

Abstract
The Hedgehog (Hh) signaling pathway regulates normal development and cell proliferation in metazoan organisms, but its aberrant activation can promote tumorigenesis. Hh-induced tumors arise from various tissues and they may be indolent or aggressive, as is the case with skin basal cell carcinoma (BCC) or cerebellar medulloblastoma (MB), respectively. Little is known about common cell-intrinsic factors that control the development of such diverse Hh-dependent tumors. Transcription factor Zfx is required for the self-renewal of hematopoietic and embryonic stem cells, as well as for the propagation of acute myeloid and T-lymphoblastic leukemias. We report here that Zfx facilitates the development of experimental BCC and MB in mice initiated by deletion of the Hh inhibitory receptor Ptch1. Simultaneous deletion of Zfx along with Ptch1 prevented BCC formation and delayed MB development. In contrast, Zfx was dispensable for tumorigenesis in a mouse model of glioblastoma. We used genome-wide expression and chromatin binding analysis in a human MB cell line to characterize direct, evolutionarily conserved targets of Zfx, identifying Dis3L and Ube2j1 as two targets required for the growth of the human MB cells. Our results establish Zfx as a common cell-intrinsic regulator of diverse Hh-induced tumors, with implications for the definition of new therapeutic targets in these malignancies.
Keywords: Zfx, Hedgehog, basal cell carcinoma, medulloblastoma, Dis3L, Ube2j1
INTRODUCTION
The Hedgehog (Hh) pathway controls cell proliferation and differentiation in response to a gradient of secreted Hh ligands (1–4). Hh proteins such as Sonic Hedgehog (Shh) bind the inhibitory receptor Patched1 (Ptch1) on the cell surface, thereby releasing Ptch1-mediated inhibition of seven-pass transmembrane protein Smoothened (Smo). Smo is an essential, non-redundant transducer of Hh signals whose activation leads to activation of GLI-type transcription factors, which in turn induce the expression of Hh target genes. These target genes include downstream transcription factors such as Mycn (5), along with core Hh pathway components including Ptch1 and Gli1. The Hh pathway controls multiple processes in normal development, including specification of motor neuron (MN) and interneuron pools in the ventral spinal cord (6, 7), expansion of granule neuron precursors (GNP) and foliation in the cerebellum (8–10), and hair follicle morphogenesis (11, 12). On the other hand, aberrant activation of the Hh pathway may induce malignant transformation of these same Hh-dependent cell types (13).
In particular, inactivating mutations in PTCH1 are present in ~90% of human sporadic basal cell carcinoma (BCC), a predominantly indolent tumor of the adult epidermis (14). Furthermore, Hh pathway activation has been associated with a distinct GNP-derived subtype of medulloblastoma (MB), a highly aggressive childhood cerebellar tumor (15–17). Both MB and BCC can be induced in mice by Hh pathway overactivation due to Ptch1 mutation (18, 19). The resulting tumors arise from the stem/progenitor compartments of the corresponding tissues, including GNP or earlier neuronal progenitors (20, 21) and hair follicle stem cells (22). Chemical inhibitors of the Hh pathway component Smo show promising results for treatment of Hh-dependent cancers such as BCC and MB (23, 24). However, the acquisition of Smo mutations by MB to escape targeted molecular therapy has been demonstrated (25). Furthermore, targeting of the core Hh pathway in pediatric MB should be done with caution, to avoid adverse effects on Hh-dependent normal cerebellar development and skeletal growth (13, 23, 26). Identification of novel cell-intrinsic regulators of Hh pathway-initiated cancers could suggest novel therapeutic targets in contexts where Hh inhibitors induce resistance or cause serious developmental side effects.
Zfx is a transcription factor that is highly conserved in vertebrates and contains a large acidic transcriptional activation domain and a C-terminal zinc finger domain (27). Zfx is encoded on the mammalian X chromosome and is expressed ubiquitously, yet acts in a cell- and tissue-specific fashion. Zfx is required for the self-renewal of pluripotent embryonic stem cells (ESC) and of adult hematopoietic stem cells (HSC) (28, 29), as well as for B lymphocyte development (30). On the other hand, Zfx is dispensable for ESC differentiation and for the growth of multiple cell types such as myeloid progenitors and embryonic fibroblasts. The role of Zfx in cancer development in vivo remains incompletely understood. A recent study employing a transposon-based screen in a murine MYC-dependent liver cancer model reported Zfx as a potential tumor suppressor gene (31). In contrast, several studies have reported that Zfx knockdown in cancer cell lines in vitro impaired their growth (32–34). Our laboratory recently reported that Zfx is necessary for the initiation and maintenance of two disparate leukemias in vivo, acute myeloid leukemia (AML) and T-cell acute lymphoblastic leukemia (T-ALL) (35).
Our analysis of Zfx target genes in stem cells revealed that Zfx directly activates the expression of Smo (J. M. G.-C., unpublished observations). Together with the ubiquitous expression of Zfx and its cell-intrinsic requirement in multiple cancer types, these data raised the possibility that Zfx might facilitate Hh-driven malignancy. We therefore explored the role of Zfx in BCC and MB, two tumors caused by Hh pathway activation in two different tissues. We now report that Zfx is required for the formation of BCC and optimal progression of MB in vivo. The analysis of Zfx genomic targets revealed a conserved set of genes, some of which were required for growth of human tumor cell lines in vitro. Collectively, these results identify Zfx as a common cell-intrinsic regulator of distinct Hh-induced tumors such as BCC and MB.
MATERIALS AND METHODS
Supplementary Methods and associated references may be found in the Supplementary Data provided online.
Animals
Zfxflox (28), Ptenflox (36), and Ptch1flox (37) conditional mice have been previously described. The hGFAP-Cre brain-wide deleter strain (38) and Rosa26-SmoM2 mice bearing a Cre-inducible, constitutively active allele of Hh signal transducer Smoothened (39) were obtained from the Jackson Laboratory. Tamoxifen (Tmx)-inducible Rosa26-CreER mice were generously provided by Dr. Thomas Ludwig, Columbia University.
