

Abstract
CK2 (official acronym for casein kinase 2 or II) is a potent suppressor of apoptosis in response to diverse apoptotic stimuli —thus its molecular downregulation or activity inhibition results in potent induction of cell death. CK2 downregulation is known to impact mitochondrial apoptotic circuitry but the underlying mechanism(s) remain unclear. Utilizing prostate cancer cell lines subjected to CK2-specific inhibitors which cause loss of cell viability, we have found that CK2 inhibition in cells causes rapid early decrease in mitochondrial membrane potential (Δψm). Cells treated with the CK2 inhibitors TBB (4,5,6,7-tetrabromobenzotriazole) or TBCA (tetrabromocinnamic acid) demonstrate changes in Δψm which become apparent within 2 h, i.e., significantly prior to evidence of activation of other mitochondrial apoptotic signals whose temporal expression ensues subsequent to loss of Δψm. Further, we have demonstrated the presence of CK2 in purified mitochondria and it appears that the effect on Δψm evoked by inhibition of CK2 may involve mitochondrial localized CK2. Results also suggest that alterations in Ca2+ signaling may be involved in the CK2 mediated regulation of Δψm and mitochondrial permeability. Thus, we propose that a key mechanism of CK2 impact on mitochondrial apoptotic circuitry and cell death involves early loss of Δψm which may be a primary trigger for apoptotic signaling and cell death resulting from CK2 inhibition.
Keywords: Apoptosis, mitochondrial membrane potential, mitochondrial permeability transition, prostate cancer, signaling
Protein kinase CK2 is a highly conserved and ubiquitous protein Ser/Thr kinase consisting of two catalytic subunits α and α′ and two regulatory β subunits with the catalytic subunits linked through the β subunits. CK2 has been found to play a role in a vast number of normal and abnormal cell functions and has emerged as a key cellular regulator with a large number of substrates [Ahmed, 1999; Guerra and Issinger, 1999; Meggio and Pinna, 2003; Pinna, 2002; St-Denis and Litchfield, 2009; Tawfic et al., 2001]. In particular, much work has been undertaken regarding its function in cancer pathobiology and it is remarkable that CK2 has been found to be uniformly dysregulated in all cancers examined [Ahmed et al., 2000; Guerra and Issinger, 2008; Tawfic et al., 2001; Trembley et al., 2009]. While it was known for a long time that CK2 was elevated in rapidly growing normal and cancer cells, its role in cancer cells was unclear. We demonstrated that CK2, in addition to its role in cell growth and proliferation, is also a potent suppressor of apoptosis; further, the CK2α catalytic subunit is responsible for the majority of the apoptosis suppression [Ahmad et al., 2008; Ahmed et al., 2002; Guo et al., 2001; Tawfic et al., 2001]. Thus, the latter characteristic of CK2 provided an important link of this kinase to the cancer cell phenotype as continued cell proliferation and resistance to cell death are two consistent features of cancer cell biology [Hanahan and Weinberg, 2011].
Much evidence has suggested that elevated CK2 levels have a broad role in cell death suppression mediated through diverse signals [Ahmad et al., 2008]. In this context, we demonstrated that one mode of CK2 mediated suppression of apoptosis involves the mitochondrial apoptotic circuitry. In these studies, we observed that downregulation of CK2 resulted in upregulation of Bax and downregulation of Bcl-2 and Bcl-xL accompanied with release of cytochrome c. These events were completely blocked when forced overexpression of CK2α was instituted [Wang et al., 2005a; Wang et al., 2006]. In subsequent studies it was observed that downregulation of CK2 activity or expression in prostate cancer cells for 6–24 h either by chemical inhibition or use of antisense to CK2α resulted in production of H2O2 hinting that this may initiate apoptotic signaling under these conditions [Ahmad et al., 2006]. However, the mechanism underlying the induction of apoptosis following downregulation of CK2 is not fully understood, especially with respect to the earliest events that occur upon inhibition of CK2.
Here we have examined the effect of CK2 inhibition on mitochondrial function and observed that, following the treatment of cells with a chemical inhibitor of CK2, a change in the mitochondrial membrane potential (Δψm) is detected as early as 2 h, thus occurring significantly prior to activation of other apoptotic signals. While CK2 is known to be localized in the nuclear and cytoplasmic fractions, we have also identified its presence in prostate cell mitochondria. Further, our results provide evidence for the first time suggesting that inhibition of mitochondrial CK2 may be involved in causing rapid loss of Δψm as a primary event that triggers the mitochondrial apoptotic circuitry under these conditions.
Materials and Methods
Materials
Sources of various chemicals and reagents used in these studies are as follows: BAPTA (Calbiochem); BSA (Thermo Fisher); Dulbecco’s PBS (Gibco Life Technologies); bovine liver catalase (Sigma Aldrich); CCCP (Calbiochem); CellTiter 96® Aqueous One solution (Promega); FBS (Atlanta Biologicals); oligomycin (Sigma Aldrich); Matrigel® (BD Biosciences); TBB, TBCA and thapsigargin (EMD Millipore); trypsin + EDTA (Gibco Invitrogen); and JC-1 (Life Technologies). The following antibodies were used for western blot analysis: AKT-1 phospho-Ser129 (1:1000, Epitomics 5508-1); AKT-1 (1:1000, Cell Signaling 9272); actin (1:1000, Santa Cruz sc-1616); cleaved caspase-3 (1:1000, Cell Signaling 9661); caspase-9 (1:1000, Cell Signaling 9508); lamin A/C (1:1000, Cell Signaling 2032); CKII α (1:3000, Bethyl Laboratories A300-197); CKII α′ (1:3000, Bethyl Laboratories A300-199); cytochrome c (1:10,000, Epitomics 2119-1); Bax (1:1000, Cell Signaling 2772); Bid (1:1000, Cell Signaling 2002); and Cox IV (1:1000, Cell Signaling 4850).
Cell culture
The cell lines employed were PC3-LN4, LNCaP and C4-2 (human prostate cancer cell lines) and BPH-1 (human benign prostate epithelial cell line), as described previously [Slaton et al., 2004]. PC3-LN4 cells were maintained in RPMI 1640 media with 5% FBS, 2 mM glutamine, and 1% penicillin-streptomycin (P-S), whereas LNCaP, C4-2 and BPH-1 cells were maintained in RPMI 1640 with 10% FBS, 2 mM glutamine, and 1% P-S [Trembley et al., 2012].
Cell fractionation
Cell pellets were suspended gently in 9 packed cell volumes of homogenization buffer A1 (10 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 25 mM KCl, 0.25 M sucrose) with phosphatase and protease inhibitors added at 1:200 just before use (Sigma Aldrich: P5726, P8340). The suspension was incubated for 10 min on ice to promote cell swelling after which the cells were ruptured using a Dounce homogenizer using 9 strokes with an “A” pestle. The suspension was centrifuged at 12,000 × g for 30 min at 4 °C to remove the mitochondria. The supernatant (cytosolic fraction) was subjected to a second centrifugation at 12,000 × g for 30 min at 4 °C. The final supernatant was filtered through a 0.2 μm Ultrafree MC filter (Millipore) by centrifuging at 12,000 × g for 2 min at 4 °C. Aliquots were flash frozen in liquid nitrogen.
Isolation of purified mitochondria and analysis of mitochondrial membrane permeability
Preparation of mitochondria from cultured prostate cells was carried out according the manufacturer’s instructions (Pierce 89874). Preparation and purification of rat liver mitochondria was performed according to a previously described procedure [Schnaitman and Greenawalt, 1968]. Analysis of mitochondrial permeability changes was carried out as described [Savage et al., 1991] utilizing the purified mitochondrial preparation resuspended in a medium consisting of 213 mM D-mannitol, 71 mM sucrose, and 3 mM HEPES buffer (pH 7.4). Details of conditions used for analysis of mitochondrial swelling are outlined in the legend for Fig. 5.
Fig. 5. Effect of CK2 inhibitors on membrane permeability transition in isolated mitochondria.
