

Abstract
Thioamides can be used as photoswitches, as reporters of local environment, as inhibitors of enzymes, and as fluorescence quenchers. We have recently demonstrated the incorporation of thioamides into polypeptides and proteins using native chemical ligation (NCL). In this protocol, we describe procedures for the synthesis of a thioamide precursor and an NCL-ready thioamide-containing peptide using Dawson’s N-acyl-benzimidazolinone (Nbz) process. We include a description of the synthesis by NCL of a thioamide-labeled fragment of the neuronal protein α-synuclein.
Keywords: Thioamide, Native Chemical Ligation, Thioester, Fmoc SPPS
Scope and Comments
The thioamide is a nearly isosteric substitution of the carbonyl oxygen in the peptide bond with a sulfur atom [1]. Conservative backbone modification with a thioamide has been used to analyze the importance of specific hydrogen bonds to the folding of secondary structure motifs and to coupled protein folding and binding processes [2,3]. Because of its enhanced proteolytic stability and modulated activity, thioamide substitution has also been applied in several biologically active oligopeptides [4-6]. For example, Fischer and coworkers showed that a thioamide-containing peptide substrate resisted hydrolysis by prolyl oligopeptidase [5]. The red-shifted absorption band of the thioamide peptide bond can be used in several different ways to study structural changes in the backbone with high spatial and temporal control. The π-to-π* transition at 270 nm and the n-to-π* transition at 340 nm give rise to thioamide-specific CD signatures that can be identified against a background of oxoamide signal at shorter wavelengths [7,8]. Wildemann et al. assembled ribonuclease S (RNAse S) through noncovalent association of a proteolytic fragment of RNAse S and a synthetic thiopeptide and turned its enzymatic activity on and off through cis/trans photoisomerization of the thioamide [9]. Our laboratory expanded the utility of thioamides by demonstrating that a thioamide can act as a quencher of various fluorophores such as endogenous Tyr or Trp, and the unnatural amino acids p-cyanophenylalanine, 7-methoxycoumarin-4-ylalanine, and acridon-2-ylalanine [10-12]. Quenching can occur through either Förster resonance energy transfer or photo-induced electron transfer mechanisms, depending on the identity of the chromophore. The placement of thioamides is expected to be tolerated in almost any position in a protein because of its small size, although the sulfur substitution may prove disruptive to hydrogen bonding in some cases [7,13-15]. Despite these attractive applications, the incorporation of thioamides has traditionally been limited to peptides made through solid phase peptide synthesis (SPPS).
Recently, we have shown that a thioamide can be incorporated into a full-length protein by exploiting the native chemical ligation (NCL) reaction [16,17]. NCL requires two fragments: one bearing an N-terminal Cys and the other bearing a C-terminal thioester [18]. The synthesis of the N-terminal Cys fragment is straightforward, but the development of an efficient strategy for synthesizing a thioamide-containing C-terminal thioester using Fmoc-based SPPS has required considerable study. Boc-based SPPS, commonly used in the NCL community, is not viable as the acidic cleavage conditions degrade the thioamide peptide. We have previously published syntheses using solution-phase thioesterification with PyBOP activation as well as on-resin thioesterification using an Aimoto-type Cys-Pro-Gly linker where the Cys is protected as a disulfide and Gly is replaced by glycolic acid (CbPGo) (Figure 1) [16,19]. However, the former method could lead to epimerization at the α-carbon of the C-terminal residue and often suffers from poor solubility of the protected peptides [20]. Although the latter method alleviates the epimerization problem, it requires one to synthesize a dipeptide with the C-terminal residue of the thioester fragment coupled to Cys, which tends to couple to the Pro on the resin in a very low yield. In unpublished work, we have used an α-hydroxycysteine (Cya) strategy developed by Muir and Botti to generate thiopeptide thioesters (Figure 1) [21,22]. While the latent thioester of Cya did ligate rapidly, the synthesis of Cya is non-trivial (11% yield over five steps) and may be a barrier for some users. In contrast to these three methods, Dawson’s N-acyl-benzimidazolinone (Nbz) strategy could be used to generate thioamide-containing thioester peptides in relatively high yields and involves a simple synthesis using commercially available components [23]. After elongation of the peptide chain on 3,4-diaminobenzoyl (Dbz) resin, the introduction of 4-nitrophenylchloroformate closes the ring to form the N-acylurea moiety, Nbz.
Figure 1.
