

Abstract
Calcific Aortic Valve Disease (CAVD) is increasingly prevalent worldwide with significant morbidity and mortality. Therapeutic options beyond surgical valve replacement are currently limited. In 2011, the National Heart Lung and Blood Institute assembled a working group on aortic stenosis. This group identified CAVD as an actively regulated disease process in need of further study. As a result, the Alliance of Investigators on CAVD was formed to coordinate and promote CAVD research, with the goals of identifying individuals at risk, developing new therapeutic approaches, and improving diagnostic methods. The group is composed of cardiologists, geneticists, imaging specialists, and basic science researchers. This report reviews the current status of CAVD research and treatment strategies with identification of areas in need of additional investigation for optimal management of this patient population.
Keywords: Calcific aortic valve disease, aortic stenosis, inflammation, calcification
Calcific aortic valve disease: Where are we now?
Calcific aortic valve disease (CAVD) is the most common valvular heart disease in the aging population of the developed world with projected disease burden expected to increase from 2.5 million in 2000 to 4.5 million in 2030.[1] In CAVD, the leaflets become thick, stiff, scarred and calcified, often covered with nodules on the surface facing the aorta. In general, symptoms of CAVD are minimal even as the valve narrows into “aortic stenosis (AS),” where pressure overload leads to progressive left ventricular hypertrophy and life-threatening symptoms – angina and/or syncope. This hemodynamic catastrophe results in heart failure and, without intervention, death within months to years. The mainstay of treatment for CAVD is surgical aortic valve replacement (AVR) with a mechanical or bioprosthetic valve.[2] Limitations include increased complications due to anticoagulation and the need for reoperation due to the limited lifespan of prosthetic valves.[3, 4] Transcatheter aortic valve replacement (TAVR) is currently reserved for patients with excess operative risk, but its indications are now expanding due to the “minimally invasive” nature of TAVR compared to surgical AVR.[5] If TAVR becomes more routine, some hope it will reduce the need for repeat surgery. Tissue engineered valve replacement, particularly in pediatric patients, holds great potential, but remains elusive after years of effort. Currently, there are no medical therapies for CAVD as alternatives to surgery, other than for complications, such as antibiotics for infections and vasodilators for acute decompensation.
How do we develop improved treatments for CAVD?
Research now implicates osteogenic processes as key mechanisms in CAVD (Figure)[4], opening two general directions for development of new therapies: 1) inhibition of pro-calcific regulators and 2) induction of pro-resorptive regulators in aortic valve tissue. Tissue targeting may be essential to avoid damage to skeletal bone; for example, if osteoclasts or nanoparticles carrying osteoclastic differentiation factors could be targeted to valve tissue, they may reverse CAVD.[6] However, even if successful, mineral resorption may leave behind damaged leaflets. Molecular imaging of these events may provide biomarkers for clinical and research monitoring far in advance of critical stenosis. Preventive measures that do not require tissue targeting are under consideration, including modification of various factors (dietary phosphorous, vitamins K and D, fetuin, warfarin, bisphosphonates, anti-inflammatory agents, osteopontin, mineralocorticoids, antioxidants, estrogen) and conditions (uremia, metabolic syndrome, hyperglycemia, hyperlipidemia, hypertension). Based on the association of CAVD with hyperlipidemia and atherosclerosis, clinical trials of “statins” were undertaken, but no benefit seen.[7, 8] It remains to be determined whether they may have benefit in earlier disease or in patients with hyperlipidemia. Additionally, while atherosclerosis and CAVD have similar risk factors, they are somewhat poor predictors of CAVD, suggesting that an additional set of unknown factors may contribute to the onset and progression of valve disease.
Figure.
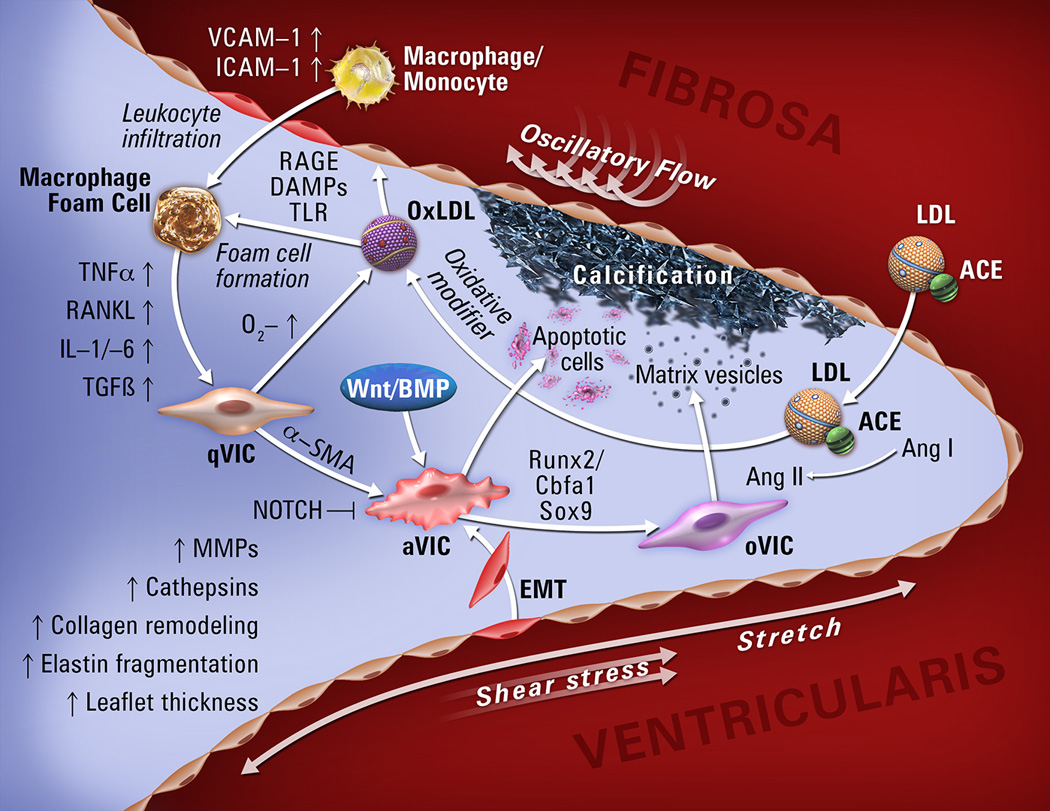
Molecular, cellular, and biomechanical mechanisms in CAVD.