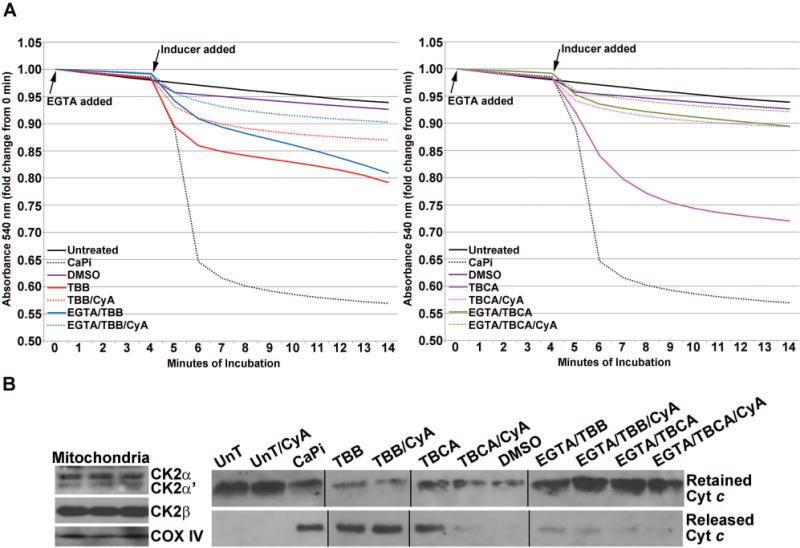
(A) Effect of TBB (left panel) or TBCA (right panel) treatment on mitochondrial permeability transition. Purified mitochondria were subjected to various treatments as shown in the graph legends. Mitochondrial respiration was maintained using 10 mM Na-succinate as the substrate. When used, EGTA or cyclosporin A was added at time zero; “inducers” were added at the start of the 5th min after mitochondria equilibration in the incubation medium and baseline absorbance (540 nm) had been captured for 4 min. Absorbance was measured for an additional 10 min following the addition of the various chemicals as indicated. The concentrations of various agents were: 80 μM TBB; 80 μM TBCA; 70 μM Ca2+, 3 mM Pi; 0.5 μM cyclosporin A; 150 μM EGTA. A volume of DMSO equivalent to the volume of TBB/TBCA was used. All other details are as described under Materials and Methods. (B) Left panel, rat CK2α, CK2α′, CK2β′ and COX IV were detected in all preparations of rat liver mitochondria by western blot analysis; 3 representative preparations are shown. Right panel, cytochrome c that was retained in the mitochondria or released in the medium corresponding to the various treatments under A (left and right panels) was detected by western blot analysis.
Western blot analysis
Whole cell and mitochondrial lysates prepared using RIPA buffer [Trembley et al., 2012] and cytosolic fractions in buffer A1 (50 μg) were subjected to SDS polyacrylamide gel electrophoresis using Tris-Glycine Laemmli gels. Proteins were transferred onto nitrocellulose membrane and 5% non-fat dairy milk in TBS/0.1% Tween 20 was used for blocking and antibody incubations.
Cell viability assay
CellTiter 96® Aqueous One Assay was used to assess cell viability following various treatments. Cells were plated in 96-well plates (4000 cells/well) and allowed to attach overnight. Time course experiments were performed with incubation of cells in complete media with 8 or 80 μM TBB for 2, 4, 6, 24 or 48 h. For experiments with TBCA, concentrations of 1, 10, 20, 40, and 80 μM were applied for 24 and 48 h. Controls included untreated and DMSO treated cells. Aqueous One assay solution was combined with complete media at a ratio of 100 μ of media plus 20 μl of the assay solution per well, and cells were incubated for 3 h at 37 °C. Absorbance was measured at 490 and 700 nm using a Molecular Devices 5 plate reader with absorbance values for media alone subtracted from the experimental values.
Crystal violet clonal survival assays
Cells were treated with 8 or 80 μM TBB or equivalent volume of DMSO for 4, 6, 8, and 24 h. Cells were trypsinized at the end of treatment period and replated (in triplicate) using complete fresh media at a concentration of 1000 cells per 35 mm plate for 7 d (media was replaced at 4 days). On day 7 cells were stained with crystal violet (1× PBS containing 1% (v/v) methanol, 1% (v/v) formaldehyde and 0.05% (w/v) crystal violet) for 20 min, the stain removed, and plates washed by immersion in water with continuous water flow. Plates were air-dried, colonies (containing at least 50 cells/colony) were counted, and plates were scanned. The data shown represent the results of 3 replicates per experiment and a minimum of three experiments.
Coating of cover slips with Matrigel
Individual sterile cover slips (22 × 22 mm) were placed in 6-well tissue culture plates and covered with 750 μl of sterile filtered Matrigel® (333 μg/ml; BD Biosciences 354234) in 1× PBS. Open plates were placed overnight to dry in a laminar flow biosafety cabinet. Plates were then sealed with paraffin film.
JC-1 fluorescence imaging
JC-1 was employed as a marker for studying Δψm changes. Cells were plated on cover slips coated with Matrigel® in 6-well plates to reach 60–70% confluence the next day. Each well was treated for 2, 4, 6, or 24 h with TBB or TBCA at varying concentrations as indicated in the figure legends. All experiments included cells treated with equivalent volumes of DMSO or untreated as controls. When used, CCCP (10 μM) and oligomycin (2.5 μg/ml) were added for 30 min prior to termination of the experiment. JC-1 was added at a concentration of 5 μg/ml in complete media and cells were incubated at 37 °C in a 5% CO2 atmosphere for the final 1 or 2 h depending on the experiment. Subsequently, the cover slips were inverted onto slides containing Slow-Fade Gold (Invitrogen) solution. Images were taken immediately at 20× and 40× magnification employing an Olympus BX60 conventional compound fluorescence microscope. Red (emission = 519 nm) and green (emission = 565,615 nm) fluorescence were captured using appropriate filters. The data presented in the tables represent counting cells in 4 fields (60 – 100 cells per field) per condition from 3 different experiments for the PC3-LN4/TBB, 2 different experiments for the PC3-LN4/TBCA, 2 different experiments for BPH-1/TBB, and 1 experiment for LNCaP/TBB.
JC-1 FACS analysis
JC-1 was added at a final concentration of 2 μM in complete media containing 8 or 80 μM TBB or equivalent volume of DMSO and the cells incubated at 37 °C in 5% CO2 for 1 h for the 2 h time-point or 2 h for the 24 h time-point. CCCP at a final concentration of 10 μM was added to untreated cells 30 min prior to termination of JC-1 labeling as a positive control. At the termination of labeling, the cells were detached using Tryple Express (Invitrogen), collected by centrifugation (5 min at 500 × g) at 25 °C, resuspended in 0.5 ml of 37 °C live cell imaging solution (Invitrogen) and immediately analyzed using a FACS ARIA III (Becton Dickinson) excitation at 488 nm, collecting data using the 530/30 nm bandpass filter (green) and 582/15 nm bandpass filter (red) with a minimum of 20,000 gated events. The data were analyzed using the FACS ARIA III software.
Intracellular calcium assay
Measurement of intracellular Ca2+ concentration was performed using the FluoForte® calcium assay kit (Enzo Life Sciences). PC3-LN4 cells were plated in complete media at 1 × 105 cells/well using 96-well black wall plates. The following day, the cells were loaded with 0.1 ml dye/HBSS solution per well for 45 min at 37 °C/5 % CO2 according to kit instructions; BAPTA was included in the dye-loading solution for the appropriate wells. The plate was removed from the incubator and placed at room temperature for 15 min. Drug or diluent was first diluted from stock solutions into Ca2+ -free PBS and then added to every well in a volume of 25 μl to final concentration as indicated in Fig. 3B. The plate was immediately placed in a microplate reader (Molecular Devices 5, excitation 490 nm, emission 525 nm, cutoff 515 nm) and data collected every 1 min for 5 min. This experiment was performed 3 times.
Fig. 3. Mitochondrial membrane potential change mediated by CK2 inhibition is blocked by pretreatment of cells with BAPTA but not catalase and is associated with transient release of intracellular Ca2+.
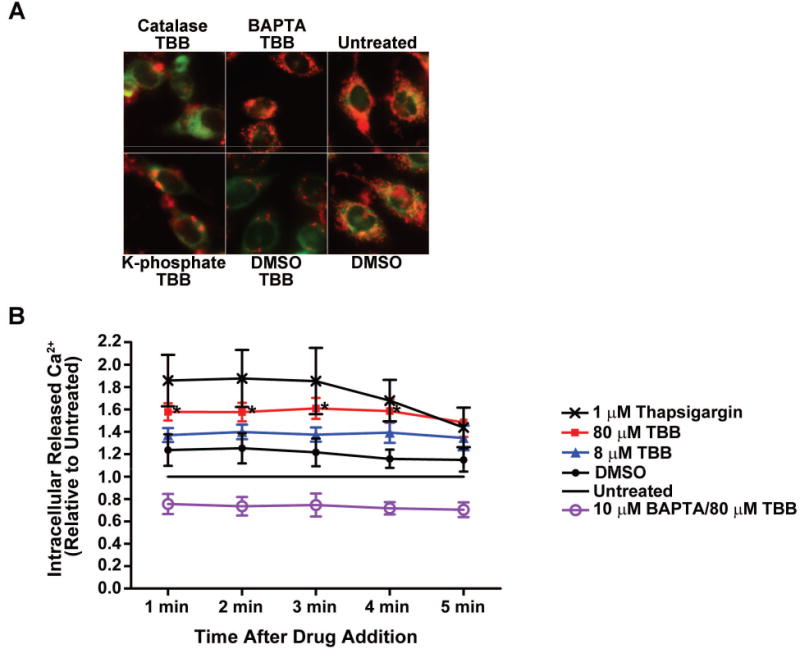
(A) PC3-LN4 cells were treated with catalase (3000 units) or BAPTA (10 μM) for 1 h followed by treatment with 80 μM TBB for 4 h. JC-1 loading was performed for the final 1 h of incubation. Controls included pretreatment for 1 h with drug solvent (DMSO for BAPTA; 50 mM potassium phosphate buffer pH 7.0 for catalase) followed by DMSO. Untreated cells were analyzed as a further control. Panels are accordingly labeled. Mitochondrial membrane potential was measured as described under materials and methods. (B) PC3-LN4 cells were loaded with FluoForte calcium assay dye, and with 10 μM BAPTA as indicated, for 1 h as described under Materials and Methods. Drug or diluent was added to each well as indicated in the legend (untreated cells received PBS diluent), and the plate immediately read once per min for 5 min. DMSO was used at the same concentration as 80 μM TBB. * p < 0.05 to untreated.