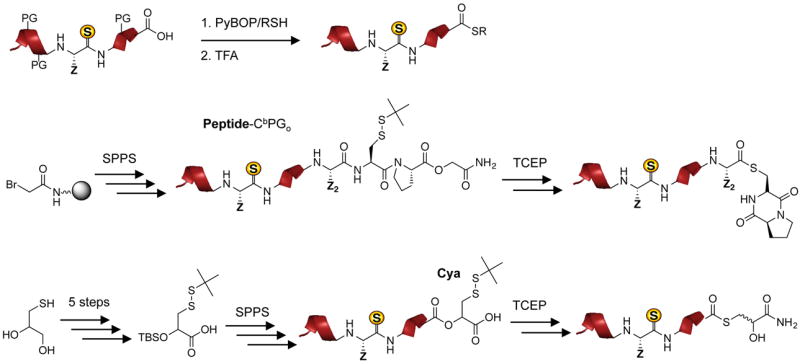
Other Fmoc-based thioesterification methods tested. Top: off-resin activation of protected peptides. Middle: on-resin synthesis of protected Cys-Pro-glycolic acid (CbPGo) linker, with off-resin activation by reduction of t-Bu disulfide. Bottom: on-resin incorporation of α-hydroxycysteine (Cya), with off-resin activation by reduction of t-Bu disulfide.
In this protocol, we describe our exploration of the compatibility of thioamides with the acylating conditions and report an optimized procedure for the synthesis of Nbz-thiopeptides. In addition, we describe the synthesis of the Fmoc-based SPPS thioamide precursor, thiocarboxybenzotriazole 4a, using 4-nitro-1,2-phenylendiamine and P4S10 (Scheme 1) [24]. The use of 4-nitro-1,2-phenylenediamine presents several advantages over N-Boc-phenylenediamine, which was used in our previous reports: (i) 4-nitro-1,2-phenylenediamine is generally more affordable than N-Boc-phenylenediamine, (ii) the removal of the Boc protecting group with TFA is omitted, simplifying the procedure slightly, and (iii) the nitro route is compatible with amino acids bearing acid-labile protecting groups such Glu or Asp. There are some advantages to the Boc route; Boc-protection suppresses the formation of a benzimidazole (BzIm) side product during the thionation step (conversion of 2b to 3b), and the lack of the nitro group in the benzotriazole products (e.g. 4b) makes them more stable. Nonetheless, the nitro route (i.e. 2a–4a) is generally preferable. The most common thionation reagents are Lawesson’s reagent (LR) and P4S10 [25,26]. Although LR is reported to have higher thionation yields, this was not observed for certain amino acids in our hands. For Ala, thionation with LR was higher yielding (89% vs 10%). However, the yield for the rest of the amino acids tested was limited to 10–35%, and recycling of the unreacted oxoanilide (2a) was necessary to obtain a reasonable thionation yield. In contrast, thionation with P4S10 proved to be more efficient and higher yielding, particularly for β-branched amino acids such as valine.
Here, we describe the preferred synthetic strategy for the thioamide precursor, an efficient method to obtain thiopeptide thioesters using Dawson’s resin, and the use of a thiopeptide thioester in an NCL reaction to synthesize a fragment of the Parkinson’s disease protein α-synuclein, Ac-αS1-19 V′3 (the prime symbol denotes a backbone thiocarbonyl at the indicated residue) [27,28]. Recently, Lee and coworkers have characterized the structure and binding affinity of this fragment to the calcium signaling protein calmodulin (CaM) [29]. Our laboratory is currently exploring the αS/CaM interaction using thioamide fluorescence quenching.
Experimental Procedure
Optimized Thiovaline Precursor Synthesis
The synthesis of thiovaline precursor 4a is presented as a general method. We chose to compare methods using Fmoc-Val-OH (1) because the thionation yield of this amino acid was particularly low using previously published methods. As a first step, the carboxyl group of Fmoc-Val-OH was amidated with 4-nitro-1, 2-phenylenediamine, a latent benzotriazole. To a solution of Fmoc-Val-OH in THF stirred in an NaCl/ice bath at −10 °C, NMM and isobutylchloroformate were added to generate Fmoc-Val-anhydride in situ, and then, 4-nitro-1,2-phenylenediamine was added. After stirring overnight at room temperature (r.t.), the reaction solvent THF was removed by rotary evaporation. The crude mixture was dissolved in DMF, and a saturated KCl solution was added, resulting in the precipitation of a yellow solid. This precipitation removes the NMM salts effectively, although organic side products precipitate too. Because of the poor solubility of compound 2a in DCM, complete purification via silica chromatography was difficult. Thus, compound 2a was isolated in 90% yield with 73% purity after KCl precipitation.