Ptch1flox and Zfxflox mice were crossed to generate doubly-conditional Ptch1flox Zfxflox mice. To model Hh pathway-dependent BCC formation in the skin, Ptch1flox and Ptch1flox Zfxflox mice were bred with Rosa26-CreER mice. To effect Tmx-induced deletion of Ptch1 with or without concomitant Zfx deletion in the skin, 7–9 week-old mice were shaved across their lower dorsal skin (from above the tail to the posterior border of the ribcage) and treated topically for five consecutive days with 1 mg tamoxifen (Sigma) in 100 μL acetone. Rosa26-SmoM2 and Rosa26-SmoM2 Zfxflox mice were analogously generated, crossed with Rosa26-CreER deleter mice, and treated topically with Tmx to induce BCC.
Induction of Hh pathway-dependent MB in mice via conditional deletion of Ptch using hGFAP-Cre has been previously described (21). Ptch1flox and Ptch1flox Zfxflox mice were bred to hGFAP-Cre+ Ptch1flox/+ mice to generate hGFAP-Cre+ Ptch1flox/flox and hGFAP-Cre+ Ptch1flox/flox Zfxflox/Y mice that delete the inhibitory Hh receptor Ptch1 with or without concomitant deletion of Zfx. Singly- and doubly-conditional pups, along with Cre− or Ptch1 heterozygote controls, were followed for survival or were euthanized at post-natal timepoints for analysis by immunohistochemistry.
Glioblastoma models were induced by stereotactic injection into brain of young Ptenflox and Ptenflox Zfxflox adult mice of retrovirus coding for PDGF-IRES-Cre, as previously described (40). All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Columbia University.
Cell Culture
The DAOY human MB cell line was obtained from the ATCC and cultured in accordance with ATCC recommended culture conditions.
Lentiviral shRNA Knockdown
Lentiviral constructs containing Zfx-specific (H2, H3, H4) and non-specific, scrambled (SCR) short hairpin RNAs (shRNAs) in the pLKO.1 backbone were purchased from Open Biosystems. For each lentiviral construct, triplicate cultures of DAOY cells were transduced with ultracentrifuge-concentrated lentiviral supernatant for 16–24 hrs. Puromycin-selected cells were collected for gene expression analysis and Western Blot 4 or 6 days after transduction.
Lentiviral particles carrying shRNA targeting DIS3L and UBE2J1 mRNA or a control shRNA targeting non-mammalian sequence (NonM) were obtained from the MISSION shRNA library (Sigma Aldrich). For each lentiviral construct (3–4 per gene), triplicate cultures of DAOY cells were transduced for 16–24 hours. Puromycin-selected cells were collected for gene expression analysis 5 days after transduction or were followed subsequently for growth curves.
Expression analysis, microarray, and ChIP-seq
Quantitative PCR was performed as described (28). RT-PCR results were calculated by ΔΔCT method and normalized for a housekeeping gene (Actb or Gapdh).
RNA for microarray analysis of DAOY human MB cells after ZFX knockdown was obtained 4 days after lentiviral transduction from one culture replicate for each of the ZFX-targeting shRNA constructs (H2, H3, H4) and non-targeting “scrambled” control (SCR). Labeled cDNA was analyzed on Affymetrix Mouse Gene 1.0 ST arrays. Pattern-matching genes following expression profile of Zfx were identified using NIA Array (41).
Sonication-sheared chromatin from DAOY human MB cells was generated and isolated using Covaris truChIP High Cell Chromatin Shearing Kit with SDS Buffer and S2 Sonicator. ZFX ChIP was performed using rabbit polyclonal antibody (28) and Protein A Dynabeads (invitrogen), and unprobed sheared chromatin was used as a control (Input). Library construction and sequencing were performed by the Yale Center for Genome Analysis (New Haven, CT). For comparison with murine ESCs, previously reported Zfx ChIP-seq data were downloaded from the NCBI Sequence Read Archive (SRX000552) (42). Sequencing reads were aligned to the human genome (UCSC hg19) using the short read aligner, Bowtie (version 2.0.0) (43). Significant ZFX binding enrichment peaks were picked using MACS (1.4.0) (44) and were visualized using DNAnexus (Mt. View, CA). Peak distance to the nearest TSS was determined using PeakAnnotator (version 1.4) (45). Unbiased motif analysis was performed on MACS-selected ChIP-seq peaks using the ChIPseeqer FIRE module of ChIPseeqer (2.0) (46).
Immunohistochemistry
Paraffin sections (5 μm) from Tmx-treated skin were stained with hematoxylin and eosin (H&E), or were immunostained with anti-Zfx (1:500) or with anti-Sox9 (Santa Cruz, 1:500) antibodies, incubated with biotinylated anti-rabbit secondary antibody (Vector Labs), visualized ABC and DAB Peroxidase Substrate Kits (Vector Labs), and counterstained with hematoxylin.
Sagittal paraffin sections (5 μm) from cerebella of P7 and P14 pups were cut at 4 levels equally spaced across half the cerebellum and subsequently stained with H&E, or immunostained with anti-Zfx antibody as described above.
Micrographs of histological sections were recorded using an AxioImager.M2 microscope with attached AxioCam MRc camera (Zeiss). Measurements of cross-section area in cerebellar paraffin sections were made using Axiovision 4.8.2.
Statistics
Hematoxylin/eosin-stained sections of BCC were scored by a pathologist who was blinded to the identity of the samples. Specimens were scored for the percentage of skin area containing BCC lesions (−, 0%; +, 1–33%; ++, 34–66%; +++, 67–100%), and the scores were analyzed using Fisher’s exact test extended to a 2×4 contingency table. Cerebellar cross-sectional areas were compared with unpaired, two-tailed Student’s t-tests. Survival curves were analyzed with the LogRank test.
