

Abstract
Objective
We previously reported the expression of the 2-pore domain K+ channel TREK-1 in lung epithelial cells and proposed a role for this channel in the regulation of alveolar epithelial cytokine secretion. In this study we focused on investigating the role of TREK-1 in vivo in the development of hyperoxia-induced lung injury.
Design
Laboratory animal experiments.
Setting
University research laboratory.
Subjects
Wild type and TREK-1 deficient mice.
Interventions
Mice were anesthetized and exposed to 1) room air, no mechanical ventilation, 2) 95% hyperoxia for 24 hours, 3) 95% hyperoxia for 24 hours followed by mechanical ventilation for 4 hours.
Measurements and Main Results
Hyperoxia exposure accentuated lung injury in TREK-1 deficient mice but not controls, resulting in increased in Lung Injury Scores (LIS), broncho-alveolar lavage (BAL) fluid cell numbers and cellular apoptosis, and a decrease in quasi-static lung compliance. Exposure to a combination of hyperoxia and injurious mechanical ventilation resulted in further morphological lung damage, increased LIS and BAL fluid cell numbers in control but not TREK-1 deficient mice. At baseline and after hyperoxia exposure BAL cytokine levels were unchanged in TREK-1 deficient mice compared to controls. Exposure to hyperoxia and mechanical ventilation resulted in an increase in BAL IL-6, MCP-1 and TNF-α levels in both mouse types, but the increase in IL-6 and MCP-1 levels was less prominent in TREK-1 deficient mice than in controls. Lung tissue MIP-2, KC and IL-1β gene expression was not altered by hyperoxia in TREK-1 deficient mice compared to controls. Furthermore, we show for the first time TREK-1 expression on alveolar macrophages and unimpaired TNF-α secretion from TREK-1 deficient macrophages.
Conclusion
TREK-1 deficiency resulted in increased sensitivity of lungs to hyperoxia but this effect is less prominent if overwhelming injury is induced by the combination of hyperoxia and injurious mechanical ventilation. TREK-1 may constitute a new potential target for the development of novel treatment strategies against hyperoxia-induced lung injury.
Keywords: TREK, lung, lung injury, acute respiratory distress syndrome, ARDS
INTRODUCTION
Acute Respiratory Distress Syndrome (ARDS) remains a challenging disease to manage in both the adult and pediatric populations[1, 2] and is still associated with significant mortality and morbidity rates[3]. The main treatment strategies against ARDS, mechanical ventilation and oxygen supplementation[4], are life-saving therapies, but unfortunately the proinflammatory effects of hyperoxia (HO) and mechanical stretch promote further lung injury and can be reproduced in both in vitro and in vivo models of ARDS[5-8]. Therefore, the development of new therapeutic strategies against ARDS represents a major research interest.
While our own group[9, 10] and others[11, 12] confirmed the detrimental effects of HO on lung tissue and documented the time course of HO-induced lung inflammation[13], the contribution of ion channels, in particular 2-pore domain potassium (K2P) channels including TREK-1, to the development of ARDS remains unclear. To date, the main function of K2P channels is thought to consist in regulation of the resting cell membrane potential[14-16] by sustaining so-called “background” or “leak” potassium currents, and little is known about other potential functions of K2P channels. We previously suggested that the K2P channel TREK-1 may play a regulatory role in the development of alveolar epithelial injury. We found that TREK-1 deficiency altered the cytokine release profile of alveolar epithelial cells upon TNF-α stimulation, resulting in decreased IL-6 and increased MCP-1 secretion[17, 18]. Although activation of several TNF-α induced signaling pathways, including p38 kinase and the PKCθ isoform, appeared altered in TREK-1 deficient epithelial cells, the specific regulatory mechanisms underlying these signaling changes remains unclear.
Since the pathological processes leading to ARDS affect not only the alveolar epithelium but rather the lung as a whole, in this study we investigated the effects of HO in vivo using a TREK-1 deficient mouse model. To the best of our knowledge, the only other studies describing K2P channels in the lung suggested a role for these channels in bronchial Na+ and Cl− transport[19] and in mucosal gland secretion[20]. We show for the first time that exposure of TREK-1 deficient mice to HO resulted in an increase in inflammatory parameters, including higher lung injury scores, worsening of quasi-static lung compliance, and increased cellular apoptosis, which was independent of BAL fluid cytokine levels and cytokine gene expression in the lung tissue. Overall, our results suggest that the K2P channel TREK-1 may play an important role in the development of HO-induced lung injury.
MATERIALS AND METHODS
Animals
C57BL/J6 wildtype (WT) mice, aged 8-10 weeks were obtained from Charles River Laboratories (Wilmington, MA). Age-matched TREK-1 ko mice were obtained from an existing colony at Baylor College of Medicine (Texas Genetic Institute)[21], and all experiments were performed in accordance with the animal protocols approved by the University of Tennessee Health Science Center and Baylor College of Medicine.
Ventilation protocol
Mice were divided into 3 experimental groups: 1) room air, no mechanical ventilation, 2) exposure to 95% HO for 24 hours using an HO chamber (BioSpherix), and a ProOx C21 O2/CO2 controller (BioSpherix), 3) exposure to 95% HO for 24 hours followed by injurious mechanical ventilation (MV) for 4 hours using the Flexivent system (SQIREC; tidal volume 25ml/kg, RR 60 breaths/min, PEEP 2mmH2O). These conditions have consistently proven to be injurious and result in a phenotype similar to the one observed in patients with ARDS[9, 10]. Adequate anesthesia was provided with an intraperitoneal injection of a ketamine/xylazine mixture (1:1, 0.15mL/10grams body weight), and 1% inhaled isofluorane using a precision gas mixer (PEGAS 400, Columbus Instruments, OH). MV was provided using the Flexivent system via a tracheostomy with an 18 gauge needle. Quasi-static lung compliance was measured under general anesthesia. Mice in groups 1) and 2) were sacrificed after quasi-static compliance was recorded, and mice from group 3 were ventilated as described above. Pressure-volume curves (P-V) were recorded at the beginning and the end of each experiment. Each set of P-V curves was preceded by two inflation maneuvers to total lung capacity to insure equal standard volumes for each experiment. Rectal temperature was maintained within the normal range using a heat lamp. At the end of mechanical ventilation a second lung compliance measurement was performed and values were calculated by fitting data derived from the P-V curves to the Salazar-Knowles equation as previously described[22]. In addition, mice were given intraperitoneal injections of 100 μl of saline every hour on the ventilator to maintain an isovolemic fluid balance by correcting for insensible losses. At the end of the experiment, mice were sacrificed by cardiac puncture and a 4% isofluorane overdose.