Thionation of Fmoc-Val-nitroanilide 2a with LR resulted in either low conversion at r.t. or formation of significant amounts of a benzimidazole side product at 70 °C. On the other hand, thionation with P4S10 was high yielding with minimal side product formation. To a solution of P4S10 and anhydrous Na2CO3 in THF, compound 2a was added and stirred at r.t under slow argon flow. The reaction was monitored by TLC to track the consumption of 2a. Upon completion, the reaction solution was filtered through Celite, washed with 5% NaHCO3, and purified on a silica column to afford pure compound 3a as yellow solid in 86% yield. Purification of P4S10 by Soxhlet extraction is often recommended. We find this to be unnecessary and obtain high yields with commercial P4S10 stored in a dessicator at r.t. and used directly.
To form the benzotriazole 4a, NaNO2 was added to compound 3a in glacial acetic acid diluted with 5% water. Nitrous acid generated in situ reacts with the primary amine of compound 3a, forming a benzotriazole through intramolecular diazonium cyclization. This reaction completed in 30 min, and compound 4a precipitated upon the addition of cold water to the reaction solution. Purification via silica chromatography afforded very pure 4a as an orange solid in a 90% yield. However, Ala and other amino acids with protected side chains such as Asp degraded in the process of chromatographic purification. Thus, we recommend minimal handling of the benzotriazoles to prevent hydrolysis and degradation through cyclization. As long as the aminothioacylanilide (e.g. 3a) is pure going into the cyclization reaction, precipitation provides sufficiently pure material for peptide coupling. After precipitation followed by filtration, the nitrobenzotriazole 4a was dried in the presence of P2O5 under vacuum at r.t. overnight and was used for peptide synthesis. The detailed procedures and 1H and 13C NMR spectra for compound 2a, 3a, and 4a are reported in the Supporting Information.
Thiopeptide-Nbz Synthesis
The thiopeptide Ac-MDV′FMKGL-Nbz (7) was synthesized on commercially available Dawson Dbz AM resin (Novabiochem®, San Diego, CA, USA). After removal of the Fmoc protecting group, the first amino acid was loaded by HATU/DIPEA activation. The peptide was elongated by standard SPPS procedures with HBTU/DIPEA activation. Thiovaline was introduced by adding the preactivated derivative Fmoc-thioval-nitrobenzotriazole (4a) with DIPEA but without HBTU. The last amino acid was loaded as Ac-Met to avoid undesired acetylation on the Dbz group when an acetylating reagent such as Ac2O is used. After assembly of peptide 5, the resin was treated with p-nitrophenyl chloroformate to form the peptidyl carbamate 6, which was then converted to peptide–Nbz resin by a subsequent treatment of DIPEA. The Nbz peptide 7 was cleaved from resin by TFA treatment and purified by preparative HPLC. The detailed procedures for peptide synthesis are provided in the Supporting Information.
Native Chemical Ligation
To a solution of 6M guanidinium hydrochloride (Gdn•HCl) and 200 mM sodium phosphate, triscarboxyethyl phosphine and thiophenol were added such that their final concentrations were 20 mM and 1% (v/v), respectively. It is crucial to adjust the pH of the solution to 7.0 and to degas it with Ar before use in ligation. Ac-αS1-8V′3-Nbz (7, 0.35 μmol) and αS9-19C9 (8, 0.51 μmol) were dissolved in NCL buffer, and the reaction was initiated upon combining the two peptides. After purging with Ar briefly, the reaction solution was placed in an incubator and shaken at 1000 rpm at 37 °C overnight. An aliquot (25 μl) was taken out periodically, quenched by the addition of 975 μl of 0.1% TFA in water, and stored at −20 °C until analysis by analytical reversed phase HPLC. Conversion to the product, Ac-αS1-19 V′3 (9), was greater than 95% by peak area (relative to unreacted 7 and hydrolyzed 7) after 30 min (Figure 2). The thiopeptide reactant and products are sufficiently stable that reactions can be allowed to stand overnight. The product was isolated in 15% yield as determined by UV/Vis spectroscopy.
Figure 2.
