

Abstract
A major barrier to successful cancer immunotherapy is the tumor’s ability to induce T-cell tolerance by exploiting host regulatory mechanisms. Having discovered the DC-HIL receptor, which inhibits T-cell responses by binding to syndecan-4 on effector T-cells, we posited the DC-HIL/syndecan-4 pathway to play an important role in cancer promotion. Among DC-HIL+ myelomonocytic cells, during growth of implanted mouse melanoma, CD11b+Gr1+ cells were the most expanded population and the most potent at suppressing T-cell activation. Deletion of the DC-HIL gene or infusion of anti-DC-HIL mAb abrogated these cells’ suppressor function and expansion, and markedly diminished melanoma growth and metastasis. IL-1β and IFN-γ were elevated in mice bearing melanoma, and concurrent exposure to both cytokines optimally induced DC-HIL expression by tumor-infiltrating CD11b+Gr1+ cells. Ligation of DCHIL transduced phosphorylation of its intracellular immunoreceptor tyrosine-based activation motif (ITAM), that in turn induced intracellular expression of IFN-γ and inducible nitric oxide synthase (iNOS), known to mediate T-cell suppression by CD11b+Gr1+ cells. Thus DC-HIL is the critical mediator of these cells’ suppressor function in melanoma-bearing mice and a potential target for improving melanoma immunotherapy.
Introduction
Despite recent advances in the treatment of metastatic melanoma, it remains the most lethal form of skin cancer, in large part because of its ability to overcome host anti-tumor immunity (Fang et al., 2008). Among immune mechanisms exploited by melanoma are regulatory T-cells, type-2 macrophages, immature dendritic cells (DC), and CD11b+Gr1+ cells (which overlap with myeloid-derived suppressor cells) (Serafini et al., 2006). These latter cells are the most potent suppressors of T-cell function.
CD11b+Gr1+ cells consist of myeloid progenitors (Gabrilovich and Nagaraj, 2009), that in healthy individuals are confined to the bone marrow (BM), where they differentiate into DC, granulocytes, or macrophages, none of which are immunosuppressive. By contrast, in cancer patients, differentiation is blocked by tumor-derived soluble factors, causing these cells’ exponential proliferation and expansion into blood and other organs, where they become immunosuppressive (Diaz-Montero et al., 2009).
CD11b+Gr1+ cells were shown to suppress T-cell function by producing inhibitory soluble factors like urea/L-ornithine (produced by arginase I) (Rodriguez and Ochoa, 2008), nitric oxide (NO) (Bingisser et al., 1998), and reactive oxygen species (ROS) (Kusmartsev and Gabrilovich, 2003). Coinhibitory pathways like CD80/86 (ligands for CTLA-4) and PD-L1 (for PD-1) were shown to mediate suppressor function (Liu et al., 2009; Liu et al., 2008; Yang et al., 2006), but inconsistently and sometimes in a contradictory manner. Moreover, the molecular underpinning for these cells’ suppressor function is not fully understood.
We discovered the DC-HIL receptor on antigen presenting cells (APC), which binds syndecan-4 (SD-4) on effector/memory (but not naive) T-cells and down-regulates T-cell receptor (TCR)-activation signals (Chung et al., 2007a; Chung et al., 2007b). We showed the DC-HIL/SD-4 pathway to inhibit inflammatory responses in mouse models of contact hypersensitivity and graft-versus-host disease (Akiyoshi et al., 2010; Chung et al., 2007a). Furthermore, DC-HIL is expressed by melanoma cells, and suppresses T-cell activation via SD-4 on cytotoxic T-cells (Tomihari et al., 2010), but had little-to-no impact on these cells’ tumor-killing activity.
Our present study heralds a melanoma-promoting role for DC-HIL expressed by myelomonocytic cells bearing the CD11b+Gr1+ phenotype. Blocking DC-HIL function via gene deletion or specific Ab abrogated these cells’ suppression of T-cell function and promotion of melanoma growth. Ligated DC-HIL activated its intracellular immunoreceptor tyrosine-based activation motif (ITAM) and induced expression of IFN-γ and iNOS (producing NO). Our findings indicate that DC-HIL is responsible for secretion of T-cell inhibitory factors by CD11b+Gr1+ cells, which render them immunosuppressive.
Results
DC-HIL gene deletion inhibited growth of melanoma
We created mice knocked-out (KO) for the DC-HIL gene with no gross abnormality nor developmental defects in lymphoid organs, except for absence of DC-HIL protein and whose DC are 2-fold greater activators of T-cells than wild-type (WT) counterparts (Supplementary Figures S1 and S2), thereby confirming DC-HIL’s inhibitory effect on T-cell activation.
B16 melanoma subcutaneously inoculated into WT vs. DC-HIL−/− mice grew aggressively in the former, but its growth was inhibited markedly in the latter (Figure 1a). Having reported DC-HIL expression by B16 cells to promote tumor growth by suppressing T-cell activation (Tomihari et al., 2010), we probed this influence in DC-HIL−/− mice, using B16 cells knocked-down for DC-HIL (KD-B16). KD-B16 melanoma growth was slower than parental B16 melanoma, and its growth markedly inhibited in KO mice (Figure 1b). We then examined lung metastasis (Figures 1c and d) following i.v. injection of B16 cells: KO mice had markedly lighter lungs, less metastatic foci, less melanin content per lung, and less melanin per metastatic focus. Thus melanoma growth was supported by tumor-associated DC-HIL and by host-derived DC-HIL.
Figure 1. Growth and metastasis of B16 melanoma are suppressed in DC-HIL−/− mice.
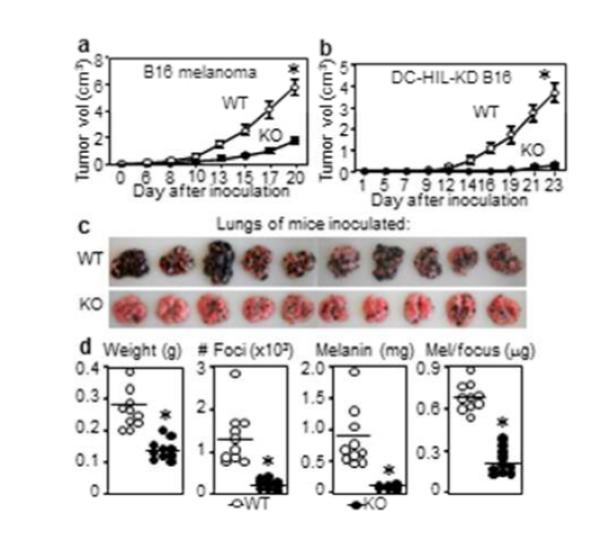
(a) Tumor volume of B16 cells implanted into WT or DC-HIL KO mice (n=5). (b) Tumor volume after implanting DC-HIL-KD-B16 cells (n=5). Lung metastasis (c) at 19 days after B16 cells injected i.v. into WT or KO mice (n=10); lung weight, number of metastatic foci, melanin content/lung, and melanin content/focus plotted (d). Representative data from 3 separate experiments, *p< 0.01 versus WT.
DC-HIL is expressed by CD11b+Gr1+ cells in mice bearing melanoma
We next addressed which DC-HIL-expressing host cells promote melanoma growth. Since DC-HIL is expressed by myelomonocytic cells (Chung et al., 2009), we identified the most expanded DC-HIL+ myeloid population from spleen of mice with melanoma (Figure 2a), which turned out to be CD11b+Gr1+ cells, representing about 10% of splenocytes (vs. CD11c+ DC or F4/80+ macrophages which were about 5% each). Forty percent of splenic CD11b+Gr1+ cells expressed DC-HIL, whereas 20% and 66-73% of these cells were respectively PD-L1+ and CD80/CD86+ (Figure 2b). For CD11b+Gr1+ cells from BM, blood, and the B16 tumor site, DC-HIL expression ranged 27-61% (Supplementary Figure S3a-c). By contrast, CD11b+Gr1+ cells from tumor-free mice did not express DC-HIL (Figure 2c). Thus melanoma induced DC-HIL expression of some CD11b+Gr1+ cells in many organs.
