. Global alignments were generated using ALIGN (Myers and Miller 1988). The statistics for global alignments were merged to represent single alignments with large gaps (up to 10 kb). Merged alignments of <1 kb and <90% identity were removed, leaving 1,026 alignments (837 interchromosomal and 189 intrachromosomal). Their mean length was 7,351 bp of genomic sequence. The means for intra- and interchromosomal alignments were 11,146 bp and 6,519 bp, respectively. The lower mean length for interchromosomal alignments most likely reflects increased fragmentation due to draft sequence on other human chromosomes, rather than any biological difference between the two types. Before the pairwise analysis, gaps (up to 50 kb) were traversed to combine the more-fractured interchromosomal draft alignments. Views of the pairwise alignments and other sequence features were generated with the graphical alignment viewer PARASIGHT (J.A.B., unpublished data). Evolutionary genetic distance was corrected for multiple substitutions using Kimura’s two-parameter model (Kimura 1980).
Isolation of Chromosome 22 Clones and Fluorescent In Situ Hybridization
Large-insert genomic BAC clones within the duplicated regions, RCPI-11 and CITD, were selected on the basis of end-sequence alignment against the human chromosome 22 reference (>99.5% sequence identity). For regions lacking identifiable BAC-end sequenced clones, we designed, labeled, and hybridized STS probes, as described elsewhere (Horvath et al. 2000a), to identify clones from the chromosome 22–specific cosmid library (LL22NC01). The positions of these clones within the chromosome 22–sequence assembly was confirmed by end-sequencing analysis. Additional clones (2336n9 and 803p16) were selected within the unique region flanking this pericentromeric duplication zone, to determine the transition between duplicated and nonduplicated sequence. FISH analysis of chromosomal metaphase spreads of lymphoblastoid lines from two different humans and four closely related primates—chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), orangutan (Pongo pygmaeus), and macaque (Macaca fascicularis)—was performed as described elsewhere (Horvath et al. 2000a, 2000b). The sequence identity among these primates is sufficiently high that genomic probes routinely cross-hybridize between species. (As a control, reciprocal experiments using baboon BAC DNA as a probe against human metaphases have been performed for several duplicated loci.) Chromosome identification was obtained by DAPI banding. When FISH signals hybridized to chromosomes not easily distinguished by banding, cohybridization experiments with appropriate probes were performed.
Somatic Cell PCR and Sequencing
Somatic cell hybrid DNA was assayed using PCR, to determine the extent and sequence identity between chromosome 14 and chromosome 22. Two sources were used: a monochromosomal somatic cell hybrid DNA panel (National Institute of General Medicine and Science [NIGMS], Human Genetic Mutant Cell Repository Mapping Panel 2) and a multichromosomal hybrid line (Coriell GM14972) containing chromosomes 2, 14, 17, 20, 21, and t(4;16). PCR primers were amplified with standard conditions as described elsewhere (Horvath et al. 2000a). The primer sequence and genomic positions are: pair A (forward, 5′-tcacagcaaattgtgagggaggac-3′, and reverse, 5′-agtgcctctatcctgacacttgtg-3′ [13,021,120–13,021,525 bp]); pair B (forward, 5′-aacaacaaggaagaggcaagtggg-3′, and reverse, 5′-tacaacaaactgagccaggcaacc-3′ [13,056,181–13,056,461 bp]); pair C (forward, 5′-tcaaggtctgctgaactctggatc-3′, and reverse, 5′-cagaagatacacaaagtggcaccag-3′ [13,100,424–13,100,842 bp]); pair D (forward, 5′-cctggtcttctctggtcttctcat-3′, and reverse, 5′-ccttacccaggttatgctaccaaac-3′ [13,100,871–13,101,287 bp]); pair F (forward, 5′-aataatcccaccactagcctccag-3′, and reverse, 5′-cagatagcactggcttaggagatg-3′ [13,102,261–13,102,641 bp]); pair G (forward, 5′-gcagcattgtggaggtcagataac-3′, and reverse, 5′-tgactatgccctcccttgaagatg-3′ [13,163,898–13,164,366 bp]); pair H (forward, 5′-tgtctgatttctggctgatgcagg-3′, and reverse, 5′-gcaataccccactgagataagagg-3′ [13,227,422–13,227,937 bp]); pair I (forward, 5′-gcatatagtgtgcagataccaggg-3′, and reverse, 5′-gcctcatcagctgtgtttttctcc-3′ [13,265,009–13,265,326 bp]); pair J (forward, 5′-tttcatactgctccagacccaagc-3′, and reverse, 5′-ttgcaatccaaggaatccctccag-3′ [13,370,510–13,370,826 bp]); and pair K (forward, 5′-ggacaggcttaggaaagacagaac-3′, and reverse, 5′-tgggagggatacagaaaggaaagg-3′ [13,575,543–13,575,961 bp]).
Module Definition and Transcriptional Potency
The assignment of modules (minimal sequence segments that demonstrate a shared evolutionary history) combined automated initial detection and detailed hand curation. Underlying our attempt to define modules, we searched for all similarities to genic sequence with intron-exon structure. We used automated BLAST analysis of all duplicated genomic sequence versus full-length NCBI Locus Link/reference sequence (RefSeq) and Unigene human transcripts. From these results, we extracted the highest-similarity transcript to any given region of chromosome 22 that showed intron-exon structure (December 2000). In the case where transcripts showed comparable sequence similarity, the full-length mRNA or longer transcript was chosen. BLASTN sequence-similarity searches were then used to define the most likely allelic locus (>99% identity) of the transcript within the genome assembly. The alignment was further refined with sim4 (Florea et al. 1998) to delineate the transcript’s intron-exon structure. The defined underlying genomic sequence was RepeatMasked and was searched by BLAST against chromosome 22 to detect putative modules. Gaps of <10 kb were joined during identification of the boundaries of the modules, to traverse large high copy repeats such as L1 elements. These data were combined with full-length mRNAs and known genes that were assigned to chromosome 22 (UCSC Genome Browser). We used PARASIGHT to examine these putative modules and transcripts, along with the underlying duplications. Each putative module was assessed by hand, and the positions of all copies were defined and recorded in a table. Modules were defined, first and foremost, with regard to definable duplication boundaries. However, within the highly duplicated pericentromeric region, duplications were often defined solely on the extent of similarity to genomic sequence underlying the intron-exon structure.
To search for new or modified transcripts, we treated interchromosomal and intrachromosomal duplications separately. For intrachromosomal duplications, we initially ascertained all of the modules that showed evidence for expression from two or more of the copies. We also included modules where single transcripts had highly similar matches (>98% identity) to two or more copies. This was done to ensure that a small EST was not subsumed in our initial survey, by a longer, less-identical full-length mRNA. For interchromosomal modules, all transcripts showing >99% identity to the chromosome 22 sequence were initially selected as possible novel genes. In total, 37 possible transcripts were identified. EST and mRNA BLAT (J. Kent, unpublished data) alignments for the duplications from the UCSC genome browser were examined to determine transcriptional support. Transcriptional support was only concluded if at least two mRNAs and/or ESTs showed support for a particular intron-exon structure. Transcripts lacking specific transcriptional support for each copy were excluded from further analysis. In the case of highly similar duplications (>99%), several transcripts were excluded on the basis of the inability to determine the transcribed locus. These criteria excluded many transcripts within interchromosomal regions that showed high similarity (>99.5%) but lacked underlying genic sequence at another locus. Also, we required that interchromosomal duplications be the recipient copy of the transposition event. Thus, many pericentromeric transcripts that showed >99.5% similarity were excluded as possible chromosome 14 transcripts. Subtelomeric RABL2B and three other transcripts that showed specific expression on chromosome 2 and 22 were excluded, since chromosome 22 was determined to be the ancestral copy.
Results
Initial Computational Detection and Analysis of Chromosome 22 Duplications
Using the finished sequence of the q arm of chromosome 22 (Dunham et al. 1999), we sought to detect both internal and external pairwise similarities on the basis of the published draft genome sequence (IHGSC 2001). To accomplish this, we used a method described elsewhere (Bailey et al. 2001), which is optimized to detect large, highly similar duplication events by spanning large gaps or deletions within the DNA (see Material and Methods section). On the basis of this in silico approach, we found 10.8% (3.672/33.786 Mb) of the 22q sequence to be involved in segmental duplications. Sequence involved in intrachromosomal and interchromosomal duplications comprised 6.85% (2.317 Mb) and 5.75% (1.945 Mb), respectively (see online-only supplements 1 and 2). The majority (68%) of duplicated bases resided within alignments ⩾10 kb. The spatial distribution of duplicated sequence is clustered (fig. 1). Interchromosomal duplications are concentrated within the most centromeric and most telomeric regions of the chromosome, whereas the majority of intrachromosomal duplications localize to the proximal third of the arm. We found a 4.4-fold enrichment in sequence assigned to both interchromosomal and intrachromosomal positions (1.74%), compared with a random expectation (0.394%), suggesting an association between interchromosomal and intrachromosomal duplications. Figure 1 also demonstrates clustered nature of interchromosomal duplications, in terms of both assignment and location within the nonhomologous chromosomes.
Figure 1.