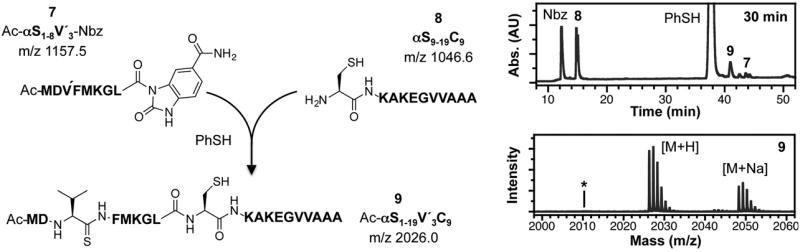
Native chemical ligation to form Ac-αS1-19 V′3 (9). Left: reaction scheme. Right: reaction monitored by HPLC (monitored at 215 nm) and MALDI MS of product (9) peak (m/z Calcd: 2026.0, Found: 2026.3). Asterisk indicates the expected mass of Ac-αS1-19 (oxoamide at position 3).
It is worthwhile to note that the same procedures can be used to obtain a full-length protein labeled with a thioamide by expressing a protein fragment (e.g. αS9-140C9) with an N-terminal Cys, revealed by treatment with Factor Xa protease or methionine aminopeptidase [17]. With either N-terminal proteolysis method, the presence of the thioamide in the product should be verified by MALDI MS, for full-length proteins trypsin-digestion and MALDI MS is recommended.
Limitations
Native chemical ligation enables the incorporation of thioamides into peptides and proteins but also leaves Cys at the ligation site, which may be undesirable. The common strategies for the removal of Cys must be evaluated for compatibility with thioamides. Radical desulfurization of Cys to form Ala can compete with desulfurization of thioamides [30,31]. Alternatively, selective masking of Cys with alkylating reagents is possible, but the alkylation reaction must be monitored closely to avoid thioamide alkylation [32].
To circumvent the branching of Dbz resin, Mahto et al. employed allyloxycarbonate (Alloc) as a protecting group on the second amino group [33]. The deprotection of the Alloc group requires the use of catalytic Pd0, which can desulfurize thioamides (unpublished results) [34]. Thus, thioamide reactivity must be considered in the use of alternative protecting groups.
Supplementary Material
Scheme 1.
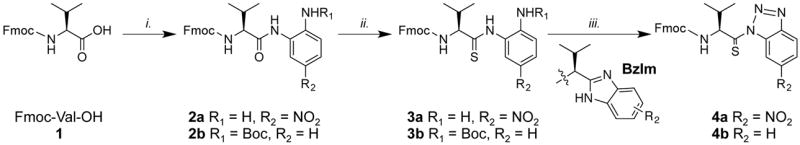
Synthesis of Fmoc-thiovaline-benzotriazole derivatives 4a. Reagents and conditions: (i) NMM, isobutylchloroformate, 4-nitro-1,2-phenylenediamine, THF, overnight, r.t. (90%), (ii), P4S10, Na2CO3, THF, r.t. (86%), and (iii) NaNO2, AcOH, H2O, r.t. (90%). Compounds 2b, 3b, and 4b are shown for discussion purposes.
Scheme 2.
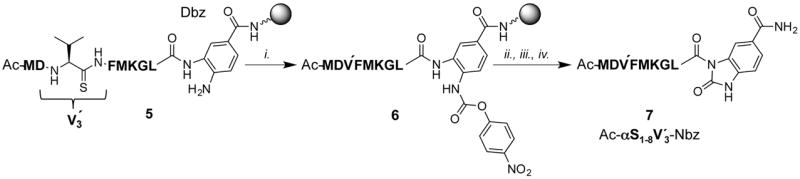
Synthesis of Ac-αS1-18 V′3-Nbz (7). Reagents and conditions: (i) SPPS on Dbz AM resin, r.t., (ii) p-nitrophenyl chloroformate, r.t., (iii) DIPEA/DMF, r.t., and (iv) TFA/TIPS/thioanisole/DCM (80 : 5 : 2.5 : 12.5), r.t.
REACTION SCHEME.
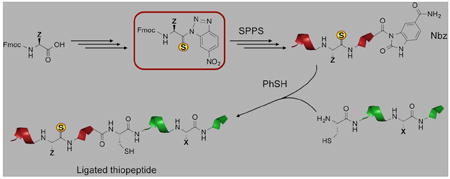
GENERAL OPTIMIZED PROCEDURE
Thioamide precursors can be synthesized by a general procedure as follows (using thiovaline as an example): Fmoc-Val-OH was coupled to 4-nitro-1,2-phenylenediamine to form an aminoacyl anilide, which was treated with P4S10 to thionate the carbonyl. NaNO2 treatment was used to form the benzotriazole for peptide coupling. The thiopeptide was then synthesized on 3,4-diaminobenzoyl (Dbz) resin, which was treated with p-nitrophenyl chloroformate to form a C-terminal N-acyl-benzimidazolinone (Nbz), activating the thiopeptide for native chemical ligation (NCL). NCL reactions were carried out under standard conditions in denaturing buffer (6 M guanidinium hydrochloride).