Figure 2. Melanoma induces DC-HIL expression by most potent CD11b+Gr1+ suppressors.
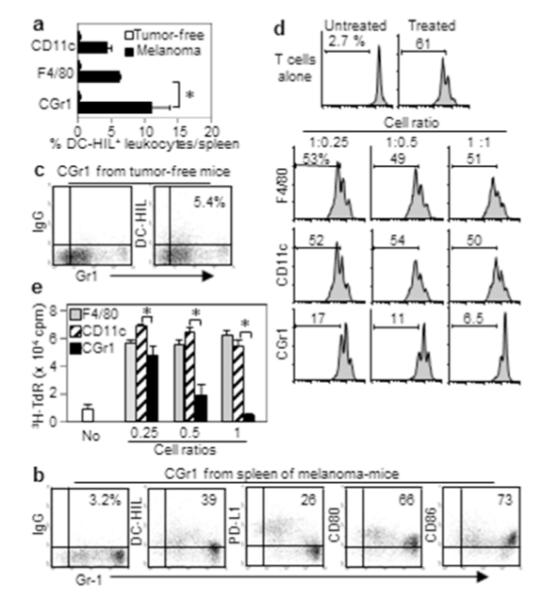
(a) Splenocytes from B16 melanoma-bearing or tumor-free mice (n=3) were assayed for % DCHIL+ cells in 3 myeloid populations. (b, c) CD11b+Gr1+ (CGr1) cells isolated from mice with (b) or without (c) melanoma were examined for expression of Gr1 and coinhibitory receptors (%). (d) These myeloid cells (increasing cell ratios) were cocultured with CFSE-labeled T-cells activated by anti-CD3/CD28 Ab. T-cell proliferation (%) was determined by FACS (histograms). (e) Purified myeloid cells were examined for T cell-stimulatory capacity. Different numbers of myeloid cells were pulsed with gp100 Ag and added to culture of CD8+ pmel-1 T-cells. Culture of T-cells with Ag served as control (No). T-cell proliferation was measured by 3H-thymidine (TdR) incorporation. *p<0.01.
Given DC-HIL’s T cell-inhibitory function, we compared the 3 subsets of DC-HIL-expressing myelomonocytic cells for suppressor function (Figure 2d). Each was similarly assayed for suppressor activity to CFSE-labeled T-cells activated by anti-CD3/CD28 Ab. F4/80+ macrophages or CD11c+ DC did not inhibit T-cell proliferation even at highest cell ratio (1:1), whereas CD11b+Gr1+ cells showed inhibition dose-dependently. We next examined their T cell-stimulatory capacity by mixing CD8+ T-cells from pmel-1 transgenic mice (in which all CD8+ T-cells express the same TCR specific for gp100 Ag) (Overwijk et al., 2003) with each of the 3 DC-HIL+ subsets pulsed with gp100 peptide. CD11c+ DC or F4/80+ macrophages induced strong T-cell proliferation at cell ratios of 1:0.25 or higher (Figure 2e), whereas CD11b+Gr1+ cells exhibited similar stimulatory capacity at 1:0.25, while inhibiting T-cell proliferation completely. Thus CD11b+Gr1+ cells in mice with melanoma were the predominant DC-HIL-expressing population and the most potent suppressors of T-cell proliferation.
DC-HIL mediated the T cell-suppressive function of CD11b+Gr1+ cells
CD11b+Gr1+ cells from melanoma-bearing mice were cocultured with pmel-1 splenocytes and gp100 peptide at a 1:1 cell ratio; anti-DC-HIL mAb or control IgG was added to block DC-HIL on CD11b+Gr1+ cells (Figure 3a). The mAb (but not control) restored pmel-1 T-cell activation dose-dependently and completely, but had no effect on T-cell activation without CD11b+Gr1+ cells. Anti-CD80, anti-CD86, or anti-PD-L1 Ab had no significant effect on CD11b+Gr1+ cells’ suppressor activity (Figure 3b). Thus among coinhibitors tested, DC-HIL was responsible solely for suppressor activity of CD11b+Gr1+ cells.
Figure 3. DC-HIL mediates T cell-suppressor activity of CD11b+Gr1+ cells.
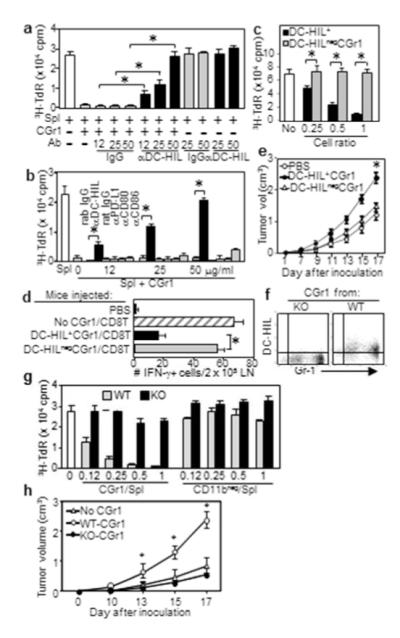
(a, b) CGr1 cells from melanoma-bearing mice were cocultured with pmel-1 splenocytes (Spl), gp100 Ag, and anti-DC-HIL mAb (a), anti-coinhibitor Ab or control IgG (b). 3H-TdR incorporation measured. (c) Undepleted (DC-HIL+) or DC-HIL-depleted (DC-HILneg) CGr1 (Supplementary method) were assayed for suppression of pmel-1 splenocyte proliferation triggered by Ag (increasing ratios). (d) Mice (n=5) injected with pmel-1 CD8+ T-cells and DC-HIL+CGr1 or DC-HILnegCGr1. Ten days after giving gp100, IFN-γ-producing cells in LN were counted. (e) Tumor growth following coinjection of DC-HIL+ or DC-HILneg CGr1 cells with B16 cells s.c. into naive mice (n=5). Using similar methods, DC-HIL−/−CGr1 cells were compared with DCHIL+/+ counterparts for DC-HIL expression by FACS (f), T-cell suppressing (g) and tumor-promoting ability (CD11bneg cells as control) (h). *p<0.01.
We next compared T-cell suppressor capacity of DC-HIL+ vs. DC-HILnegCD11b+Gr1+ cells. Undepleted CD11b+Gr1+ cells inhibited gp100-triggered T-cell activation, whereas the DC-HIL-depleted CD11b+Gr1+ fraction showed no inhibition at all doses examined (Figure 3c). In vivo suppressor ability of DC-HIL+ cells was assessed by injecting mice with pmel-1 CD8+ T-cells, followed by infusion of undepleted or DC-HIL-depleted CD11b+Gr1+ cells and by gp100 vaccination. Ten days later, mice infused with CD8+ T-cells but without CD11b+Gr1+ cells, generated a lot of activated (IFN-γ+) T-cells in LN (Figure 3d), whereas coinfusion of undepleted CD11b+Gr1+ cells led to fewer activated T-cells and coinfusion of DC-HIL-depleted CD11b+Gr1+ cells prevented suppression. An experiment using DC-HIL−/− CD11b+Gr1+ cells showed similar results (Supplementary Figure S4). Thus DC-HIL+ CD11b+Gr1+ cells were responsible for suppressor activity.