Abbreviations: qVIC, quiescent valve interstitial cell; aVIC, activated valve interstitial cell; oVIC, osteogenic valve interstitial cell; EMT, endothelial-to-mesenchymal transition; MV, matrix vesicles. VCAM-1, vascular adhesion molecule-1; ICAM-1, intracellular adhesion molecule-1; TNF-α, tumor necrosis factor-α; RANKL, receptor activator of nuclear factor kappa-B ligand; IL1/-6, interleukin1/-6; TGF-β, transforming growth factor-β; BMP, bone morphogenic protein; Runx2, runt-related transcription factor 2; ACE, angiotensin-converting enzyme; AngI/II, angiotensinI/II; LDL, low-density lipoprotein.
Whether CAVD is a manifestation of an inflammatory disease or whether inflammation is an associated process occurring secondarily in injured and repairing tissue is not entirely known. Thus, an area of interest remains in investigating whether anti-inflammatory agents may prevent and/or reduce the extent of calcification and osteogenesis, which is fundamentally triggered by inflammatory mediators. To this end, patients with mild-moderate CAVD and coronary artery disease with either diabetes or metabolic syndrome will be evaluated in the ongoing Cardiovascular Inflammation Reduction Trial (CIRT) to examine whether treatment with low dose methotrexate, an effective anti-inflammatory drug, affects progression of CAVD.[9] Evaluation of new therapeutic approaches will require systematically-collected, detailed knowledge of the processes of CAVD that include analysis of normal and diseased human aortic valve tissue, development of suitable animal models, functional assays, and clinical and population studies.
Clues to CAVD mechanisms from human population studies
Several large population cohorts have sought to identify the key epidemiological factors contributing to CAVD. The Cardiovascular Health Study (CHS), focusing on risk factors for coronary artery disease, identified age, male gender and lipoprotein (a) (LPA) levels as showing the strongest correlates of prevalent aortic valve sclerosis and stenosis; hypertension, tobacco use and LDL cholesterol levels gave weaker signals.[10, 11] Advanced age remains the leading risk factor for CAVD. In all age categories aortic valve sclerosis and stenosis were more prevalent in male subjects compared with women.[10] The Multi-Ethic Study of Atherosclerosis (MESA) demonstrated that the prevalence of CAVD was highest in non-Hispanic whites, followed by Hispanics and African Americans[12], and confirmed a significant role for age and male gender with the development of CAVD.[13] By comparison, impaired kidney function is only modestly associated with aortic valve calcification.[14] Metabolic syndrome was independently associated with the development of CAVD[15], potentially due to similar factors participating in their pathogenesis, including circulating ox-LDL and small dense LDL particle phenotype.[15] In addition, metabolic syndrome was independently associated with the progression of aortic stenosis.[16] A polymorphism in the LPA gene associated with serum apo(a) peptide levels has been identified as critical in the pathogenesis of CAVD.[17, 18] In addition, recent investigations have identified Lp-PLA2 expression in stenotic aortic valves and vulnerable plaques. Lp-PLA2 produced within the aortic valve hydrolyzes oxidized phospholipids from lipoproteins which have proinflammatory effects that promote valve mineralization.[19]. Thus this lipoprotein may mediate unique aspects of atherosclerosis and CAVD.
Mechanical, hemodynamic and genetic aspects of BAV and CAVD
Bicuspid aortic valve (BAV) is the most common congenital valvular heart disease (0.6–1.0% of the adult population), the primary cause of ~50% of isolated severe AS requiring AVR, and is frequently associated with aortic aneurysm and aortic dissection.[20] BAV patients, exhibit wide variation in the clinical progression of AS and CAVD. BAV-associated AS commonly requires AVR between the 4th–9th decades of life, although many patients never require surgery. To date, the age of presentation for AVR in BAV patients has not been associated with genetic or environmental factors, nor variants in LPA previously associated with CAVD.[17] Clinical symptoms often presage the diagnosis of BAV, thus complicating studies of the relationship between BAV and CAVD and hindering identification of effective therapies of CAVD. Tenable hypotheses for the markedly increased incidence and severity of early-onset CAVD seen in BAV patients include the abnormal mechanical strain and flow patterns across the BAV that may initiate the cellular processes of CAVD. Alternatively, the genetic or cellular differences that promote BAV may lead to CAVD, independent of the hemodynamic insult.