Sample collection
Using three 1ml syringes, bronchoalveolar lavage (BAL) was performed using 3× 1ml PBS with 0.6mM EDTA. Cytospins were prepared using BAL fluid to perform differential cell counts as previously described[9]. Total cell counts were calculated from BAL fluid using a hemocytometer after 1:1 mix with Turk's solution. From the rest of the BAL fluid, total protein concentrations were measured and the remainder was used for cytokine analysis by ELISA. The heart and lungs were removed en bloc, retrograde perfused with 5ml PBS to remove red blood cells, and photographs were taken of the lungs. Tissue samples for confocal microscopy were immediately frozen on dry ice, tissue samples for Western blot analysis were placed in RIPA buffer (50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 0.1% SDS) containing a protease inhibitor cocktail (Roche, Burlington, NC) on ice and stored at −80°C. Tissue samples for RT-PCR were frozen in RNA lysis buffer (Agilent Technologies).
Lung histology
After retrograde perfusion of the lungs, lung tissue was harvested and processed for histological examination as described previously[10]. To determine Lung Injury Scores (LIS), a pathologist blinded to the experimental conditions assigned a 5-parameter LIS (1. Interstitial edema, 2. alveolar edema, 3. neutrophil infiltration, 4. parenchymal hemorrhage and 5. perivascular-peribronchial hemorrhage) to each H&E stained lung section by examining 4 high power fields (40× magnification) per slide. Each criterion was assigned a score from 0-4 points with 0 points representing no injury and 4 points representing severe lung injury. The histological composite LIS in Figure 1C represents the mean value of the 5 parameters for each mouse.
Figure 1. TREK-1 deficient mice were more sensitive to hyperoxia (HO).
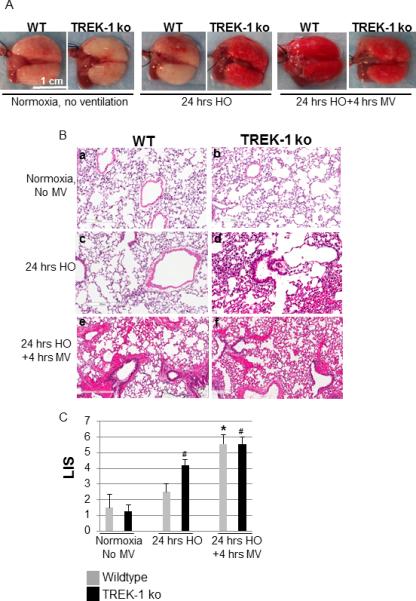
Panel A shows representative lung photographs of wildtype (WT) and TREK-1 deficient (TREK-1 ko) mice exposed to either normoxia and no mechanical ventilation (MV), 24 hours of 95% HO, or 24 hours of 95% HO followed by 4 hours of injurious MV. Similarly, Panel B shows representative H&E stained lung sections of mice exposed to the conditions described in Figure 1A. Panel C shows a summary of the composite lung injury scores (LIS) assigned to each section. (* compared to untreated WT control; # compared to untreated TREK-1 ko control; n=4-7 animals per group).
BAL fluid cytokine measurements by ELISA
TNF-α, IL-6 and MCP-1 levels in BAL fluid were quantified using BD Bioscience OptEIA species-specific ELISA kits following the manufacturer's instructions. Briefly, BAL was performed as described above by lavaging each lung 3 times with 1ml of PBS. The 3 aliquots were combined and supernatants were collected after ultracentrifugation at 8,000 rpm for 5 min. For cytokine measurements, 100μL of BAL supernatant was loaded into a 96 well ELISA plate. All samples were run in triplicates. Cytokine amounts were displayed in pg/mL.
Cytokine gene expression by RT-PCR
Total RNA was isolated from whole lung homogenates using an Absolutely RNA Miniprep kit (Agilent Technologies) according to the manufacturer's instructions. Contaminating genomic DNA was removed by treatment with DNA-free (Ambion, Austin, TX) according to the manufacturer's directions. Single-stranded DNA was synthesized from 1μg total RNA and RT-PCR was performed using a Transcriptor First Strand cDNA kit (Roche) according to the manufacturer's instructions and then amplified with Tag polymerase for 30–35 cycles in an Eppendorf Mastercycler EP Thermal Cycler using primers specific for each cytokine. The following primer sequences were used: MIP-2 forward: TGG GTG GGA TGT AGC TAG TTC C, MIP-2 reverse: AGT TTG CCT TGA CCC TGA AGC C; KC (CXCL1) forward: AAC GGA GAA AGA AGA CAG ACT, KC (CXCL1) reverse: GAC GAG ACC AGG AGA AAC AG. IL-1β forward: CAG GAT GAG GAC ATG AGC ACC, IL-1β reverse: CTC TGC AGA CTC AAA CTC CAC; The housekeeping gene β-actin was used as an internal control. After amplification, 16 μL of each reaction mixture was separated by electrophoresis on 1.8% agarose gels and the bands visualized by ethidium bromide staining. The pictures shown are representative of 3 experiments.
Lung macrophage stimulation
Alveolar macrophages (AM) were recovered from the BAL fluid from untreated mice as described previously[23]. AMs (2×105 cells/well) were stimulated with 0 or 1 ng/ml LPS and culture supernatants were collected following 24 hour incubation at 37°C for TNF-α production as measured by ELISA (BD Bioscience).
Macrophage immunofluorescence staining
AMs were collected from BAL fluid and 30,000 cells were spun onto a glass slide. Cells were washed with PBS and fixed with IC Fixation buffer (eBioscience) for 20 min at room temperature. The cells were then washed with PBST (PBS with 0.05% Tween-20) and incubated with Dako protein block (Dako, Carpinteria, CA) for 30 min at room temperature. Cells were washed again in PBST and incubated for 1 hour with an anti-TREK-1 antibody (C-20, 1:50 dilution in PBST, 1% BSA, Santa Cruz Biotech). After an additional washing step, cells were incubated for 1 hour with a secondary, biotinylated rabbit anti-goat antibody (1:150 in PBST with 1% BSA, Vector Labs) at room temperature. Thereafter, cells were washed again and then incubated for 1 hour with streptavidin-Cy3 (1:750 in PBST in 1%BSA, Jackson Immuno, West Grove, PA) at room temperature in the dark. After an additional washing step, the slides were dried and coverslips were mounted with Vectashield+DAPI (Vector labs). Images were taken on an EVOS FL Auto (Life Technologies) instrument.