Statistical analysis
The primary cell line for study of the various parameters was PC3-LN4, and most experiments using these cells were performed at least three times. The data are expressed as mean ± standard error (SE). To determine the generality of the results obtained for Δψm changes in PC3-LN4 cell line, they were confirmed in two other cell lines (LNCaP and BPH-1); in this case the experiments were performed 2 and 1 times, respectively. Statistical significance was analyzed for the Aqueous One cell viability and the intracellular Ca2+ assays using analysis of variance with Bonferroni correction, and for clonal survival using t-tests.
Results
Effect of TBB on prostate cancer PC3-LN4 cell survival
Experiments were undertaken to determine the time course of the effect of inhibition of CK2 on cell viability in the presence of the relatively specific CK2 inhibitor TBB at varying concentrations. In these experiments we determined the effects of TBB treatment for much shorter time periods than previously examined. We employed the CellTiter 96® Aqueous One assay which measures the bioreduction (thought to occur via NADPH/NADH) of an MTS tetrazolium compound to a colored formazan product whose quantity as measured by absorbance at 490 nm is directly proportional to the number of metabolically active cells [Berridge and Tan, 1993]. This assay is a snapshot of the bioreduction activity of the cells at the specific time-point during the incubation. The results in Fig. 1A demonstrate that PC3-LN4 cells treated with TBB at 80 μM begin to show a change in viability as early as 2 h following the addition of TBB. By 24 h and 48 h the respective cell viability was 40% and 22%, as indicated graphically in Fig. 1A. Similar observations were made when cells were treated with another CK2 inhibitor TBCA, although a less dramatic loss of viability at 24 and 48 h using 80 μM TBCA was observed (Fig. 1A).
Fig. 1. Cell viability and clonal survival in cells treated with inhibitors of CK2.

(A) Prostate cancer PC3-LN4 cells were treated with varying concentrations of TBB or TBCA (two inhibitors of CK2) over different periods of time (varying from 2 to 48 h) as indicated. Cell viability was determined relative to the controls in which no additions (untreated) were made. Statistical analysis of the data is as follows: * p < 0.001 to untreated, p < 0.0001 to DMSO; & p < 0.001 to untreated, p < 0.0001 to DMSO; # p = 0.04 to untreated. (B) Clonal survival of cells treated with 80 μM of TBB for varying periods of time (4 to 24 h). Representative colony formation results are shown on the right side while their quantitation is presented graphically on the left side of the panel. All other details are as described under Materials and Methods. Statistical analysis of the data is as follows: * p = 0.004 to DMSO; # p < 0.0001 to DMSO.
In order to assess the survival and proliferative capacity of the cells over a period of 7 d following TBB treatment, crystal violet clonal survival assays were carried out following 4, 6, 8, and 24 h treatment with TBB. In this assay, TBB was removed from the cells after the indicated period of treatment, and the cells were replated in complete media. The results demonstrated that 6 h of treatment with 80 μM TBB was sufficient to cause significant loss of cell proliferation and survival (Fig. 1B). Thus, the initiation of loss of cell viability occurring as early as 6 h following inhibition of CK2 suggested the presence of preceding events that triggered these changes. Concordant with this it may be noted that exposure of prostate cancer cells in culture to 80 μM TBB for 6 h shows an inhibition of cellular CK2 activity by 20–35% and this inhibition progresses with time [Wang et al., 2008].
Effect of CK2 inhibition on mitochondrial membrane potential
We have previously demonstrated that modulations in CK2 activity impact mitochondrial apoptotic circuitry observed at 24 h following changes in CK2 [Wang et al., 2005a; Wang et al., 2006]. Since Δψm is regarded as a key determinant of cell viability [Mayer and Oberbauer, 2003], we investigated the effect of CK2 inhibition on mitochondrial function by examining the effect of TBB treatment at varying concentrations over time. The results in Fig. 2A show evidence of a rapid loss of Δψm as measured by microscopic detection of JC-1 uptake and fluorescence in cells treated with TBB. Red JC-1 fluorescence capture indicates intact Δψm, whereas green JC-1 fluorescence capture indicates that membrane potential has been lost. It is particularly noteworthy that the loss of membrane potential (Δψm) is apparent as early as 2 h following treatment with 8 or 80 μM TBB, occurring in a concentration dependent manner. These experiments were also performed using TBCA. Similar to the data presented in Fig. 1A, the effects of TBCA-mediated CK2 inhibition were slightly less dramatic but essentially the same as TBB. The quantitation of these data is presented in Tables 1 and 2. We also investigated the effect of TBB on Δψm in two other prostate cell lines (LNCaP and BPH-1); representative results of these experiments are shown in Fig. 2, B and C. The response of LNCaP cancer cells was similar to that observed for PC3-LN4 cells (Fig. 2B). However, the Δψm change in the benign prostate epithelial cell line (BPH-1) was notably lower in response to TBB under similar conditions (Fig. 2C). The quantitation of these data is presented in Tables 3 and 4. These results suggest that cancer cell lines are more susceptible to inhibition of CK2 activity and respond by a dramatic change in the Δψm and loss of viability. This observation accords with our previous findings on the relative resistance of normal versus benign cells to inhibition of CK2 activity [Slaton et al., 2004].
Fig. 2. Changes in mitochondrial membrane potential (Δψm) caused by treatment of cells with the CK2 inhibitor TBB.
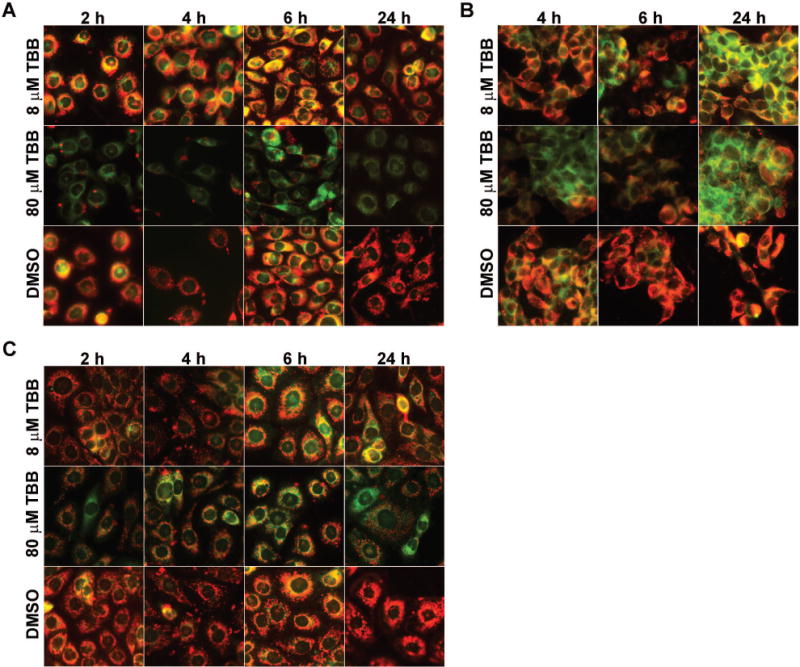
(A) PC3-LN4 cells were treated with 8 or 80 μM TBB as shown. DMSO control cells were treated with a volume of DMSO equivalent to that for 80 μM TBB. The period of treatment varied from 2 to 24 h. (B) LNCaP cells were treated with TBB at 8 or 80 μM for the periods of time varying from 4 to 24 h. (C) BPH-1 cells were treated with 8 or 80 μM TBB for the periods of time varying from 2 to 24 h. Mitochondrial membrane potential was measured using JC-1 as described under Materials and Methods.
Table 1.
Effect of TBB on mitochondrial membrane potential in PC3-LN4 cells over time
| Time of treatment (h) | Agent | Mitochondrial membrane potential (Δψm) in PC3-LN4 cellsa | ||
|---|---|---|---|---|
| High Δψm (red) |
Low Δψm (yellow) |
No Δψm (green) |
||
| 2 | None | 80.7 ± 6.9 | 18.9 ± 2.3 | 0 |
| DMSO | 64.2 ± 4.2 | 35.4 ± 6.1 | 0 | |
| 8 μM TBB | 55.1 ± 5.9 | 39.8 ± 3.6 | 11.8 ± 1.2 | |
| 80 μM TBB | 16.8 ± 1.3 | 0.6 ± 0 | 82.7 ± 2.2 | |
| 4 | None | 93.4 ± 3.5 | 5.1 ± 3.4 | 1.3 ±0 |
| DMSO | 93.9 ± 4.8 | 6.7 ± 2.7 | 0 | |
| 8 μM TBB | 66.1 ± 4.6 | 29.1 ± 4.0 | 3.7 ± 1.4 | |
| 80 μM TBB | 0.3 ± 0 | 0.1 ± 0 | 99.4 ± 8.2 | |
| 6 | None | 90.5 ± 4.5 | 9.2 ±4.5 | 0.3 ± 0 |
| DMSO | 85.5 ± 4.6 | 14.5 ± 4.6 | 0 ± 0 | |
| 8 μM TBB | 41.0 ± 4.7 | 41.2 ± 4.1 | 17.8 ± 2.6 | |
| 80 μM TBB | 0.9 ± 0 | 0 ± 0 | 99.1 ± 1.5 | |
| 24 | None | 97.0 ± 0.2 | 2.2 ± 0 | 0 ± 0 |
| DMSO | 98.9 ± 0.4 | 0.5 ± 0 | 0 ± 0 | |
| 8 μM TBB | 58.2 ± 3.1b | 13.7 ± 2.5 | 28.1 ± 2.2 | |
| 80 μM TBB | 0.7 ± 0 | 0 ± 0 | 99.2 ± 1.9 | |
The mitochondrial membrane potential Δψm is represented by red (high), yellow (low), and green (none). Data are presented as mean±SEM. The amount of DMSO as the control corresponded to the higher concentration of TBB.