Although differences in the transcriptional profile and histology of aortic aneurysms between tricuspid and bicuspid aortic valves are well-documented[21], there is no evidence for functional transcriptional differences between bicuspid and tricuspid aortic valves in end-stage CAVD. This implies that CAVD is a common-pathway resulting from initiating insults in BAV, but does not guide us as to these initiating events; be they hemodynamic, cellular or both. This also states the need for examining BAV early in the CAVD process – a difficult field of study. Increased biomechanical stress has been predicted based on modeling of human BAV versus normal tricuspid valves[22], but whether these forces contribute to accelerated CAVD has not yet been determined. Little is known regarding the developmental origins of human BAV, but animal models support the dual importance of embryonic cardiac neural crest cells and the second heart field that could underlie cellular mechanisms of CAVD.[23, 24] These models provide opportunities to test genetic and biological mechanisms of BAV in CAVD, but are limited by differences in anatomy, developmental pathways, and distribution of pathologic calcification between rodents and humans.[25] Thus, incomplete and conflicting evidence from molecular, embryologic, animal model, and clinical studies have not yet resolved the roles of biomechanic and cellular mechanisms.
Osteogenic mechanisms in CAVD
Valve endothelial cells (VECs) likely play a key role in initiation of valve injury. VECs are uniquely positioned to sense hemodynamic forces and encounter systemic circulating factors which in turn play a critical role in the paracrine regulation of valve interstitial cells (VICs).[26] VICs comprise a diverse and highly plastic population of resident cells responsible for remodeling and integrity of extracellular matrix. Various VIC phenotypes have been identified in human heart valves, including quiescent fibroblast-like VICs, which upon pathological cues can differentiate into activated myofibroblast-like VICs, smooth muscle cells located subendothelially and at the base of the leaflet[27, 28] and osteoblast-like VICs, which are responsible for the active deposition of calcium in CAVD (Figure). The initiating processes of CAVD have been proposed to stem from endothelial dysfunction where increased permeability results in the nonspecific diffusion of cytokines and cells into valve leaflets that subsequently initiate a phenotypic switch of quiescent VICs to a myofibroblast-phenotype and thereby promotes aortic valve sclerosis and stiffness.[29] However, the progression from aortic valve sclerosis to AS is not well elucidated, though it appears to relate to active processes of biomineralization and ossification of osteoprogenitor cells derived from multiple sources, including activated VICs, endothelial-mesenchymal transition of VECs, and bone marrow myeloid cells.
Histological studies report the formation of bone nodules in many stenotic aortic valves as a significant contributor to CAVD. Subsequently, regulatory pathways of bone mineralization in the skeletal system have been shown to be activated in CAVD. These include the Wnt/β-catenin and BMP signaling pathways, transcription factors Runx2, Sox9, and Msx2, as well as proteins involved directly in mineralization, such as alkaline phosphatase, Gla proteins, proteoglycans, osteopontin, and bone sialoprotein.[30] Osteogenic gene induction occurs in CAVD of distinct etiologies and may represent a final common pathway in aortic valve mineralization. Inflammation is one of the causes of osteogenic gene induction through the production of cytokines (e.g., TNFα and IL6)[31] and generation of intracellular oxidative stress, which activates both Runx2 and Msx2/Wnt signaling cascades providing a milieu conducive for mineralization and ectopic bone formation.[32] Another important mechanism by which inflammation promotes CAVD is via the formation of mineralizing matrix vesicles. The pro-inflammatory cytokine S100A9, an endogenous agonist of the receptor for advanced glycation endproducts and the toll-like receptor 4, is involved in the secretion of calcifying matrix vesicles from macrophages via formation of a phosphatylserine-annexin5-S100A9-hydroxyapatite complex.[33]
Endocrine mechanisms of CAVD
While vascular calcification and CAVD have many similarities, it is probable that they are not precisely the same in terms of the cell types or inductive mechanisms driving mineralization, which may affect optimization of treatment strategies. Likewise, similarities between CAVD and bone mineralization could be exploited therapeutically. Intriguing differences are emerging in cellular ontogeny and regulation of early-phase CAVD mineralization and skeletal mineralization. Inflammatory oxylipids, innate immune signaling and the integrated tissue response fundamentally differ between skeletal and vascular parenchymal cells with distinct origins. This difference may also extend to endocrine regulation. In diabetic, dyslipidemic LDLR−/− mice, administration of the bone anabolic hormone PTH (parathyroid hormone) increases bone formation while inhibiting arterial calcification.[34] Reductions in oxidative stress, oxylipid production, and serum phosphate signaling by PTH/PTHrP receptor (PTH1R) activation may be important underlying mechanisms.[34] Yet, the net impact of PTH1R signaling on CAVD remains unclear. Although PTH1R is expressed throughout the vasculature, and the risk for CAVD is increased in patients with primary hyperparathyroidism (pHPT),[35] the impact of chronic PTH exposure on valve PTH1R signaling is unknown. Recent studies suggest that PTH is regulated by the renin-angiotensin system (RAS) via angiotensin II that directly stimulates PTH secretion.[36] Moreover, angiotensin-converting enzyme is expressed in human aortic valve stenosis,[37] and angiotensin receptor blockade inhibits aortic valve pathology in hypercholesterolemic rabbits.[38] Clearly, the endocrine and metabolic regulation of CAVD deserves additional inquiry. Strategies focused upon understanding oxylipid signaling, tissue-specific innate immune responses and its endocrine regulation are likely to afford therapeutic opportunities.