Confocal Microscopy
After resection, lungs were immediately fixed in 4% formalin, and paraffin-embedded sections were cut into 4 μm thick tissues slices using a Microtome. TUNEL staining was performed according to the manufacturer's instructions using a fluorescence-based In Situ Cell Death Detection kit (Roche). Nuclear staining was obtained using Fluoro Gel II mounting medium containing DAPI (Electron Microscopy Sciences, Hatfield, PA). Images were acquired using the Zeiss 710 confocal imaging system available in the Department of Physiology at UTHSC. Emitted fluorescence was collected using a 20× magnification objective lens (NA 1.4 Oil), and the images were recorded using Zen 2009 Light Edition software (Zeiss).
For double stained lung sections, we used the TUNEL assay protocol as above and subsequently stained lung slices of TREK-1 deficient mice exposed to HO, or HO+MV with an anti-proSPC antibody (Millipore, Billerica, MA; 1:300; 24 hours at 4°C) to identify type II pneumocytes, or an anti-Aquaporin-5 antibody (Alamone Labs, 1:100; room temperature for 2 hours). Lung slices were then washed 3 times, and a species specific Alexa Fluor 594 secondary antibody was applied for 1 hour at room temperature. Slices were then mounted in Fluoro Gel II mounting medium with DAPI (EMS, Hatfield, PA) and analyzed as described above.
Western Blot Analysis
Frozen lung tissue was thawed on ice and whole lung homogenates were prepared using a tissue homogenizer in RIPA buffer containing the protease inhibitor cocktail. Homogenates were centrifuged at 17,000 g for 15 min at 4°C, and total protein concentrations were determined using the Bradford assay (BioRad, Hercules, CA). A total of 50 μg protein of each sample was separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) on 4-12% NuPage Bis-Tris gradient gels (Invitrogen) and transferred onto nitrocellulose membranes at 35 mV for 2 hours. Membranes were blocked in 5% non-fat dry milk in Tris-buffered saline (Bio-Rad) containing 0.1% Tween-20 (TBST) for 1 h at 37°C. The membranes were then incubated overnight with an anti-cleaved PARP-1 antibody (Cell Signaling, 1:1000) or an anti-total PARP-1 antibody (Cell Signaling, 1:1000) at 4°C. The next day, after 3 washing steps in TBST, all membranes were incubated for 1 hour at room temperature with an anti-rabbit HRP-conjugated IgG antibody (1:3000, Cell Signaling). Bands were visualized by enhanced chemoluminescence with ECL SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Rockford IL). Band densitometry measurements to determine relative quantities of protein were performed using ImageJ 1.42 software for Windows.
Statistical Analysis
All values were expressed as means±SEM. Data were analyzed using 2×3 (2 mouse types, 3 experimental conditions) ANOVA models, and pairwise comparison of the means were tested using the Tukey-Kramer method to adjust for multiple comparisons. For some of the BAL cytokines, transformations were used when residuals from the models were not normally distributed. All statistical analyses were performed using StatPlus software and SAS/STAT® software, and a p-value of p<0.05 was considered significant. (*) represents groups of mice compared to untreated WT control; (#) represents groups of mice compared to untreated TREK-1 ko control. (^) represents comparisons of WT to TREK-1 ko mice within the same treatment group.
RESULTS
Hyperoxia caused increased lung damage in TREK-1 deficient lungs
We previously proposed a role for TREK-1 in cytokine secretion from alveolar epithelial cells[17, 18] and now investigated whether TREK-1 also plays a role in an in vivo model of hyperoxia (HO)-induced lung injury. We exposed WT and TREK-1 deficient mice to either 24 hours of HO alone, or 24 hours of HO followed by 4 hours of injurious mechanical ventilation (MV) and analyzed lung morphology (Figure 1A), lung histology (Figure 1B), and a 5-point composite lung injury score (LIS) from each section (Figure 1C). We found that at baseline both untreated WT and TREK-1 deficient mice showed normal, uninjured lung morphology and histology, which was confirmed by a low LIS in both animal groups. HO exposure for 24 hours resulted in no significant lung damage in WT mice but in increased morphological and histological injury in TREK-1 deficient mice, which was also reflected by an increased LIS in HO-treated TREK-1 deficient mice when compared to untreated TREK-1 deficient mice. Since we knew from our previous studies that pre-exposure of WT mice to HO+MV resulted in additional lung injury[10], we investigated whether this would also occur in TREK-1 deficient animals. Interestingly, when compared to HO alone, exposure to HO+MV led to a further increase in morphological and histological lung injury in WT but not TREK-1 deficient mice, which was also reflected in the LIS of these groups.
HO increased BAL cell counts but not protein levels in TREK-1 deficient mice
To further characterize the proinflammatory phenotype observed in TREK-1 deficient lungs exposed to HO, we quantified total (Figure 2A) and differential BAL fluid cell counts (Figure 2B) and measured total BAL fluid protein levels (Figure 2C). Exposure of mice to HO resulted in an increase in the total number of cells in the BAL fluid recovered from TREK-1 deficient mice but not from WT mice. Exposure to HO+MV led to a further increase in the total number of cells in the BAL fluid of WT but not TREK-1 deficient mice. Analysis of BAL fluid differential cell counts showed a relative increase in the percentage of neutrophils in TREK-1 deficient but not WT mice exposed to HO. Exposure to HO+MV caused a similar relative increase in the percentage of neutrophils in both mouse types.
Figure 2. HO exposure increased the total cell number but not protein levels in the BAL fluid of TREK-1 deficient mice.
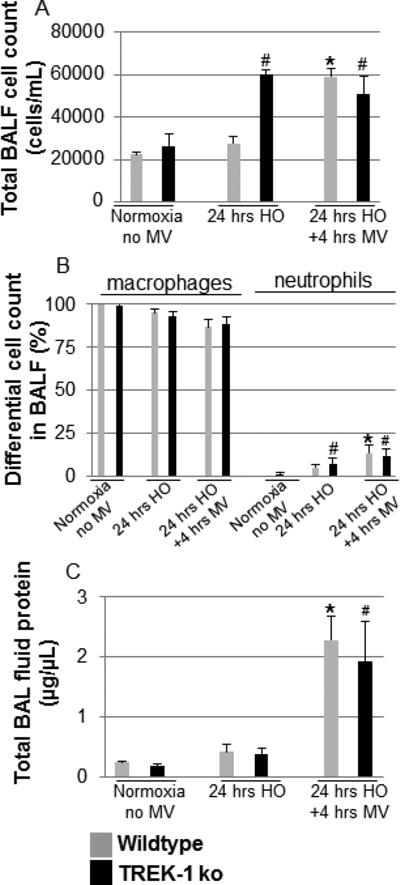
Panel A: Exposure to HO increased the total BAL cell count in TREK-1 deficient mice but not in WT mice. After exposure to HO+MV total BAL cell counts were increased to a similar degree in WT and TREK-1 deficient mice. Panel B shows analysis of differential cell counts from BAL fluid. Panel C: Exposure to HO alone did not affect BAL protein levels in either WT or TREK-1 deficient mice when compared with untreated controls. Exposure to HO+MV increased total BAL protein levels in both WT and TREK-1 deficient mice when compared to untreated or HO exposed mice. *compared to untreated WT control; # compared to untreated TREK-1 ko control; n=4-7 animals per group.