Table 2.
Effect of TBCA on mitochondrial membrane potential in PC3-LN4 cells over time
| Time of treatment (h) | Agent | Mitochondrial membrane potential (Δψm) in PC3-LN4 cellsa | ||
|---|---|---|---|---|
| High Δψm (red) |
Low Δψm (yellow) |
No Δψm (green) |
||
| 2 | None | 86.3 ± 1.3 | 13.8 ± 1.3 | 0 |
| DMSO | 90.5 ± 0.9 | 7.9 ± 0.8 | 1.6 ± 1.0 | |
| 20 μM TBCA | 35.3 ± 3.4 | 29.3 ± 1.4 | 34.3 ± 1.9 | |
| 80 μM TBCA | 20.3 ± 2.4 | 26.6 ± 1.0 | 53.1 ± 2.1 | |
| 4 | None | 90.8 ± 1.2 | 4.4 ± 0.8 | 3.0 ± 1.1 |
| DMSO | 89.2 ± 1.0 | 10.3 ± 0.7 | 0.7 ± 0.1 | |
| 20 μM TBCA | 40.9 ± 5.8 | 36.0 ± 3.7 | 23.0 ± 3.6 | |
| 80 μM TBCA | 14.0 ± 2.1 | 31.3 ± 2.0 | 54.6 ± 5.8 | |
| 6 | None | 88.8 ± 2.6 | 10.1 ± 2.0 | 1.0 ± 0 |
| DMSO | 91.4 ± 1.1 | 7.5 ± 1.2 | 1.3 ± 0.5 | |
| 20 μM TBCA | 88.5.0 ± 2.7 | 9.6 ± 2.1 | 1.9 ± 0.5 | |
| 80 μM TBCA | 13.5 ± 3.1 | 37.1 ± 1.6 | 49.3 ± 2.5 | |
| 24 | None | 90.4 ± 1.0 | 9.6 ± 1.0 | 0 ± 0 |
| DMSO | 94.8 ± 0.3 | 5.3 ± 0.3 | 0 ± 0 | |
| 20 μM TBCA | 17.6 ± 3.6 | 33.5 ± 2.5 | 48.9 ± 1.9 | |
| 80 μM TBCA | 0.6 ± 0.5 | 0 ± 0 | 99.4 ± 1.7 | |
The mitochondrial membrane potential Δψm is represented by red (high), yellow (low), and green (none). Data are presented as mean±SEM. The amount of DMSO as the control corresponded to the higher concentration of TBCA.
Table 3.
Effect of TBB on mitochondrial membrane potential in LNCaP cells over time
| Time of treatment (h) | Agent | Mitochondrial membrane potential (Δψm) in LNCaP cellsa | ||
|---|---|---|---|---|
| High Δψm (red) |
Low Δψm (yellow) |
No Δψm (green) |
||
| 4 | None | 81.5 ± 2.5 | 18.5 ± 1.5 | 0 |
| 8 μM TBB | 45.7 ± 1.5 | 10.1 ± 3.5 | 44.5 ± 1.5 | |
| 80 μM TBB | 15.6 ± 3.3 | 0 | 80.1 ± 5.0 | |
| 6 | None | 90.8 ± 1.0 | 9.1 ±1.2 | 0 |
| 8 μM TBB | 55.1 ± 1.8 | 9.4 ± 0.7 | 35.6 ± 1.2 | |
| 80 μM TBB | 19.9 ± 1.5 | 0 ± 0 | 80.9 ± 5.0 | |
| 24 | None | 87.1 ± 1.3 | 12.9 ± 0.9 | 0 ± 0 |
| DMSO | 93.6 ± 2.4 | 6.4 ± 1.2 | 0 ± 0 | |
| 8 μM TBB | 43.9 ± 5.9 | 16.2 ± 1.8 | 40.0 ± 3.0 | |
| 80 μM TBB | 23.2 ± 1.9 | 2.8 ± 0.9 | 74.1 ± 4.6 | |
The mitochondrial membrane potential Δψm is represented by red (high), yellow (low), and green (none). Data are presented as mean±SEM. The amount of DMSO as the control corresponded to the higher concentration of TBB.
Table 4.
Effect of TBB on mitochondrial membrane potential in BPH-1 cells over time
| Time of treatment (h) | Agent | Mitochondrial membrane potential (Δψm) in BPH-1 cellsa | ||
|---|---|---|---|---|
| High Δψm (red) |
Low Δψm (yellow) |
No Δψm (green) |
||
| 2 | None | 86.3 ± 5.6 | 12.5 ± 3.1 | 0 |
| DMSO | 85.1± 4.5 | 14.8 ± 4.2 | 0.12 | |
| 8 μM TBB | 81.3 ± 3.6 | 14.9 ± 1.9 | 3.8 ±2.1 | |
| 80 μM TBB | 71.7 ± 6.3 | 13.8 ± 2.8 | 12.6 ± 5.5 | |
| 4 | None | 89.8 ± 6.6 | 4.4 ± 1.3 | 5.9 ±0 |
| DMSO | 94.1 ± 3.3 | 3.9 ± 0.9 | 1.8 ± 1.1 | |
| 8 μM TBB | 54.1 ± 1.8 | 13.7 ± 2.0 | 35.4 ± 3.9 | |
| 80 μM TBB | 13.4 ± 9.6 | 16.0 ± 2.7 | 70.6 ± 4.9 | |
| 6 | None | 88.6 ± 1.9 | 11.4 ± 2.2 | 0 |
| DMSO | 85.2 ± 12.6 | 14.0 ± 1.5 | 0 | |
| 8 μM TBB | 63.1 ± | 21.5 ± 2.8 | 15.4 ± 1.1 | |
| 80 μM TBB | 30.3 ± 0 | 20.7 ± 2.6 | 49.5 ± 3.6 | |
| 24 | None | 95.5 ±2.0 | 3.4 ± 2.2 | 0 |
| DMSO | 91.6 ± 12.6 | 8.4 ± 1.5 | 0 | |
| 8 μM TBB | 66.9 ± 1.8 | 20.5 ± 1.6 | 11.4 ± 1.0 | |
| 80 μM TBB | 52.6 ± 7.2 | 15.8 ± 2.6 | 31.6 ± 3.6 | |
The mitochondrial membrane potential Δψm is represented by red (high), yellow (low), and green (none). Data are presented as mean±SEM. The amount of DMSO as the control corresponded to the higher concentration of TBB.
We used a second technique to verify loss of Δψm following CK2 inhibition with TBB. PC3-LN4 cells were treated with 1 or 80 μM TBB using equivalent volumes of DMSO as controls, and JC-1 fluorescence was analyzed by FACS after 2 or 24 h of TBB treatment. The results again demonstrate that CK2 activity inhibition by TBB results in a dramatic loss of Δψm after 2 h of treatment (Table 5). Continued incubation with 80 μM TBB for 24 h resulted in virtually all cells demonstrating a loss of Δψm, which is in agreement with the JC-1 microscopic analysis. We also used siRNA transfection to downregulate CK2αα′ protein expression in PC3-LN4 cells. At 48 h post-transfection, we observed a 21.3 ± 1.03% loss in Δψm relative to 4.2 ± 1.42% loss in the control siRNA transfected cells. Loss of about 70% of CK2αα′ protein expression was indicated by immunoblot (data not shown). This data is supportive of a CK2-specific role in maintaining Δψm; however, the extended nature of siRNA-mediated downregulation of protein expression over time compared to the kinetically precise inhibition of CK2 activity using a small molecule inhibitor is a probable explanation for the less dramatic change in Δψm at a given timepoint post-transfection (please see Discussion section also).
Table 5.
FACS analysis of the effect of TBB on mitochondrial membrane potential in PC3-LN4 cells over timea
| Agent | Time of treatment (h) |
Cells with intact Δψm (%) |
Cells with loss of Δψm (%) |
|---|---|---|---|
| None | 97.3 | 2.7 | |
| DMSOb | 2 | 96.6 | 3.4 |
| 1μM TBB | 2 | 97.7 | 2.3 |
| DMSOb | 2 | 65.4 | 34.6 |
| 80 μM TBB | 2 | 21.0 | 79.0 |
| DMSOb | 24 | 55.3 | 44.7 |
| 80 μM TBB | 24 | 0.3 | 99.7 |
Following the various treatments as described under Experimental Procedures, cells were analyzed by FACS and the percentage of cells with intact or loss of Δψm was determined by gating on the untreated control cells.