We next coinjected undepleted or DC-HIL-depleted CD11b+Gr1+ cells with B16 cells s.c. into naive mice. A week later, similarly treated CD11b+Gr1+ cells alone were infused i.v. (Figure 3e). Melanoma in mice coinjected with undepleted suppressor cells grew markedly larger than cohorts infused with B16 cells alone, whereas tumors in mice treated with DC-HIL-depleted CD11b+Gr1+ cells were similar to those given B16 alone. This outcome for DC-HIL depletion was not observed using Rag2 KO mice (Supplementary Figure S5), suggesting T-cells were involved. Experiments using DC-HIL-deficient CD11b+Gr1+ cells from KO mice did not suppress T-cell activation nor promote melanoma progression (Figure 3f-h). Thus DCHIL+CD11b+Gr1+ cells were critical suppressors of T-cells and promoters of melanoma growth.
IFN-γ and NO mediated T cell-suppressive activity of CD11b+Gr1+ cells
We addressed the contribution of soluble factors to T-cell suppression by adding specific inhibitors to cocultures of pmel-1 splenocytes and CD11b+Gr1+ cells. Neutralizing Ab to TGF-β (Filipazzi et al., 2007) or IL-10 (Wang et al., 2010) had no effect on suppressor activity, whereas anti-IFN-γ Ab blocked suppression completely (Figure 4a). Chemical inhibitors for arginase and indoleamine restored T-cell activation marginally; and catalase and superoxide dismutase for neutralizing ROS had no effect (Figure 4b). By contrast, inhibitors of all NOS molecules or specific to NOS-2 blocked suppressor function substantially or completely. Thus IFN-γ and NOS-2 independently mediated suppressor activity of CD11b+Gr1+ cells induced by melanoma.
Figure 4. Crosslinked DC-HIL on CD11b+Gr1+ cells induces tyrosine phosphorylation and IFN-γ/iNOS expression.
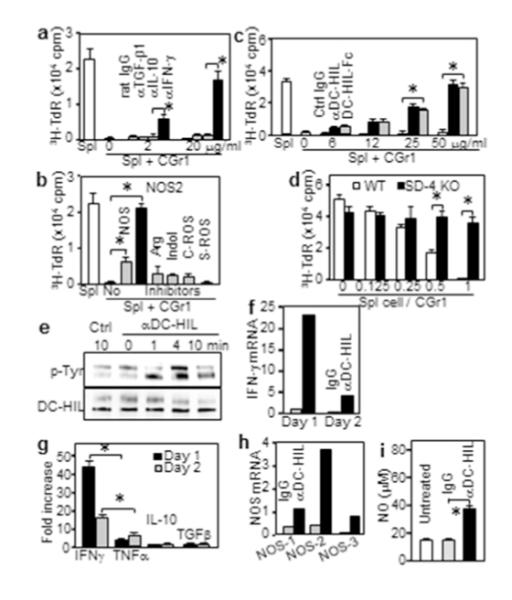
(a-c) CD11b+Gr1+ (CGr1) cells cocultured with pmel-1 splenocytes (1:1 ratio) with inhibitors; including (a) anti-cytokine Ab; (b) 5 mM L-NG-monomethyl-arginine citrate (NOSs); 0.5 mM N6-(1-iminoethyl)-L-lysine (NOS-2); 1 mM N-hydroxyl-nor-arginine (Arg); 0.2 mM 1-methyl-tryptophan (Indol); 1000 U/ml catalase (C-ROS); and 200 U/ml superoxide dismutase (S-ROS); and (c) anti-DC-HIL mAb or DC-HIL-Fc. 3H-TdR uptake was measured. (d) CGr1 cells cocultured with SD-4+/+ or SD-4−/− pmel-1 splenocytes. (e-i) At varying times after crosslinking with Ab, CGr1 cells were assayed for: tyrosine-phosphorylation (p-Tyr) on DC-HIL protein (e); cytokine mRNA and secretion (f, g); mRNA of NOS genes (h); or NO production (i). Data (mean ± sd, n=3) are shown as fold increase relative to control. *p<0.01.
T-cell expression of SD-4 is required for CD11b+Gr1+ cells’ suppressor activity
Since inhibitory function of DC-HIL on APC requires ligation of SD-4 on T-cells, a similar mechanism was likely for CD11b+Gr1+ cells. DC-HIL-Fc, which interferes with DC-HIL/SD-4 binding (Chung et al., 2007b), restored T-cell activation dose-dependently, whereas control Ig did not (Figure 4c). For SD-4 specificity, we compared effects of CD11b+Gr1+ cells on SD-4+/+ vs. SD-4−/− pmel-1 splenocytes with gp100 Ag (Figure 4d). Without CD11b+Gr1+ cells, splenocytes from both mice exhibited similar T-cell proliferation, whereas their presence inhibited SD-4+/+ (but not SD-4−/−) T-cell activation. Thus melanoma-induced CD11b+Gr1+ cells inhibited T-cell activation via binding to SD-4.
Crosslinked DC-HIL induced tyrosine phosphorylation and expression of IFN-γ and NOS-2 (iNOS) by CD11b+Gr1+ cells
Since DC-HIL has an intracellular ITAM (Shikano et al., 2001), we posited that ligation of DC-HIL would activate CD11b+Gr1+ cells’ suppressor function via ITAM. Crosslinking DC-HIL with specific Ab induced tyrosine-phosphorylation on the receptor (Figure 4e) as shown using transfection experiments (Chung et al., 2009).
Does activated DC-HIL induce IFN-γ and iNOS in CD11b+Gr1+ cells? IFN-γ mRNA in CD11b+Gr1+ cells was increased markedly a day after DC-HIL crosslinking, followed by a quick fall (Figure 4f). IFN-γ was the predominant cytokine secreted, followed by TNF-α, but neither IL-10 nor TGF-β (Figure 4g). Crosslinked DC-HIL also induced iNOS expression 4-fold greater than control (Figure 4h). Among NOS species, iNOS was most abundantly expressed, consistent with its known selective inducibility by IFN-γ (Kamijo et al., 1994), and supported by increased NO after DC-HIL crosslinking (Figure 4i). These outcomes were not seen in DC-HIL−/− CD11b+Gr1+ cells (Supplementary Figure S6).
Anti-DC-HIL mAb treatment of melanoma-bearing mice suppressed tumor growth and reduced circulating CD11b+Gr1+ cells
We next assessed effects of the mAb on melanoma growth and frequency of CD11b+Gr1+ cells in blood. Because these cells begin to accumulate in spleen 6 days after tumor inoculation (when tumors reach ~0.1 cm3) (Cheng et al., 2008), we injected anti-DC-HIL mAb i.p. then and every other day, for 6 treatments. In mice treated with control IgG, melanoma grew aggressively, in proportion to frequency of blood CD11b+Gr1+ cells. The mAb markedly suppressed subsequent melanoma growth (Figure 5a) and prevented expansion of CD11b+Gr1+ cells in blood (Figures 5b and c): the latter effect was supported by inability of KO mice to expand CD11b+Gr1+ cells (Supplementary Figure S7). It also significantly enhanced the IFN-γ response by T-cells from mice with melanoma (Figure 5d). Blocked expansion may be due to reduced tumor size with less secretion of relevant soluble factors.
Figure 5. Infusion of anti-DC-HIL mAb suppresses melanoma growth and expansion of CD11b+Gr1+ cells.
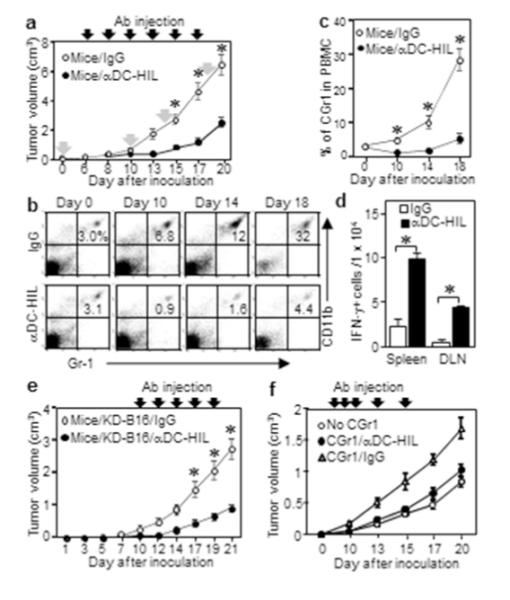
(a) Tumor volume 6 days after implanting B16 cells into WT mice (n=7), mice injected with anti-DC-HIL mAb or control IgG on indicated days (closed arrows). On days shown by gray arrows in (a), blood taken from mouse, CGr1 cells counted from PBMCs by FACS (b), and data summarized (c). (d) A day after 3 injections, IFN-γ-secreting cells in spleen or LN in each mouse (n=3) counted as number per 1 × 104 cells. (e) Tumor volume on mice treated with the 2 Abs 11 days after implanting KD-B16 cells (n=7). (f) Tumor volume on DCHIL−/− mice treated with Ab 7 days after co-injection of B16 and CGr1 cells. *p<0.001.