In contrast to cell numbers in the BAL fluid of TREK-1 deficient mice, total protein levels were not significantly increased in either WT or TREK-1 deficient mice after HO treatment. Exposure to HO+MV increased total BAL fluid protein levels to a similar degree in both WT and TREK-1 deficient mice when compared to untreated and HO-treated mice.
HO exposure decreased quasi-static lung compliance in TREK-1 deficient mice
We previously found that exposure of mice to HO+MV resulted in a decrease in quasi-static lung compliance[10], which is a measure of the elastic recoil pressure of the lungs at a given lung volume and correlates with worsening lung injury in both patients and in animal models of lung injury[24, 25]. We now investigated whether the lung damage observed in TREK-1 deficient mice exposed to HO was also reflected by a decrease in quasi-static lung compliance. We found that HO exposure decreased quasi-static lung compliance in TREK-1 deficient mice but not in WT mice (Figure 3). Exposure to HO+MV further decreased quasi-static lung compliance in both WT and TREK-1 deficient mice but no difference in quasi-static lung compliance was observed between the two mouse types under these conditions.
Figure 3. HO exposure decreased quasi-static lung compliance in TREK-1 deficient mice.

HO exposure (24 hrs) decreased quasi-static lung compliance in TREK-1 but not WT mice. Exposure to HO+MV further decreased quasi-static lung compliance in both WT and TREK-1 deficient mice to a similar level. *compared to untreated WT control; #compared to untreated TREK-1 ko control; n=6-10 animals per group.
Exposure of TREK-1 deficient mice to HO had no effect on BAL fluid TNF-α, IL-6 and MCP-1 levels
To determine whether the increased cell counts and decreased quasi-static compliance in TREK-1 deficient mice correlated with alterations in BAL fluid cytokine secretion, we measured TNF-α, IL-6 and MCP-1 levels in WT and TREK-1 deficient mice (Figure 4A). Exposure of both mouse types to HO alone had no effect on BAL fluid cytokine levels when compared to untreated animals. Exposure to HO+MV increased BAL fluid TNF-α, IL-6 and MCP-1 levels in both WT and TREK-1 deficient mice. However, while TNF-α levels increased to a similar degree in WT and TREK-1 deficient mice after exposure to HO+MV, IL-6 and MCP-1 levels increased to a lesser degree in TREK-1 deficient mice than in controls.
Figure 4. TREK-1 deficiency altered BAL fluid cytokine levels after HO+MV exposure but not after HO alone.

HO exposure alone did not affect TNF-α, IL-6 or MCP-1 secretion in either WT or TREK-1 deficient mice (Panel A). Although exposure to HO+MV increased BAL fluid levels of all 3 cytokines, the increase in TNF-α was equal between WT and TREK-1 deficient mice, whereas the increase in IL-6 and MCP-1 was diminished in TREK-1 deficient mice compared to WT mice. *compared to untreated WT control; #compared to untreated TREK-1 ko control; n=4-8 animals per group. Neither HO nor HO+MV affected MIP-2, KC or IL-1β gene expression in whole lung homogenates using PCR (Panel B). Interestingly, TREK-1 deficient mice but not WT mice expressed IL-1β at baseline (representative picture of 4-5 experiments).
Exposure of TREK-1 deficient mice to HO had no effect on whole lung MIP-2, KC and IL-1β gene expression levels
Using conventional RT-PCR, we measured gene expression of the inflammatory cytokines MIP-2, KC and IL-1β in whole lung homogenates of WT and TREK-1 deficient mice (Figure 4B). We detected neither MIP-2 nor KC mRNA at baseline or after 24 hours of HO exposure in WT or TREK-1 deficient mice. Exposure of both WT and TREK-1 deficient mice to 24 hours HO followed by 4 hours of MV induced equal levels of MIP-2 and KC gene expression in both mouse types. In contrast, we detected IL-1β gene expression in untreated TREK-1 deficient animals but not WT mice. However, after exposure of mice to HO or HO+MV no further differences in IL-1β gene expression were observed between WT and TREK-1 deficient mice.
Macrophage activation was intact in TREK-1 deficient mice
To evaluate whether the increase in lung injury observed in TREK-1 deficient mice was related to alterations in macrophage activation in these animals, we isolated alveolar macrophages (AMs) from WT and TREK-1 deficient mice (Figure 5). We first investigated whether AMs from untreated WT mice expressed TREK-1 protein using immunofluorescence staining (Figure 5A). In fact, AMs showed both intracellular and membrane bound TREK-1 protein staining. In addition, since we knew from our in vitro[18] as well as our in vivo studies (Figure 4) that HO is a weak stimulus for cytokine release, we stimulated AMs with LPS and measured TNF-α secretion (Figure 5B). Baseline secretion of TNF-α was low in WT and TREK-1 deficient animals but after LPS stimulation both WT and TREK-1 deficient mice showed and an equal increase in TNF-α release. Based on these results, AM activation appeared intact in TREK-1 deficient mice.
Figure 5. Alveolar macrophages express TREK-1 protein and secrete normal amounts of TNF-α.
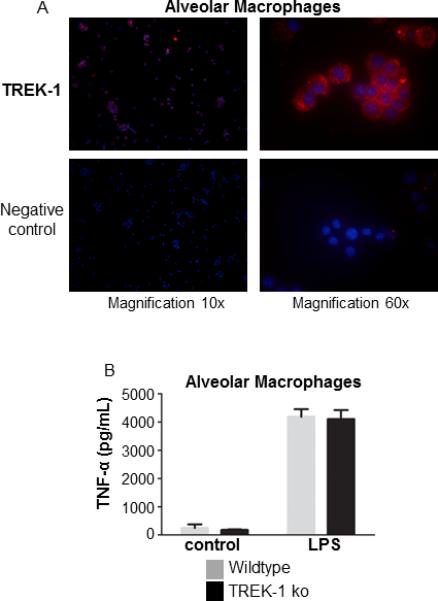
Isolated alveolar macrophages express TREK-1 protein by immunofluorescence staining (Panel A). Alveolar macrophages from WT and TREK-1 deficient mice secrete equal amounts of TNF-α upon LPS stimulation (Panel B); n=5 animals per group.