The amount of DMSO as control corresponds to the volume of the TBB solution added.
Effect of catalase on CK2 inhibition mediated mitochondrial membrane potential
It was previously observed that inhibition of CK2 activity by TBB or downregulation of CK2 expression levels by antisense CK2α in prostate cancer cells promoted production of ROS such as H2O2 which became apparent between 4 to 8 h after treatment [Ahmad et al., 2006]. Subsequently, similar results were observed in the human leukemia Cem cell line treated with TBB. Compared with prostate cancer cell lines employed in the present work, leukemia Cem cells are significantly more sensitive to inhibition of CK2 such that in the presence of 50 μM TBB less than 40% cell survival was observed at 24 h post-treatment, and accordingly under these experimental conditions the production of ROS was shown to occur at 3 h [Hanif et al., 2009]. In these experiments, treatment of cells with the H2O2 scavenger catalase for 1 h prior to treatment with TBB resulted in a 50% reduction in the production of H2O2 confirming the nature of the ROS produced on inhibition of CK2 in Cem cells. We had originally suggested that a possible mechanism of induction of apoptosis upon inhibition or downregulation of CK2 was via the upstream production of H2O2 [Ahmad et al., 2006]. However, as described above, we have now observed that an effect on the Δψm is apparent at an even earlier time (2 h in the case of prostate cancer cells employed here). If production of ROS preceded changes in Δψm in these cells, then treatment of cells with a ROS scavenger should block the changes in Δψm. Thus, we investigated if Δψm was effected when PC3-LN4 cells were treated with 3000 and 6000 units of catalase for 1 h prior to inhibition of CK2 with 80 μM TBB for 4 h. The results indicated that there was no effect of catalase treatment on Δψm under these conditions, suggesting that the Δψm change in prostate cancer cells occurred prior to the production of H2O2 (Fig. 3, left panels).
Effect of BAPTA on CK2 inhibition mediated mitochondrial membrane potential
Since it is well known that Δψm and mitochondria permeability are both regulated by cellular Ca2+ (for a review see, e.g., [Perry et al., 2011]), we investigated the effect of treatment of cells with the Ca2+ chelator BAPTA (10 μM) for 1 h prior to treatment of cells with 80 μM TBB for 4 h. As shown in Fig. 3, center panels, pre-treatment of cells with BAPTA prevented Δψm loss suggesting a role of Ca2+ in promoting these changes in response to CK2 inhibition. These results predict the involvement of Ca2+ signaling as an early response to inhibition of CK2 activity in promoting loss of Δψm.
Release of intracellular Ca2+ stores upon CK2 inhibition
Because we observed that BAPTA treatment protected cells from Δψm loss after inhibition of CK2, we tested whether there is a detectable release of intracellular Ca2+ upon addition of TBB to PC3-LN4 cells. Intracellular Ca2+ was measured at 2 h, 1 h, 30 min, 15 min and immediately after addition of TBB. A significant change in detectable Ca2+ was observed upon treatment with 80 μM TBB immediately after its addition (Fig. 3B). Thapsigargin is known to cause a rapid increase in intracellular free Ca2+ concentration by release of intracellular Ca2+ stores and was used as positive control. A lesser amount of Ca2+ release was detected following 8 μM TBB treatment (Fig. 3B). No difference between untreated and TBB treated cells was detected at the other time points, suggesting the transient nature of the Ca2+ signal response to CK2 inhibition (data not shown).
Demonstration of presence of CK2 in mitochondria
CK2 is distributed in diverse compartments within the nucleus and cytoplasm of the cell. Previous reports had indicated CK2-like activity in the mitochondria of mammalian organs, including rat liver and bovine kidney [Damuni and Reed, 1988; Sarrouilhe and Baudry, 1996]. Here we provide evidence on the presence of CK2 in human prostate mitochondria using various cell lines. We prepared highly purified mitochondria from several prostate cell lines including LNCaP, C4-2, BPH-1, and PC3-LN4. All of them showed the presence of CK2 by immunoblot analysis. Fig. 4 shows the representative results on three different prostate cell lines (LNCaP, C4-2, and BPH-1) which indicate the distribution of the various CK2 subunits in different cell fractions, including the presence of all three subunits of CK2 in the mitochondrial fraction. The purity of these mitochondrial preparations was established by incubating the blots with antibodies that detect specific markers for mitochondrial (Cox IV) and non-mitochondrial fractions (lamin A/C and β-tubulin). Note the presence of Cox IV in mitochondria and its absence in the cytosol, the absence of lamin A/C in both the mitochondrial and cytosolic fractions, and the absence of β-tubulin in mitochondria. As shown subsequently, we also studied if the mitochondrial localized CK2 might be involved in the rapid response of cells to inhibition of CK2.
Fig. 4. Presence of CK2 in mitochondria.
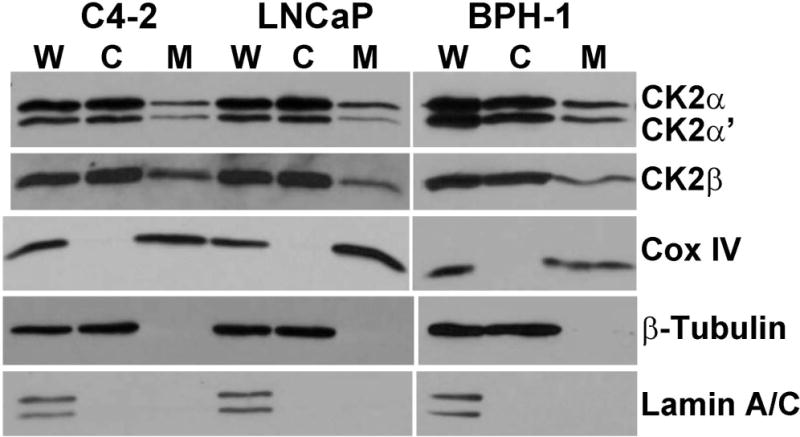
The results demonstrate the presence of CK2 in the purified mitochondrial fractions from prostate cells C4-2, LNCaP, and BPH-1. Markers for nuclear, cytosolic, or mitochondrial fractions were analyzed in whole cell lysate (W), cytosolic fraction (C), and mitochondria (M). Markers include Cox IV for mitochondria; β-tubulin for cytosol; and lamin A/C for nuclei.
Effect of CK2 inhibitors TBB and TBCA on isolated mitochondria
The results on the effect of CK2 inhibition on Δψm changes that were blocked by prior treatment of cells with the Ca2+ chelator BAPTA suggested that rapid mitochondrial permeability changes may be involved in the Δψm changes. Mitochondrial permeability transition (MPT) can occur associated with dissipation of the difference in voltage across the inner mitochondrial membrane, i.e., Δψm. Further, it is generally recognized that a complex interaction between Ca2+ shuttling, mitochondrial membrane permeability transition, and mitochondrial membrane potential exists in relation to induction of cell death, although this relation can vary with the cell type [Isenberg and Klaunig, 2000; Lemasters et al., 1998; Lemasters et al., 2009; Stavrovskaya and Kristal, 2005]. In view of these various considerations, our observation on the presence of CK2 in the mitochondrial fraction raised the question of the possible involvement of intrinsic CK2 in influencing mitochondrial function pertinent to membrane permeability transition.
To address this question, we employed rat liver mitochondria using procedures that yield highly pure preparations and evaluated the effects of endogenous CK2 inhibition on MPT as measured by mitochondrial swelling [Schnaitman and Greenawalt, 1968]. Cyclosporin A was added as a control to block the permeability transition, whereas Ca2+ and Pi combined were added as positive control inducers to cause permeability. The results in Fig. 5A show the effect of two CK2 inhibitors, TBB and TBCA, tested at 80 μM concentrations. When added to purified mitochondria these inhibitors produced a rapid (within 5 min) significant change in permeability transition resulting in mitochondrial swelling. This was also accompanied by cytochrome c release (Fig. 5B, right panel). Cyclosporin A partially inhibited the mitochondrial swelling in response to CK2 inhibition with either TBB or TBCA, indicating involvement of the mitochondrial permeability transition pore (PTP), whereas it did not significantly inhibit the cytochrome c release (Fig. 5B, right panel). The swelling of the mitochondria after CK2 inhibition was rescued more effectively by the addition of excess EGTA (chelator of Ca2+), implying that Ca2+ plays a pivotal role in the CK2-dependent process. Further, in the presence of EGTA the release of cytochrome c was also significantly mitigated (Fig. 5B, right panel). Of particular note is the observation that comparison of the effect of TBB or TBCA with that of EGTA+TBB or TBCA demonstrates a clear reduction in the effect of TBB or TBCA when EGTA is included. However, an additive effect was observed when EGTA and cyclosporin A were combined, resulting in almost complete inhibition of mitochondrial swelling and prevention of cytochrome c release. The presence of endogenous CK2 in the mitochondria used for these assays was verified by immunoblot analysis (Fig. 5B, left panel). These observations agree with the data presented in Fig. 2 and Fig. 3, reinforcing that early changes in mitochondrial function relate to involvement of the mitochondrial transition pore and Ca2+ mediated mitochondrial membrane permeability coupled with loss of Δψm. Taken together, it appears that these early events are critical for initiation of the apoptotic machinery in response to inhibition of CK2. Further, our results hint that mitochondrial associated CK2 may be the primary site of initiation of these events.