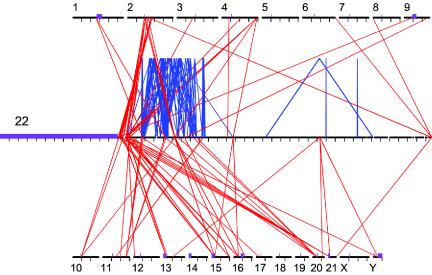
Spatial distribution of large segmental duplications between chromosome 22 and other human chromosomes. A scaled (50×) version of chromosome 22, surrounded by the other chromosomes, shows lines representing interchromosomal (red) and intrachromosomal (blue) alignments (⩾10 kb). The majority of chromosome 22 pericentromeric duplications localize to the pericentromeric regions of other chromosomes. Likewise, the majority of subtelomeric duplications localize to subtelomeric regions of nonhomologous chromosomes. There is little cross-hybridization between subtelomeric and pericentromeric duplications. Chromosomes 2 and 20 share the largest amount of sequence with chromosome 22, whereas chromosomes 5, 7, 14, 18, 19, and X do not share any duplications with chromosome 22 that are >10 kb in size. The coordinates are based on the published UCSC human genome assembly. For chromosome 22, each tick mark represents a 1-Mb interval. For the other chromosomes, tick marks represent 50-Mb intervals. Purple boxes represent the unsequenced centromeres, acrocentric p arms, and Y heterochromatin. Gaps are denoted by white space. The program PARASIGHT was used to generate this diagram.
To provide a first approximation of the evolutionary timing of these duplications, we calculated the number of substitutions per base pair for each intrachromosomal and interchromosomal pairwise alignment (Kimura’s two-parameter estimate of genetic distance, K; see fig. 2) (Kimura 1980). Under the assumption of a constant neutral mutation rate (Goodman 1999; Chen and Li 2001), the genetic distance separating two sequences should be directly proportional to the evolutionary time since their divergence. When the genetic distance is estimated as a function of the number of aligned base pairs (fig. 2a), interchromosomal alignments show a noticeable mode (K=0.03 to 0.04). As the precise duplication events themselves are unknown, we also used the count of pairwise alignments as an event surrogate (fig. 2b). The only noticeable difference between interchromosomal and intrachromosomal duplications is the relative lack of highly similar alignments (K<0.02) for interchromosomal duplications when compared to intrachromosomal duplications. Excluding this possible reduction in the number of most-recent (<5 million years ago) interchromosomal duplications, both interchromosomal and intrachromosomal alignments showed a relatively consistent number of alignments across all remaining bins of divergence with no more than two-fold deviation from the mean. Thus, it appears that segmental duplications have been occurring continuously (although possibly not at a constant rate) over the past 35 million years of human evolution.
Figure 2.

Pairwise sequence distance (K) of chromosome 22 duplications. The two histograms show the distribution of genetic distance in terms of the number of aligned base pairs (a) and the number of alignments (b). Alignments are separated into interchromosomal (gray) and intrachromosomal (black). Distance (K) is the number of substitutions per 100 bp aligned and was corrected for multiple substitutions (see Material and Methods section).
Patterns of Interchromosomal Duplications
In addition to previous analyses that have characterized complex intrachromosomal duplications (Edelmann et al. 1999a, 1999b; Shaikh et al. 2000), our in silico analysis also predicts a remarkably complex pattern of interchromosomal duplications on chromosome 22. To further characterize these interchromosomal patterns, we targeted three of the most duplicated regions for further experimental analysis by FISH (see Material and Methods section). Each of the clones spanning the in silico duplications demonstrated hybridization to multiple human chromosomes including human chromosome 22. The results of this analysis are summarized (fig. 3 and the “HSA” column of table 1).
Figure 3.

Interchromosomal duplications of the pericentromeric region of chromosome 22. The combined results of in silico and FISH duplication detection are displayed for the most proximal 2 Mb of 22q. Labeled dark gray boxes above the tick-marked sequence denote the positions of chromosome 22 clones used for FISH analysis. Below the sequence, light gray boxes represent positive FISH signals to particular chromosomes. Black bars show the in silico positions of duplicated alignments, on the basis of comparison of the chromosome 22 reference sequence to the rest of the human genome. The majority of the paralogous segments mapped to pericentromeric positions on the other chromosomes. CER denotes a region containing a 150-kb expanse of centromeric-associated repeat. Blank spaces represent sequence gaps. “UK” denotes sequence with unknown chromosome assignments.
Table 1.
Human and Comparative Primate FISH Results for Interchromosomal Duplications[Note]
|
Boundary |
Chromosome(s) Showing FISH Signals in |
|||||||
| Region andClone | Library | Beginning(kb) | Ending(kb) | Humana | Chimpanzee | Gorilla | Orangutan | Macaqueb |
| Pericentromeric: | ||||||||
| 235d20 | RPCI-11 | 13,028 | 13,130 | 22, 2q21, 9, 13, 14, 15, 21 | 22, 2p, 13, 14, 15, 18, 21 |
2p, 9, 13, 14, 15 |
2q, 13, 14, 15, 21 |
10 |
| 140m6 | RPCI-11 | 13,079 | 13,229 | 22, 2q21, 14, 15, 18, 21 |
2q, 14, 15 |
2q, 13, 14, 15 |
2q, 14, 21 |
10, 20 |
| 354f21 | RPCI-11 | 13,217 | 13,401 | 22, 2q21, 9qter, 14 |
2q, 14 |
2q, 14 |
2q, 21 |
2q, 20 |
| 134c5 | RPCI-11 | 13,323 | 13,464 | 22, 2q21, 14 |
2q, 14, 15 |
2q, 14 |
2q, 3, 13, 14, 15, 20, 21 |
10 |
| 27c11 | LL22NC01 | 13,487 | 13,529 | 22 (multiple), 14, 21 | 22, 22pter, 13, 14, 18 | 22 | 22 (multiple) | No signal |
| 3087k20 | CIT-HSP | 13,625 | Within gap | 22, 2q21, 9, 13, 14, 15, 18, 21 | 22, 2q, 9, 10, 13, 14, 15, 18, 21 | 10, 14, 15, 21 | 22, 10, 15, 21 |
10 |
| 394j3 | RPCI-11 | 13,823 | 14,028 | 22, 1, 2, 4q22, 7qter, 9, 10, 14, 15, 16, Y | 22, 2p, 10, 14, 16, 18, Y |
2p, 15, 16 |
22, 4q22, 7qter, 21 | 22, 4 |
| 164d11 | RPCI-11 | 13,913 | 14,098 | 22c, 1q21, 2p12, 2q13, 9p12, 9q13, 10p11.2, 13, 14, 15, 16, 21, Y | 22, 2p, 9, 10, 13, 14, 15, 16, 21 | 22, 2p, 7, 9, 13, 15, 16, 18, 21 | 22, 7, 13, 15, 21 | 22, 7qter, 20 |
| 104f9 | RPCI-11 | 14,111 | 14,294 | 22, 2q13, 3p12, 12p13, 13q12, 20 | 22, 3, 9, 12, 20, Y | 22, 3, 12, 15 | 22, 2p, 3, 12, 13, 17 | 22, 1 |
| 66F9 | RPCI-11 | 14,325 | 14,501 | 22, 2, 9, 13, 14, 21 | 22, 2p, 10, 15 | 22, 9c, 13, 15, 18, 21 | 22q | 22 |
| 2336n9 | CIT-HSP | 14,541 | 14,697 | 22 (multiple) | 22 (multiple) | 22 (multiple) | 22 | 22 (multiple) |
| 803p16 | CIT-A | 14,758 | 14,914 | 22 | 22 | 22 | 22 | 22 |
| VCFS: | ||||||||
| 379n11 | RPCI-11 | 18,264 | 17,349 | 22q11, 1, 2, 13 | 22, 1 | 22, 1, 2p, 9, 13, 15, 18, 21 | 22 (multiple) | 22, 1 |
| 291k7 | RPCI-11 | 18,171 | 18,326 | 22, 1p12, 2, 5p13, 5q12, 13, 13qter, 20 | 22, 1, 2p, 13qter, 20 | 22, 1, 2p, 9, 13qter, 15, 18, 20, 21 | 22 (multiple), 1, 13qter | 22, 1, 13qter |
| Subtelomeric: | ||||||||
| 22b22 | LL22NC01 | 47,611 | 47,661 | 22qter, 2q13 | 22;2p | 22, 2p, 2q, 4q2.5, 7q36, 10p2.6 | 22qter | 22 |
Note.— All FISH images are available at the Rocchi Lab Web site. Underlining and boldface type indicate the lack of chromosome 22 signals. Signals are pericentromeric, unless otherwise noted. For nonhuman primates, Arabic numerals represent phylogenetic chromosomes (2p and 2q represent IIp and IIq chromosomes). “(Multiple)” denotes two or more signals by metaphase or interphase on chromosome 22.
Two independent experiments for each clone were performed on different human individuals.
Phylogenetic chromosome 22 in MMU is part of a large chromosome (MMU 13) resulting from the fusion of phylogenetic chromosome 22 and 20 (Wienberg et al. 1992)
Within the pericentromeric region, FISH confirms that the entire proximal 1.5-Mb region of chromosome 22q is highly duplicated (each BAC hybridized, on average, to six other chromosomes). Not a single probe hybridizes uniquely to human chromosome 22. Figure 3 shows a direct comparison between the in silico predicted patterns of duplication (black bars) and the multisite pattern observed by FISH (gray overlay). A complex pattern of duplication is readily apparent within the pericentromeric region, where certain sequences are found on multiple chromosomes. Remarkably long stretches (>150 kb) of sequence appear to be shared between certain chromosomes such as chromosome 2 and 14. It is also apparent that the in silico and FISH results, on occasion, show poor correlation, suggesting the lack of or misassignment of sequence (Bailey et al. 2001; IHGSC 2001).
