

Abstract
Gene-mapping studies routinely rely on checking for Mendelian transmission of marker alleles in a pedigree, as a means of screening for genotyping errors and mutations, with the implicit assumption that, if a pedigree is consistent with Mendel’s laws of inheritance, then there are no genotyping errors. However, the occurrence of inheritance inconsistencies alone is an inadequate measure of the number of genotyping errors, since the rate of occurrence depends on the number and relationships of genotyped pedigree members, the type of errors, and the distribution of marker-allele frequencies. In this article, we calculate the expected probability of detection of a genotyping error or mutation as an inheritance inconsistency in nuclear-family data, as a function of both the number of genotyped parents and offspring and the marker-allele frequency distribution. Through computer simulation, we explore the sensitivity of our analytic calculations to the underlying error model. Under a random-allele–error model, we find that detection rates are 51%–77% for multiallelic markers and 13%–75% for biallelic markers; detection rates are generally lower when the error occurs in a parent than in an offspring, unless a large number of offspring are genotyped. Errors are especially difficult to detect for biallelic markers with equally frequent alleles, even when both parents are genotyped; in this case, the maximum detection rate is 34% for four-person nuclear families. Error detection in families in which parents are not genotyped is limited, even with multiallelic markers. Given these results, we recommend that additional error checking (e.g., on the basis of multipoint analysis) be performed, beyond routine checking for Mendelian consistency. Furthermore, our results permit assessment of the plausibility of an observed number of inheritance inconsistencies for a family, allowing the detection of likely pedigree—rather than genotyping—errors in the early stages of a genome scan. Such early assessments are valuable in either the targeting of families for resampling or discontinued genotyping.
Introduction
Microsatellites, or short-tandem-repeat polymorphisms (STRPs), and single-nucleotide polymorphisms (SNPs) are the markers of choice for gene-mapping studies. Analytic strategies often entail the genotyping of hundreds of STRPs in hundreds or thousands of individuals, followed by the investigation of interesting regions by use of additional STRPs and SNPs. The sheer volume of genotypes is large enough that genotyping errors are inevitable. This problem is only exacerbated in fine-mapping studies, for which markers are often chosen according to their chromosomal location, rather than because of either their reliability or ease of genotyping (Ewen et al. 2000).
Genotyping errors can arise for a number of reasons, including laboratory errors (e.g., errors in allele calling) and incorrect data interpretation or entry. Mutations, which are not uncommon for STRPs (Weber and Wong 1993), may also manifest themselves as apparent genotyping errors. It is important to distinguish genotyping errors and mutations versus pedigree errors. Pedigree errors involve the incorrect specification of familial relationships and systematically affect more than one genotype; examples include unknown adoptions, nonpaternity, and sample mixups. Both genotyping errors and pedigree errors can adversely impact the power of a gene-mapping study.
Given sufficient pedigree and marker data, many genotyping and pedigree errors can be detected on the basis of apparent inconsistencies with Mendelian inheritance; for example, a parent and offspring may fail to share an allele at one or more genetic markers. Still, the probability of observing an inheritance inconsistency depends on the pedigree members who are genotyped, the type of error and in whom it occurs, and the marker-allele frequency distribution. For example, a genotyping error in a homozygous parent that affects only one allele cannot be detected as an inheritance inconsistency in a nuclear family. Similarly, for biallelic markers such as most SNPs, a genotyping error cannot, in the absence of parental genotype data, be detected in sibships of any size, since only three genotypes are possible and since, even with parental data, such errors are often difficult to detect.
In this article, we calculate the expected frequency of detectable genotyping errors—that is, errors that are detectable on the basis of inheritance inconsistencies—for nuclear-family data. Under the assumption of correctly specified familial relationships and a random-allele–error model, we calculate the frequency of detectable error, as a function of both the number of genotyped parents and offspring and the marker-allele frequency distribution. Through computer simulation, we explore the impact that other genotyping-error models have on the detection rate. Our calculations permit assessment of the likelihood that genotyping errors and mutations could be responsible for an observed number of failures of Mendelian inheritance in a given family if the reported relationships are correct. Such assessments are valuable during the early stages of a genome scan, when relatively few marker genotypes are available for evaluation of the presence of pedigree errors. Families for which the observed number of errors is much greater than expected might be either targeted for resampling or excluded from further genotyping. In addition, numerical results from our analytic investigations suggest that at least a quarter of genotyping errors are undetectable on the basis of inheritance inconsistencies, under even the most favorable circumstances (i.e., fully polymorphic markers and completely genotyped nuclear families). These results underscore the importance of identification—for example, through multipoint analysis (Ehm and Kimmel 1995; Ehm et al. 1996; Douglas et al. 2000; Sobel et al. 2002 [in this issue])—of genotyping errors that are consistent with Mendel’s laws.