Temporal relation of CK2 inhibition and mitochondrial membrane potential changes compared with other markers of apoptosis
The presence of the CK2 inhibitor TBB promoted loss of prostate cancer cell viability that becomes apparent around 4–6 h as shown in Fig. 1. To further identify the temporal response of various mitochondrial apoptotic signals to inhibition of CK2, we examined several markers of apoptosis over time following treatment of cells with TBB. The results in Fig. 6 show the effect of treating PC3-LN4 cells with 80 μM TBB for varying periods of time. In cytosolic lysates in which the mitochondria have been removed by centrifugation, loss of full length Bid compared to DMSO controls was apparent from 4 h through 24 h. In the same cytosolic lysates, release of cytochrome c and Cox IV from the mitochondria into the cytosol was evident at 24 h (Fig. 6A). Analysis of other signals associated with apoptosis such as lamin A/C, caspase-9, and caspase-3 cleavage was performed in whole cell lysates and indicated their appearance at 24 h (Fig. 6B). The effectiveness of TBB as inhibitor of CK2 was also analyzed by examining the phosphorylation status of AKT-1. The inhibition of phosphorylation at Ser129 which is a specific target of CK2 was indicated at 24 h and further increased at 48 h following treatment with TBB (Fig. 6B). Concurrent loss of the general AKT-1 signal was also observed and was specific to TBB and not etoposide treatment. These results further suggest that the activation of these various cellular effects and apoptotic signals are downstream of the above described changes in Δψm most likely triggered by altered mitochondrial Ca2+ flux in response to inhibition of CK2 activity, as discussed subsequently.
Fig. 6. Temporal response of CK2 substrate and apoptotic signals following TBB mediated inhibition of CK2 in PC3-LN4 cells.
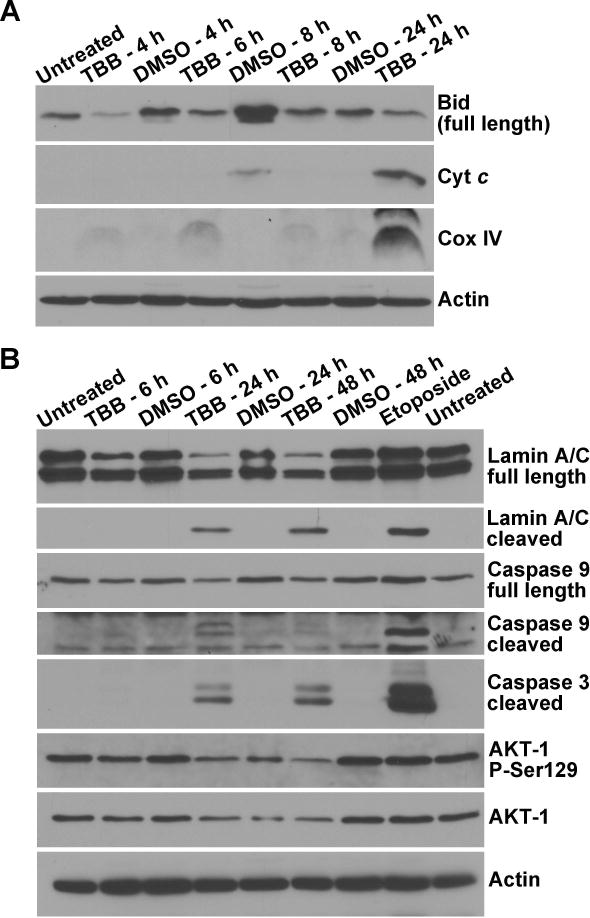
(A) Effect of 80 μM TBB treatment of cells for varying times on apoptotic signals measured in the cytosol. Cells were treated with TBB for 4, 6, 8 and 24 h. Analysis of Bid, cytochrome c, and Cox IV was carried out in the purified mitochondria-free cytosolic fractions. (B) Effect of 80 μM TBB treatment of cells for varying times in whole cell lysates. Cells were treated with TBB for 6, 24, and 48 h as shown, with DMSO as the corresponding control. Full length and cleaved lamin A/C, full length and cleaved caspase 9, cleaved caspase 3, total and phospho-Ser129 AKT-1 were analyzed. Etoposide (100 μM for 24 h) was employed as the positive control for induction of apoptosis. All other details were as described under Materials and Methods.
Discussion
Much work has established CK2 to be involved in the regulation of a wide range of cellular activities both in normal cells and in many disease processes [Guerra and Issinger, 2008]. In particular, CK2 has emerged as a particularly important signal in cancer biology [Ahmed et al., 2000; Ahmed et al., 2002; Trembley et al., 2010; Trembley et al., 2009]. Over time, it has been observed that CK2 is markedly elevated at the protein level in all cancers that have been examined [Tawfic et al., 2001]. Several hallmarks of cancer have been described [Hanahan and Weinberg, 2011], and there is considerable evidence of functional association of CK2 in several of these hallmarks [Trembley et al., 2010; Trembley et al., 2009; Trembley et al., 2013]. Among the hallmarks of cancer two features are consistently noted, namely deregulated growth and proliferation and altered resistance to cell death. The involvement of CK2 in both normal and abnormal cell growth was recognized for a long time; however, its relation to the cancer cell phenotype became apparent with recognition that CK2 not only promotes cell growth and proliferation but also suppresses apoptosis [Ahmad et al., 2008; Ahmed et al., 2002; Guo et al., 2001]. A general concept that emerged from our studies was that nuclear CK2 dynamics were intimately related to cell growth and cell death so that removal of growth stimuli resulted in the loss of CK2 from the nuclear structures (chromatin and nuclear matrix) which preceded the induction of apoptosis [Ahmed et al., 1993; Guo et al., 1999; Yu et al., 2001]. Further, our demonstration that prior forced overexpression of CK2 in cells protected them from undergoing apoptosis mediated by etoposide and diethylstilbestrol provided direct evidence of the ability of CK2 to suppress apoptosis [Ahmed et al., 2002; Guo et al., 2001]. The now accepted role of CK2 as a suppressor of apoptosis is underscored by its impact on apoptosis mediated by diverse stimuli such as loss of survival factors (hormones, growth factors), chemical agents (drugs such as etoposide, diethylstilbestrol, inhibitors of CK2), death receptor mediated signals (such as TNFα, TRAIL, FasL), physical agents (such as heat, radiation), and molecular downregulation of CK2 [Ahmad et al., 2008; Ahmed et al., 2002; Davis et al., 2002; Guo et al., 1999; Guo et al., 2001; Trembley et al., 2012; Trembley et al., 2011; Trembley et al., 2013; Wang et al., 2005a; Wang et al., 2006; Wang et al., 2008; Wang et al., 2001; Yamane and Kinsella, 2005]. These various observations prompted us to suggest that CK2 could serve as a cancer therapy target [Ahmad et al., 2005; Slaton et al., 2004; Unger et al., 2004; Wang et al., 2005b; Wang et al., 2001]; the potential of CK2 as a target continues to gain broad acceptance with mounting evidence of its druggability for cancer therapy [Pinna and Allende, 2009; Sarno and Pinna, 2008].
Several factors and pathways that may play a role in the apoptotic process triggered by the inhibition of CK2 have been examined, including IAPs, Bcl family proteins, ROS, and caspases [Ahmad et al., 2006; Duncan et al., 2011; McDonnell et al., 2008; Ponce et al., 2011; Tapia et al., 2006; Turowec et al., 2011; Wang et al., 2006; Wang et al., 2008; Wang et al., 2001]. These activities point to the temporal progression of apoptotic signaling that occurs in response to inhibition or downregulation of CK2. However, the nature of the primary trigger(s) for the initiation of apoptotic signaling has been unclear. To address this issue, we have presented novel observations that suggest a potential mechanism for the initiation and progression of apoptosis after CK2 inhibition. We have shown that when cells are treated with the relatively specific CK2 inhibitors TBB or TBCA the initiation of cell survival loss occurs between 4 to 6 h following inhibitor treatment, implying the existence of events preceding this time frame. Accordingly, we have found that under the present experimental conditions, loss of Δψm is apparent as early as 2 h following treatment with CK2 inhibitors. Further, our results hint that this alteration in mitochondrial physiology involves changes in the permeability transition pore as well as Ca2+ dynamics since prior treatment with the specific Ca2+ chelator BAPTA blocked the change in membrane permeability and Δψm.