An example of this lack of correlation is the lack of chromosome 10 sequence underlying BACs 394j3 and 164d11, for which strong chromosome 10 FISH signals are present. We have extensively studied this region from the perspective of chromosome 2 and have used chromosome-specific nucleotide variants generated from monochromosomal hybrids to assign sequences to specific chromosomes (Horvath et al. 2000a). From sequence-similarity searches of these chromosome-specific variants, it is clear that the chromosome 10 copy is indeed present in the assembly, being misassigned to the pericentromeric region of chromosome 4 (data not shown). These results highlight the difficulties associated with automated sequence assembly of such highly duplicated regions and emphasize the need for experimental validation of their organization.
Comparative Primate FISH
To further analyze the evolutionary history of the interchromosomal duplications, we performed interspecific FISH, using closely related primates—chimpanzee, gorilla, orangutan, and macaque (table 1; images at the Rocchi Lab Web site). These species are hypothesized to have shared a common ancestor with Homo sapiens ∼5, ∼7, ∼14, and ∼23 million years ago, respectively (Kumar and Hedges 1998; Goodman 1999; Chen and Li 2001) and to have an average sequence divergence, compared with that in humans, of 1.2%, 1.6%, 3.1%, and 5.5%, respectively (Chen and Li 2001; E.E.E., unpublished data). Using this approach, we compared the duplication patterns of chromosome 22 probes between humans and primates (table 1).
In general, the probes show differences in copy number and location among the different primates. Macaques have fewer localizations, whereas the great apes almost always have multiple pericentromeric signals, akin to the pattern observed for humans—although a few exceptions were noted. The localizations among the great apes, however, are not always consistent, because of either lineage-specific deletions or subsequent duplications (table 1). This general pattern of apparent loss and gain of hybridization signal implies that duplications/deletions in these regions may be an ongoing process in great ape–chromosome evolution. As an example, human clone BAC 134c5 has hybridization signals on chromosome II and XIV in all great apes. However, additional signals in orangutan suggest either that secondary duplication events have occurred or that multiple deletions eliminated these copies in the common ancestor of humans and African apes. These quantitative and qualitative differences among closely related primates and the general reduction in the number of signals observed among the chromosomes of more divergent species are consistent with the predicted evolutionary timing of pericentromeric duplications, on the basis of sequence comparisons (see online-only supplement 1).
Similarly, the analysis of the most subtelomeric clones from chromosome 22 showed conservation of signal between chromosomes II and XXII among all African apes (table 1). The chromosome XXII copy is the sole signal detected for the macaque and orangutan species. Given the high degree of identity between the human chromosome 2 and 22 duplications (98.9%), these data are consistent with a duplication of this region in the common ancestor of chimpanzees, humans, and gorillas. Although there is good correlation between in silico estimates of duplication timing and FISH data, some exceptions were noted. For example, comparative FISH analysis of the VCFS region revealed a long-standing pattern of interchromosomal hybridization between chromosomes I and XXII. Cross-hybridization signals between these two chromosomes are observed among all primate species examined. The degree of sequence identity between human I and XXII duplications (97.5%) is significantly greater than the genomic average between macaque and human genomic DNA (94.5%) suggesting duplicative transposition from I to XXII. Furthermore, the additional chromosomal localizations observed in gorilla (table 1) may imply secondary, lineage-specific interchromosomal duplication events within this species.
A Pericentromeric Gradient of Duplications
The most striking observation was the absence of chromosome 22 signals among nonhuman primates for the four most centromeric human chromosome 22 probes (235d20, 140m6, 354f21, and 134c5). This corresponds to a ∼450-kb region extending from sequence map positions 13.028–13.465 Mb, (underlining, table 1). Figure 4 shows representative hybridizations that demonstrate the absence of chromosome 22 signals among the great apes and their specificity for human chromosome 22. For these clones, the only positive chromosome 22 localization detected in a nonhuman primate is found for the most proximal clone, 235d20. On the basis of the degree of overlap between 235d20 and 140m6, this chromosome 22 signal is limited to a 51-kb segment of chromosome 22 (13.028–13.078 Mb). Within the next region (13.487–13.625 Mb), strong chromosome 22 conservation is observed among the great apes but not in macaques, suggesting the possibility of a great ape–specific event. Conservation of chromosome 22 signal among all primates is observed for all probes distal to 394j3 (14.028 Mb). Since our in silico detection is limited to 90%, we altered the parameters to detect more-divergent sequences within the proximal 3 Mb of chromosome 22 (data not shown). This analysis uncovered more-divergent duplicated regions (<90% sequence identity) believed to contain recently evolved genes (Footz et al. 2001). These more-divergent duplicated segments are located at the most-distal end of the pericentromeric duplications (between 14.2 and 14.5 Mb; fig. 3). This region lies proximal to a well-conserved mouse syntenic region (Footz et al. 2001). In summary, both in silico and comparative FISH analysis suggest the stepwise evolutionary accretion of proximal chromosome 22. The most recently duplicated sequence appears to lie most proximal to the centromere, whereas, presumably, more-ancient duplications (based on sequence divergence) lie more distal.
Figure 4.
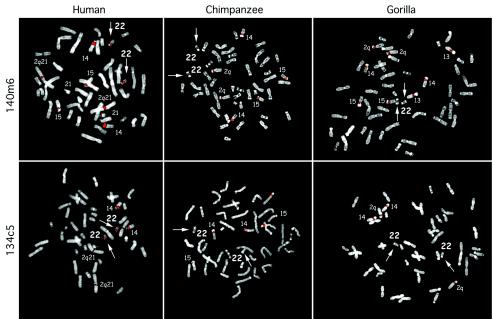
Comparative FISH of a human-specific duplication. Human, chimpanzee, and gorilla comparative FISH results are shown for human chromosome 22 probes 140m6 (a) and 134c5 (b). Both BACs lack any signal to chromosome 22 among the nonhuman primates.
A Human-Specific Duplicative Transposition from 14q11 to 22q11
Our in silico analysis indicates a conspicuous absence of paralogous sequence for human chromosome 14, despite FISH signals which suggest that a chromosome 14 duplicated segment may span up to the first megabase (13–14 Mb) of the sequenced 22q pericentromere. Within this entire region, only one small pairwise comparison (5 kb) was found to chromosome 14. This segment showed 99% sequence identity to chromosome 22. Interspecific FISH results indicated the absence of the most proximal 400 kb in other nonhuman primate species. We hypothesized that the proximal region of chromosome 22 was a human-specific duplication that originated from chromosome 14. Sequencing or assembly of the putative chromosome 14 region may have been overlooked because of the high degree of sequence identity. To confirm the presence of the duplication and to assess its integrity on chromosome 14, we designed 10 PCR amplicons within the most-proximal 600 kb of chromosome 22. Monochromosomal hybrid DNA from chromosomes 14 and 22 was used as a template for PCR, and the resulting products were sequenced (fig. 5). Comparison between the 3,215 bases sequenced from each chromosome hybrid showed >99.4% nucleotide identity. Interestingly, close inspection of the chromosome 14 sequences revealed the presence of heterogeneous nucleotide sites. Such variant sites are unexpected from a monochromosomal resource material and usually indicate the presence of multiple of copies of sequence within the chromosome. Of the 3,215 bases of high-quality sequence aligned, we found five fixed variants and 13 heterogeneous sites (with one of the bases the same as in chromosome 22) between monochromosomal 14 and 22 cell lines. Since phase cannot be assigned to the monochromosomal 14 copies, the lowest and highest possible sequence identity for the chromosome 14 duplicates to chromosome 22 range from 99.4% (5+13 differences/3,215 bases examined) to 99.8% (5 fixed differences/3,215 bases examined). It is interesting to note that the heterogeneous sites correlate with the most proximal region underlying 235d20, which showed the sole primate 22 signal in chimpanzee. This may indicate an earlier chromosomal exchange within the common ancestor of chimpanzees and humans. To eliminate the possibility of chromosome 22 contamination within the chromosome 14 hybrid, we verified our results using a second hybrid containing a single copy of chromosome 14 (Coriell GM14972).
Figure 5.

PCR analysis of a 550-kb duplication between human chromosomes 14 and 22. The figure shows the position on chromosome 22 of oligonucleotides that were designed to amplify paralogous sequences from chromosomes 14 and 22. Ten PCR products (A-K) were designed, spanning ∼550 kb. Products were amplified and sequenced from both chromosome 14 and chromosome 22. The total high-quality sequence (SEQ TOTAL), the number of sites with fixed differences (SEQ fixed) between 22 and 14, and the number of heterogeneous sites (SEQ hetero) for each product are shown. These heterogeneous sites in the most centromeric products suggest multiple copies for this region within chromosome 14. The average sequence identity between chromosome 14 and 22 for all 3,215 bases was 99.4%–99.8% (with and without heterogeneous sites).