Methods
Here we calculate the probability of detection of a genotyping error or mutation as an inheritance inconsistency for nuclear-family data, as a function of both the number of genotyped parents and offspring and the marker-allele frequency distribution. We say that an error is detectable if it results in an inheritance inconsistency. On the basis of these analytic calculations and an assumed genotyping-error rate, we determine the expected number of inheritance inconsistencies per family, as well as the expected number of families in a sample displaying a fixed number of inheritance inconsistencies.
Data, Notation, and Assumptions
Assume that, for a nuclear family, genotype data are observed at a single genetic marker with n alleles and that familial relationships are correctly specified. Let pi denote the frequency of allele i, and let s⩾1 denote the number of genotyped offspring. For the sake of illustration, we present calculations for zero or two genotyped parents; calculations for nuclear families with only one genotyped parent are similar. We perform our analytic calculations for the random-allele–error model, in which an allele is randomly replaced by another allele in a manner proportional to allele frequencies; for example, allele i is replaced by allele j, with probability pj/(1-pi). Although this model is not entirely realistic, it simplifies the calculations and provides a sense of the consequences of errors that may occur. Moreover, simulation results under other error models suggest that the error-rate estimates from this model are useful to approximate those for the other error models. We further assume that there is exactly one genotyping error per family, with the error equally likely to occur in any family member’s genotype. Given acceptable genotyping-error rates and typically sized nuclear families, the one-error assumption has a negligible impact on the resulting probabilities (see the “Discussion” section).
Probability of Inheritance Inconsistency
To calculate the probability of detection of an error as an inheritance inconsistency (E), we condition on the mating type, G=gf×gm, of the father and mother of a nuclear family; in practice, 0, 1, or 2 of the parental genotypes may be observed. By the law of total probability, P(E)=ΣGP(E|G)P(G). Under the assumptions of random mating and Hardy-Weinberg equilibrium, P(G) is easily calculated as a function of the parental genotypes. The calculation of P(E) can be further simplified by noting that there are only seven distinct classes of parental mating types. These mating-type classes and their corresponding frequencies, P(G), are given in table 1 (Thompson 1975). Note that, for a marker with n alleles, calculation of P(E) requires summation over n terms for parental mating-type class ii×ii; n(n-1) for ii×ij; n(n-1)/2 for ii×jj and ij×ij; n(n-1)(n-2)/2 for ii×jk and ij×ik; and n(n-1)(n-2)(n-3)/8 for ij×kl. The conditional probability of detectable error P(E|G) is a function of the number of genotyped parents and offspring, the error model, and the marker-allele frequency distribution.
Table 1.
Conditional Probability of Detectable Error, Given Two Genotyped Parents and s>2 Genotyped Offspring[Note]
|
P(E|G) for Genotyping Error in |
|||
| ParentalMating Type G | P(G) | Offspring | Parent |
| ii×ii | p4i | 1 | 0 |
| ii×ij | 4p3ipj | 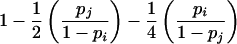 |
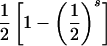 |
| ii×jj | 2p2ip2j | 1 | 0 |
| ii×jk | 4p2ipjpk | 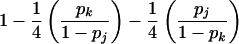 |
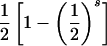 |
| ij×ij | 4p2ip2j | 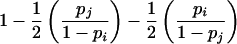 |
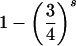 |
| ij×ik | 8p2ipjpk | 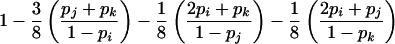 |
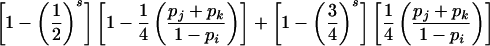 |
| ij×kl | 8pipjpkpl | 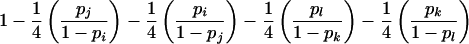 |
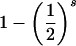 |
Note.— Assume the random-allele–error model and exactly one genotyping error per family; i, j, k, and l are distinct alleles with frequencies pi, pj, pk, and pl.
Conditional Probability of Inheritance Inconsistency: Two Parents Genotyped
Under the assumption that both parents are genotyped, P(E|G), given mating type G, can be calculated easily. For example, consider parental mating-type class ii×ij with possible offspring-genotype set {ii,ij}. If the error occurs in an offspring with genotype ii, then the error is detectable unless an i allele is mistaken for a j allele. Under the random-allele–error model, this latter event occurs with probability pj/(1-pi). Similarly, if the error occurs in an offspring with genotype ij, then the error is detectable unless the j allele is mistaken for an i allele; any change in the i allele is necessarily detectable. Therefore, under a random-allele–error model, for parental mating-type class ii×ij, an offspring error is detectable with probability
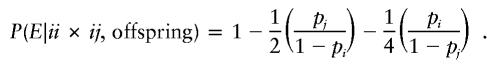 |
In contrast, if a random-allele error occurs in the parent with genotype ii, then it cannot be detected as an inheritance inconsistency, since the parent will continue to share the remaining i allele with all offspring. If the error occurs in the parent with genotype ij, then it is detectable if and only if there is at least one ii or ij genotype among the s offspring, depending on whether allele i or allele j is mistaken, respectively. Therefore, a parental random-allele error is detectable with probability
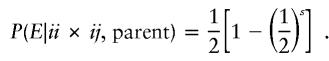 |
If the random occurrence of exactly one genotyping error per family is assumed, then the conditional probability of detectable error for mating-type class ii×ij in a family with t genotyped parents and s genotyped offspring is given by
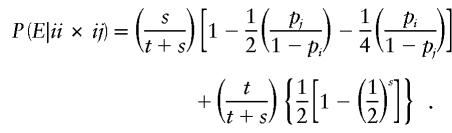 |
Conditional probabilities of detectable error for the remaining parental mating types can be calculated in the same way; results are given in table 1.