Inhibited CD11b+Gr1+ cell function accounted for beneficial effects of anti-DC-HIL mAb on melanoma
Because DC-HIL is expressed by B16 cells, APC, and CD11b+Gr1+ cells, we compared their respective contributions via effects of anti-DC-HIL mAb. For DC-HIL on melanoma itself, we implanted DC-HIL-knocked-down B16 cells (KD-B16) into WT mice, while injecting the mAb as before (Figure 5e). In this assay, in which CD11b+Gr1+ cells and APC were both DC-HIL+, KD-B16 tumor hardly grew in mice treated with anti-DC-HIL mAb. Thus DC-HIL on melanoma was not critical to the overall effect of anti-DC-HIL mAb. When DC-HIL+ DC were infused into DC-HIL−/− mice bearing DC-HIL+ B16 tumor, mAb treatment had no significant effect on the APC function (Supplemental Figure S8). Finally, we gave the mAb to DC-HIL−/− mice inoculated with B16 cells and CD11b+Gr1+ cells (both DC-HIL+), and it blocked tumor promotion (Figure 5f), indicating that its beneficial effects are due primarily to neutralizing CD11b+Gr1+ cells’ function.
DC-HIL was not required for growth of EL-4 lymphoma and LL2 lung carcinoma
Does DC-HIL contribute to growth of other malignancies? EL-4 or LL2 cells implanted s.c. into DCHIL−/− or WT mice showed no significant difference in growth (Figure 6a, p>0.05). Neither tumor was associated with significant DC-HIL expression on CD11b+Gr1+ cells (Figure 6b). Using the CFSE-dilution assay (Figure 6c), CD11b+Gr1+ cells from spleen of mice with LL2 or EL-4 tumor showed some suppressor activity that was significantly weaker than from melanoma. Thus DC-HIL did not contribute to growth of EL-4 and LL2 malignancies.
Figure 6. IFN-γ and IL-1β induce DC-HIL expression by CD11b+Gr1+ cells.
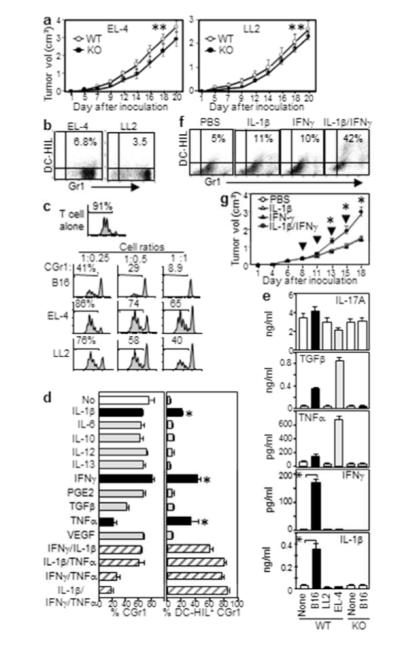
(a) Growth of EL-4 or LL2 tumor in WT or DC-HIL−/− mice (n=5, **p>0.05). CGr1 cells from those mice were examined for: Gr1 vs. DC-HIL expression (b) and suppression of CFSE-labeled T-cells activated by anti-CD3/CD28 (c). (d) Linneg BM cells cultured with cytokines were assayed for % CGr1 or DC-HIL+ cells among CD11c+/CD19+-depleted or total CGr1 cells, respectively (mean ± sd, n=3). (e) Blood from WT or DC-HIL KO mice (n=5) with or without (None) tumor measured for cytokines. (f) DC-HIL vs. Gr1 expression by CGr1 cells within LL2 tumor on mice one day after the second intratumoral injection of cytokines. (g) Tumor volume after cytokines injected intratumorally (arrows) into LL2 tumor-bearing mice. *p<0.01.
Inducible DC-HIL expression by CD11b+Gr1+ cells required IL-1β and IFN-γ
To identify inducers of DC-HIL expression on CD11b+Gr1+ cells, we examined soluble factors known to activate these cells from lineage marker-negative (Linneg) BM cells cultured in GM-CSF/IL-4 (Cheng et al., 2008). GM-CSF/IL-4 treatment alone generated 80% CD11b+Gr1+ cells, without significant DC-HIL expression. IL-1β, IFN-γ, and TNF-α each induced DC-HIL expression by 20-40% (Figure 6d). Combined IL-1β and IFN-γ treatment amplified DC-HIL expression to 60%. TNF-α enhanced effects of IL-1β and of IFN-γ, while suppressing CD11b+Gr1+ cell proliferation. Thus IL-1β and IFN-γ synergistically triggered DC-HIL expression by CD11b+Gr1+ cells.
Could differential secretion of cytokines account for the distinctive behavior of B16 melanoma vs. other tumors? Because B16 cells in vitro did not secrete IL-1β or IFN-γ (Supplementary Figure S9), we assayed blood cytokines from mice bearing B16, LL2, or EL-4 tumor (Figure 6e). Serum IL-1β and IFN-γ were elevated significantly in mice with melanoma, but not with other tumors, tumor-free mice, nor KO mice with melanoma. No other cytokines tested were expressed preferentially by mice with melanoma, thereby linking melanoma to IL-1β/IFN-γ secretion by host cells.
Do IL-1β and IFN-γ induce CD11b+Gr1+ cells to express DC-HIL? We injected both cytokines into LL2 tumors on mice and examined tumor-infiltrating CD11b+Gr1+ cells for DC HIL expression (Figure 6f). Injection of either cytokine induced marginal expression by LL2-infiltrating CD11b+Gr1+ cells. Coinjection of both cytokines enhanced DC-HIL expression, and repeated concurrent injections of both IL-1β and IFN-γ (but not just either cytokine or PBS) promoted LL2 tumor growth (Figure 6g). Thus, melanoma stromal cells may secrete IL-1β and IFN-γ that activate the DC-HIL pathway.
Discussion
The detrimental effect of an expanding population of T cell-suppressors in cancer patients is well established, but the mechanism underlying these cells’ profound immunosuppression remains unclear. We now show that melanoma is associated with an autocrine loop in which the T cell-inhibitory receptor DC-HIL induced on CD11b+Gr1+ myelomonocytic cells promotes melanoma growth. Binding of DC-HIL on CD11b+Gr1+ cells to SD-4 on effector T-cells inactivates the latter, while also transducing ITAM signals in the former that nudge the IFN-γ/iNOS axis into secreting NO. Although this mechanism did not apply to models of lung carcinoma and lymphoma, it is premature to consider it distinct for melanoma since studies involving other malignancies are ongoing.
Researchers have used the term myeloid-derived suppressor cells (MDSC) for cells coexpressing CD11b and Gr1 and exerting T cell-inhibitory activity. We have avoided the MDSC label because of phenotypic and functional heterogeneity among these cells. In fact, we sorted melanoma-CD11b+Gr1+ cells into Ly6ChighGr1low, Ly6CmedGr1hgih, Ly6CmedGr1low, and Ly6ClowGr1low subsets, with Ly6ChighGr1low monocytes as highest expressors of DC-HIL and sole suppressors of T-cells (Supplementary Figure S3d-e).