Assignment of Duplicated Sequence to Modules
The structure of regions harboring segmental duplications is highly complex, because of the large number of successive segmental transposition, juxtaposition, and rearrangement events (Eichler et al. 1996; Jackson et al. 1999; Guy et al. 2000; Horvath et al. 2000a). As one approach to dissecting this complexity, we attempted to define modules or sequence blocks in terms of segments that likely shared the same evolutionary history—that is, segments that can be traced to a common ancestral sequence. We defined these modules using two different approaches. The first and preferred method was to define the junctional boundaries of a minimal shared paralogous segment. Using the program PARASIGHT, we graphically displayed all optimal global alignments to chromosome 22 sequences and identified shared duplication breakpoints with other chromosomal regions. If this was not possible, as was the case with complex mosaic regions, then the extent of gene-sequence coverage was used to delineate the modules. The use of expressed gene sequence to define common duplicated blocks has been used extensively in the study of mosaic duplications such as ALD, CTR (Eichler et al. 1996), and VCFS-region duplications (Shaikh et al. 2000). Experimentally, it has been shown that many of the derived duplications contain only partial intron-exon structure, such that functional expressed copies of the gene often correspond to the ancestral locus. Because of the conservative nature of our approach and a lack of well-defined boundaries or gene homology, roughly a quarter of the duplicated segments remained uncharacterized. In total, we defined 78 modules, with a grand total of 157 copies on chromosome 22 (fig. 6 and online-only supplement 3) that were distributed inter- and intrachromosomally. The number of copies on chromosome 22 for the individual modules ranged from 1 (solely interchromosomal) to 11 copies (DKFZp434P211/BCR). We defined 64 modules on the basis of the extent of shared intron-exon structure and 14 modules solely on the basis of well-demarcated junctional boundaries. Of these 14, 11 had no identifiable sequence features (defined as “unknown”), whereas 3 included processed pseudogene markers (defined as “ppseudo”). Duplicated sequences lacking module assignments were located predominantly within the pericentromeric and subtelomeric regions, where multiple copies and the draft nature of interchromosomal comparisons made it particularly difficult to define junctional boundaries. Within the pericentromeric region, 30 different modules were identified. Of these, 4 modules showed evidence for expression, whereas 16 of the remaining modules containing intron-exon structure lacked evidence of expression (unprocessed pseudogenes). These unprocessed pseudogenes, arising from recent interchromosomal duplications, help to explain earlier observations of a dense clustering of pseudogenes in this region (Dunham et al. 1999). Our analysis indicates a mosaic structure with the most divergent copies (presumed ancestral locus) originating from diverse regions of the genome that are often non-pericentromeric (online-only supplement 3).
Figure 6.

The modular structure of segmental duplications on chromosome 22. The position and size of the 78 defined modules are shown along the entire chromosome 22 sequence (black line; each line = 1 Mb). Modules are arbitrarily colored, except that gray and black are used for interchromosomal duplications. Arrows indicate orientation relative either to a defining transcript or to the most proximal copy. The positions of interchromosomal (red bars) and intrachromosomal (blue bars) duplications are shown overlapping the sequence line. Tick marks represent 100 kb. Gaps (white space) in the sequence are drawn to scale. The program PARASIGHT was used to generate this diagram.
Duplications and Transcript Formation
As the majority of duplicons harbor intron-exon structure, these segmental duplications have the potential to evolve novel transcripts. This may be mediated through the creation of whole-gene duplications or through the juxtaposition of different modules to create mosaic transcripts (similar to exon shuffling and domain accretion). To investigate this role, we examined all duplicated sequence for evidence that a duplication event has generated or altered a transcript (fig. 7). Stringent evidence was required (see Material and Methods section). First, at least two copies of a module had to show evidence of transcripts that utilized the same underlying sequence region within the duplications. Second, there must exist multiple ESTs or mRNAs supporting transcriptional potency of each putative copy. Supporting transcripts were assigned on the basis of best genomic location, and, thus, any individual transcript could not support both copies. Third, in the case of interchromosomal duplications, evidence was required that the new/modified transcript was created on chromosome 22.
Figure 7.

Transcripts created or modified through segmental duplication. We identified 11 transcripts that have been created or modified via the process of segmental duplication. This was a comprehensive and stringent search of chromosome 22 duplications, to identify overlapping regions of transcriptional activity. Transcriptional activity was based on finding two or more spliced cDNA sequences that had been placed to their best genomic location (see Material and Methods section). Eight examples illustrating the intron-exon structure, as well as the underlying duplications, are shown for the new (top) and putative ancestral (bottom) transcripts. Positions within the genome assembly are given in kb. Exons are positioned approximately, but exon size is not shown to scale. a, AL001299, a full-length transcript (1,625 bases) that originates from mosaic modules within the pericentromeric region. It has a putative ORF of 98 aa. The intron-exon structure spans ∼100 kb (14,027–14,124 kb), with each exon originating from a different module. Two modules underlying the gene show expressed genes suggesting the ancestral origin of these modules: solute carrier family 25 member 15 (SLC25A15), for the 13q14 module, and von Willebrand factor (vWF), for the 12p11 module. Thus, the pericentromeric juxtaposition of these modules leads to the formation of AL001299. Exon 2 does not contain any exon sequence from SLC25A15. Exon 3 is composed of vWF exon sequence, albeit in the reverse orientation. b, Partial-gene duplication of the proximal seven exons of lipoprotein receptor–related protein 5 (LRP5) from 11q13. Alignment of five transcripts suggests multiple transcriptional start sites or alternative splicing. Both AL137651 and AI972731 utilize exon sequence from LRP5, including exons 1, 4, 5, 6, 7, 8, and 9. The best ORFs are 252 aa for AL137651 and 77 aa for BE396696. c, Whole-gene duplication (ancestral copy undetermined) leading to the formation of DGCR6 and DGCR6L genes. The duplication also includes a whole-gene duplication of proline dehydrogenase (PRODH), which forms an unprocessed pseudogene (PRODHΨ) in the distal copy. DGCR6 and DGCR6L transcripts have conserved intron-exon and coding structure (220 aa). The transcripts have been experimentally verified and show expression from multiple tissues, with differential expression between the two copies (Edelmann et al. 2001). Function is unknown. d, Partial-gene duplication of the seven terminal exons (17–23) of BCR (NM_021574) that has led to the creation of a fusion transcript in one of the distal copies. The full-length transcript (NM_014549) has seven exons and is in the reverse orientation, compared to BCR. Exon 1 is derived from the flanking distal chromosome 22 sequence and exons 2–7 are derived from the duplicated sequence. These terminal exons incorporate the reverse sequence of the BCR exons 19, 20, and part of 22. NM_014549 contains a putative ORF of 428 aa. e, Partial-gene duplication of the last three exons of AK024854, a phorbolin-related gene, has lead to the formation of a five-exon fusion transcript. Exon 1 is derived from adjacent chromosome 22 sequence, whereas the terminal 4 exons are derived from the three duplicated exons of AK024854. Exon 2 and 3 correspond to exons 5 and 6 of AK024854. Exon 4 and 5 correspond to exon 7 of AK024854. f, Another partial gene duplication of a phorbolin-related transcript AF18240 (exons 1–4) has created a transcript represented at its 3′ end by EST AI092348. AI092348 has two exons with an ORF of at least 77 aa, extending in a 5′ orientation and terminating within the 3′ exon. The penultimate exon is derived from exon 2 of AF18240. g, Partial-gene duplication of the last three exons of crystallin beta B2 (CRYBB2) has lead to the formation of a new gene, represented by EST AW190323. Three exons of AF18240 are utilized in the new transcript, with the addition of two additional 5′ exons from the adjacent unduplicated sequence and a putatively new 3′ terminal exon from previously nonexonic sequence. The ESTs have ORFs ranging from 88–105 aa, compared to 205 aa for CRYBB2. h,ESTs supporting potential whole-gene duplication, with representative transcripts from both copies (AI669658 and AA228976). The most proximal transcript, AI669658, contains two exons with a predicted ORF of >90 aa. The distal transcript, AA228976, contains three exons with a predicted ORF of >59 aa. Both transcripts appear to extend in a 5′ orientation, with an undetermined intron-exon structure. The three transcripts not shown in this figure have been previously described: (1) multiple partial-gene duplications within the immunoglobulin lambda (IGL) locus (Kawasaki et al. 1997), and (2) whole-gene duplications of ret-like finger proteins (RLPF1, RLPF2, and RLPF3), creating two new genes with conserved intron-exon and coding structure (Seroussi et al. 1999).
In total, 11 transcripts met these criteria (fig. 7). These duplications include the internal modification of existing genes, whole-gene duplications with well-maintained structure and mosaic transcripts composed of modules from multiple chromosomes. For example, a pericentromeric mosaic, AK001299, consisted of three exons each from a different module (fig. 7a). Exon 1 is from an undefined duplication (7q36), exon 2 shares paralogy with the transmembrane phosphate with tensin homology (TPTE) module, and exon 3 is paralogous to von Willebrand factor (vWF) exon (albeit transcribed in the reverse orientation). The other interchromosomal duplication with transcriptional potency originates from a duplication of the first nine exons of low-density lipoprotein receptor–related protein 5 (LRP5) of chromosome 5 (fig. 7b). The copy on chromosome 22 shows multiple transcripts, one of which (AL137651) incorporates sequence from LRP5 exons 5–9 and encodes a putative 252–amino acid (aa) protein. The well-characterized duplication of BCR has a copy that generates a transcript (DKFZp434P211), transcribed in the opposite orientation with a reading frame of 428 aa (fig. 7d). The immunoglobulin lambda (IGL) locus has been modified by extensive duplication (Kawasaki et al. 1997), and we find evidence for the creation of at least five new variable regions and one new constant region within the parameters of this study. In terms of whole-gene duplications giving rise to new family members, several examples exist, including DiGeorge critical region 6 (DGCR6) genes (Edelmann et al. 2001), and RET-finger protein–like (RFLP) genes (Seroussi et al. 1999). Additional members of the phorbolin and crystallin gene families (figs. 7e and 7g, respectively) appear to be forming through partial gene duplications that are remodeled by alternative splicing and the addition of new exons. It is interesting to note that few of these clusters show signs of direct tandem duplication; instead, the duplications giving rise to majority of these families are interspersed.