RESULTS
The loss of Zfx prevents Hh-induced BCC formation
We first asked whether Zfx was required for BCC formation after Hh pathway hyperactivation in vivo. To model Hh-induced BCC, mice with a conditional null allele of Ptch1 (Ptch1flox) with or without conditional null allele of Zfx (Zfxflox) were crossed with the Rosa26-CreERT2 (R26-CreER) strain expressing tamoxifen (Tmx)-inducible Cre recombinase. Male mice carrying the Y chromosome and Zfxflox allele on their single X chromosome (Zfxflox/y) were used to target Zfx. Topical application of Tmx enables deletion of Ptch1 alone in R26-CreER+ Ptch1flox/flox (Ptch1) mice or concomitant deletion of Ptch1 and Zfx in R26-CreER+ Ptch1flox/flox Zfxflox/y (Ptch1-Zfx) mice.
To test the consequences of Zfx loss on Hh-induced BCC formation, Tmx was applied to the lower dorsal skin of Ptch1, Ptch1-Zfx or control CreER–negative (Ctrl) mice for five consecutive days. Successful Cre-mediated deletion and the loss of Zfx protein from the basal layer of epidermis and hair follicle bulge of Ptch1-Zfx skin was confirmed by immunostaining of skin sections isolated 3 days after Tmx treatment (Fig. 1). Within 8–9 weeks after treatment, Ptch1 mice lost hair and developed gross induration in the treated skin patch, whereas Ptch1-Zfx mice retained hair and intact skin (Fig. 2A). Histological analysis of Ptch1 skin at 8 wk post-Tmx revealed frank BCC with massive skin infiltration and multiple dermal cysts in lieu of the intact hair follicles (Fig. 2B). These BCC lesions were positive for Zfx (Fig. 2B) and for Sox9 (Fig. S1), a marker of the hair follicle stem cell compartment that is also a marker of BCC (47). In contrast, Ptch1-Zfx skin showed occasional aberrant hair follicles but no overt BCC. Importantly, the occasional aberrant follicles in Ptch1-Zfx skin were comprised of Zfx-expressing cells (Fig. 2B), suggesting that they arose exclusively from cells that escaped Zfx deletion. The analysis of dorsal skin sections confirmed that the extent of BCC outgrowth was significantly reduced in Ptch1-Zfx mice, with most Ptch1-Zfx sections (11/13) receiving the lowest score above the wild-type baseline (Fig. 2C). Thus, Zfx is required for BCC formation caused by Ptch1 loss in the epidermis.
Figure 1. Topical tamoxifen treatment successfully ablates Zfx in the skin.

Ptch1flox/flox Zfx+/y R26-CreER+ (Ptch1) and Ptch1flox/flox Zfxflox/y R26-CreER+ (Ptch1-Zfx) mice, along with Ptch1flox/flox R26-CreER− controls (Ctrl), were treated topically with tamoxifen (Tmx) to induce deletion of Ptch1 alone or of both Ptch1 and Zfx in the skin. Shown is Zfx expression in the skin 3 days after Tmx treatment. Representative micrographs of IHC staining for Zfx in sections from treated dorsal skin are shown. Scale bars represent 100 μm (upper panel) and 50 μm (lower panel). Epidermis (E), dermis (D), and hair follicle (F) are labeled, and the boundary between epidermis/follicles and dermis is marked (dotted line; upper panel). Zoomed insets show decreased nuclear staining for Zfx in the hair follicle (lower panel).
Figure 2. Zfx loss prevents BCC formation after Ptch1 deletion in vivo.

As described in Figure 1, mice carrying tamoxifen (Tmx)-inducible Cre recombinase and conditional alleles of Ptch1 alone (Ptch1) or of both Ptch1 and Zfx (Ptch1-Zfx), as well as Cre− controls (Ctrl), were treated with Tmx on shaved lower dorsal skin.
(A) Representative photographs showing dorsal skin of mice of the indicated genotypes following euthanasia 8 weeks after Tmx treatment.
(B) BCC development in the mice of indicated genotypes. Shown are sections of treated dorsal skin isolated 8 weeks after Tmx treatment and stained with H&E or with anti-Zfx antibody. Scale bars represent 100 μm.
(C) Pathology scores of BCC in treated dorsal skin 8–9 weeks after Tmx treatment. Shown are the fractions of indicated BCC severity scores in Ctrl, Ptch1 and Ptch1-Zfx mice (n = 5, 9, and 13, respectively).
We also tested the consequences of Zfx loss in an alternative model of Hh pathway-dependent BCC initiated by Cre-mediated induction of the Rosa26-SmoM2flox-Stop-flox (R26-SmoM2) transgene, which expresses a constitutively active version of the Hh pathway signal transducer Smo (39). We treated shaved lower dorsal skin of R26-SmoM2+ Zfxflox/y R26-CreER+ (SmoM2-Zfx) and R26-SmoM2+ R26-CreER+ (SmoM2) mice with topical tamoxifen for five consecutive days, to induce SmoM2 expression and BCC formation, with or without Zfx co-deletion. Although less severe than the phenotype observed in Ptch1 mice, SmoM2 mice exhibited hair loss and gross induration in the treated skin patch by 8–10 weeks post-Tmx, whereas treated SmoM2-Zfx dorsal skin patches retained hair follicles and largely intact skin (Fig. S2A). Frank BCC observable in histological sections from SmoM2 dorsal skin was absent in sections from SmoM2-Zfx dorsal skin (Fig. S2B). As with Ptch1-Zfx mice in Fig. 2B, the residual hair follicle cells in Tmx-treated SmoM2-Zfx mice expressed Zfx, revealing a strong selection for occasional non-deleter cells (Fig. S2C). SmoM2-Zfx mice also showed a significant reduction in the extent of skin infiltration by BCC lesions (Fig. S2D). Thus, Zfx is also required for formation of an alternative BCC model initiated by expression of constitutively active Smo.