Dysregulated mineral metabolism and osteoclast deficiency in CAVD
Patients with end stage kidney disease have accelerated progression of CAVD compared to patients with early stage chronic kidney disease (CKD) or the general population.[39] This increased risk has been attributed to non-traditional risk factors related to mineral imbalances, including abnormalities in serum Pi, Ca and vitamin D that may drive bone formative processes.[40] Indeed, therapies aimed at normalizing mineral and mineral-regulating hormone balance, such as phosphate binders and low phosphate diets, vitamin D receptor agonists, and calcimimetics have been shown to have some benefit in reducing vascular and valvular calcification in human and experimental studies.[41] In fact, in several studies, vascular calcification actually decreased from baseline with treatment, suggesting that mechanisms for ectopic mineral regression do exist.[42, 43] Other factors that may also contribute to the high rate of CAVD in these patients are high levels of phosphate that promote apoptosis via the Pit1 signaling pathway,[44, 45] undercarboxylated Matrix Gla-Protein (MGP) potentially as a result of vitamin K deficiency, fetuin deficiency, and abnormalities in FGF23/klotho and RANK/RANKL/OPG systems.[39]
The RANK/RANKL/OPG system plays a major role in regulating bone resorption. Receptor Activator of NF-κB ligand (RANKL) binding to RANK stimulates osteoclast formation and skeletal bone resorption, while the RANK decoy receptor osteoprotegerin (OPG) inhibits this process. Although bone-like anabolic processes appear to be turned on, there is a paucity of data for active, bone-like catabolic processes in CAVD, since few osteoclasts have been observed in these lesions.[46] Mounting evidence suggests that the inflammatory milieu in CAVD actually favors inhibition of osteoclastogenesis, since OPG is upregulated early in disease progression in valve tissue and in serum, where it predicts mortality in symptomatic CAVD patients.[47] This finding is also true in late stage CKD patients, where OPG levels are elevated and directly correlate with the presence of vascular calcification and the risk of cardiovascular mortality.[48] Together, these data suggest that enhancing osteoclast-like mineral resorptive activities might represent a novel approach to inhibiting and potentially regressing CAVD. In support of this, osteoclasts derived from rat bone marrow were capable of removing mineral from calcified elastin in vitro and limited elastin calcification in rat subdermal implants.[49] Furthermore, new techniques to engineer monocytes that conditionally differentiate into OPG-resistant osteoclasts have been recently developed.[6] Thus, approaches to control osteoclast differentiation and/or locally deliver autologous osteoclasts represent potential novel therapeutic modalities to treat or even regress CAVD.
Developmental signaling and gene regulatory mechanisms in CAVD
Human genetic analyses and experimental models have demonstrated that multiple cell signaling pathways active in valve development also are active in CAVD.[50, 51] The Notch signaling pathway was among the first pathways associated with CAVD based on the association of mutations in human NOTCH1 with BAV and CAVD.[52] Indeed, Notch signaling protects against valve mineralization in studies in mice and cultured VICs.[53, 54] In contrast, Wnt/β-catenin signaling is induced in human CAVD and has been hypothesized to promote progression of disease through activation of osteogenic diferentiation.[55, 56] BMP signaling, evident in increased ligand expression and expression of phospho-Smad1/5/8, is activated in human and mouse CAVD.[57] However, the mechanisms of BMP2 induction are not known, and the potential for BMP signaling inhibition in the treatment of CAVD has not yet been demonstrated. The intersection of Notch and BMP signaling is crucial during the early stages of embryonic valvulogenesis and also in aortic VIC calcification in culture.[53, 58] Thus, multiple signaling pathways linked to valve and/or bone development likely act to promote or inhibit CAVD in distinct valve cell populations under different physiological or genetic conditions.
Mouse models have been useful in identifying specific regulatory mechanisms of CAVD. ApoE−/− and LDLR−/− mice fed a high fat diet model hyperlipidemic cardiovascular disease in the context of cardiovascular inflammation and lipid deposition.[59] Notch signaling-deficient mice exhibit aortic valve calcification, but, unlike humans, there is little or no association with BAV.[54] Klotho-deficient mice exhibit aortic valve calcification associated with premature aging and hyperphosphatemia due to kidney disease.[60] These various mouse models may be useful in dissecting mechanisms of CAVD with distinct causes and progressive mechanisms. Currently the cellular origins of CAVD are not well defined. Thus lineage tracing may reveal the specific cellular origins and contributions to CAVD using mouse models. Such studies will likely be useful in developing new-targeted pharmacological approaches for CAVD based on specific molecular or cellular mechanisms.
MicroRNAs as biomarkers and potential therapeutics for CAVD
MiRNAs control the signaling proteins involved in CAVD and have diagnostic, prognostic, and therapeutic potential.[61] The stable extracellular miRNAs found in plasma, urine, and saliva are excellent biomarkers that reflect the physiological or pathological conditions of specific tissues. Although miRNA expression profiling is being widely investigated in coronary diseases, specific data regarding CAVD is limited.[61] Unique miRNA signatures have been reported within the valves themselves[62, 63] or in aortic aneurysms and plasma from patients with BAV.[64] Distinct flow and side-related miRNA patterns are found when endothelial cells isolated from the fibrosa, which is prone to calcification, are compared with those from the ventricularis side of the valve.[65] Understanding miRNA patterns associated with CAVD resulting from different etiologies may provide predictive clues or biomarkers of the pathophysiology of each patient’s disease. In addition, miRNAs, singly or in combination, could have therapeutic potential through inhibition of signals driving CAVD. For example, the miR-34 family of miRNAs, which repress the pro-calcific BMP2[66], improved heart function in models of cardiomyopathies and could be applied to CAVD treatment. It remains to be determined if miRNA profiles distinguish between patients with the common risk factors for both atherosclerosis and CAVD.