Because CD80, CD86 and PD-L1 deliver negative signals to T-cells through corresponding receptors (Egen et al., 2002; Keir et al., 2007), each might mediate CD11b+Gr1+ cells’ effects in melanoma-bearing mice. Indeed, these cells expressed CD80, CD86, and PD-L1 at levels similar or greater than DC-HIL, but only DC-HIL was responsible for suppressor activity. This distinct DC-HIL property may be due its transduction of bidirectional signals: binding to SD-4 activates a protein tyrosine phosphatase CD148 in T-cells (Chung et al., 2011) while tyrosine phosphorylation of its ITAM motif in CD11b+Gr1+ cells leads to IFN-γ and iNOS expression. Our preliminary results suggest that ligated DC-HIL on CD11b+Gr1+ cells activates Syk kinase, a major signal mediator for ITAM (Lowell, 2011). Unlike DC-HIL, CD80/CD86 and PD-L1 lack tyrosine-based signal motifs, restricting them to unidirectional signaling via respective T-cell receptors (Carreno and Collins, 2002; Sharpe et al., 2007).
CD11b+Gr1+ cells differ from APC in the mechanism(s) for suppressing T-cell function. The latter appears focused on down-regulation of TCR signals, whereas the former may employ the same pathway plus other avenues. Thus classic coinhibitors may not be as critical for CD11b+Gr1+ cells’ suppressor activity.
IFN-γ might have simultaneously opposing effects on cancers. Its well-known anti-tumor benefits (Kerkar et al., 2011; Takeda et al., 2011) are mediated via activation and proliferation of T-cells and NK cells, while its pro-tumor effects are transmitted via MDSC. We showed that IFN-γ’s deleterious pro-tumor effects arise not only by inducing DC-HIL expression on MDSC, but also by MDSC expressing the cytokine to mediate MDSC’ T-cell suppressor function. These data are consistent with MDSC from IFN-γ−/− mice with colon carcinoma that lost their suppressor activity (Gallina et al., 2006). Indeed, we showed IFN-γ−/− CD11b+Gr1+ cells to be poor tumor-promoters, similar to DC-HIL−/− CD11b+Gr1+ cells (Supplementary Figure S10).
MDSC’s phenotypic and functional plasticity to microenvironmental conditions may apply to different cancers in which divergent soluble factors produced by the tumors or their stromal cells drive MDSC differentiation toward particular subsets (Condamine and Gabrilovich, 2011). IL-1β-overexpressing breast cancer honed MDSC differentiation towards the Gr1highLy6Cneg phenotype with highest ability to suppress NK cell activity (Elkabets et al., 2010), consistent with our DC-HIL-expressing fraction among CD11b+Gr1+ cells responsible for melanoma-induced immunosuppression. The DC-HIL phenotype may be nurtured by IL-1β and IFN-γ secreted by melanoma stromal cells as elicited by our experimental administration of both cytokines into a non-melanoma LL2 carcinoma. Highly elevated IL-1β and IFN-γ expression was reported in skin and LN bearing spontaneous melanoma in a transgenic mouse model (Meyer et al., 2011).
High DC-HIL expression on blood CD14+HLA-DRno/low cells (the equivalent of mouse CD11b+Gr1+ cells) in melanoma patients and the restoration of suppressed T-cell function in those patients using anti-DC-HIL mAb (see accompanying submitted manuscript) provide further rationale for exploring DC-HIL-expressing myelomonocytic suppressor cells as a target for improving melanoma immunotherapy.
Materials and Methods
Reagents, animal, cell culture, and leukocyte preparations
Ab were purchased from eBioscience and Upstate Biotechnology; recombinant cytokines from Pepro Tech; and chemical inhibitors from Sigma-Aldrich. We generated 1E4 rat anti-mouse DCHIL (for immunoblotting) and UTX103 rabbit anti-mouse DC-HIL (for flow cytometric analysis and functional blocking) (Chung et al., 2009). DC-HIL-Fc fusion protein was produced (Chung et al., 2007b). C57BL/6 mice and pmel-1 TCR transgenic mice were purchased from Harlan and Jackson Laboratory, respectively. The DC-HIL−/− mice were generated by the deletion of exons 2 and 3 of the gene, resulting in depletion specifically of the DC-HIL protein (Supplementary Figure S1). SD-4−/− mice were obtained from Dr. Kojima (Nagoya University) (Ishiguro et al., 2000), and SD-4−/− pmel-1 mice were produced by breeding between these strains.
All tumor cell lines were purchased from the American Type Culture Collection. CGr1 and/or CD11bneg cells were isolated from spleen, blood, or tumor-infiltrating cells of mice (2-3 wks after implanting with tumor cells) (Chung et al., 2014). Depletion of DC-HIL+ cells from the CGr1 preparation was performed using UTX103 mAb-coated magnetic beads; and BM-DC, splenic CD11c+ or F4/80+ cells were prepared (Chung et al., 2014).
Tumor growth assays
Tumor cells (5 × 105) were injected s.c. into the right shaved flank of mice. Tumor volume was measured (Tomihari et al., 2010). Eight days after implanting LL2 cells, PBS or IL-1β and/or IFN-γ (50 ng/tumor) was injected into the tumor every 2 d for 4 times. CD11b+Gr1+ (CGr1) cells were purified from tumor the day after second injection. For lung metastasis, B16 cells (1 × 106) were injected into mice via the lateral tail veil. Lungs were harvested 18 or 19 d post-injection, and their weight, metastatic foci, and melanin content determined (Tomihari et al., 2010).
T-cell assays
Myeloid cells purified from splenocytes of tumor-bearing mice (2-3 wks after implanting s.c.) were cocultured with WT-T cells or pmel-1 splenocytes (1 ×105 cells/well) at varying cell ratios with anti-CD3/CD28 Ab or gp100 peptide (each 1 μg/ml), respectively, for 3 d. Inhibitors were added to splenocytes/CGr1 cells (1:1 ratio). CGr1 cells were also cocultured with SD-4+/+ or SD-4−/− pmel-1 splenocytes. For T cell-stimulatory assays, myeloid cells were pulsed with 1 μg/ml hgp100 peptide and cocultured with pmel-1 CD8+ T-cells at increasing cell ratios. After 2 d culture, T-cell activation was measured by CFSE-dilution assay or 3H-thymidine uptake (Chung et al., 2007b).
Crosslinking and soluble factor assays
CGr1 cells (5 × 106) were crosslinked with anti-DC-HIL mAb and assayed for tyrosine phosphorylation (Chung et al., 2009). CGr1 cells were also cultured in 96 well-plates (2 × 105 cells/well) precoated with anti-DC-HIL mAb or control IgG (10 μg/ml). After 1 or 2 d of culture, the culture supernatant and cell pellets were collected, respectively, for cytokine secretion using ELISA and whole cell extracts for measuring NO production using Griess method (De Santo et al., 2005)) or total RNA for measuring IFN-γ and NOS mRNA using quantitative RT-PCR.
Ab treatment
On day 6 or 11 after inoculating 5 × 105 B16 or KD-B16 cells s.c., mice were sorted to 2 groups with similar tumor volume (~0.1 cm3) and injected i.p. with Ab (200 μg/mouse) every other day for 5-6 injections. At varying times, blood was taken from tails of 5 representative mice/group, depleted of red blood cells, pulsed with OVA257-264 peptide (1 μg/ml), and examined by FACS for % CGr1 in total H-2Kb+ cells.
Generation of CGr1 cells
Linneg BM cells isolated using Lineage cell depletion kit (Miltenyi, Auburn, CA) were cultured (5 × 105 cells/well) for 5 d with GM-CSF/IL-4 (each 10 ng/ml) plus cytokines (10-20 ng/ml). Cultured cells were depleted of CD11c+/CD19+ cells (10-15% in total cells), and examined for % CGr1 cells and DC-HIL expression.