Conditional Probability of Inheritance Inconsistency: No Parents Genotyped
If neither parent is genotyped, calculation of P(E|G) conditional on mating type G is more complicated but still tractable. Note that an inheritance inconsistency will be detected only for sibships with s>2 sibs and that inconsistencies can be detected if and only if there are either (1) more than four distinct alleles in the sibship, (2) more than three distinct alleles in the sibship with a homozygous sib, (3) more than two distinct alleles in the sibship with two different homozygous sibs, or (4) more than three distinct alleles in the sibship with three heterozygous sibs who share an allele in common. Hence, for the random-allele–error model, inheritance inconsistencies can be detected for, at most, four of the seven parental mating types (table 2).
Table 2.
Conditional Probability of Detectable Error, Given No Genotyped Parents and s>2 Genotyped Offspring[Note]
| ParentalMating Type G | P(E|G) |
| ii×jk | 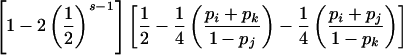 |
| ij×ij | 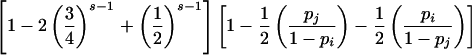 |
| ij×ik |
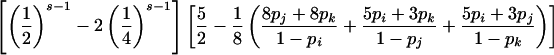 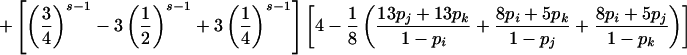 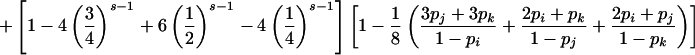
|
| ij×kl |
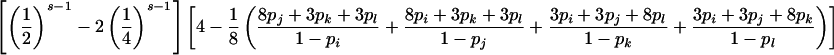 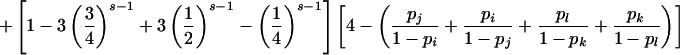
|
Note.— Data are as described in the footnote to table 1.
P(E|G) is straightforward to calculate for parental mating-type classes ii×jk and ij×ij. For example, consider parental mating-type class ij×ij with possible offspring genotype set {ii,ij, jj}. By observation (3) in the preceding paragraph, an error is detected if and only if homozygous genotypes ii and jj each appear at least once among s-1 of the offspring and the error is introduced into the genotype of the remaining offspring in such a way that neither an i allele is mistaken for a j nor a j allele is mistaken for an i. The probability that genotypes ii and jj appear at least once among s-1 of the offspring is given by
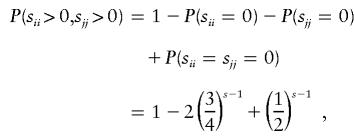 |
where sii denotes the number of s-1 offspring with genotype ii. Therefore, for parental mating-type class ij×ij, an error is detectable with probability
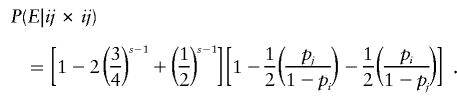 |
An analogous argument based on observation (4) in the preceding paragraph gives the conditional probability of detectable error for parental mating-type class ii×jk (table 2).
P(E|G) is considerably more complicated for parental mating-type classes ij×ik and ij×kl but can be calculated by enumerating all possible combinations of genotypes represented at least once among s-1 of the offspring. For example, consider parental mating type ij×ik with possible offspring genotype set {ii,ij, ik,jk}. If C denotes the combination of genotypes represented at least once among s-1 of the offspring, then, by the law of total probability,
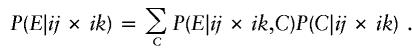 |
For parental mating-type class ij×ik, C includes the 15 nonempty subsets of {ii,ij,ik,jk}. As an illustration, consider the combination in which only genotypes ij and ik appear at least once among s-1 of the offspring. This event occurs with probability
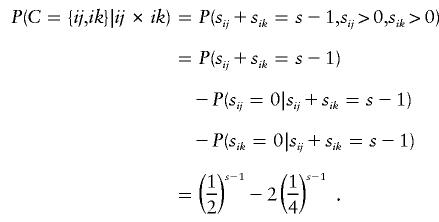 |
Now, if C={ij,ik}, an error will be detected if and only if the remaining offspring has genotype ij or ik and the error is introduced into his or her genotype in such a way that neither the j allele is mistaken for an i or a k nor the k allele is mistaken for an i or a j, which occurs with probability
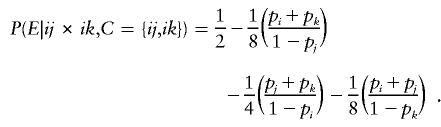 |
Summation over all such genotypic combinations C gives the probability of detectable error for parental mating-type class ij×ik. Calculation of the conditional probability of detectable error, given mating-type class ij×kl, is analogous (table 2).