TREK-1 deficiency promoted apoptosis in HO exposed lungs
We previously reported that exposure of lungs to HO+MV activated pro-apoptotic signaling cascades resulting in increased levels of cleaved poly [ADP-ribose] polymerase-1 (PARP-1)[9]. To determine whether cellular apoptosis was increased in TREK-1 deficient mice, we performed Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) staining of lung sections and measured PARP-1 activation in whole lung homogenates. HO exposure induced cellular apoptosis as indicated by an increase in TUNEL positive cells in TREK-1 deficient but not in WT mice when compared to untreated mice (Figure 6A). Exposure to HO+MV increased cellular apoptosis in both WT and TREK-1 deficient mice, but the effect was more pronounced in TREK-1 deficient mice. Quantification of the number of TUNEL positive cells per field is summarized in Figure 6B. To determine the identity of TUNEL positive cells, we double-stained lung sections of TREK-1 deficient animals exposed to HO and HO+MV using the TUNEL assay and an anti-pro-SPC antibody to label type II pneumocytes (Figures 7A and B). In addition, we also double stained lung sections of TREK-1 deficient animals exposed to HO+MV with TUNEL assay and an Aquaporin-5 antibody, a marker of alveolar type I pneumocytes (Figure 7C). The inserts of the confocal images, cell size and location demonstrate both apoptotic type II cells (TUNEL and pro-SPC positive) and type I cells (TUNEL and Aquaporin-5 positive). However, we cannot exclude the possibility that some of the TUNEL positive but pro-SPC or Aquaporin-5 negative cells were endothelial cells or fibroblasts.
Figure 6. HO increased cellular apoptosis in TREK-1 deficient lungs.

HO exposure increased the number of apoptotic cells as shown by TUNEL staining in lungs of TREK-1 ko mice but not in WT mice. Exposure to HO+MV resulted in a further increase in apoptotic cells in both WT and TREK-1 ko mice, but the effect was more pronounced in TREK-1 deficient mice. Panel A: green fluorescent, TUNEL positive cells are indicated by white arrows. Panel B: Quantification of TUNEL positive cells. *compared to untreated WT control, #compared to untreated TREK-1 ko control, ^comparison between WT and TREK ko mice within the same treatment group.
Figure 7. Both type I and type II AECs undergo apoptosis.
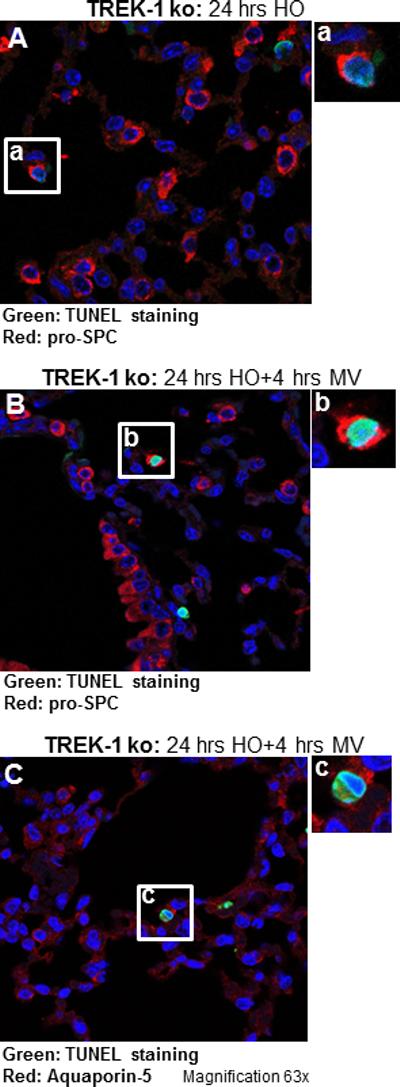
Double staining of lung sections from TREK-1 deficient mice exposed to HO (Panel A) and HO+MV (Panel B) using the TUNEL assay and an anti-pro-SPC antibody, or the TUNEL assay and an anti-Aquaporin-5 antibody (Panel C). Inserts (a) and (b) show TUNEL and pro-SPC double positive cells representing type II pneumocytes, whereas insert c shows a TUNEL and Aquaporin-5 double positive cell representing a type I pneumocyte. Magnification 63×. Images are representative of 4 lung fields from 3 different mice per group.
Similarly to TUNEL staining, HO exposure induced PARP-1 cleavage in TREK-1 deficient but not in WT mice when compared to untreated mice (Figure 8A). Exposure to HO+MV caused a further increase in PARP-1 activation in both WT and TREK-1 deficient mice. Quantification of cleaved to total PARP-1 ratios by band densitometry is summarized in Figure 8B.
Figure 8. HO exposure induced PARP-1 activation in TREK-1 deficient mice.
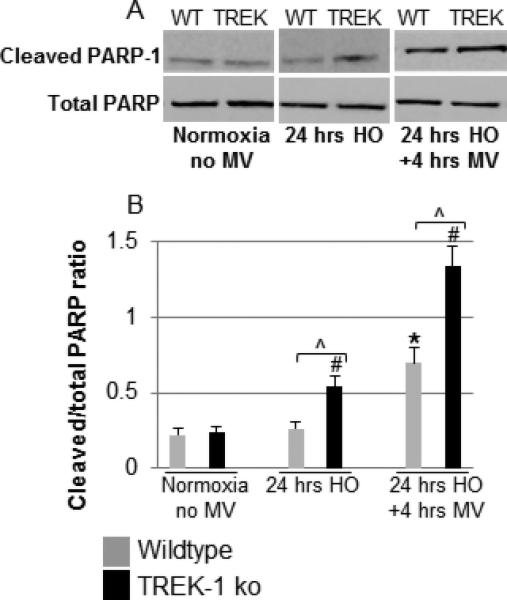
Panel A shows a representative Western blot showing an increase in cleaved PARP-1 levels in HO exposed lungs of TREK-1 ko but not WT when compared to untreated mice. Exposure to HO+MV caused a further increase in cleaved PARP-1 levels in both WT and TREK-1 ko mice, but the effect was more pronounced in TREK-1 ko mice. Panel B shows band densitometry analysis of cleaved to total PARP-1 ratios. *compared to untreated WT control; #compared to untreated TREK-1 ko control; ^comparison between WT and TREK ko mice within the same treatment group; n=4-7 animals per group.