Existing gaps in imaging/characterization of CAVD
By the time CAVD comes to clinical attention, it is often in advanced disease stages. Aortic valve abnormalities are often diagnosed incidentally on echocardiograms performed for unrelated clinical indications. Based on current management guidelines, these subjects are followed with intermittent imaging studies until symptoms appear. This approach is based on the absence of an appropriate medical therapy to slow or reverse the disease progression in asymptomatic subjects. The development of an approach to identify the subjects who are at high risk for CAVD can facilitate the development and validation of such medical therapies and help track the effectiveness of therapeutic interventions in individual subjects. Besides detecting the extent of calcification, a modest determinant of progression,[13] classical imaging modalities (e.g., echocardiography) do not provide any information on CAVD propensity for progression. In addition, long clinical trials with interventional monitoring of disease progression are largely impractical. By detecting and quantifying the earliest molecular and cellular events involved in CAVD development, molecular imaging can address this important gap in clinical care. Furthermore, molecular imaging can help address aspects of biology that are not readily detectable through common biological techniques, and thus advance our understanding of basic CAVD pathobiology. The feasibility of molecular imaging of CAVD in humans has been shown in a few recent clinical studies using FDA-approved radiotracers, 18F-fluorodeoxyglucose (FDG)[67] and 18F sodium fluoride (NaF).[68] In parallel to inflammation and calcification, it is important to focus on other relevant aspects of valvular pathobiology (e.g., proteolytic activity), which could provide complementary information regarding the pathogenesis and progression of CAVD.[69, 70]
Unanswered questions and future directions
Despite recent progress in clinical, genetic, and animal studies, many important questions remain in terms of optimal diagnosis, treatment strategies, and mechanistic understanding of CAVD. The causes of CAVD are not fully understood, but valve pathogenesis can occur with congenital malformation, atherosclerotic disease, advanced kidney disease, diabetes, or aging. Additional factors that influence the incidence of CAVD include sex and race, but exactly how they affect CAVD initiation or progression is unknown. It seems likely that the molecular mechanisms and cellular contributions of CAVD differ depending on the initial pathogenic stimulus and comorbidities. For example, the specific localization and molecular progression of CAVD resulting from hyperphosphatemia versus that resulting from inflammation may not be the same. For congenitally malformed valves, such as BAV, biomechanics may be a driving force in the disease process, but the specific stimuli and molecular mechanisms by which this occurs are in need of further investigation. The relationship between vascular and valvular calcification is unclear; each may result from different etiologies and molecular mechanisms depending on patient comorbidities. In addition, it is recognized that, in the human population, it is likely that an individual may have multiple risk factors for CAVD, further affecting the heterogeneity of disease manifestation and progression, making determination of optimal treatment strategies a challenge.
It is difficult to identify patients at risk for CAVD before symptom onset, but the population of patients with congenital BAV, who are highly vulnerable to CAVD, may serve as a valuable study group. The current standard of care is surgical valve replacement, but this is usually a temporary solution. Thus, development of improved bioprosthetic valves and methods of delivery such as TAVR, though costly, are priorities. Current clinically important questions include the determination of when a stenotic valve should be replaced and how best to manage the risk of replacement. Emerging molecular imaging technologies and identification of biomarkers that detect the earliest stages of valve mineralization will be important in the early diagnosis and monitoring of progression of CAVD. Studies in animal models support the reversibility of CAVD in some cases. With improved diagnosis and mechanistic insights into CAVD, the development of new therapeutic approaches that will eliminate the need for surgical replacement in patients with CAVD may be possible.
Significance.
Calcific aortic valve disease (CAVD) is the most common valvular heart disease in the aging population with a disease burden of millions worldwide. Despite recent progress in clinical, genetic, and animal studies, many important questions remain in terms of optimal diagnosis, treatment strategies, and mechanistic understanding of CAVD. Inflammatory, endocrine, osteogenic, biomechanic, and developmental factors contribute to CAVD progression and heterogeneity related to the initial pathogenic stimulus and comorbidities. Clinically important questions include the determination of when a stenotic valve should be replaced and how best to manage the risk of replacement. Emerging molecular imaging technologies and identification of biomarkers that detect the earliest stages of valve mineralization will be important in the early diagnosis and monitoring of progression of CAVD. With improved diagnosis and mechanistic insights into CAVD, the development of new therapeutic approaches that will eliminate the need for surgical replacement in patients with CAVD may be possible.
Acknowledgements
We thank Frank Evans of the NHLBI for his valuable suggestions.
Sources of Funding
R01 HL114682 to K.E.Y., R01 HL114709 to L.L.D., R01 HL114823 to S.C.B., R01 HL114794 to G.S.H., R01 HL114806 to D.A.T., R01 HL114611 to C.M.G., R01 HL114821 to M.A.H., R01 HL114751 to D.P.M. and M.B.R., R01 HL114703 to M.M.S., and R01 HL114805 to E.A.
Abbreviations
- AS
aortic stenosis
- AVR
aortic valve replacement
- BAV
bicuspid aortic valve
- CAVD
calcific aortic valve disease
- ECM
extracellular matrix
- LPA
lipoprotein (a)
- TAVR
transcatheter aortic valve replacement
- VIC
valvular interstitial cell
- VEC
valvular endothelial cell
Footnotes
Disclosures: D.A.T is a paid consultant for Eli Lilly and Merck & Co.