Adoptive transfer
For T-cell suppression, mice were injected i.v. with pmel-1 CD8+ T-cells (1 × 106 cells/mouse); 3 d later injected i.v. with undepleted or DC-HIL-depleted CGr1 (1 × 106 cells/mouse) and vaccinated with Freund’s adjuvant/gp100 peptide (1 mg/ml); and 10 d later LN cells were restimulated by culturing for 2 d with gp100 peptide in ELISPOT wells, and IFN-γ-secreting cells counted. For melanoma-promotion, 2 × 106 CGr1 and 2 × 105 B16 cells were co-injected s.c. into mice. A week later, mice were injected again with CGr1 cells. Some mice were injected i.p. with Ab (200 μg/mouse) starting on day 7, every other day, for 5 injections.
Serum cytokines
Two weeks post-implantation of tumor cells (5 × 105 cells/mouse), sera were taken, and albumin removed by passing through the Swell Blue albumin removal spin column (Pierce, Rockford, IL) before ELISA assays for cytokines.
Statistical analysis
Statistical analyses were performed using two-sided student’s t test, with p (<0.01) considered significant. All data shown are representative of at least 2 independent experiments.
Supplementary Material
Supplementary Figure S1. Characterization of DC-HIL-deficient APC. (a) WT allele (consisting of 11 exons) of C57BL/6 background and targeted KO allele are represented schematically. (b) Mouse DNA samples were PCR-amplified with 3 PCR primers (shown on the map): (1) intron between exons 1 and 2 (669 bp PCR band by primers #1 and #2); and (2) region spanning the intron to Neo gene (952 bp by primers #1 and #3), and separated on 1.5% agarose gel. WT and KO allele produces bands of 669 and 952 bp, respectively; heterozygote showed a mixed pattern. (c) Total RNA isolated from BM-derived DC of a KO or WT mouse was analyzed by RT-PCR using 2 primer sets to amplify exons 2-3 (E2-3, 416 bp) and exons 5-9 (E5-9, 673 bp). β-actin mRNA was also PCR-amplified. (d) BM-DC were used to immunoblot DC-HIL and β-actin proteins (20 μg of crude protein/lane) using 1E4 anti-DC-HIL and anti-β-actin Ab. Total proteins were also stained by Coomassie blue. (e-f) BM-DC or macrophages (MΦ) from WT or KO mice were examined by FACS for expression of DC-HIL and CD11c (e) or CD11b (f). (g) Varying numbers of BM-DC from WT or KO mice were cocultured with CD4+ or CD8+ T cells (from OT-II or OT-I transgenic mice, respectively) with OVA peptide, and IL-2 and/or IFN-γ secretion measured. (h) DC preparations were assayed for surface expression of CD80 and CD86 on CD11c+ cells. Two other KO and WT mice showed similar results. *Students’ t-test (p<0.001) between WT and KO.
Supplementary Figure S2. Leukocytes in lymphoid organs and bone marrow of WT and DC-HIL KO mice. Single cell suspensions prepared from LN (a), spleen (b) and BM (c) of WT and KO naive mice (7 week-old female) were stained fluorescently with cell surface marker Ab and analyzed by flow cytometry for frequency (%) of each leukocyte (shown in dot-plots). Other two pairs (7 and 10 week-old females) showed similar results.
Supplementary Figure S3. DC-HIL is expressed by some CD11b+Gr-1+ cells from several tissues. Three weeks after implanting B16 melanoma cells in mice, CD11b+Gr1+ (CGr1) cells were purified from different tissues and examined for DCHIL expression by FACS. (a) BM cells were prepared from the femur, from which CGr1 cells were purified and fluorescently stained with anti-Gr1 and anti-DC-HIL mAb (or control IgG). (b) Peripheral blood was collected from mouse tail veins. (c) Tumor-infiltrating cells were prepared from B16 tumor (~2 cm3): The tumor was minced in PBS, treated with digestive enzymes, and applied to Ficoll-gradient to remove debris. % DC-HIL+ cells among total CGr1 cells are shown in dot-plots. (d) CGr1 cells purified from splenocytes of mice with B16 melanoma were analyzed for expression of Gr-1 vs. Ly6C. Based on differential expression of these markers, CD11b+Gr-1+ cells sorted into 4 different fractions: Fr. 1 (Ly6ChighGr1low); Fr. 2 (Ly6CmedGr1hgih); Fr. 3 (Ly6CmedGr1low); and Fr. 4 (Ly6ClowGr1low). Each fraction was examined for DC-HIL expression: open and gray-filled histograms show anti-DC-HIL mAb and control IgG staining, respectively. Fr. 1 showed highest expression of DC-HIL. (e) CGr1 subsets (Fr. 1 through Fr. 4) were purified by FACS sorting and cocultured with pmel-1 splenocytes/Ag at different cell ratios. T cell activation was assessed by proliferation, and the suppressive ability of each Fr or unfractionated CGr1 is expressed as % suppression (1 – cpm of CGr1-added culture/ cpm of T cell alone × 100%). *p<0.001 between Fr.1 and Fr.2. Data are representative of 3 experiments.
Supplementary Figure S4. DC-HIL−/− CGr1 cells are defective in suppressing proliferation of CD8+ T cells in vivo. Mice (n=5) were injected i.v. with pmel-1 CD8+ T-cells (1 × 106 cells/mouse); 3 days later injected i.v. with CGr1 cells isolated from WT or DC-HIL KO mice bearing melanoma (1 × 106 cells/mouse) and vaccinated with CFA/gp100 peptide; and 10 days later LN cells were harvested and cultured for 2 d in ELISPOT wells with gp100 peptide, and IFN-γ-secreting cells counted. *p<0.01 between WTCGr1 and KO-CGr1. Data are representative of 2 experiments.
Supplementary Figure S5. Tumor-promoting ability of DC-HIL+ CD11b+Gr1+ cells is diminished in Rag2 KO mice. On day 0, undepleted (DC-HIL+) or DC-HIL-depleted CD11b+Gr1+ cells (DC-HILneg CGr1) (2 × 106 cells/mouse) were coinjected with B16 cells (2 × 105 cells) s.c. into naive Rag2 KO mice (n=5). On day 6, similarly purified cells alone were injected i.v. into corresponding mice. Control mice were injected with B16 cells alone (no CGr1 cells). Tumor volume is shown (mean ± sd), with insignificant (p>0.1) difference between mice injected with B16 cells + DC-HIL+ and B16 cells + DC-HILneg CGr1 cells. Data shown are representative of 2 independent experiments. NS stands for not significant between DC-HIL+ and DC-HILneg CGr1.
Supplementary Figure S6. DC-HIL−/− MDSC are unable to induce expression of IFN-γ and iNOS following DC-HIL-crosslinking. CGr1 cells isolated from WT or DC-HIL KO mice (n=3) bearing melanoma were DC-HIL-crosslinked and assayed (a) for IFN-γ production (by ELISA) and (b) for iNOS mRNA expression (by real time PCR): the former was measured individually by MDSC (mean ± sd, n=3); and the latter by pooled MDSC. Data are shown as fold increase (anti-DC-HIL-treated culture vs. control IgG). Data are representative of 2 experiments. *p<0.001 between WT and KO.
Supplementary Figure S7. Expansion of MDSC in WT and DC-HIL KO mice following implantation of B16 melanoma. Spleen cells isolated from WT or DC-HIL KO mice (n=3) with (B16) or without (No) B16 melanoma were examined by FACS for frequency (%) of CD11b+Gr1+ (CGr1) cells. Representative FACS data are shown (a), and data are calculated and summarized as % CGr1 cells (b) or absolute number in spleen (c). Data are representative of 3 separate experiments. *p<0.001 between WT and KO mice with melanoma.