Expected Number of Inheritance Inconsistencies
The probability P(E) of detection of an inheritance inconsistency, given exactly one genotyping error, can be used to determine the expected number of inheritance inconsistencies per family, as a function of the genotyping-error rate, e. For example, when two parents and s offspring are genotyped for a single marker, exactly one genotyping error occurs with probability (2+s)e(1-e)1+s, and, therefore, an inheritance inconsistency, I, is observed with probability P(I)=P(E)(2+s)e(1-e)1+s. Consequently, for M markers, exactly m (⩽M) inheritance inconsistencies are observed with probability 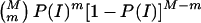 , and the expected number of inheritance inconsistencies per family is given by
, and the expected number of inheritance inconsistencies per family is given by 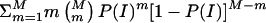 . Alternatively, for a sample of N families (each with two parents and s offspring genotyped on a common set of M markers), the expected number of families with m inheritance inconsistencies is given by
. Alternatively, for a sample of N families (each with two parents and s offspring genotyped on a common set of M markers), the expected number of families with m inheritance inconsistencies is given by 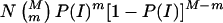 .
.
Numerical Calculation and Computer Simulation
On the basis of the analytic calculations noted above, we wrote a computer program to calculate the expected probability of detection of a genotyping error as an inheritance inconsistency, under the assumption of a random-allele–error model. We computed the expected probability of detectable error for nuclear families with 0, 1, or 2 genotyped parents and s=2, 3, 6, or 9 genotyped offspring, assuming exactly one genotyping error per family. To examine the impact that marker-allele frequencies and marker heterozygosity, H, have on the ability to detect errors, we considered markers with 2 (H=0.50), 4 (H=0.75), and 10 (H=0.90) equally frequent alleles, as well as markers with 2 (0.90 and 0.10; H=0.18) and 7 (0.40, 0.20, 0.20, 0.05, 0.05, 0.05, and 0.05; H=0.75) non–equally frequent alleles.
To evaluate the sensitivity of our results to the assumptions of our analytic calculations—in particular, the assumption of a random-allele–error model and the presence of exactly one genotyping error per family—we performed computer simulations. Specifically, to assess how well the detection rate for a random-allele–error model approximates the rate for other types of genotyping-error models, we simulated genetic data under three additional genotyping-error models: (i) random-genotype error, (ii) heterozygote-to-homozygote genotype error, and (iii) homozygote-to-heterozygote genotype error. Under random-genotype error, a genotype was randomly replaced by another genotype, in a manner proportional to genotype frequencies under the assumption of Hardy-Weinberg equilibrium. Under heterozygote-to-homozygote genotype error, one of the alleles present in the heterozygous genotype was randomly replaced by the other allele; in practice, PCR-amplification failure might generate errors of this kind. Under homozygote-to-heterozygote genotype error, a homozygous genotype was randomly replaced by an adjacent-allele heterozygous genotype; these types of errors might arise from the presence of stutter bands after PCR-amplification failure. For each of these error mechanisms and under the assumption of Hardy-Weinberg equilibrium, we simulated genotype data, at a single marker, for 10,000 nuclear families, introducing exactly one genotyping error per family. To assess the impact of the assumption of a single genotyping error per family, we simulated, for a subset of the simulations, more than one genotyping error per family; in addition to the first error, genotyping errors were introduced at a rate of 1%–5% for all other family members. Using MENDEL (Sobel et al. 2002 [in this issue]) version 4, we determined the fraction of errors detectable as inheritance inconsistencies for each simulated data set.
Results
Probability of Detectable Error
For two genotyped parents and s=2–9 genotyped offspring, table 3 displays the probability of detection of an error as an inheritance inconsistency, as a function of the number of equally frequent marker alleles and under the assumptions of the random-allele–error model and exactly one genotyping error per family. As expected, the probability of detectable error increases with increasing numbers of alleles. For example, for four-person nuclear families, average rates of detectable error are 34%, 59%, and 77% for markers with 2, 4, and 10 equally frequent alleles, respectively. The rate of detectable error, however, is not always an increasing function of marker heterozygosity H. For example, the rate of detectable error for a biallelic marker is greater when alleles are non–equally frequent (H=0.18) than when alleles are equally frequent (H=0.50); for a four-person nuclear family, the detection rate is 44% (data not shown) instead of 34%, a finding previously reported by Gordon et al. (2000). Note that, in this case, the corresponding rates of detectable error for offspring and parents are 75% and 13%, respectively (data not shown). The rate of detectable error for a marker with seven non–equally frequent alleles (H=0.75) is 63% (data not shown), which is comparable to the rate of 59% for a marker with four equally frequent alleles (H=0.75). Not surprisingly, for small numbers of genotyped offspring, error-detection rates are higher when the error occurs in the genotype of an offspring rather than in the genotype of a parent, since all offspring alleles must be observed in the parents but not vice versa. Moreover, genotyping of additional offspring modestly increases the rate of detectable error when the error occurs in a parent but not when the error occurs in an offspring. For example, for a marker with four equally frequent alleles, the rate of detectable error in parents is 51%, 61%, 71%, and 74% when two, three, six, and nine offspring are genotyped, respectively.