References
- 1.Aikawa E, Schoen FJ. In: Calcific and degenerative heart valve disease, in Cellular and molecular pathobiology of cardiovascular disease. Stone J, Homeister JM, Willis MS, editors. Elsevier; 2014. pp. 161–181. [Google Scholar]
- 2.Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP, Guyton RA, O'Gara PT, Ruiz CE, Skubas NJ, Sorajja NJ, Sundt TM, Thomas J. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association task force on practical guidelines. Circulation. 2014;129:2440–2492. doi: 10.1161/CIR.0000000000000029. [DOI] [PubMed] [Google Scholar]
- 3.Lindman BR, Bonow RO, Otto CM. Current management of calcific aortic stenosis. Circ Res. 2013;113:223–237. doi: 10.1161/CIRCRESAHA.111.300084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O'Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: Not simply a degenerative process. Circulation. 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desai CS, Roselli EE, Svensson LG, Bonow RO. Transcatheter aortic valve replacement: Current status and future directions. Semin Thoracic Surg. 2013;25:193–196. doi: 10.1053/j.semtcvs.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Rementer CW, Wu M, Buranaphatthana W, Yang HY, Scatena M, Giachelli CM. An inducible, ligand-independent receptor activator of NF-kB gene to control osteoclast differentiation from monocytic precursors. PLoS One. 2013;8:e84465. doi: 10.1371/journal.pone.0084465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 8.Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121:306–314. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- 9.Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, Zaharris E, Gupta M, Clearfield M, Libby P, Hasan AA, Glynn RJ, Ridker PM. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199–207. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM. Clinical factors associated with calcific aortic valve disease. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 11.Arsenault BJ, Boekholdt SM, Dube MP, Rheaume E, Wareham NJ, Khaw KT, Sandhu MS, Tardif JC. Lipoprotein(a) levels, genotype and incident aortic valve stenosis. Circ Cardiovasc Genet, 2014;7:304–310. doi: 10.1161/CIRCGENETICS.113.000400. [DOI] [PubMed] [Google Scholar]
- 12.Nasir K, Katz R, Takasu J, Shavelle DM, Detrano R, Lima JA, Blumenthal RS, O'Brien KD, Budoff MJ. Ethnic differences between extra-coronary measures on cardiac computed tomography: multi-ethnic study of atherosclerosis (MESA) Atherosclerosis. 2008;198:104–114. doi: 10.1016/j.atherosclerosis.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Owens DS, Katz R, Takasu J, Kronmal R, Budoff MJ, O'Brien KD. Incidence and progression of aortic valve calcium in the Multi-ethnic Study of Atherosclerosis (MESA) Am J Cardiol. 2010;105:701–708. doi: 10.1016/j.amjcard.2009.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ix JH, Shlipak MG, Katz R, Budoff MJ, Shavelle DM, Probstfield JL, Takasu J, Detrano R, O'Brien KD. Kidney function and aortic valve and mitral annular calcification in the Multi-Ethnic Study of Atherosclerosis (MESA) Am J Kidney Dis. 2007;50:412–420. doi: 10.1053/j.ajkd.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 15.Katz R, Budoff MJ, Takasu J, Shavelle DM, Bertoni A, Blumenthal RS, Ouyang P, Wong ND, O'Brien KD. Relationship of metabolic syndrome with incident aortic valve calcium and aortic valve calcium progression: the Multi-Ethnic Study of Atherosclerosis (MESA) Diabetes. 2009;58:813–819. doi: 10.2337/db08-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Briand M, Lemieux I, Dumesnil JG, Mathieu P, Cartier A, Despres JP, Arsenault M, Couet J, Pibarot P. Metabolic syndrome negatively influences disease progression and prognosis in aortic stenosis. J Am Coll Cardiol. 2006;47:2229–2236. doi: 10.1016/j.jacc.2005.12.073. [DOI] [PubMed] [Google Scholar]
- 17.Thanassoulis G, Campbell C, Owens D, Smith J, Smith A, Peloso G, Kerr K, Pechlivanis S, Budoff M, Harris T, Malhotra R, O'Brien K, Kamstrupp P, Nordestgaard B, Tybjaerg-Hansen A, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamstrup P, Tybjaerg-Hansen A, Nordestgaard B. Elevated liproprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 19.Mahmut A, Boulanger M, El Husseini D, Fournier D, Bouchareb R, Despres J, Pibarot P, Bosse Y, Mathieu P. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease. J Am Coll Cardiol. 2014;63:460–469. doi: 10.1016/j.jacc.2013.05.105. [DOI] [PubMed] [Google Scholar]
- 20.Lemaire S, McDonald M, Guo D, Russell L, Miller C, Johnson R, Bekheirnia M, Franco L, Nguyen M, Pyeritz R, Bavaria J, Devereux R, Maslen C, Holmes K, Eagle K, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leone O, Biagini E, Pacini D, Zagnoni S, Ferlito M, Graziosi M, Di Bartolomeo R, Rapezzi C. The elusive link between aortic wall histology and echocardiographic anatomy in bicuspid aortic valve: implications for prophylactic surgery. Eur J Cardiothorac Surg. 2012;41:322–327. doi: 10.1016/j.ejcts.2011.05.064. [DOI] [PubMed] [Google Scholar]