Discussion
We have detected and characterized segmental duplications on chromosome 22, providing the first systematic and detailed chromosome-wide view of segmental duplications. We found that over one-tenth (10.8%) of the sequence was involved in such duplications (⩾90% identity and ⩾1 kb). This is in marked contrast to chromosome-painting studies that suggest chromosome 22 has been well conserved, with only one rearrangement since the common primate ancestor (Muller et al. 1999). These data suggest that significant chromosomal structural changes have occurred on a much smaller scale. The segmental duplications showed spatial biases consistent with previous reports (Eichler et al. 1996; Trask et al. 1998a; Guy et al. 2000; Horvath et al. 2000a; Bailey et al. 2001). Interchromosomal duplications clustered within the pericentromeric and subtelomeric regions, whereas intrachromosomal duplications clustered within the proximal third of the chromosome arm. We verified and further studied the poorly characterized interchromosomal duplications by means of comparative FISH. In general, the comparative analysis conformed to estimates based on the degree of sequence identity among different duplicates. In particular, our analysis revealed a human-specific ∼400-kb duplication event from chromosome 14 to chromosome 22. We attempted to organize the duplicated sequence into minimal evolutionary shared segments (modules) and to examine the transcriptional and coding potential of the duplications, in a search to identify recent transcript innovations. Our analysis yielded the following important insights into the nature of segmental duplications on chromosome 22.
First, segmental duplication appears to be an ongoing process that has been active throughout recent primate evolution. This is supported by the distribution of pairwise genetic-distance estimates (K values) and interspecific FISH of the duplicated regions. Using pairwise counts as a surrogate for duplication events, the relatively even distribution implies the occurrence of duplications at various time points over the past 35 million years of evolution. However, our analysis cannot preclude the occurrence of a punctuated event, which may be suggested by the large amount of interchromosomal sequence between K=0.03 and K=0.04 (fig. 2). A recent comparative study of noncoding primate sequences estimated the genetic distance between human and orangutan at K=0.031 (Chen and Li 2001). This suggests that a large amount (∼61%) of the interchromosomally duplicated sequences on chromosome 22 arose near the time of the separation of African and Asian ape lineages (∼14 million years ago). Such a possible punctuated event may be a result of increases in the size or the number of duplication events. At present, the best surrogate for events (the number of pairwise alignments) does not support a dramatic increase in the number of events, which may suggest that the genomic segments may have been larger at this particular epoch during primate evolution. Not only will finished sequence be required to definitively answer such questions of timing, but sophisticated phylogenetic analysis must also be developed to model the complex forces of both duplication and deletion (Lynch and Conery 2000). The results of this study provide an important framework for such future studies. It is noteworthy that, despite the fragmentary nature of the working draft sequence, our study has detected the presence of relatively large-scale (>100 kb) duplications. The majority of these larger alignments tend to localize to the pericentromeric region of chromosome 22. The larger segments are often composed of multiple smaller duplication modules of diverse evolutionary origin. This lends support to the previously purposed two-step hypothesis that smaller duplications accrue within pericentromeric regions and then are subsequently distributed as larger mosaic blocks among nonhomologous pericentromeric regions (Eichler et al. 1997; Horvath et al. 2000a; Luijten et al. 2000).
Second, our results demonstrate a gradient of pericentromeric duplication. We observed that the most recent duplication (<2 million years ago) to chromosome 22 was localized to the most centromeric position. Comparative FISH indicated that this event was specific to the genus Homo, confirming the recent origin. Sequencing of multiple PCR products from monochromosomal hybrids of chromosome 14 and 22 indicate a sequence identity of ⩾99.4%. Such a high degree of sequence identity spanning >400 kb makes this the largest and most recent interchromosomal duplication yet defined for any human autosome. It will be interesting to determine whether this large duplication is polymorphic within the human population. Further in silico analysis was used to detect more-divergent duplications on chromosome 22, revealing a pocket of duplications (<90% identity for the most similar pairwise alignments) within the most distal part of the pericentromeric region (14.2 Mb). Regions in the middle of the pericentromeric region show intermediate levels of identity for the most similar pairwise alignments. Here, our comparative FISH analysis generally supported the presence of great ape–specific duplication events. These data suggest a model in which large blocks of mosaic sequence integrate next to the centromere. As more events occur, previously inserted sequence is pushed outward from the centromere to a more distal location within the pericentromeric region. At a mechanistic level, such a model implies that exchange may be linked to the centromeric repeats themselves (α-satellite), providing a focal point for conversion and exchange between nonhomologous chromosomes. This hypothesis is supported by the observation of virtually identical (99.5%) α-satellite between chromosomes 14 and 22 (Jorgensen et al. 1988).
Third, intrachromosomal duplications within the VCFS region appear to have a complex and ongoing evolutionary history that also includes interchromosomal duplication events. FISH analyses with two BAC probes from this region confirm interchromosomal localizations to chromosomes 1, 2, 13, 15, and 20. Interestingly, the association between chromosomes I and XXII (table 1) is well conserved among most primates, including the macaque. However, the degree of sequence identity between human chromosome 1 and 22 within this region is much higher (97.5%) than the average genomic identity between macaque and human noncoding sequences (94.5%). This suggests either an interchromosomal gene conversion since the divergence of macaques and humans or that a portion of chromosome 1 sequence has been transposed to chromosome XXII in the lineage leading to humans. If the latter scenario is true, significant restructuring of the LCR22s should be expected in comparisons of Old World and hominoid species. Although the complex history of the VCFS region makes it difficult to unambiguously derive the evolutionary history, the most recent and human-specific events—on the basis of sequence identity—include an inverted duplication event (module BA; 99.5% identity) and the duplication of a block of modules that represents the largest region of similarity between the proximal (LCR22-2) and distal (LCR22-4) duplicon (160 kb and 99.1% identity). Rearrangements between these LCR22s are associated with 85%–90% of all VCF rearrangements and are thought to be mediated by aberrant recombination (Edelmann et al. 1999a; Shaikh et al. 2000). Resolution of the more distant evolutionary history will require large-scale comparative sequencing among primates, as well as detailed phylogenetic reconstruction.
Fourth, we identified 11 new or altered transcripts arising from both interchromosomal and intrachromosomal duplications. These transcripts have been created and modified in a wide variety of ways—including whole-gene duplications with a well-conserved open reading frame (RFLP), partial gene duplication modifying the existing gene (IGL-V and C), and mosaic transcripts with exons taken from different duplications (AK022914). The majority of these transcripts, particularly mosaic ones, show poor coding potential and therefore are likely failed evolutionary experiments. We therefore expect fewer functional genes to arise from the hodgepodge of pericentromeric duplications, where we observe mosaic transcripts with little apparent function. However, such juxtapositions may offer unique evolutionary avenues for the creation of new genes. We have found evidence for the emergence of novel transcripts composed of diverse duplication modules or transcripts that traverse unique and duplicated sequence. These combinations of novel promoters and exon-encoding sequences are unlikely to arise through single–base-pair mutational events.
Although many of these transcripts require further experimental confirmation, it is instructive to extrapolate our results to the entire genome. Given that we have observed 11 transcripts on a chromosome representing ∼1% of the euchromatic genome, we estimate that ∼1,100 transcripts may have been created or modified from duplicated sequence in the past 35 million years. This is based on the observation that segmental duplications have been identified on all human chromosomes (IHGSC 2001; J.A.B., unpublished data). A more conservative estimate, considering only the five transcripts that contain well-conserved ORFs with experimentally verified expression, yields an estimate of ∼500. If we assume that gene evolution is a relatively constant process and is equally active in chimpanzees, then this estimate suggests that human and chimpanzee genomes may differ by an estimated 150–350 transcripts. Such differences in the transcriptome may provide another avenue for the generation of the phenotypic differences between man and the great apes, for which significant sequence difference has long been lacking among the majority of identified genes (King and Wilson 1975).
Supplementary Material
Acknowledgments
We would like to thank Laurie Christ, Zheng Sun, Gang Zheng, Corey Valley, and Christine O’Keefe for technical assistance, and Tamim Shaikh for helpful comments in the preparation of this manuscript. This work was supported by National Institutes of Health grants GM58815 and HG002385, by Department of Energy grant ER62862-1013741-0005006 (to E.E.E.) and support from Associazone Italiana Ricerca sul Cancro, Centro di Eccellenza di Genomica in campo Biomedico e Agrario, Ministero della Università e della Ricerca Scientifica e Tecnologica, and Téléthon (to N.A. and M.R.). J.A.B. was supported, in part, by a Medical Sciences Training Program Grant. The financial support of the W. M. Keck Foundation, the Charles B. Wang Foundation, and a Howard Hughes Medical Institute grant to Case Western Reserve University's School of Medicine also are gratefully acknowledged.
Online-Only Supplement 1
Percentage breakdown of all pairwise alignments along chromosome 22 (doc1.pdf). This figure shows the distribution of all interchromosomal (red) and intrachromosomal (blue) alignments, in terms of their position and percent identity (90%–100%). For percent identity, color denotes chromosome (see supplement 2 for key). Each line represents 1 Mb, with tick mark values in megabases. The first 13 Mb of ambiguous nucleotides, representing unsequenced p arm and centromere, is not shown. Gaps are shown as white space.
Online-Only Supplement 2
Chromosome assignment of all pairwise alignments along chromosome 22 (doc2.pdf). This figure shows the distribution of all interchromosomal (red) and intrachromosomal (blue) chromosomal alignments, in terms of their position along chromosome 22 and chromosomal assignment. Color denotes chromosome assignment of other copy. Each line represents 1 Mb, with tick-mark values in megabases. The first 13 Mb of ambiguous nucleotides, representing unsequenced p arm and centromere, is not shown. Gaps are shown as white space.