The loss of Zfx delays Hh-induced medulloblastoma development
To test whether Zfx is also required for the development of Hh-induced MB, we used brain-specific Ptch1 deletion mediated by the hGFAP-Cre deleter strain (38). The hGFAP-Cre+ Ptch1flox/flox mice show early expansion of the GNP-containing external granular layer (EGL) of the cerebellum, develop MB shortly after birth, and succumb to it by 4 weeks of age (21). To delete Ptch1 alone or concomitantly with Zfx, we generated hGFAP-Cre+ Ptch1flox/flox (hGFAP-Ptch1) and hGFAP-Cre+ Ptch1flox/flox Zfxflox/y (hGFAP-Ptch1-Zfx) mice, respectively.
All hGFAP-Ptch1 pups at P7 and P14 showed disrupted cerebellar architecture with expanded EGL and minimal foliation (Fig. 3A). hGFAP-Ptch1-Zfx pups also showed EGL expansion comprised of cells that lacked Zfx expression (Fig. 3B). Thus, in contrast to the inducible BCC model, Zfx deletion did not preclude transformation or favor the emergence of non-deleter cells. However, hGFAP-Ptch1-Zfx pups showed better preservation of cerebellar architecture and reduced EGL expansion (Fig. 3A), as confirmed by significantly smaller cross-sectional EGL areas in P7 cerebella (Fig. 3C). This was not due to developmental defects of Zfx deletion, because hGFAP-Cre+ Zfxflox/y mice exhibit normal cerebella throughout life (Fig. S3). Nearly all hGFAP-Ptch1 mice died by 25 days, which corresponds to 4–6 days post-weaning and suggests a failure to thrive independent of the dam (Fig. 3D). The majority of hGFAP-Ptch1-Zfx mice (~70%) also died around the same time, likely due to the similar inability to survive weaning (Fig. 3D). However, ~30% of hGFAP-Ptch1-Zfx mice survived past weaning and ~20% lived for >50 days, resulting in a significant (P<0.01) improvement in survival. All hGFAP-Ptch1-Zfx mice developed and eventually succumbed to tumors, suggesting that the loss of Zfx does not prevent MB initiation but impairs tumor propagation.
Figure 3. Zfx loss delays MB development after Ptch1 deletion in vivo.

MB development was followed in mice with CNS-specific hGFAP-Cre and conditional alleles of Ptch1 alone (Ptch1) or of both Ptch1 and Zfx (Ptch1-Zfx), as well as in Cre− control mice (Ctrl).
(A) H&E-stained sagittal sections of cerebella from P7 and P14 mice of the indicated genotypes. Scale bars represent 500 μm.
(B) Anti-Zfx-stained sagittal sections of P7 cerebella from mice of the indicated genotypes. Scale bars represent 100 μm.
(C) Cross-section area of external granular layer (EGL) in sagittal sections from P7 cerebella (mean ± SD of 6–7 mice).
(D) Kaplan-Meier survival plot of Ptch1 versus Ptch1-Zfx mice.
The observed effects of Zfx loss in MB development could reflect a specific role of Zfx in Hh-driven MB, or a more general role in brain tumorigenesis. Genome-wide expression data on murine MB subtypes (48) show a prominent and specific overexpression of Zfx in Hh-driven compared to Myc-driven MB or to normal neural stem cells (Fig. S4A). To directly test the role of Zfx in other types of brain tumors, we used model of glioblastoma (GBM) in adult mice. We performed retroviral delivery of platelet-derived growth factor (PDGF) and Cre recombinase into the brains of mice with conditional null Pten allele (Ptenflox/flox), which results in aggressive Pten-deficient GBM of the proneural subtype (40). In contrast to the MB model (Fig. 3D), concomitant deletion of Zfx in Ptenflox/flox Zfxflox/y mice significantly accelerated rather than delayed mortality (Fig. 4A). This was not due to inefficient deletion of Zfx, as the resulting tumors were completely Zfx-negative (Fig. 4B). Thus, the role of Zfx in Hh-driven MB formation does not reflect its general requirement for tumorigenesis in the brain.
Figure 4. Loss of Zfx does not impair Pten-dependent glioblastoma development.
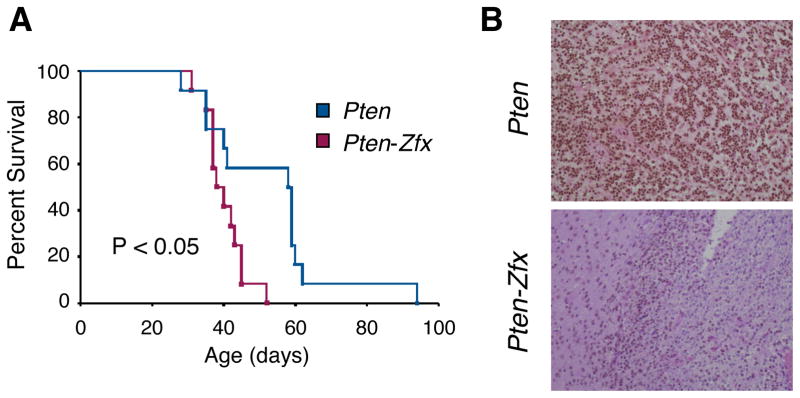
(A) Kaplan-Meier survival curve for retrovirus-injected Pten and Pten-Zfx mice. Glioblastoma was induced via stereotactic injection of retrovirus encoding PDGF and Cre recombinase into brains of young adult mice with conditional null alleles of Pten only (Pten) or of both Pten and Zfx (Pten-Zfx).
(B) Immunohistochemical staining for Zfx in sections of glioblastomas from retrovirus-injected Pten and Pten-Zfx mice.