Table 3.
Probability of Detectable Error, Given Two Genotyped Parents[Note]
|
Probability of Detectable Error, When Random-Allele Error Occurs in |
|||
| No. of GenotypedOffspring andNo. of Allelesa | Offspring | Parent | Either |
| 2: | |||
| 2 | .38 | .30 | .34 |
| 4 | .67 | .51 | .59 |
| 10 | .88 | .66 | .77 |
| 3: | |||
| 2 | .38 | .36 | .37 |
| 4 | .67 | .61 | .65 |
| 10 | .88 | .78 | .84 |
| 6: | |||
| 2 | .38 | .45 | .39 |
| 4 | .67 | .71 | .68 |
| 10 | .88 | .88 | .88 |
| 9: | |||
| 2 | .38 | .48 | .39 |
| 4 | .67 | .74 | .68 |
| 10 | .88 | .90 | .89 |
Note.— Assume the random-allele–error model and exactly one genotyping error per family.
Equally frequent alleles.
Table 4 gives the probability of detectable error when parents are not genotyped, as a function of the number of genotyped offspring. The rate of detectable error more than doubles as the number of genotyped offspring increases from three to six. Still, for markers with 75% heterozygosity and families with three to nine genotyped offspring, only 10%–38% of errors are detectable on the basis of inheritance inconsistencies. The rate of detectable error is substantially increased only when the number of genotyped offspring is large (i.e., at least six) and the marker heterozygosity is high (i.e., ⩾0.90). Table 5 gives the probability of detectable error as a function of the number of genotyped parents for s=2–9 genotyped offspring. For multiallelic markers, the rate of detectable error increases notably when the number of genotyped parents increases from zero to one and from one to two; for SNPs, the rate of detectable error more than doubles when the number of genotyped parents increases from one to two.
Table 4.
Probability of Detectable Error, Given No Genotyped Parents[Note]
|
Probability of Detectable ErrorWhen No. of Genotyped Offspring Is |
|||
| No. of Alleles(Heterozygosity) | 3 | 6 | 9 |
| 4a (.75) | .10 | .28 | .32 |
| 7b (.75) | .13 | .34 | .38 |
| 10a (.90) | .31 | .69 | .75 |
Note.— Assume the random-allele–error model and exactly one genotyping error per family.
Equally frequent alleles.
Non–equally frequent alleles (0.40, 0.20, 0.20, 0.05, 0.05, 0.05, and 0.05).
Table 5.
Probability of Detectable Error, Given Two, One, or Zero Genotyped Parents[Note]
|
Probability of Detectable ErrorWhen No. of Genotyped Parents Is |
|||
| No. of GenotypedOffspring andNo. of Allelesa | 2 | 1 | 0 |
| 2: | |||
| 2 | .34 | .14 | .00 |
| 4 | .59 | .25 | .00 |
| 10 | .77 | .32 | .00 |
| 3: | |||
| 2 | .37 | .15 | .00 |
| 4 | .65 | .38 | .10 |
| 10 | .84 | .65 | .31 |
| 6: | |||
| 2 | .39 | .15 | .00 |
| 4 | .68 | .50 | .28 |
| 10 | .88 | .82 | .69 |
| 9: | |||
| 2 | .39 | .14 | .00 |
| 4 | .68 | .51 | .32 |
| 10 | .89 | .82 | .75 |
Note.— Assume the random-allele–error model and exactly one genotyping error per family, with the error equally likely to occur in any family member’s genotype.
Equally frequent alleles.
Probability of Detectable Error: Other Error Models
To assess the impact of assuming a random-allele–error model in our analytic calculations, we simulated genetic data at a single marker and estimated the probability of detectable error under four genotyping-error models. In tables 6 and 7, results are given for nuclear families with two and zero genotyped parents, respectively. For two-genotyped-parent families and multiallelic markers, the probability of detectable error is highest for a random-genotype error, somewhat less for heterozygous-to-homozygous error and random-allele error, and noticeably less for homozygous-to-heterozygous error, whereas, for these same families and biallelic markers, the probability of detectable error is highest for heterozygous-to-homozygous error. For families in which parents are not genotyped, the rates of detectable error are higher under a random-genotype–error model and comparable across allele-error models, with differences that generally are <10%. Note that, in most cases, the probability of detectable error for the random-allele–error model is approximately equal to the average probability over all four error models. Also observe that, under the random-allele–error model, the rates of detectable error, as estimated by simulation (column 3 of tables 6 and 7), are equivalent to and confirm the rates estimated by our analytic calculations (tables 3 and 4), with differences of ⩽1%.