Acknowledgments
This work was supported by funding from the University of Pennsylvania, including a grant from the Institute on Aging and the National Institutes of Health (NIH) (NIH NS081033 to E.J.P.). Instruments supported by the National Science Foundation and NIH include the following: High resolution mass spectrometer (HRMS) (NIH RR-023444), MALDI MS (NSF MRI-0820996), and NMR (NIH RR-022442).
Footnotes
This article is published in Journal of Peptide Science as part of the Special Issue devoted to contributions presented at the Chemical Protein Synthesis Meeting, April 3–6, 2013, Vienna, edited by Christian Becker (University of Vienna, Austria).
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web site.
References
- 1.Hoeg-jensen T. Review: endothiopeptides alias peptide thioamides. Phosphorus Sulfur Silicon Relat Elem. 1996;108:257–278. [Google Scholar]
- 2.Culik RM, Jo H, DeGrado WF, Gai F. Using thioamides to site-specifically interrogate the dynamics of hydrogen bond formation in β-sheet folding. J Am Chem Soc. 2012;134:8026–8029. doi: 10.1021/ja301681v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bachmann A, Wildemann D, Praetorius F, Fischer G, Kiefhaber T. Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proc Natl Acad Sci U S A. 2011;108:3952–3957. doi: 10.1073/pnas.1012668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessler H, Matter H, Geyer A, Diehl H-J, Köck M, Kurz G, Opperdoes FR, Callens M, Wierenga RK. Selective inhibition of trypanosomal triosephosphate isomerase by a thiopeptide. Angew Chem Int Ed. 1992;31:328–330. [Google Scholar]
- 5.Schutkowski M, Jakob M, Landgraf G, Born I, Neubert K, Fischer G. Probing substrate backbone function in prolyl oligopeptidase catalysis. Eur J Biochem. 1997;245:381–385. doi: 10.1111/j.1432-1033.1997.00381.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhang W, Li J, Liu L-W, Wang K-R, Song J-J, Yan J-X, Li Z-Y, Zhang B-Z, Wang R. A novel analog of antimicrobial peptide polybia-MPI, with thioamide bond substitution, exhibits increased therapeutic efficacy against cancer and diminished toxicity in mice. Peptides. 2010;31:1832–1838. doi: 10.1016/j.peptides.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 7.Miwa JH, Pallivathucal L, Gowda S, Lee KE. Conformational stability of helical peptides containing a thioamide linkage. Org Lett. 2002;4:4655–4657. doi: 10.1021/ol027056d. [DOI] [PubMed] [Google Scholar]
- 8.Hollósi M, Kollát E, Kajtár J, Kajtár M, Fasman GD. Chiroptical labeling of folded polypeptide conformations: the thioamide probe. Biopolymers. 1990;30:1061–1072. [Google Scholar]
- 9.Wildemann D, Schiene-Fischer C, Aumuller T, Bachmann A, Kiefhaber T, Lucke C, Fischer G. A nearly isosteric photosensitive amide-backbone substitution allows enzyme activity switching in ribonuclease S. J Am Chem Soc. 2007;129:4910–4918. doi: 10.1021/ja069048o. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg JM, Batjargal S, Petersson EJ. Thioamides as fluorescence quenching probes: minimalist chromophores to monitor protein dynamics. J Am Chem Soc. 2010;132:14718–14720. doi: 10.1021/ja1044924. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg JM, Speight LC, Fegley MW, Petersson EJ. Minimalist probes for studying protein dynamics: thioamide quenching of selectively excitable fluorescent amino acids. J Am Chem Soc. 2012;134:6088–6091. doi: 10.1021/ja3005094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldberg JM, Wissner RF, Klein AM, Petersson EJ. Thioamide quenching of intrinsic protein fluorescence. Chem Commun. 2012;48:1550–1552. doi: 10.1039/c1cc14708k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miwa JH, Patel AK, Vivatrat N, Popek SM, Meyer AM. Compatibility of the thioamide functional group with beta-sheet secondary structure: incorporation of a thioamide linkage into a beta-hairpin peptide. Org Lett. 2001;3:3373–3375. doi: 10.1021/ol0166092. [DOI] [PubMed] [Google Scholar]