Zfx knockdown impairs growth in a human MB cell line in vitro
We investigated whether ZFX was also required in human MB cells in vitro, using a system for ZFX knockdown with short hairpin RNAs (shRNAs) recently validated in human ESC (29). The human MB cell line DAOY was transduced with lentiviral vectors encoding ZFX-specific shRNAs (H2, H3, H4) or a control scrambled (SCR) shRNA. Two ZFX-specific shRNAs (H2, H3) strongly reduced Zfx protein (Fig. 5A) and transcript (Fig. 5B), whereas the third (H4) caused a more moderate reduction (Fig. 5B). These shRNAs impaired the growth of DAOY cells proportionately to the degree of ZFX knockdown (Fig. 5C), suggesting that ZFX is required for optimal growth of human MB in vitro. Notably, genome-wide expression data on human MB samples divided into molecular subtypes (49) reveal a significant enrichment of ZFX in the SHH subtype compared to two common Hh-independent subtypes (Fig. S4B). Unlike murine Zfx, human ZFX escapes X inactivation, and its reduced dosage in male somatic cells is compensated by the Y-chromosomal gametolog ZFY. Within male MB samples, ZFY was significantly enriched in the SHH subtype (Fig. S4B). Collectively, these data suggest that the role of Zfx in Hh-driven MB might be conserved between mice and humans.
Figure 5. Zfx knockdown impairs cell growth in a human medulloblastoma cell line in vitro.
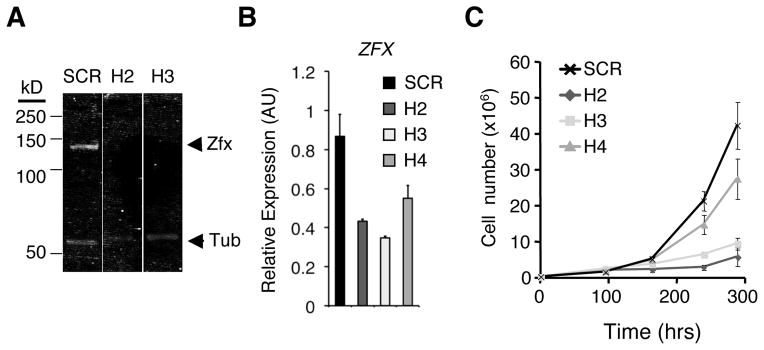
The human MB cell line DAOY was transduced in vitro with lentiviruses encoding short hairpin RNAs (shRNAs) targeting ZFX transcript (H2, H3, H4) or a scrambled control shRNA (SCR). Transduced cells were selected for by growth in puromycin-containing medium.
(A) Western blot for ZFX and tubulin control in DAOY cells transduced with control (SCR) or ZFX-targeting (H2, H3) lentivirus.
(B) Expression of ZFX in DAOY cells after shRNA knockdown. Shown are ZFX expression levels 4 days after transduction, as determined by qPCR (mean ± SD of triplicate parallel cultures; representative of three independent experiments).
(C) Growth curves for DAOY cells transduced with lentivirus expressing control (SCR) or ZFX-targeting (H2, H3, H4) shRNA (mean ± SD of triplicate parallel cultures, representative of three independent experiments).
Identifying novel functional targets of ZFX in human MB cells in vitro
We hypothesized that an evolutionarily conserved set of target genes may contribute to the role of Zfx in Hh-induced tumors. To identify potential functional targets of Zfx, we first performed microarray-based gene expression analysis of the DAOY cell line at an early time point after transduction with lentiviral particles bearing ZFX-targeting shRNAs. We searched for genes whose RNA expression levels behaved similarly to that of ZFX, exhibiting strong reduction by qPCR with shRNAs H2 and H3 and more moderate reduction with shRNA H4. A set of 163 genes whose expression level patterns matched that of ZFX in transduced DAOY cells has been identified (Fig. 6A, Dataset S1).
Figure 6. Identification of conserved direct targets of ZFX in human medulloblastoma cells.

(A) Downregulated genes (n= 163) identified in pattern matching analysis of Affymetrix Gene ST 1.0 array data from DAOY cells after shRNA knockdown. Full gene list is presented in Dataset S1.
(B) Conserved ZFX transcriptional targets. Shown are numbers of pattern-matching genes from ZFX KD microarray in (A) that had enrichment peaks >1kb from TSS in ZFX ChIP-seq from DAOY cells, from murine embryonic stem cells (mESC), from both cell types (Overlap), or from neither cell type (N/A). DAOY, mESC, and Overlap gene lists are presented in full in Dataset S2.
(C) Zfx binding to Dis3L in murine and human ChIP-seq. Shown are sequencing-read enrichment peaks near TSS of Dis3L in anti-Zfx ChIP-seq from murine ESC (Chen et al., 2008) (left), and in anti-ZFX ChIP-seq and sheared nuclear lysate control (Input) from DAOY human MB cells (right).
(D) Zfx binding to Ube2j1 in murine and human ChIP-seq. Shown are sequencing-read enrichment peaks near TSS of Ube2j1 in anti-Zfx ChIP-seq from murine ESC (Chen et al., 2008) (left), and in anti-ZFX ChIP-seq and sheared nuclear lysate control (Input) from DAOY human MB cells (right).
We then compared this gene set with direct binding targets of Zfx in mouse and human. Direct targets of Zfx in murine ESC have been previously identified via chromatin precipitation followed by massively parallel sequencing (ChIP-seq) (42). To identify direct targets of ZFX in human MB, we performed anti-ZFX ChIP-seq on the human DAOY cell line. Then, we compared the list of 163 pattern-matching genes with overlapping ZFX direct binding targets that were within 1 kb of transcriptional start sites (TSS) of their nearest gene and were conserved ChIP-seq in both mouse ESC and human DAOY MB cells (Dataset S2). Of 163 genes identified by pattern-matching analysis of microarray gene expression data, 68 genes (42%) were bound by Zfx in DAOY cells, and 30 genes (18%) were bound in both DAOY and murine ESC (Fig. 6B, Dataset S2).