Table 6.
Probability of Detectable Error, under Four Genotyping-Error and Mutation Models, Given Two Genotyped Parents[Note]
|
Probability of Detectable Error When Error Model Is |
||||
| No. of GenotypedOffspring andNo. of Allelesa | Random Genotype | Random Allele | Het→Homb | Hom→Hetc |
| 2: | ||||
| 2 | .45 | .33 | .51 | .07 |
| 4 | .73 | .59 | .70 | .25 |
| 10 | .93 | .78 | .81 | .41 |
| 3: | ||||
| 2 | .48 | .37 | .55 | .08 |
| 4 | .77 | .65 | .74 | .29 |
| 10 | .95 | .84 | .87 | .49 |
| 6: | ||||
| 2 | .50 | .39 | .55 | .11 |
| 4 | .78 | .68 | .75 | .38 |
| 10 | .96 | .88 | .90 | .61 |
| 9: | ||||
| 2 | .49 | .39 | .53 | .11 |
| 4 | .78 | .68 | .74 | .40 |
| 10 | .96 | .88 | .89 | .66 |
Note.— Data were simulated at a single genetic marker for 10,000 nuclear families. According to the specified error model, exactly one genotyping error was introduced into the genotype of either a parent or an offspring (with proportionate probability) for each replicate family.
Equally frequent alleles.
Heterozygous genotype changed to incorrect homozygous genotype.
Homozygous genotype changed to incorrect heterozygous genotype.
Table 7.
Probability of Detectable Error under Four Genotyping-Error and Mutation Models, Given No Genotyped Parents[Note]
|
Probability of Detectable Error When Error Model Is |
||||
| No. of GenotypedOffspring andNo. of Allelesa(Heterozygosity) | Random Genotype | Random Allele | Het→Homa | Hom→Hetb |
| 3: | ||||
| 4c (.75) | .17 | .10 | .10 | .06 |
| 7d (.75) | .23 | .13 | .12 | .12 |
| 10c (.90) | .49 | .30 | .24 | .16 |
| 6: | ||||
| 4c (.75) | .39 | .28 | .27 | .22 |
| 7d (.75) | .48 | .34 | .32 | .31 |
| 10c (.90) | .84 | .68 | .60 | .61 |
| 9: | ||||
| 4c (.75) | .43 | .32 | .31 | .26 |
| 7d (.75) | .52 | .38 | .37 | .36 |
| 10c (.90) | .87 | .74 | .67 | .69 |
Note.— Data were simulated at a single genetic marker for 10,000 nuclear families. According to the specified error model, exactly one genotyping error was introduced into the genotype of one offspring per replicate family.
Heterozygous genotype changed to incorrect homozygous genotype.
Homozygous genotype changed to incorrect heterozygous genotype.
Equally frequent alleles.
Non–equally frequent alleles (0.40, 0.20, 0.20, 0.05, 0.05, 0.05, and 0.05).
Evaluating the Presence of Pedigree Error
Like genotyping errors, pedigree errors or misspecification of familial relationships may be detected on the basis of apparent inconsistencies with Mendelian inheritance. Accurate inference of the correct relationships, however, often requires that many genetic markers be typed in the relevant family members. Hence, in the early stages of a genome scan, it is often difficult to distinguish, on the basis of an observed number of inheritance inconsistencies, between pedigree errors and genotyping errors. However, given a prespecified genotyping-error rate, our analytic results for the rate of detectable error permit assessment of the plausibility of an observed number of inheritance inconsistencies, when it is assumed that relationships are correctly specified. For example, table 8 gives the expected number of families with one or more inheritance inconsistencies, for a sample of 100 four-person nuclear families genotyped for 20 markers, each with seven non–equally frequent alleles (75% heterozygosity), and a genotyping-error rate of 1%–5%. In this case, for a genotyping-error rate of 1%, no more than three inheritance inconsistencies would be expected in any of the 100 families, and even for a 5% genotyping-error rate, no more than six inconsistencies would be expected in any of the 100 families. Thus, under reasonable genotyping-error rates (i.e., ⩽5%), pedigree error, rather than genotyping error, is more likely for any family in the sample displaying more than six inheritance inconsistencies. Note that, in this example, the expected numbers of inheritance inconsistencies per family are 0.49, 1.47, and 2.45 for genotyping-error rates of 1%, 3%, and 5%, respectively.
Table 8.