- 14.Reiner A, Wildemann D, Fischer G, Kiefhaber T. Effect of thioxopeptide bonds on alpha-helix structure and stability. J Am Chem Soc. 2008;130:8079–8084. doi: 10.1021/ja8015044. [DOI] [PubMed] [Google Scholar]
- 15.Choudhary A, Raines RT. An evaluation of peptide-bond isosteres. ChemBioChem. 2012;12:1801–1807. doi: 10.1002/cbic.201100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batjargal S, Wang YJ, Goldberg JM, Wissner RF, Petersson EJ. Native chemical ligation of thioamide-containing peptides: development and application to the synthesis of labeled alpha-synuclein for misfolding studies. J Am Chem Soc. 2012;134:9172–9182. doi: 10.1021/ja2113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wissner RF, Batjargal S, Fadzen CM, Petersson EJ. Labeling proteins with fluorophore/thioamide Förster resonant energy transfer pairs by combining unnatural amino acid mutagenesis and native chemical ligation. J Am Chem Soc. 2013;135:6529–6540. doi: 10.1021/ja4005943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dawson PE, Muir TW, Clarklewis I, Kent SBH. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 19.Kawakami T, Aimoto S. The use of a cysteinyl prolyl ester (CPE) autoactivating unit in peptide ligation reactions. Tetrahedron. 2009;65:3871–3877. [Google Scholar]
- 20.Nagalingam AC, Radford SE, Warriner SL. Avoidance of epimerization in the synthesis of peptide thioesters using Fmoc protection. Synlett. 2007:2517–2520. [Google Scholar]
- 21.Botti P, Villain M, Manganiello S, Gaertner H. Native chemical ligation through in situ O to S acyl shift. Org Lett. 2004;6:4861–4864. doi: 10.1021/ol0481028. [DOI] [PubMed] [Google Scholar]
- 22.George EA, Novick RP, Muir TW. Cyclic peptide inhibitors of staphylococcal virulence prepared by Fmoc-based thiolactone peptide synthesis. J Am Chem Soc. 2008;130:4914–4924. doi: 10.1021/ja711126e. [DOI] [PubMed] [Google Scholar]
- 23.Blanco-Canosa JB, Dawson PE. An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew Chem Int Ed. 2008;47:6851–6855. doi: 10.1002/anie.200705471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shalaby MA, Grote CW, Rapoport H. Thiopeptide synthesis. Alpha-amino thionoacid derivatives of nitrobenzotriazole as thioacylating agents. J Org Chem. 1996;61:9045–9048. doi: 10.1021/jo961245q. [DOI] [PubMed] [Google Scholar]
- 25.Cava MP, Levinson MI. Thionation reactions of Lawesson reagents. Tetrahedron. 1985;41:5061–5087. [Google Scholar]
- 26.Scheeren JW, Ooms PHJ, Nivard RJF. General procedure for conversion of a carbonyl group into a thione group with tetraphosporous decasulfide. Synthesis. 1973:149–151. [Google Scholar]
- 27.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. [alpha]-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 29.Gruschus JM, Yap TL, Pistolesi S, Maltsev AS, Lee JC. NMR structure of calmodulin complexed to an N-terminally acetylated alpha-synuclein peptide. Biochemistry. 2013;52:3436–3445. doi: 10.1021/bi400199p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan LZ, Dawson PE. Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization. J Am Chem Soc. 2001;123:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 31.Hendrickson TL, Imperiali B. Metal-ion dependence of oligosaccharyl transferase – implications for catalysis. Biochemistry. 1995;34:9444–9450. doi: 10.1021/bi00029a020. [DOI] [PubMed] [Google Scholar]
- 32.Hopkins CE, Hernandez G, Lee JP, Tolan DR. Aminoethylation in model peptides reveals conditions for maximizing thiol specificity. Arch Biochem Biophys. 2005;443:1–10. doi: 10.1016/j.abb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 33.Mahto SK, Howard CJ, Shimko JC, Ottesen JJ. A reversible protection strategy to improve Fmoc-SPPS of peptide thioesters by the N-acylurea approach. ChemBioChem. 2011;12:2488–2494. doi: 10.1002/cbic.201100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grieco P, Gitu PM, Hruby VJ. Preparation of ‘side-chain-to-side-chain’ cyclic peptides by allyl and Alloc strategy: potential for library synthesis. J Peptide Res. 2001;57:250–256. doi: 10.1111/j.1399-3011.2001.00816.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.