Online-Only Supplement 3
Chromosome 22 Module Summary
|
Proximal Copya |
||||||||||||
| Module | Nameb | Boundary Definitionc | Previous Namesd | Total Number of Copies on Chromosome 22e | GenBank Reference Transcript(s)f | Average Copy Identityg | Duplicated Exonsh | Copies on Other Chromosomesi | Most Diverged Copyj | Begin (bp) | End (bp) | Size (bp) |
| A | AL133604 | … | 1 | AL133604* | 94.7% | 1 | 13 | 13q13 | 13,050,572 | 13,053,285 | 2,713 | |
| C | Hs.214410 | … | 1 | AW139736* | 90.2% | 2 | 10 | 10q11 | 13,104,074 | 13,109,415 | 5,341 | |
| D | Hs.290347 | … | 1 | AW291290, AA52826* | 93.7% | 5 | 2, 10, 13, 15 | 13q12 | 13,185,960 | 13,149,680 | 36,280 | |
| E | Hs.128246tandem | JB | … | 2 | … | 91.7% | … | 2 | … | 13,193,842 | 13,188,961 | 4,881 |
| F | Hs.299072 | … | 1 | BF676987 | 92.4% | 3 | 2*, 10, 15 | 15q11 | 13,225,028 | 13,232,347 | 7,319 | |
| G | NF1 | JB | … | 1 | NM_000267 | 94.0% | 10 | 2, 15, 17*, 22 | 12q12, 17q11, 15q11 | 13,288,801 | 13,306,549 | 17,748 |
| H | Hs.128246 | … | 1 | AL043002 | 93.8% | 1 | 2*, 7 | 2q21 | 13,354,660 | 13,344,151 | 10,509 | |
| I | Hs.297211 | … | 1 | BE973696* | 94.3% | 3 | 2 | 2q21 | 13,361,668 | 13,364,152 | 2,484 | |
| J | Hs.131740 | … | 1 | AI701161 | 93.5% | 2 | 2, 12 | 12q11 | 13,380,907 | 13,378,422 | 2,485 | |
| K | unknown_chr15 | JB | … | 1 | … | 95.6% | … | 15 | 15q11 | 13,388,421 | 13,403,906 | 15,485 |
| L | Hs.289121 | JB | 4q24† | 1 | AL163202 | 94.5% | 2 | 2, 10, 13*, 21 | 21q11 | 13,569,561 | 13,619,760 | 50,199 |
| M | ABCD1 | JB | ALD† | 1 | NM_000033 | 96.7% | 4 | 2, 4, 16, 22, X* | Xq28 | 13,812,578 | 13,822,401 | 9,823 |
| N | Hs.135840 | JB | … | 1 | AW135265 | 96.0% | 4 | 2, 4*, 16, 21, 22 | 4q24 | 13,903,945 | 13,829,495 | 74, 450 |
| O | AY026350 | … | 1 | AY026350* | 96.0% | 1 | 1, 2, 10 | 1q11 | 13,912,130 | 13,913,449 | 1,319 | |
| P | CHK2 | … | 2 | NM_007194 | 96.3% | 3 | 2, 4, 15, 16 | 22q | 13,927,907 | 13,934,713 | 6,806 | |
| Q | NF1_CAGGG | … | 2 | NM_00267 | 95.5% | 0 | 2, 4, 5, 15, 16, 21, Y, UK | 21q11 | 13,949,904 | 13,956,768 | 6,864 | |
| R | Hs.127268_CAGGG | JB | … | 2 | N71154 | 96.0% | 2 | 2*, 5, 15, 16, UK | 2q12/3 | 13,949,904 | 13,983,199 | 33, 295 |
| S | PIK3CA | … | 1 | NM_006218 | 98.0% | 5 | 3* | 3q26 | 13,993,711 | 14,000,304 | 6,593 | |
| T | KCNMB3 | … | 1 | NM_014407 | 95.0% | 2 | 3* | 3q36 | 14,005,892 | 14,007,920 | 2,028 | |
| U | KCNMB3L | … | 1 | NM_014406 | 97.5% | 1 | 7 | 7q36 | 14,015,958 | 14,017,919 | 1,961 | |
| W | TPTE | JB | … | 2 | NM_013315 | 97.1% | 3 | 13, 20, 21*, Y | 13q14 | 14,035,464 | 14,066,457 | 30,993 |
| X | SLC25A15 | … | 1 | NM_014252 | 9530.0% | 4 | 13*, Y | 13q14 | 14,077,536 | 14,070,318 | 7,218 | |
| Y | vWF | … | 1 | NM_000522 | 97.0% | 12 | 12* | 12q13 | 14,130,197 | 14,106,396 | 23,801 | |
| Z | unknown_chr3 | JB | … | 1 | … | 96.3% | … | 3 | 3p12 | 14,130,583 | 14,168,872 | 38,289 |
| AA | Hs.164116 | … | 1 | BE891998, AI433316 | 97.1% | 3 | 12, 20 | 12q13 | 14,173,256 | 14,172,283 | 973 | |
| AB | unknownE | JB | … | 1 | … | 93.1% | … | 4, 9, 12, 15, 17, 19, UL | 15q25, 4q35 | 14,173,903 | 14,191,171 | 17,268 |
| AC | unknown_chr21 | JB | … | 1 | … | 97.8% | … | 5, 13, 21 | 21p11 | 14,439,206 | 14,463,685 | 24,479 |
| AD | USP18 | GRR1* | 2 | NM_017414* | 98.1% | 8 | None | … | 15,580,506 | 15,600,158 | 19,652 | |
| AE | USP18/Hs.99551 | … | 4 | USP18/Hs.99551* | NA | 1 | None | … | 15,686,829 | 15,686,206 | 623 | |
| AF | Hs.99551/Hs.153817 | JB | GRR2* | 6 | DKFZp434P2413, BE041610* | 97.5% | 5, 2 | 11, 21 | 11 | 15,629,343 | 15,600,878 | 28,465 |
| AG | ppseudo_NM_014811 | JB | GRR3* | 1 | NM_014811 (ppseudo) | 99.5% | Ppseudo | 9 | 9 | 15,633,285 | 15,651,614 | 18,329 |
| AH | HsPOX2/DGCR6 | GRR8* | 2 | NM_016335*, NM_005675* | 96.0% | 14, 5 | None | … | 15,831,034 | 15,904,391 | 73,357 | |
| AI | DKFZp434P211/BCR | GRR7* | 11 | NM_014549*, NM_021574* | 96.0% | 5, 7 | 20, 21 | … | 15,690,727 | 15,691,149 | 422 | |
| AJ | GGT1/GGTLA1 | JB | GRR4* | 6 | NM_005265*, NM_004121* | 97.6% | 14, 1 | 2, 20 | … | 15,721,006 | 15,690,727 | 30,279 |
| AL | Hs.4791 | GRR6* | 6 | AB002374* | 99.3% | 2 | 2, 20 | … | 15,773,187 | 15,768,701 | 4,486 | |
| AN | PIK4CA | GRR11* | 2 | NM_002650* | 98.2% | 23 | None | … | 17,740,993 | 17,703,261 | 26,490 | |
| AO | KIAA1292 | JB | GRR10* | 2 | AB033118* | 92.5% | 7 | None | … | 17,068,765 | 17,094,560 | 10,934 |
| AP | DDT/GSTT2 | JB | … | 3 | NM_001355, NM_000854* | 95.2% | 2,5 | None | … | 20,928,169 | 20,957,821 | 12,281 |
| AQ | HS322B1A | … | 3 | NM_015371* | 94.1% | 4 | None | 20,980,558 | 20,998,192 | 37,732 | ||
| AR | P2RXL1 | … | 2 | NM_005446* | 91.0% | 3 | None | … | 18,013,416 | 18,021,242 | 25,795 | |
| AS | CRYBB2/ADRBK2 | … | 2 | NM_000496*, NM_005160* | 95.0% | 3,2 | None | … | 22,268,774 | 22,356,306 | 78,713 | |
| AT | LRRP5/Hs.27317 | JB | … | 1 | NM_02335, AL137651* | 91.0% | 7,4 | 11q13* | 11q13 | 22,392,944 | 22,456,486 | 17,634 |
| AU | Hs.27317 | … | 2 | AL137651* | 94.0% | Intronic | None | … | 20,582,788 | 20,633,387 | 34,560 | |
| AV | BK65A6.2 | … | 2 | NM_019601 | 95.2% | 4 | None | … | 18,370,532 | 18,374,837 | 20,294 | |
| AW | ppseudo_E62F | JB | GRR5* | 3 | NM_001952 (ppseudo) | 98.2% | Ppseudo | 1 | 1 | 15,721,012 | 15,768,702 | 63,542 |
| AX | ppseudo_IGSF3 | JB | … | 2 | NM_001542 (ppseudo) | 98.1% | Ppseudo | 2 | 2 | 15,722,081 | 15,722,460 | 8,409 |
| AZ | IGL-C | JB | … | 2 | IGL* | 90.8% | C2 | None | … | 19,887,138 | 19,892,423 | 4,305 |
| BA | ppseudo_AL133030 | JB | GRR12* | 2 | AL133030 | 99.5% | Ppseudo | None | … | 18,375,708 | 18,445,977 | 47,690 |
| BB | unknownA | JB | … | 2 | … | 94.8% | … | None | … | 15,407,398 | 15,421,223 | 379 |
| BD | RFPL | JB | … | 3 | NM_021026*, NM_006605*, NM_006604* | 94.0% | 2 | None | … | 26,476,678 | 26,485,944 | 6,560 |
| BE | AP1B1 | JB | … | 3 | NM_001129* | 92.0% | 2 | None | … | 26,402,305 | 26,411,017 | 29,028 |
| BF | APOL | JB | … | 2 | NM_003661 | 91.4% | 2 | None | … | 33,185,264 | 33,178,032 | 3,986 |
| BG | phorbolinA | … | 2 | U03891*, NM_004900*, AK024854* | 94.8% | 4,3 | None | … | 35,882,357 | 35,888,674 | 1,612 | |
| BH | phorbolinB | … | 2 | U38435*, AF182420* | 91.6% | 3 | None | … | 35,955,134 | 35,971,058 | 6,204 | |
| BI | CYPDP6 | … | 2 | NM_000106*, AL157851* | 92.9% | 9 | None | … | 39,052,724 | 39,057,183 | 7,328 | |
| BK | unknownD | JB | … | 2 | … | 92.9% | … | None | … | 29,285,752 | 29,335,886 | 60,735 |
| BL | BK126B4.1 | JB | … | 2 | NM_014509*, AL157851* | 93.2% | 11,7 | None | … | 39,359,506 | 39,432,742 | 35,097 |
| BM | SBF1 | … | 1 | U93181* | 90.3% | 22 | 8 | 8q14 | 47,315,782 | 47,324,458 | 70,269 | |
| BN | ACR | JB | … | 1 | NM_001097* | 97.8% | 5 | 2* | 2q14 | 47,594,744 | 47,601,824 | 13,825 |
| BO | U2snRNP | … | 1 | AJ130972, X13482 | 97.7% | 1 | 2* | 2q14 | 47,610,963 | 47,611,993 | 9,266 | |
| BQ | RABL2B | … | 1 | NM_007081* | 98.0% | 9 | 2* | 2q14 | 47,640,124 | 47,624,033 | 8,712 | |
| BR | AL365511 | … | 1 | AL365511* | 98.0% | 4 | 2*,7,8,15,19 | 4p16 | 47,640,320 | 47,645,877 | 7,232 | |
| BS | BE783165 | … | 1 | BE783165* | 98.0% | 4 | 2*,4,5,6,7,8,15,19 | 2q14,4p16 | 47,613,783 | 47,655,405 | 6,317 | |
| BT | unknown_chr3b | JB | … | 1 | … | 93.0% | … | 3 | 3q26 | 34,003,810 | 34,020,558 | 15,924 |
| BU | unknown_chr1 | JB | … | 1 | … | 90.3% | … | 1 | 1p31 | 33,391,691 | 33,405,480 | 4,459 |
| BV | unknown_chr4 | JB | … | 1 | … | 99.3% | … | 4 | 4p14 | 25,125,734 | 25,135,972 | 3,716 |
| BW | unknown_chrX | JB | … | 1 | … | 92.4% | … | X | Xq28 | 38,561,111 | 38,588,639 | 50,134 |
| CA | DGCR5a | JB | … | 2 | X91348.1* | 95.3% | 2 | None | … | 15,877,901 | 15,904,391 | 16,748 |
| CB | DGCR5b | JB | … | 2 | X91348.1* | 91.8% | 2 | None | … | 15,927,351 | 15,938,285 | 15,102 |
| CC | DGCR5c | JB | GRR9* | 2 | X91348.1* | 97.9% | 1 | None | … | 15,950,879 | 15,963,160 | 10,238 |