Supplementary Figure S8. Effect of anti-DC-HIL treatment on APC function of DC in melanoma-bearing mice. Two weeks after implanting B16 cells into DC-HIL−/− mice (n=4), CFSE-labeled pmel-1 Thy1.1+ splenocytes plus unpulsed DC (un-DC) or Ag-pulsed DC (pul-DC) were injected i.v. into mice, followed by i.p. injection of anti-DC-HIL mAb or control IgG (2 injections). One day after the last injection, spleen or LN cells were examined for CFSE fluorescence intensity on Thy1.1+ cells. Representative dot-plots with % of proliferated cells (shown in red-lined box): 16 ± 1.2% in spleen and 6.9 ± 2.1% in LN of mice injected with un-DC; 24 ± 1% and 19 ± 2% with pul-DC/αDC-HIL; and 24 ± 0.5% and 18 ± 1% with pul-DC/IgG. Representative data of 2 independent experiments.
Supplementary Figure S9. Secretion of cytokines by different tumor cell lines. Exponentially growing tumor cells (B16 melanoma, LL2 lung carcinoma, and EL-4 lymphoma) were harvested and replated onto culture dishes (5 × 105 cells/30 mm dish). After culture for 5 d, the supernatant (Sup) was harvested, spun for 10 min at 4°C, and amount of indicated cytokines were determined by ELISA. *p<0.01 between B16 and EL-4.
Supplementary Figure S10. DC-HIL−/− CGr1 cells were as poor tumor-promoters as IFN-γ−/− CGr1. On day 0, CGr1 cells from WT, DC-HIL KO, or IFN-γ KO mice with melanoma were co-injected with B16 cells s.c. into naive WT mice (n=5). On day 6, similarly purified CGr1 cells alone were injected i.v. into corresponding mice. Control mice were injected with B16 cells alone (no CGr1 cells). *p<0.001 between WT and DC-HIL/IFN-γ KO. Data shown are representative of 2 experiments.
Acknowledgements
We thank Irene Dougherty and Megan Randolph for technical and administrative assistance, respectively. This research was supported by National Institute of Health grant (AI064927-05).
Abbreviations used
- BM
bone marrow
- CGr1 cells
CD11b+Gr1+ cells
- iNOS
inducible nitric oxide synthase
- ITAM
immunoreceptor tyrosine-based activation motif
- KO
knocked out
- MDSC
myeloid-derived suppressor cells
- SD-4
syndecan-4
Footnotes
Conflict of interest The authors state no conflict of interest.
References
- Akiyoshi H, Chung JS, Tomihari M, et al. Depleting syndecan-4+ T lymphocytes using toxin-bearing dendritic cell-associated heparan sulfate proteoglycan-dependent integrin ligand: a new opportunity for treating activated T cell-driven disease. J Immunol. 2010;184:3554–61. doi: 10.4049/jimmunol.0903250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingisser RM, Tilbrook PA, Holt PG, et al. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729–34. [PubMed] [Google Scholar]
- Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- Cheng P, Corzo CA, Luetteke N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–49. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JS, Cruz PD, Jr., Ariizumi K. Inhibition of T-cell activation by syndecan-4 is mediated by CD148 through protein tyrosine phosphatase activity. Eur J Immunol. 2011;41:1794–9. doi: 10.1002/eji.201041233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JS, Dougherty I, Cruz PD, Jr., et al. Syndecan-4 mediates the coinhibitory function of DC- HIL on T cell activation. J Immunol. 2007a;179:5778–84. doi: 10.4049/jimmunol.179.9.5778. [DOI] [PubMed] [Google Scholar]
- Chung JS, Sato K, Dougherty II, et al. DC-HIL is a negative regulator of T lymphocyte activation. Blood. 2007b;109:4320–7. doi: 10.1182/blood-2006-11-053769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JS, Tamura K, Akiyoshi H, et al. The DC-HIL/syndecan-4 pathway regulates autoimmune responses through myeloid-derived suppressor cells. J Immunol. 2014;192:2576–84. doi: 10.4049/jimmunol.1301857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JS, Yudate T, Tomihari M, et al. Binding of DC-HIL to dermatophytic fungi induces tyrosine phosphorylation and potentiates antigen presenting cell function. J Immunol. 2009;183:5190–8. doi: 10.4049/jimmunol.0901319. [DOI] [PubMed] [Google Scholar]
- Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santo C, Serafini P, Marigo I, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U S A. 2005;102:4185–90. doi: 10.1073/pnas.0409783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Montero CM, Salem ML, Nishimura MI, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin- cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–8. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- Elkabets M, Ribeiro VS, Dinarello CA, et al. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. 2010;40:3347–57. doi: 10.1002/eji.201041037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Lonsdorf AS, Hwang ST. Immunotherapy for advanced melanoma. J Invest Dermatol. 2008;128:2596–605. doi: 10.1038/jid.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipazzi P, Valenti R, Huber V, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony- stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546–53. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina G, Dolcetti L, Serafini P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–90. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K, Kadomatsu K, Kojima T, et al. Syndecan-4 deficiency impairs focal adhesion formation only under restricted conditions. J Biol Chem. 2000;275:5249–52. doi: 10.1074/jbc.275.8.5249. [DOI] [PubMed] [Google Scholar]
- Kamijo R, Harada H, Matsuyama T, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–5. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. 2007;19:309–14. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Kerkar SP, Goldszmid RS, Muranski P, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121:4746–57. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol. 2003;74:186–96. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yu Y, Yang S, et al. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol Immunother. 2009;58:687–97. doi: 10.1007/s00262-008-0591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zeng B, Zhang Z, et al. B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin Immunol. 2008;129:471–81. doi: 10.1016/j.clim.2008.07.030. [DOI] [PubMed] [Google Scholar]
- Lowell CA. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Sevko A, Ramacher M, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A. 2011;108:17111–6. doi: 10.1073/pnas.1108121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Sharpe AH, Wherry EJ, Ahmed R, et al. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–45. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- Shikano S, Bonkobara M, Zukas PK, et al. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J Biol Chem. 2001;276:8125–34. doi: 10.1074/jbc.M008539200. [DOI] [PubMed] [Google Scholar]
- Takeda K, Nakayama M, Sakaki M, et al. IFN-γ production by lung NK cells is critical for the natural resistance to pulmonary metastasis of B16 melanoma in mice. J Leukoc Biol. 2011;90:777–85. doi: 10.1189/jlb.0411208. [DOI] [PubMed] [Google Scholar]
- Tomihari M, Chung JS, Akiyoshi H, et al. DC-HIL/glycoprotein Nmb promotes growth of melanoma in mice by inhibiting the activation of tumor-reactive T cells. Cancer Res. 2010;70:5778–87. doi: 10.1158/0008-5472.CAN-09-2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu JQ, Talebian F, et al. Tumor expression of CD200 inhibits IL-10 production by tumor- associated myeloid cells and prevents tumor immune evasion of CTL therapy. Eur J Immunol. 2010;40:2569–79. doi: 10.1002/eji.201040472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Cai Z, Zhang Y, et al. CD80 in immune suppression by mouse ovarian carcinoma- associated Gr-1+CD11b+ myeloid cells. Cancer Res. 2006;66:6807–15. doi: 10.1158/0008-5472.CAN-05-3755. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Characterization of DC-HIL-deficient APC. (a) WT allele (consisting of 11 exons) of C57BL/6 background and targeted KO allele are represented schematically. (b) Mouse DNA samples were PCR-amplified with 3 PCR primers (shown on the map): (1) intron between exons 1 and 2 (669 bp PCR band by primers #1 and #2); and (2) region spanning the intron to Neo gene (952 bp by primers #1 and #3), and separated on 1.5% agarose gel. WT and KO allele produces bands of 669 and 952 bp, respectively; heterozygote showed a mixed pattern. (c) Total RNA isolated from BM-derived DC of a KO or WT mouse was analyzed by RT-PCR using 2 primer sets to amplify exons 2-3 (E2-3, 416 bp) and exons 5-9 (E5-9, 673 bp). β-actin mRNA was also PCR-amplified. (d) BM-DC were used to immunoblot DC-HIL and β-actin proteins (20 μg of crude protein/lane) using 1E4 anti-DC-HIL and anti-β-actin Ab. Total proteins were also stained by Coomassie blue. (e-f) BM-DC or macrophages (MΦ) from WT or KO mice were examined by FACS for expression of DC-HIL and CD11c (e) or CD11b (f). (g) Varying numbers of BM-DC from WT or KO mice were cocultured with CD4+ or CD8+ T cells (from OT-II or OT-I transgenic mice, respectively) with OVA peptide, and IL-2 and/or IFN-γ secretion measured. (h) DC preparations were assayed for surface expression of CD80 and CD86 on CD11c+ cells. Two other KO and WT mice showed similar results. *Students’ t-test (p<0.001) between WT and KO.