Out of 30 conserved direct targets of ZFX, we have selected 6 genes (DIS3L, ETS2, FZD6, LRRC41, TNFAIP6 and UBE2J1) based on their likely involvement in cell growth control, and tested them by shRNA-mediated knockdown in DAOY cells (data not shown). The knockdown of four genes (ETS2, FZD6, LRRC41, and TNFAIP6) by multiple shRNAs had no consistent effect on the growth of DAOY cells, whereas the knockdown of DIS3L and UBE2J1 consistently inhibited cell growth. DIS3L encodes a cytoplasmic RNA exonuclease with a possible role in mRNA degradation (50, 51). Ube2j1 encodes an E2 ubiquitin-conjugating enzyme that is thought to participate in the endoplasmic reticulum-associated degradation of misfolded proteins (52). ChIP-seq analysis of DAOY cells showed ZFX binding near the TSS of both genes in murine ESC and human DAOY cells (Fig. 6C,D). The expression of Dis3L (formerly known as AV340375) strongly depends on Zfx in all examined murine cells including HSCs and ESCs (Fig. S5)(28) and activated B lymphocytes (30). The expression of Ube2j1 is also Zfx-dependent in HSCs (28) and B lymphocytes (30), although not in ESCs (Fig. S5B)(28). These data suggested Dis3L and Ube2j1 as candidate genes with potential functional relevance downstream of Zfx in both mouse and human.
Knockdown of ZFX targets DIS3L and UBE2J1 impairs human medulloblastoma growth in vitro
We tested whether the expression levels of Dis3L and Ube2j1 were affected in the context of BCC and MB following Zfx loss. We confirmed that DIS3L and UBE2J1 expression levels were decreased after shRNA-mediated ZFX knockdown in DAOY cells (Fig. 7A). Moreover, both transcripts were reduced ~2-fold after tamoxifen-induced Zfx co-deletion in epidermis and hair follicles of treated dorsal skin from the Ptch1-dependent BCC model in vivo (Fig. 7B). Together with the ChIP-seq binding data, these data suggest Zfx can directly regulate Dis3L and Ube2j1 expression in the contexts of BCC and MB models in vivo and in vitro.
Figure 7. Knockdown of ZFX targets DIS3L and UBE2J1 impairs growth of human DAOY medulloblastoma cells in vitro.

(A) Expression of DIS3L and UBE2J1 in DAOY cells after ZFX knockdown. Shown are DIS3L and UBE2J1 expression levels by qPCR 4 days after transduction with lentivirus coding for ZFX-targeting (H2, H3, H4) or scrambled control (SCR) shRNA (mean ± SD of triplicate parallel cultures; representative of three independent experiments).
(B) Expression levels of Dis3L and Ube2j1 in bulk epidermis and hair follicles from shaved lower dorsal skin 3 days after topical Tmx treatment in Ptch1, Ptch1-Zfx, and Cre− (Ctrl) mice (as described in Figure 1). Shown are normalized expression levels relative to Ctrl skin as determined by qPCR (mean ± SD of 2–4 mice; representative of four independent experiments).
(C) DAOY human MB cells were transduced in vitro with Sigma MISSION lentiviruses encoding shRNAs targeting DIS3L, UBE2J1, or a non-mammalian control gene (NonM). Shown are projected cell numbers for untreated DAOY cells versus cells transduced with NonM control virus or 3–4 shRNA lentiviruses targeting DIS3L (SH1–SH4) or UBE2J1 (SH3–SH5) (mean + SD of triplicate parallel cultures; beginning 2 days post- puromycin selection and 4 days post-transduction).
(D) Expression of DIS3L and UBE2J1 in DAOY cells after shRNA knockdown. Shown are DIS3L and UBE2J1 expression levels by qPCR in DAOY cells 5 days after transduction with Sigma MISSION lentiviruses coding for DIS3L- or UBE2J1-targeting shRNA, or for NonM control shRNA (mean ± SD of triplicate reactions; normalized to expression in untreated DAOY cells).
To determine whether DIS3L or UBE2J1 themselves are required for the growth of a human MB cell line in vitro, we transduced DAOY cells with lentiviral vectors encoding several shRNAs targeting each gene. Compared to untreated cells and a control shRNA targeting non-mammalian sequence (NonM), all tested shRNAs against DIS3L (4 shRNAs) or UBE2J1 (3 shRNAs) impaired the growth of DAOY cells (Fig. 7C). Successful knockdown of the respective targeted genes by all shRNAs was confirmed by qPCR (Fig. 7D). Thus, DIS3L and UBE2J1 represent conserved direct targets of ZFX that are required for the optimal growth of DAOY human MB cells in vitro.
DISCUSSION
Our results demonstrate that Zfx expression is required for the development of two Hh pathway-dependent model tumors in vivo. In both BCC and MB, Zfx appears to act in a cell-intrinsic manner to facilitate tumor propagation, although a role in the initiation of BCC could not be ruled out. This is in agreement with a recently reported cell-intrinsic role of Zfx in the propagation of two molecularly distinct leukemias, AML and T-ALL (35). In contrast, Zfx appears dispensable for progression of a glioblastoma model in vivo, suggesting that a requirement for Zfx is not generalizable across all tumor models. Zfx expression is enriched in Hh-driven MB compared to other MB subtypes in both human patients and animal models (Fig. S4), suggesting a preferential role for Zfx in this tumor subtype. It is possible that Zfx may act downstream of Hh signaling, for example via transcriptional induction by or cooperation with the GLI proteins. A more likely possibility is that Zfx may activate the expression of key Hh pathway components such as Smo. Indeed, Zfx directly binds to the promoter of Smo in both murine ESCs (42) and in DAOY cells (Dataset S2) and facilitates Smo expression in hematopoietic progenitors and differentiating ESCs (our unpublished data). However, the requirement for Zfx in SmoM2-induced BCC argues against Zfx-induced Smo expression as a sole mechanism. Most likely, Zfx exerts its role through a combination of targets specific for Hh-driven tumors as well as targets common for multiple tumor types, as discussed below.