| VA | IGL-Va | JB | … | 2 | IGL* | 94.3% | V1-19 | None | … | 19,323,652 | 19,330,212 | 27,528 |
| VB | IGL-Vb | JB | … | 2 | IGL* | 90.4% | V2-17,V1-7 | None | … | 19,661,525 | 19,690,553 | 8,676 |
| VC | IGL-Vc | JB | … | 3 | IGL* | 90.1% | V1-7 | None | … | 19,686,567 | 19,690,553 | 7,080 |
| VD | IGL-Vd | JB | … | 2 | IGL* | 94.9% | … | None | … | 19,668,908 | 19,670,520 | 1,030 |
| VE | IGL-Ve | JB | … | 2 | IGL* | 90.7% | ZIP | None | … | 19,714,464 | 19,721,792 | 16,091 |
| VF | IGL-Vf | JB | … | 5 | IGL* | 90.7% | V1-5 | None | … | 19,723,797 | 19,728,675 | 5,557 |
| VG | IGL-Vg | JB | … | 4 | IGL* | 91.2% | V2-18 | None | … | 19,749,652 | 19,751,384 | 1,743 |
| VH | IGL-Vh | JB | … | 2 | IGL* | 91.5% | V2-15 | None | … | 19,695,240 | 19,701,444 | 41,622 |
The position and size of the proximal copy are given as a representative example.
Names were defined on the basis of each module's genic sequence, which is transcribed or contains intron-exon structure unless denoted as a processed pseudogene (“ppseudo”).
“JB” denotes that the module's boundaries were defined by the junctional boundaries of pairwise alignments. Empty cells were defined on the basis of the extent of intron-exon structure.
For previous nomenclature used to define these modules, an asterisk (*) denotes Dunham et al. (1999); a dagger (†) denotes Horvath et al. (2000a).
The total number of copies found on chromosome 22 for the given module.
Reference transcripts for genic sequences within modules: an asterisk (*) denotes expressed transcript; “ppseudo” denotes a processed pseudogene. IGL has no reference, because of the unique nature of this locus.
Average percent identity of pairwise alignments for module.
Number of exons within the module. More exons may be outside of the module. IGL coding sequence was used as defined elsewhere (Kawasaki et al. 1997)
Chromosomes containing one or more copies of the module. An asterisk (*) denotes an expressed locus.
Most divergent copy, compared to the chromosome 22 copies. (Not calculated for intrachromosomal duplications with multiple chromosome 22 copies.)
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for SMS [MIM 182290], PWS [MIM 176270], AS [MIM 105830], NF1 [MIM 162200], VCFS [MIM 192430], DGS [MIM 188400], CES [MIM 115470], and CMT1A [MIM 118220])
- RefSeq Database, http://www.ncbi.nlm.nih.gov/LocusLink/refseq.html (for accessions of the format NM_#####)
- RepeatMasker, http://repeatmasker.genome.washington.edu/
- Rocchi Lab Web site, http://www.biologia.uniba.it/22-paper/ (for all FISH images)
- UniGene database, http://www.ncbi.nlm.nih.gov/UniGene/ (for cluster accessions of the format Hs.#####)
- University of California Santa Cruz (UCSC) Human Genome Assembly Web site, http://genome.ucsc.edu/ (for published assembly and genome browser)
References
- Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD (1999) Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 65:370–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE (2001) Segmental duplications: organization and impact within the current human genome project assembly. Genome Res 11:1005–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FC, Li WH (2001) Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet 68:444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault A, Lee C, Lupski J (1997) Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 17:154–163 [DOI] [PubMed] [Google Scholar]
- Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K (2000) NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet 9:35–46 [DOI] [PubMed] [Google Scholar]
- Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, Bruskiewich R, et al (1999) The DNA sequence of human chromosome 22. Nature 402:489–495 [DOI] [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Morrow BE (1999a) Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet 64:1076–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, Chaganti RS, Magenis E, Shprintzen RJ, Morrow BE (1999b) A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet 8:1157–1167 [DOI] [PubMed] [Google Scholar]
- Edelmann L, Stankiewicz P, Spiteri E, Pandita RK, Shaffer L, Lupski JR, Morrow BE (2001) Two functional copies of the DGCR6 gene are present on human chromosome 22q11 due to a duplication of an ancestral locus. Genome Res 11:208–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE (2001) Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet 17:661–669 [DOI] [PubMed] [Google Scholar]
- Eichler EE, Budarf ML, Rocchi M, Deaven LL, Doggett NA, Baldini A, Nelson DL, Mohrenweiser HW (1997) Interchromosomal duplications of the adrenoleukodystrophy locus: a phenomenon of pericentromeric plasticity. Hum Mol Genet 6:991–1002 [DOI] [PubMed] [Google Scholar]
- Eichler EE, Hoffman SM, Adamson AA, Gordon LA, McCready P, Lamerdin JE, Mohrenweiser HW (1998) Complex beta-satellite repeat structures and the expansion of the zinc finger gene cluster in 19p12. Genome Res 8:791–808 [DOI] [PubMed] [Google Scholar]
- Eichler EE, Lu F, Shen Y, Antonacci R, Jurecic V, Doggett NA, Moyzis RK, Baldini A, Gibbs RA, Nelson DL (1996) Duplication of a gene-rich cluster between 16p11.1 and Xq28: a novel pericentromeric-directed mechanism for paralogous genome evolution. Hum Mol Genet 5:899–912 [DOI] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W (1998) A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res 8:967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Footz TK, Brinkman-Mills P, Banting GS, Maier SA, Riazi MA, Bridgland L, Hu S, et al (2001) Analysis of the cat eye syndrome critical region in humans and the region of conserved synteny in mice: a search for candidate genes at or near the human chromosome 22 pericentromere. Genome Res 11:1053–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M (1999) The genomic record of humankind's evolutionary roots. Am J Hum Genet 64:31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratacos M, Nadal M, Martin-Santos R, Pujana MA, Gago J, Peral B, Armengol L, Ponsa I, Miro R, Bulbena A, Estivill X (2001) A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell 106:367–379 [DOI] [PubMed] [Google Scholar]
- Guy J, Spalluto C, McMurray A, Hearn T, Crosier M, Viggiano L, Miolla V, Archidiacono N, Rocchi M, Scott C, Lee PA, Sulston J, Rogers J, Bentley D, Jackson MS (2000) Genomic sequence and transcriptional profile of the boundary between pericentromeric satellites and genes on human chromosome arm 10q. Hum Mol Genet 9:2029–2042 [DOI] [PubMed] [Google Scholar]
- Halford S, Lindsay E, Nayudu M, Carey A, Baldini A, Scambler P (1993) Low-copy-number repeat sequences flank the DiGeorge/velo-cardio-facial syndrome loci at 22q11. Hum Mol Genet 2:191–196 [DOI] [PubMed] [Google Scholar]
- Horvath J, Schwartz S, Eichler E (2000a) The mosaic structure of a 2p11 pericentromeric segment: a strategy for characterizing complex regions of the human genome. Genome Res 10:839–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath J, Viggiano L, Loftus B, Adams M, Rocchi M, Eichler E (2000b) Molecular structure and evolution of an alpha/non-alpha satellite junction at 16p11. Hum Mol Genet 9:113–123 [DOI] [PubMed] [Google Scholar]
- Hughes AL, da Silva J, Friedman R (2001) Ancient genome duplications did not structure the human hox-bearing chromosomes. Genome Res 11:771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IHGSC (International Human Genome Sequencing Consortium) (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- Jackson MS, Rocchi M, Thompson G, Hearn T, Crosier M, Guy J, Kirk D, Mulligan L, Ricco A, Piccininni S, Marzella R, Viggiano L, Archidiacono N (1999) Sequences flanking the centromere of human chromosome 10 are a complex patchwork of arm-specific sequences, stable duplications, and unstable sequences with homologies to telomeric and other centromeric locations. Hum Molec Genet 8:205–215 [DOI] [PubMed] [Google Scholar]