Supplementary Figure S2. Leukocytes in lymphoid organs and bone marrow of WT and DC-HIL KO mice. Single cell suspensions prepared from LN (a), spleen (b) and BM (c) of WT and KO naive mice (7 week-old female) were stained fluorescently with cell surface marker Ab and analyzed by flow cytometry for frequency (%) of each leukocyte (shown in dot-plots). Other two pairs (7 and 10 week-old females) showed similar results.
Supplementary Figure S3. DC-HIL is expressed by some CD11b+Gr-1+ cells from several tissues. Three weeks after implanting B16 melanoma cells in mice, CD11b+Gr1+ (CGr1) cells were purified from different tissues and examined for DCHIL expression by FACS. (a) BM cells were prepared from the femur, from which CGr1 cells were purified and fluorescently stained with anti-Gr1 and anti-DC-HIL mAb (or control IgG). (b) Peripheral blood was collected from mouse tail veins. (c) Tumor-infiltrating cells were prepared from B16 tumor (~2 cm3): The tumor was minced in PBS, treated with digestive enzymes, and applied to Ficoll-gradient to remove debris. % DC-HIL+ cells among total CGr1 cells are shown in dot-plots. (d) CGr1 cells purified from splenocytes of mice with B16 melanoma were analyzed for expression of Gr-1 vs. Ly6C. Based on differential expression of these markers, CD11b+Gr-1+ cells sorted into 4 different fractions: Fr. 1 (Ly6ChighGr1low); Fr. 2 (Ly6CmedGr1hgih); Fr. 3 (Ly6CmedGr1low); and Fr. 4 (Ly6ClowGr1low). Each fraction was examined for DC-HIL expression: open and gray-filled histograms show anti-DC-HIL mAb and control IgG staining, respectively. Fr. 1 showed highest expression of DC-HIL. (e) CGr1 subsets (Fr. 1 through Fr. 4) were purified by FACS sorting and cocultured with pmel-1 splenocytes/Ag at different cell ratios. T cell activation was assessed by proliferation, and the suppressive ability of each Fr or unfractionated CGr1 is expressed as % suppression (1 – cpm of CGr1-added culture/ cpm of T cell alone × 100%). *p<0.001 between Fr.1 and Fr.2. Data are representative of 3 experiments.
Supplementary Figure S4. DC-HIL−/− CGr1 cells are defective in suppressing proliferation of CD8+ T cells in vivo. Mice (n=5) were injected i.v. with pmel-1 CD8+ T-cells (1 × 106 cells/mouse); 3 days later injected i.v. with CGr1 cells isolated from WT or DC-HIL KO mice bearing melanoma (1 × 106 cells/mouse) and vaccinated with CFA/gp100 peptide; and 10 days later LN cells were harvested and cultured for 2 d in ELISPOT wells with gp100 peptide, and IFN-γ-secreting cells counted. *p<0.01 between WTCGr1 and KO-CGr1. Data are representative of 2 experiments.
Supplementary Figure S5. Tumor-promoting ability of DC-HIL+ CD11b+Gr1+ cells is diminished in Rag2 KO mice. On day 0, undepleted (DC-HIL+) or DC-HIL-depleted CD11b+Gr1+ cells (DC-HILneg CGr1) (2 × 106 cells/mouse) were coinjected with B16 cells (2 × 105 cells) s.c. into naive Rag2 KO mice (n=5). On day 6, similarly purified cells alone were injected i.v. into corresponding mice. Control mice were injected with B16 cells alone (no CGr1 cells). Tumor volume is shown (mean ± sd), with insignificant (p>0.1) difference between mice injected with B16 cells + DC-HIL+ and B16 cells + DC-HILneg CGr1 cells. Data shown are representative of 2 independent experiments. NS stands for not significant between DC-HIL+ and DC-HILneg CGr1.
Supplementary Figure S6. DC-HIL−/− MDSC are unable to induce expression of IFN-γ and iNOS following DC-HIL-crosslinking. CGr1 cells isolated from WT or DC-HIL KO mice (n=3) bearing melanoma were DC-HIL-crosslinked and assayed (a) for IFN-γ production (by ELISA) and (b) for iNOS mRNA expression (by real time PCR): the former was measured individually by MDSC (mean ± sd, n=3); and the latter by pooled MDSC. Data are shown as fold increase (anti-DC-HIL-treated culture vs. control IgG). Data are representative of 2 experiments. *p<0.001 between WT and KO.
Supplementary Figure S7. Expansion of MDSC in WT and DC-HIL KO mice following implantation of B16 melanoma. Spleen cells isolated from WT or DC-HIL KO mice (n=3) with (B16) or without (No) B16 melanoma were examined by FACS for frequency (%) of CD11b+Gr1+ (CGr1) cells. Representative FACS data are shown (a), and data are calculated and summarized as % CGr1 cells (b) or absolute number in spleen (c). Data are representative of 3 separate experiments. *p<0.001 between WT and KO mice with melanoma.
Supplementary Figure S8. Effect of anti-DC-HIL treatment on APC function of DC in melanoma-bearing mice. Two weeks after implanting B16 cells into DC-HIL−/− mice (n=4), CFSE-labeled pmel-1 Thy1.1+ splenocytes plus unpulsed DC (un-DC) or Ag-pulsed DC (pul-DC) were injected i.v. into mice, followed by i.p. injection of anti-DC-HIL mAb or control IgG (2 injections). One day after the last injection, spleen or LN cells were examined for CFSE fluorescence intensity on Thy1.1+ cells. Representative dot-plots with % of proliferated cells (shown in red-lined box): 16 ± 1.2% in spleen and 6.9 ± 2.1% in LN of mice injected with un-DC; 24 ± 1% and 19 ± 2% with pul-DC/αDC-HIL; and 24 ± 0.5% and 18 ± 1% with pul-DC/IgG. Representative data of 2 independent experiments.
Supplementary Figure S9. Secretion of cytokines by different tumor cell lines. Exponentially growing tumor cells (B16 melanoma, LL2 lung carcinoma, and EL-4 lymphoma) were harvested and replated onto culture dishes (5 × 105 cells/30 mm dish). After culture for 5 d, the supernatant (Sup) was harvested, spun for 10 min at 4°C, and amount of indicated cytokines were determined by ELISA. *p<0.01 between B16 and EL-4.
Supplementary Figure S10. DC-HIL−/− CGr1 cells were as poor tumor-promoters as IFN-γ−/− CGr1. On day 0, CGr1 cells from WT, DC-HIL KO, or IFN-γ KO mice with melanoma were co-injected with B16 cells s.c. into naive WT mice (n=5). On day 6, similarly purified CGr1 cells alone were injected i.v. into corresponding mice. Control mice were injected with B16 cells alone (no CGr1 cells). *p<0.001 between WT and DC-HIL/IFN-γ KO. Data shown are representative of 2 experiments.