- 22.Conti CA, Della Corte A, Votta E, Del Viscovo L, Bancone C, De Santo LS, Redaelli A. Biomechanical implications of the congenital bicuspid aortic valve: a finite element study of aortic root function from in vivo data. J Thorac Cardiovasc Surg. 2010;140:890–896. doi: 10.1016/j.jtcvs.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Dyer L, Kirby M. The role of secondary heart field in cardiac development. Dev Biol. 2009;336:137–144. doi: 10.1016/j.ydbio.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirby M, Waldo K. Role of neural crest in congenital heart disease. Circulation. 1990;82:332–340. doi: 10.1161/01.cir.82.2.332. [DOI] [PubMed] [Google Scholar]
- 25.Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. Int J Inflam. 2011;2011:364310. doi: 10.4061/2011/364310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gould ST, Srigunapalan S, Simmons CA, Anseth KS. Hemodynamic and cellular response feedback in calcific aortic valve disease. Circ Res. 2013;113:186–197. doi: 10.1161/CIRCRESAHA.112.300154. [DOI] [PubMed] [Google Scholar]
- 27.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 28.Latif N, Sarathchandra P, Chester A, Yacoub M. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur Heart J. 2014 doi: 10.1093/eurheartj/eht547. In press. [DOI] [PubMed] [Google Scholar]
- 29.Poggianti E, Venneri L, Chubuchny V, Jambrik Z, Baroncini LA, Picano E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J Am Coll Cardiol. 2003;41:136–141. doi: 10.1016/s0735-1097(02)02622-0. [DOI] [PubMed] [Google Scholar]
- 30.Bostrom K, Rajamannan NM, Towler DA. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res. 2011;109:564–577. doi: 10.1161/CIRCRESAHA.110.234278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galeone A, Paparella D, Colucci S, Grano M, Brunetti G. The role of TNF-a and TNF superfamily members in the pathogenesis of calcific aortic valvular disease. Scientific World Journal. 2013;2013:875363. doi: 10.1155/2013/875363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-derived matrix vesicles: an alternative mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113:72–77. doi: 10.1161/CIRCRESAHA.113.301036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng S, Shao J, Halstead L, Distelhorst K, Sierra O, Towler D. Activation of vascular smooth muscle parathyroid hormone receptor inhibits Wnt/beta-catenin signaling and aortic fibrosis in diabetic arteriosclerosis. Circ Res. 2010;107:271–282. doi: 10.1161/CIRCRESAHA.110.219899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwata S, Walker MD, Di Tullio MR, Hyodo E, Jin Z, Liu R, Sacco RL, Homma S, Silverberg SJ. Aortic valve calcification in mild primary hyperparathyroidism. J. Clin Endocrinol Metab. 2012;97:132–137. doi: 10.1210/jc.2011-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown JM, Vaidya A. Interactions between adrenal-regulatory and calcium-regulatory hormones in human health. Curr Opin Endocrinol Diabetes Obes. 2014;21:193–201. doi: 10.1097/MED.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Brien KD, Shavelle DM, Caulfield MT, McDonald TO, Olin-Lewis K, Otto CM, Probstfield JL. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106:2224–2230. doi: 10.1161/01.cir.0000035655.45453.d2. [DOI] [PubMed] [Google Scholar]
- 38.Arishiro K, Hoshiga M, Negoro N, Jin D, Takai S, Miyazaki M, Ishihara T, Hanafusa T. Angiotensin receptor-1 blockade inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hyperchlolesterolemic rabbits. J Am Coll Cardiol. 2007;49:1482–1489. doi: 10.1016/j.jacc.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 39.Rattazzi M, Bertacco E, Del Vecchio A, Puato M, Faggin E, Pauletto P. Aortic valve calcification in chronic kidney disease. Nephrol Dial Transplant. 2013;28:2968–2976. doi: 10.1093/ndt/gft310. [DOI] [PubMed] [Google Scholar]
- 40.Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697–711. doi: 10.1161/CIRCRESAHA.110.234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu M, Rementer C, Giacelli CM. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int. 2013;93:365–373. doi: 10.1007/s00223-013-9712-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chertow GM, Burke SK, Raggi P. Sevelamer attenuates the progression of coronary and aortic calcification. Kidney Int. 2002;62:245–252. doi: 10.1046/j.1523-1755.2002.00434.x. [DOI] [PubMed] [Google Scholar]
- 43.Raggi P, Bommer J, Chertow GM. Valvular calcification in hemodialysis patients randomized to calcium based phosphorus binders or sevelamer. J Heart Valve Dis. 2004;13:134–141. [PubMed] [Google Scholar]
- 44.Salaun C, Leroy C, Rousseau A, Boitez V, Beck L, Friedlander G. Identification of a novel transport-independent function of PiT1/SLC20A1 in the regulation of TNF-induced apotosis. J Biol Chem. 2010;285:34408–34418. doi: 10.1074/jbc.M110.130989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El Husseini D, Boulanger M, Fournier D, Mahmut A, Bosse Y, Pibarot P, Mathieu P. High expression of the Pi-transporter SLC20A1/Pit1 in calcific aortic valve disease promotes mineralization through regulation of Akt-1. PLoS One. 2013;8:e53393. doi: 10.1371/journal.pone.0053393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of 'degenerative' valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 47.Ueland T, Aukrust P, Dahl CP, Husebye T, Solberg OG, Tonnessen T, Aakhus S, Gullestad L. Osteoprotegerin levels predict mortality in patients with symptomatic aortic stenosis. J Intern Med. 2011;270:452–460. doi: 10.1111/j.1365-2796.2011.02393.x. [DOI] [PubMed] [Google Scholar]