- Jauch A, Wienberg J, Stanyon R, Arnold N, Tofanelli S, Ishida T, Cremer T (1992) Reconstruction of genomic rearrangements in great apes and gibbons by chromosome painting. Proc Natl Acad Sci USA 89:8611–8615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Eichler EE, Schwartz S, Nicholls RD (2000) Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res 10:597–610 [DOI] [PubMed] [Google Scholar]
- Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, Rocchi M, Eichler EE (2001) Positive selection of a gene family during the emergence of human and African apes. Nature 413:514–519 [DOI] [PubMed] [Google Scholar]
- Jorgensen AL, Kolvraa S, Jones C, Bak AL (1988) A subfamily of alphoid repetitive DNA shared by the NOR-bearing human chromosomes 14 and 22. Genomics 3:100–109 [DOI] [PubMed] [Google Scholar]
- Kawasaki K, Minoshima S, Nakato E, Shibuya K, Shintani A, Schmeits J, Wang J, Shimizu N (1997) One-megabase sequence anlaysis of the human immunogloblulin lambda gene locus. Genome Res 7:250–261 [DOI] [PubMed] [Google Scholar]
- Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120 [DOI] [PubMed] [Google Scholar]
- King M, Wilson A (1975) Evolution at two levels in humans and chimpanzees. Science 188:107–116 [DOI] [PubMed] [Google Scholar]
- Kumar S, Hedges SB (1998) A molecular timescale for vertebrate evolution. Nature 392:917–920 [DOI] [PubMed] [Google Scholar]
- Loftus BJ, Kim UJ, Sneddon VP, Kalush F, Brandon R, Fuhrmann J, Mason T, Crosby ML, Barnstead M, Cronin L, Deslattes Mays A, Cao Y, Xu RX, Kang HL, Mitchell S, Eichler EE, Harris PC, Venter JC, Adams MD (1999) Genome duplications and other features in 12 Mbp of DNA sequence from human chromosome 16p and 16q. Genomics 60:295–308 [DOI] [PubMed] [Google Scholar]
- Luijten M, Wang Y, Smith BT, Westerveld A, Smink LJ, Dunham I, Roe BA, Hulsebos TJ (2000) Mechanism of spreading of the highly related neurofibromatosis type 1 (NF1) pseudogenes on chromosomes 2, 14 and 22. Eur J Hum Genet 8:209–214 [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155 [DOI] [PubMed] [Google Scholar]
- Muller S, Stanyon R, O'Brien PC, Ferguson-Smith MA, Plesker R, Wienberg J (1999) Defining the ancestral karyotype of all primates by multidirectional chromosome painting between tree shrews, lemurs and humans. Chromosoma 108:393–400 [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Stanyon R, O'Brien SJ (2001) Evolution of mammalian genome organization inferred from comparative gene mapping. Genome Biol 2:5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Miller W (1988) Optimal alignments in linear space. Comput Appl Biosci 4:11–17 [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Sankoff D (1998) Counting on comparative maps. Trends Genet 14:495–501 [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Taylor BA (1984) Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci USA 81:814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien SJ, Menotti-Raymond M, Murphy WJ, Nash WG, Wienberg J, Stanyon R, Copeland NG, Jenkins NA, Womack JE, Marshall Graves JA (1999) The promise of comparative genomics in mammals. Science 286:458–462, 479–481 [DOI] [PubMed] [Google Scholar]
- Ohno S (1970) Evolution by gene duplication. Springer Verlag, Berlin/Heidelberg/New York [Google Scholar]
- Ohno S, Wolf U, Atkin N (1968) Evolution from fish to mammals by gene duplication. Hereditas 59:169–187 [DOI] [PubMed] [Google Scholar]
- Patthy L (1996) Exon shuffling and other ways of module exchange. Matrix Biol 15:301–312 [DOI] [PubMed] [Google Scholar]
- Regnier V, Meddeb M, Lecointre G, Richard F, Duverger A, Nguyen VC, Dutrillaux B, Bernheim A, Danglot G (1997) Emergence and scattering of multiple neurofibromatosis (NF1)–related sequences during hominoid evolution suggest a process of pericentromeric interchromosomal transposition. Hum Mol Genet 6:9–16 [DOI] [PubMed] [Google Scholar]
- Ruault M, Trichet V, Gimenez S, Boyle S, Gardiner K, Rolland M, Roizes G, De Sario A (1999) Juxta-centromeric region of human chromosome 21 is enriched for pseudogenes and gene fragments. Gene 239:55–64 [DOI] [PubMed] [Google Scholar]
- Seroussi E, Kedra D, Pan HQ, Peyrard M, Schwartz C, Scambler P, Donnai D, Roe BA, Dumanski JP (1999) Duplications on human chromosome 22 reveal a novel ret finger protein–like gene family with sense and endogenous antisense transcripts. Genome Res 9:803–814 [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Lupski JR (2000) Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet 34:297–329 [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS (2000) Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 9:489–501 [DOI] [PubMed] [Google Scholar]
- Skrabanek L, Wolfe KH (1998) Eukaryote genome duplication—where's the evidence? Curr Opin Genet Dev 8:694–700 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Park SS, Inoue K, Lupski JR (2001) The evolutionary chromosome translocation 4;19 in Gorilla gorilla is associated with microduplication of the chromosome fragment syntenic to sequences surrounding the human proximal CMT1A-REP. Genome Res 11:1205–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teglund S, Olsen A, Khan W, Frangsmyr L, Hammarstrom S (1994) The pregnancy-specific glycoprotein (PSG) gene cluster on human chromosome 19: fine structure of the 11 PSG genes and identification of 6 new genes forming a third subgroup within the carcinoembryonic antigen (CEA) family. Genomics 23:669–684 [DOI] [PubMed] [Google Scholar]
- Tomlinson IM, Cook GP, Carter NP, Elaswarapu R, Smith S, Walter G, Buluwela L, Rabbitts TH, Winter G (1994) Human immunglobulin VH and D segments on chromosomes 15q11.2 and 16p11.2. Hum Mol Genet 3:853–860 [DOI] [PubMed] [Google Scholar]
- Trask B, Friedman C, Martin-Gallardo A, Rowen L, Akinbami C, Blankenship J, Collins C, Giorgi D, Iadonato S, Johnson F, Kuo W, Massa H, Morrish T, Naylor S, Nguyen O, Rouquier S, Smith T, Wong D, Younglbom J, van den Engh G (1998a) Members of the olfactory receptor gene family are contained in large blocks of DNA duplicated polymorphically near the ends of human chromosomes. Hum Mol Genet 7:13–26 [DOI] [PubMed] [Google Scholar]
- Trask BJ, Massa H, Brand-Arpon V, Chan K, Friedman C, Nguyen OT, Eichler EE, van den Engh G, Rouquier S, Shizuya H, Giorgi D (1998b) Large multi-chromosomal duplications encompass many members of the olfactory receptor gene family in the human genome. Hum Mol Genet 7:2007–2020 [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, et al (2001) The sequence of the human genome. Science 291:1304–1351 [DOI] [PubMed] [Google Scholar]
- Wienberg J, Stanyon R, Jauch A, Cremer T (1992) Homologies in human Macaca fascata chromosomes revealed by in situ hybridization with human chromosome specific DNA libraries. Chromosoma 101:265–270 [DOI] [PubMed] [Google Scholar]
- Wienberg J, Stanyon R, Nash WG, O'Brien PC, Yang F, O'Brien SJ, Ferguson-Smith MA (1997) Conservation of human vs. feline genome organization revealed by reciprocal chromosome painting. Cytogenet Cell Genet 77:211–217 [DOI] [PubMed] [Google Scholar]
- Wong AC, Shkolny D, Dorman A, Willingham D, Roe BA, McDermid HE (1999) Two novel human RAB genes with near identical sequence each map to a telomere-associated region: the subtelomeric region of 22q13.3 and the ancestral telomere band 2q13. Genomics 59:326–334 [DOI] [PubMed] [Google Scholar]
- Yunis JJ, Prakash O (1982) The origin of man: a chromosomal pictorial legacy. Science 215:1525–1530 [DOI] [PubMed] [Google Scholar]
- Zimonjic D, Kelley M, Rubin J, Aaronson S, Popescu N (1997) Fluorescence in situ hybridization analysis of keratinocyte growth factor gene amplification and dispersion in evolution of great apes and humans. Proc Natl Acad Sci USA 94:11461–11465 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.