Zfx is required in several cell types that undergo self-renewal, such as murine and human ESC and murine HSC (28, 29). At the same time, Zfx is dispensable in multiple cell types in which proliferation is followed by differentiation (e.g. myeloid progenitors). Thus, Zfx may control a specific genetic program that mediates prolonged self-renewal and therefore may become essential in malignant cells that acquire abnormal self-renewal capacity. In this context, Zfx would represent a clear example of the “non-oncogene addiction” of tumors to an otherwise normal and largely dispensable endogenous gene (53).
We describe a broad Zfx-dependent genetic program that may maintain the self-renewal of multiple normal and malignant cells, including Hh-dependent tumors. We have focused on two functional components of this Zfx-dependent program, Dis3L and Ube2j1, identified as direct transcriptional targets of Zfx in both mouse and human. The knockdown of DIS3L and UBE2J1 demonstrated that each is required for optimal growth of human MB cells in vitro (Fig. 7). These genes have not been previously implicated in cancer, although the DIS3L homolog DIS3L2 has been implicated in Perlman overgrowth syndrome and an associated kidney tumor (Wilm’s tumor)(54), supporting consideration of an RNA exonuclease for a possible role in cancer. The functions of Dis3L and Ube2j1, in cytoplasmic RNA exonuclease activity (50, 51) and ER-associated protein degradation (52), respectively, suggest that disregulation of either gene could potentially impact stress pathways, a hallmark of the phenomenon of non-oncogene addiction in tumors (53). While any requirements for DIS3L, UBE2J1, or other conserved direct targets of ZFX remain to be established in Hh-induced and other tumor models in vivo, they nevertheless highlight novel candidates for cell-intrinsic regulation and molecular targeting of Hh-dependent cancers.
We report here that the transcription factor Zfx is required for the development of Hh-induced BCC and MB in vivo, two cancers that are highly distinct in their tissue of origin, growth rate and clinical prognosis. Our results suggest that this effect may be partly due to direct regulation of a Zfx-dependent genetic program including conserved target genes like DIS3L and UBE2J1. A more complete understanding of the relevant functional targets of Zfx in the context of Hh-dependent MB and BCC in vivo could facilitate molecular targeting of these malignancies. Identification of the precise downstream targets with functional relevance in a Zfx-defined cell-intrinsic program shared by these two cancers could yield potentially druggable molecules for treatment of both BCC and the Hh-dependent subtype of MB. Future targeting of such molecules, as alternatives to targeting core components of Hh pathway like SMO, offers the promise of molecular therapies specific to Hh-dependent tumors that can help to bypass resistance to SMO inhibitors or spare normal Hh-dependent development in infants or children with Hh-dependent MB.
Supplementary Material
Acknowledgments
We thank C. Henderson, H. Wichterle and A. Christiano for helpful discussions and advice, F. Doetsch, S. Ghosh and T. Ludwig for equipment and reagents, and the Skin Phenotyping Core Facility of the NIH Skin Diseases Research Center (P30 AR044535) for skin analysis. This work was supported by the NIH grants HL084353 (B.R.), AR044535 (B.R.) and NS066955 (P.C.), NIH training grant HD055165 (C.J.P. and S.P.W.), American Cancer Society (B.R. and S.P.W.), Leukemia and Lymphoma Society (B.R.) and Project A.L.S (G.F.C.).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–17. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 2.Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 3.Lum L, Beachy PA. The Hedgehog Response Network: Sensors, Switches, and Routers. Science. 2004;304:1755–9. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 4.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes & Development. 2008;22:2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 5.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 6.Wijgerde M, McMahon JA, Rule M, McMahon AP. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes & Development. 2002;16:2849–64. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–503. doi: 10.1242/dev.009324. [DOI] [PubMed] [Google Scholar]
- 8.Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–21. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- 9.Dahmane N, Ruiz-i-Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 10.Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Developmental Biology. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Chiang C, Swan RZ, Grachtchouk M, Bolinger M, Litingtung Y, Robertson EK, et al. Essential Role for Sonic hedgehog during Hair Follicle Morphogenesis. Developmental Biology. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- 12.St-Jacques B, Dassule HR, Karavanova I, Botchkarev VA, Li J, Danielian PS, et al. Sonic hedgehog signaling is essential for hair development. Current Biology. 1998;8:1058–69. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- 13.Ng JMY, Curran T. The Hedgehog’s tale: developing strategies for targeting cancer. Nat Rev Cancer. 2011;11:493–501. doi: 10.1038/nrc3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743–54. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–9. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics Identifies Medulloblastoma Subgroups That Are Enriched for Specific Genetic Alterations. Journal of Clinical Oncology. 2006;24:1924–31. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 18.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered Neural Cell Fates and Medulloblastoma in Mouse patched Mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 19.Adolphe C, Hetherington R, Ellis T, Wainwright B. Patched1 Functions as a Gatekeeper by Promoting Cell Cycle Progression. Cancer Research. 2006;66:2081–8. doi: 10.1158/0008-5472.CAN-05-2146. [DOI] [PubMed] [Google Scholar]