DISCUSSION
We recently reported the expression of K2P channels in alveolar epithelial cells (AECs) and found that TNF-α stimulation of TREK-1 deficient AECs caused decreased IL-6 secretion but increased MCP-1 secretion in vitro[17, 18]. Therefore, we were interested in determining whether in vivo TREK-1 deficiency would ultimately promote a pro- or anti-inflammatory phenotype. In this study we show for the first time in an in vivo model of ARDS that TREK-1 may play an important role in the development of hyperoxia (HO)-induced lung injury. We found that exposure of TREK-1 deficient mice to HO resulted in increased lung injury at morphological and histological levels, which translated into a decrease in quasi-static lung compliance and activation of proapoptotic signaling pathways in TREK-1 deficient animals. Interestingly, the combination of HO and mechanical ventilation (MV) caused further injury in WT animals but not in TREK-1 deficient mice, and the differences in Lung Injury Score (LIS), BAL fluid cell count and lung compliance observed between control and TREK-1 deficient mice exposed to HO were no longer visible once severe injury was induced by mechanical ventilation. Therefore, it appears that TREK-1 may play a more prominent role in HO-than in stretch-induced lung injury, but the role of TREK-1 in isolated stretch-induced injury clearly needs further evaluation. However, a clinical scenario where a patient with ARDS would be ventilated with room air is unlikely, since hypoxia is one of the defining criteria of ARDS[26]. Therefore, for the purpose of this study, we did not pursue the isolated effects of mechanical ventilation further. We chose the 24 hour time point of HO exposure based on published literature reporting an oxidative stress-induced inflammatory response similar to ours in a lung injury model[13].
Our studies are the first to link TREK-1 to the development of lung injury, but the main question remaining is how TREK-1 deficiency promotes HO-induced injury in vivo. The increased number of inflammatory cells found in the BAL fluid of HO-exposed TREK-1 deficient mice without an increase in BAL fluid protein suggests increased inflammatory cell infiltration without a significant loss of alveolar barrier function (Figure 2). An increased number of inflammatory cells was also observed in the lung interstitium of HO-exposed TREK-1 deficient mice by histology (Figure 1). The majority of cells entering the BAL fluid were neutrophils but we also saw increased numbers of macrophages in BAL fluid of TREK-1 deficient mice after HO exposure. In addition, we observed small numbers of lymphocytes and occasional epithelial cells in the BAL fluid from the TREK-1 deficient mice (data not shown). It is likely that these cells, as well as monocytes and dendritic cells, contribute to the total BAL cell number in TREK-1 deficient mice compared to WT mice.
Interestingly, in contrast to inflammatory cell infiltration, BAL fluid levels of TNF-α, IL-6 and MCP-1, as well as gene expression of MIP-2, KC and IL-1β, were not altered in HO-exposed TREK-1 deficient mice (Figures 4A and B). Several reasons may explain why we observed an increase in neutrophils, and to a lesser degree of macrophages and lymphocytes, without changes in the cytokines tested. It is certainly possible that the recruitment of neutrophils and other inflammatory cells may be stimulated by BAL fluid or interstitial cytokines not measured in this study (such as GRO-α, G-CSF, C5a, LTB4, NAP-2, CINC-2α, IL-17 for neutrophils, and RANTES, MIP-1a/b, MIP-3 for macrophages)[27-30]. In addition, changes in cytokine levels may have occurred prior to the lung damage observed after 24 hours of HO exposure. Furthermore, whole lung homogenates and BAL fluid may severely underestimate the local production and secretion of cytokines and the inflammatory changes occurring in the micro-milieu of the alveoli and the lung interstitium. We addressed some of these questions by isolating alveolar macrophages and studying their activation profile (Figure 5). Importantly, we also showed for the first time expression of TREK-1 in macrophages. The weak cytokine response to HO exposure is similar to our in vitro findings in alveolar epithelial cells, where HO alone did not induce significant IL-6, MCP-1 or RANTES release[18]. Consistent with our previous in vitro findings in Trek-1 deficient AECs[17], injury caused by HO+MV resulted in decreased IL-6 levels in the BAL fluid of Trek-1 deficient mice. However, MCP-1 levels were also decreased in the BAL fluid of Trek-1 deficient mice, whereas in vitro MCP-1 secretion was increased from Trek-1 deficient AECs[31]. Altogether, these data suggest that 1) TREK-1 deficiency may have a different effect on cytokine secretion from cultured AECs than in whole lung tissue, and 2) TREK-1 may play a role in the regulation of inflammatory cell infiltration upon HO exposure without significant changes in alveolar barrier function and BAL cytokine levels, whereas after exposure to HO+MV TREK-1 deficiency regulates BAL fluid IL-6 and MCP-1 but not TNF-α concentrations. Similar to our previous study using cultured alveolar epithelial cells[18] we found no effect of TREK-1 deficiency on KC production despite observing increased neutrophil influx after HO treatment Again, it is possible that KC gene expression peaked much earlier than the 24 hour time point, and thus KC could still be important for neutrophil recruitment. In the future, a more detailed time course of KC expression could aid in determining the role of this chemokine in the observed lung injury in TREK-1 deficient mice.
Interestingly, the decrease in BAL fluid IL-6 and MCP-1 levels was associated with increased cellular apoptosis in TREK-1 deficient mice exposed to HO+MV. Previous studies have suggested that IL-6 is a multifunctional cytokine with time-, dose-and organ-specific functions[32]. For example, IL-6 is known to decrease T cell apoptosis and promote epithelial survival in the GI system[33], and to ameliorate lung injury by re-establishing surfactant levels[34]. Similarly, MCP-1 is known to play a protective role in bleomycin-induced lung injury[35]. It is possible that the increased levels of cellular apoptosis observed after HO+MV are related to the decreased IL-6 and MCP-1 levels in TREK-1 deficient mice but certainly, as mentioned above, several other mediators not tested in this study could also be involved. Ideally, direct comparisons between BAL fluid and interstitial cytokine levels would aid in answering these difficult questions, but such experiments are technically challenging. To start addressing these questions, we studied gene expression of MIP-2, KC and IL-1β in whole lung tissue and found no significant differences between WT and TREK-1 deficient mice after HO or HO+MV exposure. Interestingly, we found IL-1β expression in untreated TREK-1 deficient mice but not in WT mice, potentially suggesting a baseline activation of the inflammasome in these mice. Although we cannot exclude some type of systemic immunodeficiency in TREK-1 deficient mice resulting in decreased IL-6 and MCP-1 levels and increased sensitivity to HO, LPS–induced activation of alveolar macrophages appeared intact in TREK-1 deficient animals. Whether this also holds true for interstitial macrophages is currently under investigation. In addition to macrophages, T cell mediated responses could be altered by systemic TREK-1 deficiency. Furthermore, while we are the first group to show TREK-1 expression in macrophages, the presence of TREK-1-related channels (TREK-2 and TASK-2) on lymphocytes has been reported previously [36, 37]. Furthermore, isolation of type II pneumocytes from TREK-1 deficient mice will allow us in future experiments to determine whether alterations in epithelial cell activation are contributing to the increased sensitivity to HO in these mice. Another important point to address in future studies is whether the effects of TREK-1 deficiency are related to alterations in cellular K+ handling, or whether TREK-1 can exert functions that are unrelated to its role as a K+ channel as for example described for CFTR[38, 39]. In a recently published study[40] we showed that TREK-1 regulates the mechanobiological properties of AECs by altering F-actin metabolism and overall cellular stiffness. We are also in the early stages of developing a conditional TREK-1 knockout model to study the effects of this channel specifically in the alveolar epithelium, and to integrate our in vivo data with the results obtained with cultured AECs[17, 18].