It is well known that CK2 is localized in the nuclear and cytoplasmic fractions and that its level in different loci is of a dynamic nature depending on the cell state. We have documented here that highly purified mitochondria from prostate cells also harbor some CK2, and our results hint that the mitochondrial CK2 may have a role in the regulation of mitochondrial permeability and hence the membrane potential. In addition, a previous documentation of the localization of CK2 activity in the isolated rat liver mitochondria inner membrane and intermembrane space is consistent with inhibition of its activity being able to directly affect the PTP as well as the mitochondrial apoptosis channel (MAC). Previously, it was also suggested that production of H2O2 may be the proximal trigger for initiation of apoptosis on downregulation or inhibition of CK2 in prostate cancer cells; however, this event occurred at around 4 to 6 h following CK2 inhibition [Ahmad et al., 2006]. Further, we have observed that prior treatment of cells with the ROS scavenger catalase did not influence the rapid changes in Δψm caused by CK2 inhibition. This argues against H2O2 being the initiator of apoptotic signaling in response to CK2 inhibition in prostate cancer cells. Additional analysis of the temporal response for several of the apoptotic signals on inhibition of CK2 suggested changes in Bid to occur at 4 to 6 h while other signals of apoptosis such as the cytosolic appearance of cytochrome c, activation of caspases, and production of lamin A/C cleavage products to be among the later events. Finally, the JC-1 microscopy and FACS data together with the results using isolated mitochondria also imply a direct role for CK2 activity in maintaining the mitochondrial membrane potential. The effect of TBB and TBCA on isolated mitochondria suggests that these drugs are able to cross the mitochondrial membranes.
In previous work, we have shown that manipulation of CK2 expression levels in cells influences the mitochondrial apoptotic circuitry [Wang et al., 2006]. However, in the present work we observed that mitochondrial membrane potential loss was less dramatic following siRNA treatment for 48 h to downregulate CK2 protein levels than following TBB mediated inhibition of CK2 activity at early time points. We suggest that this may be due to the almost immediate effect that TBB or TBCA may have on mitochondrial located CK2, whereas loss of CK2 protein due to cytosolic siRNA downregulation of expression would likely take a much longer time to have impact on the mitochondrial CK2 population. Essentially, use of siRNA to CK2 produces an effect on CK2 level that does not become significant until about 48 h and even then a notable amount of CK2 protein would be expected to be available since the half-life of CK2 protein is relatively long. Thus, the induction of apoptosis by molecular downregulation of CK2 becomes apparent at later time points and in an asynchronous manner making it difficult to undertake a precise analysis of the initiating events. It is also plausible that the method of modulating CK2 (i.e., rapid activity inhibition versus slow loss of protein expression) may co-opt different cell death pathways. These aspects of CK2 regulated cell death are also focus of our future work. To reiterate, we emphasize that the goal of the present work was to identify the earliest preceding events that lead to the orchestration of the mitochondrial apoptotic pathway and induction of cell death on reduction of CK2 activity.
Based on the various aforementioned observations, we present the following scheme of progression of events subsequent to downregulation of CK2 activity in the cell (Fig. 7). We propose that one of the earliest events in response to CK2 inhibition in the cells is rapid intracellular release as well as altered Ca2+ flux into or within mitochondria which promotes a change in mitochondrial permeability and loss of mitochondrial membrane potential. It is conceivable that in response to CK2 inhibition, there is also ER stress and consequent release of Ca2+ from the ER, as is suggested by our detection of transiently increased intracellular Ca2+ after CK2 inhibition. While these studies on the regulation of various cellular Ca2+ pumps are currently being pursued in our laboratory, it is noteworthy that our data using isolated rat liver mitochondria employing buffers containing Ca2+ chelating agents suggest that only a small Ca2+ flux change may be needed in response to CK2 inhibition to cause cell death signaling. Thus, the above-described early changes in Ca2+, mitochondrial membrane permeability, and Δψm upon inhibition of CK2 are likely to represent the “point of no return” for cell fate decision so that loss of survival begins at 4 to 6 h. Associated with this time point are changes in Bid translocation as evidenced by reduction of cytosolic full length Bid (as early as 4 to 6 h) and production of H2O2 (around 4 to 6 h) followed by the previously described changes in the Bcl family proteins and activation of caspases which represent terminal phase events leading to cell death. The mechanism of change(s) in the Ca2+ dynamics in mitochondria in response to loss of CK2 signaling, the relative role of CK2 inhibition in the cell cytoplasm compared with that in the mitochondria, and the nature of conditions that determine the level of CK2 in mitochondria are currently under investigation in our laboratory.
Fig. 7. A cartoon depicting the proposed sequence of events in cells treated with CK2 inhibitors that lead to induction of apoptosis.
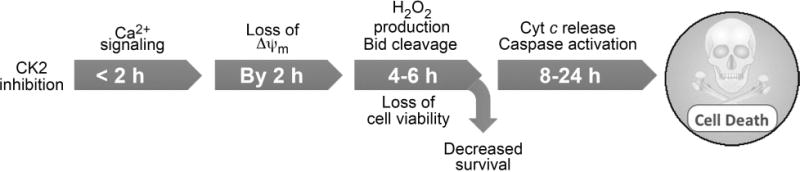
It is proposed that among the earliest changes in response to treatment of cells with CK2 inhibitors is the effect on mitochondrial membrane permeability transition associated with Ca2+ flux (before 2 h) and loss of mitochondrial membrane potential which occur as early as 2 h following the treatment of cells with TBB (earliest practical time measurements in this experimental model). The subsequent series of events that become apparent between 6 to 24 h following inhibition of CK2 appear to be production of ROS, loss of cell viability, and loss of proliferative capacity (by 4–6 h) followed by other apoptotic signals such as activation of caspases, cleavage of lamin A/C, loss of phospho-Ser129 and total AKT-1, release of cytochrome c and Cox IV from mitochondria, and loss of IAPs expression observed by 24 h [Tapia et al., 2006; Wang et al., 2008].
In summary, while it has been well documented that inhibition or downregulation of CK2 promotes apoptosis in cells through diverse mechanisms depending on the nature of the cell death signal, the underlying triggering mechanism that initiates the mitochondrial apoptotic signaling pathway was not well understood. In this report we have documented, for the first time, that downregulation of CK2 activity evokes an early change in mitochondrial Ca2+ dynamics associated with altered mitochondrial permeability and loss of Δψm as a novel mechanism of how CK2 may be involved in regulation of cell apoptosis.
Acknowledgments
We thank Omar Cespedes-Gomez and Alexandra Rex for their assistance in the laboratory.
Contract grant sponsor: U.S. Department of Veterans Affairs Merit Review Program; Contract grant number: I01BX001731.
Contract grant sponsor: National Cancer Institute; Contract grant numbers: R01CA150182 (KA), R21CA158730 (BTK).
Abbreviations
- CK2
official acronym for the inappropriate former name casein kinase 2 or II
- TBB
4,5,6,7-tetrabromobenzotriazole
- TBCA
tetrabromocinnamic acid
- BAPTA
1,2-bis(o-aminophenoxy)ethane- N,N,N′,N′-tetraacetic acid
- BSA
bovine serum albumin
- P-S
penicillin-streptomycin
- PBS
phosphate buffered saline
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazone
- FBS
fetal bovine serum
- TRAIL
tumor necrosis factor–related apoptosis inducing ligand
- ROS
reactive oxygen species
- TNFα
Tumor necrosis factor α
- FasL
Fas ligand
- IAPs
inhibitor of apoptosis proteins
- FACS
fluorescent activated cell sorting
- RIPA
radioimmunoprecipitation assay buffer
- TBS
Tris buffered saline, pH 7.4
- MPT
mitochondrial permeability transition
- PTP
permeability transition pore
- MAC
mitochondrial apoptosis-channel
Footnotes
Conflict of Interest: No conflicts to declare.
Publisher's Disclaimer: Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the U.S. Department of Veterans Affairs or the U.S. government.