- 20.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han Y-G, et al. Acquisition of Granule Neuron Precursor Identity Is a Critical Determinant of Progenitor Cell Competence to Form Shh-Induced Medulloblastoma. Cancer Cell. 2008;14:123–34. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Z-J, Ellis T, Markant SL, Read T-A, Kessler JD, Bourboulas M, et al. Medulloblastoma Can Be Initiated by Deletion of Patched in Lineage-Restricted Progenitors or Stem Cells. Cancer Cell. 2008;14:135–45. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang GY, Wang J, Mancianti M-L, Epstein EH., Jr Basal Cell Carcinomas Arise from Hair Follicle Stem Cells in Ptch1+/− Mice. Cancer Cell. 2011;19:114–24. doi: 10.1016/j.ccr.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of Medulloblastoma with Hedgehog Pathway Inhibitor GDC-0449. New England Journal of Medicine. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the Hedgehog Pathway in Advanced Basal-Cell Carcinoma. New England Journal of Medicine. 2009;361:1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 25.Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura H, Ng JMY, Curran T. Transient Inhibition of the Hedgehog Pathway in Young Mice Causes Permanent Defects in Bone Structure. Cancer Cell. 2008;13:249–60. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Schneider-Gadicke A, Beer-Romero P, Brown LG, Mardon G, Luoh S-W, Page DC. Putative transcription activator with alternative isoforms encoded by human ZFX gene. Nature. 1989;342:708–11. doi: 10.1038/342708a0. [DOI] [PubMed] [Google Scholar]
- 28.Galan-Caridad JM, Harel S, Arenzana TL, Hou ZE, Doetsch FK, Mirny LA, et al. Zfx Controls the Self-Renewal of Embryonic and Hematopoietic Stem Cells. Cell. 2007;129:345–57. doi: 10.1016/j.cell.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harel S, Tu EY, Weisberg S, Esquilin M, Chambers SM, Liu B, et al. ZFX Controls the Self-Renewal of Human Embryonic Stem Cells. PLoS ONE. 2012;7:e42302. doi: 10.1371/journal.pone.0042302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arenzana TL, Smith-Raska MR, Reizis B. Transcription factor Zfx controls BCR-induced proliferation and survival of B lymphocytes. Blood. 2009;113:5857–67. doi: 10.1182/blood-2008-11-188888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Donnell KA, Keng VW, York B, Reineke EL, Seo D, Fan D, et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proceedings of the National Academy of Sciences. 2012;109:E1377–E86. doi: 10.1073/pnas.1115433109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang J, Yu Z, Lian M, Ma H, Tai J, Zhang L, et al. Knockdown of zinc finger protein, X-linked (ZFX) inhibits cell proliferation and induces apoptosis in human laryngeal squamous cell carcinoma. Molecular and Cellular Biochemistry. 2012;360:301–7. doi: 10.1007/s11010-011-1069-x. [DOI] [PubMed] [Google Scholar]
- 33.Jiang H, Zhang L, Liu J, Chen Z, Na R, Ding G, et al. Knockdown of zinc finger protein X-linked inhibits prostate cancer cell proliferation and induces apoptosis by activating caspase-3 and caspase-9. Cancer Gene Ther. 2012;19:684–9. doi: 10.1038/cgt.2012.53. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y, Su Z, Huang Y, Sun T, Chen S, Wu T, et al. The Zfx gene is expressed in human gliomas and is important in the proliferation and apoptosis of the human malignant glioma cell line U251. Journal of Experimental & Clinical Cancer Research. 2011;30:114. doi: 10.1186/1756-9966-30-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weisberg Stuart P, Smith-Raska Matthew R, Esquilin Jose M, Zhang J, Arenzana Teresita L, Lau Colleen M, et al. ZFX Controls Propagation and Prevents Differentiation of Acute T-Lymphoblastic and Myeloid Leukemia. Cell Reports. 2014;6:528–40. doi: 10.1016/j.celrep.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten Dose Dictates Cancer Progression in the Prostate. PLoS Biol. 2003;1:e59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellis T, Smyth I, Riley E, Graham S, Elliot K, Narang M, et al. Patched 1 conditional null allele in mice. genesis. 2003;36:158–61. doi: 10.1002/gene.10208. [DOI] [PubMed] [Google Scholar]
- 38.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 39.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A Novel Somatic Mouse Model to Survey Tumorigenic Potential Applied to the Hedgehog Pathway. Cancer Research. 2006;66:10171–8. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, et al. Glioblastoma Models Reveal the Connection between Adult Glial Progenitors and the Proneural Phenotype. PLoS ONE. 2011;6:e20041. doi: 10.1371/journal.pone.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharov AA, Dudekula DB, Ko MSH. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics. 2005;21:2548–9. doi: 10.1093/bioinformatics/bti343. [DOI] [PubMed] [Google Scholar]
- 42.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, et al. Integration of External Signaling Pathways with the Core Transcriptional Network in Embryonic Stem Cells. Cell. 2008;133:1106–17. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 43.Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Liu T, Meyer C, Eeckhoute J, Johnson D, Bernstein B, et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salmon-Divon M, Dvinge H, Tammoja K, Bertone P. PeakAnalyzer: Genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics. 2010;11:415. doi: 10.1186/1471-2105-11-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giannopoulou E, Elemento O. An integrated ChIP-seq analysis platform with customizable workflows. BMC Bioinformatics. 2011;12:277. doi: 10.1186/1471-2105-12-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vidal VPI, Ortonne N, Schedl A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. Journal of Cutaneous Pathology. 2008;35:373–9. doi: 10.1111/j.1600-0560.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- 48.Pei Y, Moore Colin E, Wang J, Tewari Alok K, Eroshkin A, Cho Y-J, et al. An Animal Model of MYC-Driven Medulloblastoma. Cancer Cell. 2012;21:155–67. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, et al. Medulloblastoma Comprises Four Distinct Molecular Variants. Journal of Clinical Oncology. 2011;29:1408–14. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Staals RHJ, Bronkhorst AW, Schilders G, Slomovic S, Schuster G, Heck AJR, et al. Dis3-like 1: a novel exoribonuclease associated with the human exosome. EMBO J. 2010;29:2358–67. doi: 10.1038/emboj.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomecki R, Kristiansen MS, Lykke-Andersen S, Chlebowski A, Larsen KM, Szczesny RJ, et al. The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J. 2010;29:2342–57. doi: 10.1038/emboj.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burr ML, Cano F, Svobodova S, Boyle LH, Boname JM, Lehner PJ. HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proceedings of the National Academy of Sciences. 2011;108:2034–9. doi: 10.1073/pnas.1016229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Astuti D, Morris MR, Cooper WN, Staals RHJ, Wake NC, Fews GA, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet. 2012;44:277–84. doi: 10.1038/ng.1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.