In conclusion, although we have previously suggested a potential role for TREK-1 in the development of alveolar epithelial injury in vitro, this is the first report showing that TREK-1 deficiency accentuates HO-induced lung injury in vivo. This lung injury is evidenced by decreased compliance, increased pulmonary inflammatory infiltrates including neutrophils, macrophages, and lymphocytes, and an increase in apoptotic cells. It appeared that with the combination of 24 hours HO + 4 hours injurious MV maximal lung injury was achieved and differences between WT and TREK-1 deficient mice were less readily apparent. Clinically, this suggests that in vivo TREK-1 may play an important role in preventing or modulating moderate HO-induced lung injury but is insufficient to protect against severe lung injury.
ACKNOWLEDGMENTS
We would like to acknowledge Sherry C. Phillips (Baylor College of Medicine) for facilitating our work with the TREK-1 knockout mice. These mice were generated by the Texas Genetic Institute as previous described[21]. We would also like to thank Dr. Tamekia Jones and Dr. Sunny Anand for their help with the statistical analysis of the data.
GRANTS: This study was supported by a grant from the Le Bonheur Children's Medical Center Research Foundation of the University of Tennessee Health Science Center, and the National Institutes of Health Grants HL094366 (CMW), R01AI090059, R01ES015050, and P42ES013648 (SAC), 5K12HD047349 (AS), and HL098921and NS046666 (RMB).
Dr. Schwingshackl received support for article research from the National Institutes of Health (NIH) and the ALA. His institution received grant support and support for travel from the NIH and the ALA. Dr. Balasz is employed by the department of pathology. Dr. Bryan received support for article research from NIH (RO1 NS46666 and R21 HL098921). Dr. Lloyd received support for article research from the NIH. His institution received grant support. Dr. Waters received support for article research from the NIH.
Footnotes
Copyright form disclosures:
The remaining authors have disclosed that they do not have any potential conflicts of interest.
REFERENCES
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. The New England journal of medicine. 2005;353(16):1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 2.Zimmerman JJ, Akhtar SR, Caldwell E, Rubenfeld GD. Incidence and outcomes of pediatric acute lung injury. Pediatrics. 2009;124(1):87–95. doi: 10.1542/peds.2007-2462. [DOI] [PubMed] [Google Scholar]
- 3.Kallet RH, Jasmer RM, Pittet JF, Tang JF, Campbell AR, Dicker R, Hemphill C, Luce JM. Clinical implementation of the ARDS network protocol is associated with reduced hospital mortality compared with historical controls. Crit Care Med. 2005;33(5):925–929. doi: 10.1097/01.ccm.0000162382.59289.9c. [DOI] [PubMed] [Google Scholar]
- 4.Frank JA, Matthay MA. Science review: mechanisms of ventilator-induced injury. Crit Care. 2003;7(3):233–241. doi: 10.1186/cc1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frohlich S, Boylan J, McLoughlin P. Hypoxia-induced inflammation in the lung: a potential therapeutic target in acute lung injury? Am J Respir Cell Mol Biol. 2013;48(3):271–279. doi: 10.1165/rcmb.2012-0137TR. [DOI] [PubMed] [Google Scholar]
- 6.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L379–399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altemeier WA, Sinclair SE. Hyperoxia in the intensive care unit: why more is not always better. Curr Opin Crit Care. 2007;13(1):73–78. doi: 10.1097/MCC.0b013e32801162cb. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Lopez A, Garcia-Prieto E, Batalla-Solis E, Amado-Rodriguez L, Avello N, Blanch L, Albaiceta GM. Lung strain and biological response in mechanically ventilated patients. Intensive Care Med. 2012;38(2):240–247. doi: 10.1007/s00134-011-2403-1. [DOI] [PubMed] [Google Scholar]
- 9.Makena PS, Gorantla VK, Ghosh MC, Bezawada L, Balazs L, Luellen CL, Parthasarathi K, Waters CM, Sinclair SE. Lung injury caused by high tidal volume mechanical ventilation and hyperoxia is dependent on oxidant-mediated c-Jun NH2-terminal kinase (JNK) activation. J Appl Physiol. 2011 doi: 10.1152/japplphysiol.00539.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makena PS, Luellen CL, Balazs L, Ghosh MC, Parthasarathi K, Waters CM, Sinclair SE. Pre-Exposure to Hyperoxia Causes Increased Lung Injury and Epithelial Apoptosis in Mice Ventilated with High Tidal Volumes. Am J Physiol Lung Cell Mol Physiol. 2010 doi: 10.1152/ajplung.00072.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF. Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol. 1998;19(4):573–581. doi: 10.1165/ajrcmb.19.4.3173. [DOI] [PubMed] [Google Scholar]
- 12.Dos Santos CC. Hyperoxic acute lung injury and ventilator-induced/associated lung injury: new insights into intracellular signaling pathways. Crit Care. 2007;11(2):126. doi: 10.1186/cc5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagato AC, Bezerra FS, Lanzetti M, Lopes AA, Silva MA, Porto LC, Valenca SS. Time course of inflammation, oxidative stress and tissue damage induced by hyperoxia in mouse lungs. International journal of experimental pathology. 2012;93(4):269–278. doi: 10.1111/j.1365-2613.2012.00823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayliss DA, Barrett PQ. Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci. 2008;29(11):566–575. doi: 10.1016/j.tips.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiological reviews. 2010;90(2):559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 16.Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000;279(5):F793–801. doi: 10.1152/ajprenal.2000.279.5.F793. [DOI] [PubMed] [Google Scholar]