References
- Ahmad KA, Wang G, Ahmed K. Intracellular hydrogen peroxide production is an upstream event in apoptosis induced by down-regulation of casein kinase 2 in prostate cancer cells. Mol Cancer Res: MCR. 2006;4:331–8. doi: 10.1158/1541-7786.MCR-06-0073. [DOI] [PubMed] [Google Scholar]
- Ahmad KA, Wang G, Slaton J, Unger G, Ahmed K. Targeting CK2 for cancer therapy. Anticancer Drugs. 2005;16:1037–43. doi: 10.1097/00001813-200511000-00001. [DOI] [PubMed] [Google Scholar]
- Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2–a key suppressor of apoptosis. Adv Enzyme Regul. 2008;48:179–87. doi: 10.1016/j.advenzreg.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed K. Nuclear matrix and protein kinase CK2 signaling. Crit Rev Eukaryot Gene Expr. 1999;9:329–36. doi: 10.1615/critreveukargeneexpr.v9.i3-4.170. [DOI] [PubMed] [Google Scholar]
- Ahmed K, Davis AT, Wang H, Faust RA, Yu S, Tawfic S. Significance of protein kinase CK2 nuclear signaling in neoplasia. J Cell Biochem Suppl Suppl. 2000;35:130–5. doi: 10.1002/1097-4644(2000)79:35+<130::aid-jcb1136>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Ahmed K, Gerber DA, Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends in Cell Biology. 2002;12:226–30. doi: 10.1016/s0962-8924(02)02279-1. [DOI] [PubMed] [Google Scholar]
- Ahmed K, Yenice S, Davis A, Goueli SA. Association of casein kinase 2 with nuclear chromatin in relation to androgenic regulation of rat prostate. Proc Natl Acad Sci U S A. 1993;90:4426–30. doi: 10.1073/pnas.90.10.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys. 1993;303:474–82. doi: 10.1006/abbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- Damuni Z, Reed LJ. Purification and properties of a protamine kinase and a type II casein kinase from bovine kidney mitochondria. Arch Biochem Biophys. 1988;262:574–84. doi: 10.1016/0003-9861(88)90408-0. [DOI] [PubMed] [Google Scholar]
- Davis AT, Wang H, Zhang P, Ahmed K. Heat shock mediated modulation of protein kinase CK2 in the nuclear matrix. J Cell Biochem. 2002;85:583–91. doi: 10.1002/jcb.10158. [DOI] [PubMed] [Google Scholar]
- Duncan JS, Turowec JP, Duncan KE, Vilk G, Wu C, Luscher B, Li SS, Gloor GB, Litchfield DW. A peptide-based target screen implicates the protein kinase CK2 in the global regulation of caspase signaling. Science Signaling. 2011;4:ra30. doi: 10.1126/scisignal.2001682. [DOI] [PubMed] [Google Scholar]
- Guerra B, Issinger OG. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis. 1999;20:391–408. doi: 10.1002/(SICI)1522-2683(19990201)20:2<391::AID-ELPS391>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Guerra B, Issinger OG. Protein kinase CK2 in human diseases. Curr Med Chem. 2008;15:1870–86. doi: 10.2174/092986708785132933. [DOI] [PubMed] [Google Scholar]
- Guo C, Yu S, Davis AT, Ahmed K. Nuclear matrix targeting of the protein kinase CK2 signal as a common downstream response to androgen or growth factor stimulation of prostate cancer cells. Cancer Res. 1999;59:1146–51. [PubMed] [Google Scholar]
- Guo C, Yu S, Davis AT, Wang H, Green JE, Ahmed K. A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J Biol Chem. 2001;276:5992–9. doi: 10.1074/jbc.M004862200. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hanif IM, Ahmad KA, Ahmed K, Pervaiz S. Involvement of reactive oxygen species in apoptosis induced by pharmacological inhibition of protein kinase CK2. Annals New York Acad Sci. 2009;1171:591–9. doi: 10.1111/j.1749-6632.2009.04916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg JS, Klaunig JE. Role of the mitochondrial membrane permeability transition (MPT) in rotenone-induced apoptosis in liver cells. Toxicol Sci : an official journal of the Society of Toxicology. 2000;53:340–51. doi: 10.1093/toxsci/53.2.340. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–96. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer B, Oberbauer R. Mitochondrial regulation of apoptosis. News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society. 2003;18:89–94. doi: 10.1152/nips.01433.2002. [DOI] [PubMed] [Google Scholar]
- McDonnell MA, Abedin MJ, Melendez M, Platikanova TN, Ecklund JR, Ahmed K, Kelekar A. Phosphorylation of murine caspase-9 by the protein kinase casein kinase 2 regulates its cleavage by caspase-8. J Biol Chem. 2008;283:20149–58. doi: 10.1074/jbc.M802846200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques. 2011;50:98–115. doi: 10.2144/000113610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinna LA. Protein kinase CK2: a challenge to canons. J Cell Sci. 2002;115:3873–8. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- Pinna LA, Allende JE. Protein kinase CK2 in health and disease: Protein kinase CK2: an ugly duckling in the kinome pond. Cell Mol Life Sci. 2009;66:1795–9. doi: 10.1007/s00018-009-9148-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce DP, Yefi R, Cabello P, Maturana JL, Niechi I, Silva E, Galindo M, Antonelli M, Marcelain K, Armisen R, Tapia JC. CK2 functionally interacts with AKT/PKB to promote the beta-catenin-dependent expression of survivin and enhance cell survival. Mol Cell Biochem. 2011;356:127–32. doi: 10.1007/s11010-011-0965-4. [DOI] [PubMed] [Google Scholar]
- Sarno S, Pinna LA. Protein kinase CK2 as a druggable target. Mol Biosyst. 2008;4:889–94. doi: 10.1039/b805534c. [DOI] [PubMed] [Google Scholar]
- Sarrouilhe D, Baudry M. Evidence of true protein kinase CKII activity in mitochondria and its spermine-mediated translocation to inner membrane. Cell Mol Biol. 1996;42:189–97. [PubMed] [Google Scholar]
- Savage MK, Jones DP, Reed DJ. Calcium- and phosphate-dependent release and loading of glutathione by liver mitochondria. Arch Biochem Biophys. 1991;290:51–6. doi: 10.1016/0003-9861(91)90590-f. [DOI] [PubMed] [Google Scholar]
- Schnaitman C, Greenawalt JW. Enzymatic properties of the inner and outer membranes of rat liver mitochondria. J Cell Biol. 1968;38:158–75. doi: 10.1083/jcb.38.1.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaton JW, Unger GM, Sloper DT, Davis AT, Ahmed K. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol Cancer Res. 2004;2:712–21. [PubMed] [Google Scholar]
- St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66:1817–29. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radical Biol & Med. 2005;38:687–97. doi: 10.1016/j.freeradbiomed.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Tapia JC, Torres VA, Rodriguez DA, Leyton L, Quest AF. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc Natl Acad Sci U S A. 2006;103:15079–84. doi: 10.1073/pnas.0606845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfic S, Yu S, Wang H, Faust R, Davis A, Ahmed K. Protein kinase CK2 signal in neoplasia. Histol Histopathol. 2001;16:573–82. doi: 10.14670/HH-16.573. [DOI] [PubMed] [Google Scholar]
- Trembley JH, Chen Z, Unger G, Slaton J, Kren BT, Van Waes C, Ahmed K. Emergence of protein kinase CK2 as a key target in cancer therapy. BioFactors. 2010;36:187–95. doi: 10.1002/biof.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley JH, Unger GM, Korman VL, Tobolt DK, Kazimierczuk Z, Pinna LA, Kren BT, Ahmed K. Nanoencapsulated anti-CK2 small molecule drug or siRNA specifically targets malignant cancer but not benign cells. Cancer Lett. 2012;315:48–58. doi: 10.1016/j.canlet.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley JH, Unger GM, Tobolt DK, Korman VL, Wang G, Ahmad KA, Slaton JW, Kren BT, Ahmed K. Systemic administration of antisense oligonucleotides simultaneously targeting CK2alpha and alpha′ subunits reduces orthotopic xenograft prostate tumors in mice. Mol Cell Biochem. 2011;356:21–35. doi: 10.1007/s11010-011-0943-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley JH, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell Mol Life Sci. 2009;66:1858–67. doi: 10.1007/s00018-009-9154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley JH, Wu J, Unger GM, Kren BT, Ahmed K. CK2 suppression of apoptosis and its implications in cancer biology and therapy. In: Pinna LA, editor. The Wiley-IUBMB Series on Biochemistry and Molecular Biology: Protein Kinase CK2. 2013. pp. 319–333. [Google Scholar]
- Turowec JP, Duncan JS, Gloor GB, Litchfield DW. Regulation of caspase pathways by protein kinase CK2: identification of proteins with overlapping CK2 and caspase consensus motifs. Mol Cell Biochem. 2011;356:159–67. doi: 10.1007/s11010-011-0972-5. [DOI] [PubMed] [Google Scholar]
- Unger GM, Davis AT, Slaton JW, Ahmed K. Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr Cancer Drug Targets. 2004;4:77–84. doi: 10.2174/1568009043481687. [DOI] [PubMed] [Google Scholar]
- Wang G, Ahmad KA, Ahmed K. Modulation of death receptor-mediated apoptosis by CK2. Mol Cell Biochem. 2005a;274:201–5. doi: 10.1007/s11010-005-2952-0. [DOI] [PubMed] [Google Scholar]
- Wang G, Ahmad KA, Ahmed K. Role of protein kinase CK2 in the regulation of tumor necrosis factor-related apoptosis inducing ligand-induced apoptosis in prostate cancer cells. Cancer Res. 2006;66:2242–9. doi: 10.1158/0008-5472.CAN-05-2772. [DOI] [PubMed] [Google Scholar]
- Wang G, Ahmad KA, Harris NH, Ahmed K. Impact of protein kinase CK2 on inhibitor of apoptosis proteins in prostate cancer cells. Mol Cell Biochem. 2008;316:91–7. doi: 10.1007/s11010-008-9810-9. [DOI] [PubMed] [Google Scholar]
- Wang G, Unger G, Ahmad KA, Slaton JW, Ahmed K. Downregulation of CK2 induces apoptosis in cancer cells–a potential approach to cancer therapy. Mol Cell Biochem. 2005b;274:77–84. doi: 10.1007/s11010-005-3077-1. [DOI] [PubMed] [Google Scholar]
- Wang H, Davis A, Yu S, Ahmed K. Response of cancer cells to molecular interruption of the CK2 signal. Mol Cell Biochem. 2001;227:167–74. [PubMed] [Google Scholar]
- Yamane K, Kinsella TJ. CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res. 2005;65:4362–7. doi: 10.1158/0008-5472.CAN-04-3941. [DOI] [PubMed] [Google Scholar]
- Yu S, Wang H, Davis A, Ahmed K. Consequences of CK2 signaling to the nuclear matrix. Mol Cell Biochem. 2001;227:67–71. [PubMed] [Google Scholar]