- 48.Sigrist MK, Levin A, Er L, McIntyre CW. Elevated osteoprotegerin is associated with all-cause mortality in CKD stage 4 and 5 patients in addition to vascular calcification. Nephrol Dial Transplant. 2009;24:3157–3162. doi: 10.1093/ndt/gfp253. [DOI] [PubMed] [Google Scholar]
- 49.Simpson CL, Lindley S, Eisenberg C, Basalyga DM, Starcher BC, Simionescu DT, Vyavahare NR. Toward cell therapy for vascular calcification: osteoclast-mediated demineralization of calcified elastin. Cardiovasc Pathol. 2007;16:29–37. doi: 10.1016/j.carpath.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Wirrig EE, Yutzey KE. In: Developmental pathways in CAVD, in Calcific Aortic Valve Disease. Aikawa E, editor. InTech: Croatia; 2013. pp. 59–104. [Google Scholar]
- 51.Wirrig EE, Yutzey KE. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arterioscler Thromb Vasc Biol. 2014;34:737–741. doi: 10.1161/ATVBAHA.113.302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 53.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nus M, MacGrogan D, Martinez-Poveda B, Benito Y, Casanova JC, Fernandez-Aviles F, Bermejo J, de la Pompa JL. Diet-induced aortic valve disease in mice haploinsufficient for the Notch pathway effector RBPJK/CSL. Arterioscler Thromb Vasc Biol. 2011;31:1580–1588. doi: 10.1161/ATVBAHA.111.227561. [DOI] [PubMed] [Google Scholar]
- 55.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. 2005;112:I229–I234. doi: 10.1161/01.CIRCULATIONAHA.104.524306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, Jo H. Preferential activation of SMAD1/5/8 on the fibrosa endothelium in calcified human aortic valves - Association with low BMP antagonists and SMAD6. PLoS One. 2011;6:e20969. doi: 10.1371/journal.pone.0020969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, Adams RH, Perez-Pomares JM, de la Pompa JL. Integration of a Notch-dependent mesenchymal gene program and BMP2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. 2010;120:3493–3507. doi: 10.1172/JCI42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: Methods, models, and mechanisms. Circ Res. 2011;108:1392–1412. doi: 10.1161/CIRCRESAHA.110.234138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheek JD, Wirrig EE, Alfieri CM, James JF, Yutzey KE. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. J Mol Cell Cardiol. 2012;52:689–700. doi: 10.1016/j.yjmcc.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goettsch C, Hutcheson JD, Aikawa E. MicroRNA in cardiovascular calcification: focus on targets and extracellular vesicle delivery mechanisms. Circ Res. 2013;112:1073–1084. doi: 10.1161/CIRCRESAHA.113.300937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nigam V, Sievers HH, Jensen BC, Sier HA, SImpson PC, Srivastava D, Mohamed SA. Altered microRNAs in bicuspid aortic valve: a comparison between stenotic and insufficient valves. J Heart Valve Dis. 2010;19:459–465. [PMC free article] [PubMed] [Google Scholar]
- 63.Yanagawa B, Lovren F, Pan Y, Garg V, Quan A, Tang G, Singh KK, Shukla PC, Kalra NP, Peterson MD, Verma S. miRNA-141 is a novel regulator of BMP-2-mediated calcification in aortic stenosis. J Thorac Cardiovasc Surg. 2012;144:256–262. doi: 10.1016/j.jtcvs.2011.10.097. [DOI] [PubMed] [Google Scholar]
- 64.Ikonomidis JS, Ivey CR, Wheeler JB, Akerman AW, Rice A, Patel RK, Stroud RE, Shah AA, Hughes CG, Ferrari G, Mukherjee R, Jones JA. Plasma biomarkers for distinguishing etiologic subtypes of thoracic aortic aneurism disease. J Thorac Cardiovasc Surg. 2013;145:1326–1333. doi: 10.1016/j.jtcvs.2012.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holliday CJ, Ankeny RF, Jo H, Nerem RM. Discovery of shear-and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301:H856–H867. doi: 10.1152/ajpheart.00117.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fotinos A, Nagarajan N, Martins AS, Fritz DT, Garsetti DE, Lee AT, Hong CC, Rogers MB. Bone Morphogenetic Protein-focused strategies to induce cyototoxicity in lung cancer cells. Anticancer Research. 2014;34:2095–2104. [PMC free article] [PubMed] [Google Scholar]
- 67.Marincheva-Savcheva G, Subramanian S, Qadir S, Figueroa A, Truong Q, Vijayakumar J, Brady TJ, Hoffmann U, Tawakol A. Imaging of the aortic valve using fluorodeoxyglucose position emission tomography increased valvular fluorodeoxyglucose uptake in aortic stenosis. J Am Coll Cardiol. 2011;57:2507–2515. doi: 10.1016/j.jacc.2010.12.046. [DOI] [PubMed] [Google Scholar]
- 68.Dweck MR, Jone C, Joshi NV, Fletcher AM, Richardson H, White A, Marsden M, Pessotto R, Clark JC, Wallace WA, Salter DM, McKillop G, van Beek EJ, Boon NA, Rudd JH, Newby DE. Assessment of valvular fluorodeoxyglucose uptake in aortic stenosis. Circulation. 2012;125:76–86. doi: 10.1161/CIRCULATIONAHA.111.051052. [DOI] [PubMed] [Google Scholar]
- 69.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 70.Aikawa E, Otto CM. Look more closely at the valve: imaging calcific aortic valve disease. Circulation. 2012;125:9–11. doi: 10.1161/CIRCULATIONAHA.111.073452. [DOI] [PubMed] [Google Scholar]