Expected Number of Families with One or More Inheritance Inconsistencies[Note]
|
Expected No. of Families in Which No. of Inheritance Inconsistencies Is |
||||||||
| Random-Allele–Error Rate | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 1% | 30.52 | 7.25 | 1.09 | .12 | .01 | .00 | .00 | .00 |
| 3% | 35.51 | 24.94 | 11.06 | 3.47 | .82 | .15 | .02 | .00 |
| 5% | 24.69 | 28.34 | 20.54 | 10.54 | 4.07 | 1.23 | .30 | .06 |
Note.— Assume a sample of 100 four-person nuclear families, each with two parents and two offspring genotyped at 20 genetic markers, each with seven non–equally frequent alleles (0.40, 0.20, 0.20, 0.05, 0.05, 0.05, and 0.05). Expected numbers were calculated under the assumptions of a random-allele–error model and no more than one genotyping error per family at each marker.
Discussion
Gene-mapping studies typically rely on checking for the presence of inheritance inconsistencies in a pedigree, through both visual inspections and diagnostic programs (Stringham and Boehnke 1996; O’Connell and Weeks 1998), to identify genotyping errors and mutations. Researchers using these diagnostic checks often explicitly or implicitly assume that all or most genotyping errors are identified, in spite of the fact that little is known about the frequency with which genotyping errors consistent with Mendelian transmission are present in a pedigree. To date, systematic evaluation of the sensitivity of inheritance-error checking has been limited to nuclear families with one to three offspring and biallelic markers (Gordon et al. 1999, 2000). Our analytic investigations suggest that a substantial fraction of errors still may go undetected on the basis of inheritance checking, even for fully genotyped nuclear families and multiallelic markers.
Without question, the probability of detection of genotyping errors and mutations through violations of Mendelian inheritance depends strongly on the number and relationships of genotyped family members, the marker-allele frequency distribution, and the type of errors and in whom they occur. In our study, the number of genotyped parents, for example, had a much greater impact on the rate of detectable error than did the number of genotyped offspring. Furthermore, we found that errors were especially difficult to detect for biallelic markers, even when parents were genotyped, a finding consistent with the results of a study by Gordon et al. (1999, 2000). Even so, for multiallelic markers and families with genotyped parents, average rates of detectable error were never >89%, and most were never >77%. For example, for a typical STRP (H=0.75), an average of 37%–41% of errors were undetectable for fully genotyped four-person nuclear families.
Given incomplete error detection, even for multiallelic markers, it is clear that the true genotyping-error rate will be almost certainly underestimated in any gene-mapping study that relies solely on Mendelian-inheritance checking. For example, suppose that the average rate of detectable error is estimated to be 60% for the markers and families under consideration. Then, any apparent rate of error from inheritance-inconsistency checking would underestimate the true genotyping-error rate by a factor of 1.7. If the apparent error rate were estimated to be 0.5%, 1%, or 2%, then the true error rate would be 0.8%, 1.7%, or 3.3%, respectively. Clearly, the effect of underestimation becomes disconcerting for apparent error rates much greater than 1%.
Given these results, researchers might consider using additional methods, beyond simple inheritance-consistency checking, to assure the quality of genetic data. Depending on factors such as cost and time, partial duplicate genotyping may be one possibility, although this approach cannot detect mutations. Alternatively, multipoint analysis can be a useful strategy, particularly when marker density is high (Douglas et al. 2000). For the sake of illustration, consider a four-person nuclear family genotyped for a map of 21 markers equally spaced at 1-cM intervals. Assume that, at the middle marker, a random-genotype error is introduced into the genotype of a parent or offspring. Using a multipoint approach (Sobel et al. 2002 [in this issue]), we estimate that 50%–79% and 80%–98% of such errors would be detected for markers with two and four equally frequent alleles, respectively, for false-positive rates of 0.001–0.0001 (data not shown). Recall that only 34% and 59% of these errors would be detected on the basis of inheritance inconsistencies for markers with two and four equally frequent alleles, respectively (see table 3). In this case, the rate of error detection more than doubles for biallelic markers and is nearly complete for multiallelic markers.
In our analytic calculations, we assumed exactly one genotyping error per family. To verify the robustness of our findings to this assumption, we allowed, in a subset of our simulations, for more than one genotyping error per family. Specifically, in addition to the first error, genotyping errors were introduced at a rate of 1%–5% for all other family members. For the cases that we considered, the rate of detectable error never increased by >4% (data not shown). This is not surprising, given genotyping-error rates that are ⩽5% and average rates of detection that are ⩽80%. In practice, even with a genotyping-error rate as high as 5%, the probability, at a single marker, of two or more genotyping errors per four-person nuclear family is <1.5%.