- 17.Schwingshackl A, Teng B, Ghosh MC, Lim KG, Tigyi GJ, Narayanan D, Jaggar JH, Waters CM. Regulation of Interleukin-6 Secretion by the Two-Pore-Domain Potassium (K2P) Channel Trek-1 in Alveolar Epithelial Cells. Am J Physiol Lung Cell Mol Physiol. 2012 doi: 10.1152/ajplung.00299.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwingshackl A, Teng B, Ghosh M, West AN, Makena P, Gorantla V, Sinclair SE, Waters CM. Regulation and function of the two-pore-domain (K2P) potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L93–L102. doi: 10.1152/ajplung.00078.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao KQ, Xiong G, Wilber M, Cohen NA, Kreindler JL. A role for two-pore K(+) channels in modulating Na(+) absorption and Cl(−) secretion in normal human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L4–L12. doi: 10.1152/ajplung.00102.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis KA, Cowley EA. Two-pore-domain potassium channels support anion secretion from human airway Calu-3 epithelial cells. Pflugers Arch. 2006;451(5):631–641. doi: 10.1007/s00424-005-1505-4. [DOI] [PubMed] [Google Scholar]
- 21.Namiranian K, Brink CD, Goodman JC, Robertson CS, Bryan RM., Jr. Traumatic brain injury in mice lacking the K channel, TREK-1. J Cereb Blood Flow Metab. 2011;31(3):e1–6. doi: 10.1038/jcbfm.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salazar E, Knowles JH. An alaysis of pressure-volume characteristics of the lungs. J Appl Physiol. 1964;19:97–104. doi: 10.1152/jappl.1964.19.1.97. [DOI] [PubMed] [Google Scholar]
- 23.Fitzpatrick AM, Teague WG, Burwell L, Brown MS, Brown LA. Glutathione oxidation is associated with airway macrophage functional impairment in children with severe asthma. Pediatr Res. 2011;69(2):154–159. doi: 10.1203/PDR.0b013e3182026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Z, Guttmann J, Moller K. Assessment of a volume-dependent dynamic respiratory system compliance in ALI/ARDS by pooling breathing cycles. Physiological measurement. 2012;33(8):N61–67. doi: 10.1088/0967-3334/33/8/N61. [DOI] [PubMed] [Google Scholar]
- 25.Vanoirbeek JA, Rinaldi M, De Vooght V, Haenen S, Bobic S, Gayan-Ramirez G, Hoet PH, Verbeken E, Decramer M, Nemery B, et al. Noninvasive and invasive pulmonary function in mouse models of obstructive and restrictive respiratory diseases. Am J Respir Cell Mol Biol. 2010;42(1):96–104. doi: 10.1165/rcmb.2008-0487OC. [DOI] [PubMed] [Google Scholar]
- 26.Thompson BT, Moss M. A new definition for the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2013;34(4):441–447. doi: 10.1055/s-0033-1351162. [DOI] [PubMed] [Google Scholar]
- 27.Michael BD, Elsone L, Griffiths MJ, Faragher B, Borrow R, Solomon T, Jacob A. Post-acute serum eosinophil and neutrophil-associated cytokine/chemokine profile can distinguish between patients with neuromyelitis optica and multiple sclerosis; and identifies potential pathophysiological mechanisms - a pilot study. Cytokine. 2013;64(1):90–96. doi: 10.1016/j.cyto.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 28.Kaur M, Singh D. Neutrophil chemotaxis caused by chronic obstructive pulmonary disease alveolar macrophages: the role of CXCL8 and the receptors CXCR1/CXCR2. J Pharmacol Exp Ther. 2013;347(1):173–180. doi: 10.1124/jpet.112.201855. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Filgueiras LR, Wang S, Serezani AP, Peters-Golden M, Jancar S, Serezani CH. Leukotriene B4 Enhances the Generation of Proinflammatory MicroRNAs To Promote MyD88-Dependent Macrophage Activation. J Immunol. 2014 doi: 10.4049/jimmunol.1302982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trujillo G, Habiel DM, Ge L, Ramadass M, Cooke NE, Kew RR. Neutrophil recruitment to the lung in both C5a- and CXCL1-induced alveolitis is impaired in vitamin D-binding protein-deficient mice. J Immunol. 2013;191(2):848–856. doi: 10.4049/jimmunol.1202941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwingshackl A, Teng B, Ghosh M, Waters CM. Regulation of Monocyte Chemotactic Protein-1 secretion by the Two-Pore-Domain Potassium (K2P) channel TREK-1 in human alveolar epithelial cells. American journal of translational research. 2013;5(5):530–542. [PMC free article] [PubMed] [Google Scholar]
- 32.Zarogoulidis P, Yarmus L, Darwiche K, Walter R, Huang H, Li Z, Zaric B, Tsakiridis K, Zarogoulidis K. Interleukin-6 cytokine: a multifunctional glycoprotein for cancer. Immunome research. 2013;9(62):16535. doi: 10.1186/2090-5009-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waldner MJ, Neurath MF. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Seminars in immunology. 2014 doi: 10.1016/j.smim.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Thacker S, Moran A, Lionakis M, Mastrangelo MA, Halder T, Huby MD, Wu Y, Tweardy DJ. Restoration of lung surfactant protein D by IL-6 protects against secondary pneumonia following hemorrhagic shock. The Journal of infection. 2013 doi: 10.1016/j.jinf.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang J, Jung Y, Tighe RM, Xie T, Liu N, Leonard M, Gunn MD, Jiang D, Noble PW. A macrophage subpopulation recruited by CC chemokine ligand-2 clears apoptotic cells in noninfectious lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L933–940. doi: 10.1152/ajplung.00256.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng H, Nam JH, Pang B, Shin DH, Kim JS, Chun YS, Park JW, Bang H, Kim WK, Earm YE, et al. Identification of the large-conductance background K+ channel in mouse B cells as TREK-2. Am J Physiol Cell Physiol. 2009;297(1):C188–197. doi: 10.1152/ajpcell.00052.2009. [DOI] [PubMed] [Google Scholar]
- 37.Nam JH, Shin DH, Zheng H, Lee DS, Park SJ, Park KS, Kim SJ. Expression of TASK-2 and its upregulation by B cell receptor stimulation in WEHI-231 mouse immature B cells. Am J Physiol Cell Physiol. 2011;300(5):C1013–1022. doi: 10.1152/ajpcell.00475.2010. [DOI] [PubMed] [Google Scholar]
- 38.Suaud L, Yan W, Carattino MD, Robay A, Kleyman TR, Rubenstein RC. Regulatory interactions of N1303K-CFTR and ENaC in Xenopus oocytes: evidence that chloride transport is not necessary for inhibition of ENaC. Am J Physiol Cell Physiol. 2007;292(4):C1553–1561. doi: 10.1152/ajpcell.00064.2006. [DOI] [PubMed] [Google Scholar]
- 39.Marino GI, Kotsias BA. Cystic fibrosis transmembrane regulator (CFTR) in human trophoblast BeWo cells and its relation to cell migration. Placenta. 2013 doi: 10.1016/j.placenta.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Roan E, Waters CM, Teng B, Ghosh M, Schwingshackl A. The 2-pore domain potassium channel TREK-1 regulates stretch-induced detachment of alveolar epithelial cells. PLoS One. 2014;9(2):e89429. doi: 10.1371/journal.pone.0089429. [DOI] [PMC free article] [PubMed] [Google Scholar]