To assess the robustness of our analytic calculations to the underlying error model, we simulated genetic data and estimated the probability of detectable error under three additional error models. Not surprisingly, for multiallelic markers, the number of errors detected as inheritance inconsistencies was greater under the random-genotype–error model than under any of the allele-error models. In contrast, rates of error detection were lowest under the homozygous-to-heterozygous error model, regardless of marker type. This latter result can be largely explained by the fact that an error in a homozygous parent that affects only one allele cannot be detected as an inheritance inconsistency in a nuclear family. When parents were not genotyped, rates of detectable error were comparable across all allele-error models, with differences generally being <10%. For biallelic markers, rates of detection depended strongly on both the allele frequency and whether the error occurred in a parent or offspring. For example, when alleles were not equally frequent (0.10, 0.90), random-genotype errors were detected with probability 68% or 17%, depending on whether the error occurred in an offspring or a parent, respectively, of a four-person nuclear family (data not shown). Such large discrepancies are not surprising, given that genotypes were replaced in a manner proportional to genotype frequencies and that all offspring alleles must be observed in the parents but not vice versa. On the basis of these findings, the rate of detectable error under the random-allele–error model, as a first approximation, is likely representative of the variety of error types that might occur in practice.
Accurate inference of many pedigree relationships is possible after much or all of a genome scan has been completed (Boehnke and Cox 1997; Göring and Ott 1997; Epstein et al. 2000; McPeek and Sun 2000). However, when one is faced with inheritance inconsistencies in the early stages of a genome scan, it is often difficult to distinguish between pedigree errors and genotyping errors. At this early stage, which, for large studies, may last many months, a researcher may wish to investigate the likelihood that genotyping errors and mutations could be responsible for an observed number of failures of Mendelian inheritance; if not, he or she may conclude that pedigree error is present. Given our analytic results, it is possible to calculate the approximate distribution of genotyping errors resulting in inheritance inconsistencies for a family or sample, given a set of genotyped markers and a presumed genotyping-error rate. This, in turn, will allow a researcher to assess whether an observed number of marker inconsistencies is plausible if the reported relationships are correct. Pedigree error, rather than genotyping error, is more likely in families in which the observed number of errors is too large. Such families can be either targeted for resampling or excluded from further genotyping.
In conclusion, we have derived analytic formulae and have simulated data to estimate the expected rate at which genotyping errors will appear as inheritance inconsistencies in nuclear families. These results should be valuable for distinguishing between pedigree errors and genotyping errors when genetic data are too scarce to make accurate relationship inferences. These results can also be used to estimate the true genotyping-error rate, on the basis of data for a sample of families. Both applications have the advantage of identifying problems at the earliest point in time, allowing adjustments to be made in the sampling and genotyping strategy. In the long run, such measures can optimize the amount of usable data and, therefore, improve the power of any gene-mapping study.
Acknowledgments
We thank Kenneth Lange for making MENDEL (version 4) available to us. This research was supported, in part, by a University of Michigan Rackham Predoctoral Fellowship (to J.A.D.) and by National Institutes of Health grants T32 HG00040 (to A.D.S.) and R01 HG00376 (to M.B.).
References
- Boehnke M, Cox NJ (1997) Accurate inference of relationships for sib-pair linkage studies. Am J Hum Genet 61:423–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas JA, Boehnke M, Lange K (2000) A multipoint method for detecting genotyping errors and mutations in sibling-pair linkage data. Am J Hum Genet 66:1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehm MG, Kimmel M (1995) Error detection in genetic linkage data for human pedigrees using likelihood ratio methods. J Biol Syst 3:13–25 [Google Scholar]
- Ehm MG, Kimmel M, Cottingham RW Jr (1996) Error detection for genetic data using likelihood methods. Am J Hum Genet 58:225–234 [PMC free article] [PubMed] [Google Scholar]
- Epstein MP, Duren WL, Boehnke M (2000) Improved inference of relationship for pairs of individuals. Am J Hum Genet 67:1219–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewen KR, Bahlo M, Treloar SA, Levinson DF, Mowry B, Barlow JW, Foote SJ (2000) Identification and analysis of error types in high-throughput genotyping. Am J Hum Genet 67:727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Heath SC, Ott J (1999) True pedigree errors more frequent than apparent errors for single nucleotide polymorphisms. Hum Hered 49:65–70 [DOI] [PubMed] [Google Scholar]
- Gordon D, Leal SM, Heath SC, Ott J (2000) An analytic solution to single nucleotide polymorphism error-detection rates in nuclear families: implications for study design. Pac Symp Biocomput 663–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göring HH, Ott J (1997) Relationship estimation in affected sib pair analysis of late-onset diseases. Eur J Hum Genet 5:69–77 [PubMed] [Google Scholar]
- McPeek MS, Sun L (2000) Statistical tests for detection of misspecified relationships by use of genome screen data. Am J Hum Genet 66:1076–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel E, Papp J, Lange K (2002) Detection of genotyping errors. Am J Hum Genet 70:496–508 (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringham HM, Boehnke M (1996) Identifying marker typing incompatibilities in linkage analysis. Am J Hum Genet 59:946–950 [PMC free article] [PubMed] [Google Scholar]
- Thompson EA (1975) The estimation of pairwise relationships. Ann Hum Genet 39: 173–188 [DOI] [PubMed] [Google Scholar]
- Weber JL, Wong C (1993) Mutation of human short tandem repeats. Hum Mol Genet 2:1123–1128 [DOI] [PubMed] [Google Scholar]