

Abstract
Staphylococcus aureus is a major cause of severe pneumonia. Multiple mechanisms of proinflammatory signaling are activated to recruit immune cells into the airway in response to S. aureus. We found that IL-16, a T cell cytokine that binds CD4, is potently activated by S. aureus, specifically by protein A (SpA), and to a much greater extent than by Gram negative pathogens or LPS. IL-16 production involved multiple signals including ligation of TNFR family members or EGFR, both receptors for SpA and generation of Ca2+ fluxes to activate calpains and caspase-3. Although human airway epithelial cells, vascular endothelial cells, THP-1 and Jurkat T cells released IL-16 in response to S. aureus in vitro, in a murine model of pneumonia, CD4+ cells were the major source of IL-16 suggesting the involvement of an autocrine signaling pathway. The production of IL-16 contributed to lung damage as neutralization of IL-16 enhanced S. aureus clearance and resulted in diminished lung pathology in S. aureus pneumonia. Our results suggest that the ability of S.aureus to activate TNFR1 and Ca2+/calpain signaling contribute to T cell activation and excessive inflammation in the setting of acute pneumonia.
INTRODUCTION
Staphylococcus aureus, particularly the epidemic USA300 methicillin-resistant S. aureus (MRSA), is a major cause of pneumonia1. Common in health care associated settings, MRSA is a frequent cause of ventilator-associated pneumonia2 and superinfection complicating influenza3. Even though there are antimicrobials with good activity against MRSA, there remains significant morbidity and mortality associated with this pathogen1,4. A substantial literature suggests that specific staphylococcal virulence factors are directly responsible for lung injury such as α-hemolysin (hla)5 and Panton-Valentine leukocidin (PVL)6. However, much of the pathology associated with MRSA pneumonia can be attributed to the intensity of the host inflammatory response. S. aureus activates multiple redundant proinflammatory signaling cascades and murine models of S. aureus pneumonia lacking specific components of the innate immune signaling pathway such as type 1 interferon receptor7, inflammasome protein NLRP38 and TNF receptor 1 (TNFR1)9 have improved bacterial clearance. S. aureus infection is accompanied by a significant TNF response and the abundant S. aureus surface component protein A (SpA), directly activates TNFR1 signaling10.
The recruitment of neutrophils and macrophages in response to chemokine and cytokine expression in the lung is a critical component of innate immune signaling in response to S. aureus11,12. The contribution of T cells in the setting of acute staphylococcal infection is less well characterized, but is likely to be important13 as seen in Rag2−/− mice, which exhibited resistance to S. aureus infection in a sepsis model14. Not only are T cells essential to coordinate an adaptive immune response, they can be directly activated by S. aureus superantigens and are a major source of proinflammatory cytokines15, such as TNF. Activated T cells contribute to pulmonary pathology in the setting of acute lung injury. CXCR3 is preferentially expressed in TH 1 cells16, and is increased in both infectious and noninfectious models of acute lung injury11,12.
Overexpression of the T cell chemokine CXCL10, a CXCR3 ligand, induced airway inflammation16 and infected CXCL10 KO mice had decreased lung pathology11. In acute bacterial infection, multiple T cell chemokines and cytokines are produced such as IL-1217 and IL-1618, which serve to recruit T cells and stimulate the expression of surface receptors that mediate their proliferation and cytokine production.
IL-16 is a multifunctional cytokine with a single PDZ domain initially characterized as a product of human peripheral blood mononuclear cells and described as a lymphocyte chemoattractant19 (previously named lymphocyte chemoattractant factor). Transcribed as pro-IL-16, its production is regulated both at the level of transcription and caspase-3 dependent processing in various cell types18. IL-16 induces chemotaxis of CD4+ cells such as lymphocytes, eosinophils and dendritic cells by ligating CD4 directly at a site distinct from other ligands18,19. Among its multiple functions, IL-16 is a T cell chemoattractant involved in TH1 inflammatory responses and the regulation of both T cell growth and responsiveness to regulatory cytokines20. Chemotaxis of T cells induced by the supernatant of TNF-stimulated epithelial cells is inhibited by anti-IL-16 antibody21, indicating a close association between IL-16 and TNF signaling. The processing of pro-IL-16 to its active form is mediated by caspase-322 via caspase-8. These caspases are activated following ligation of TNFR123, the target of SpA10. In inflammatory conditions characterized by excessive TNF signaling, such as inflammatory bowel disease, IL-16 contributes significantly to pathology24, thus we postulate that in the setting of robust TNF signaling, as occurs during S. aureus pneumonia, IL-16 similarly contributes to pathology.
In the experiments detailed herein, we describe the participation of IL-16 in S. aureus pneumonia and suggest that the unique ability of this organism to directly activate TNFR1, as well as the Ca2+/calpain/caspase cascade, results in release of this T cell cytokine.
RESULTS
S. aureus induces IL-16 in the murine lung
Significantly higher levels of IL-16 were recovered from the bronchoalveolar lavage fluid (BALF) of WT mice infected with 107 cfu methicillin resistant S. aureus USA300 (MRSA) strain (p=0.0001 compared to PBS) or S. aureus 502A, a representative methicillin sensitive strain. Lower levels were retrieved from mice infected with 107 cfu of Gram negative organisms such as Klebsiella pneumoniae ST 258 and Pseudomonas aeruginosa PAK (p<0.05 compared to MRSA) (Figure 1a). To determine if human cells similarly had differential IL-16 responses to S. aureus versus Gram negative stimuli the human monocytic cells, THP-1s were stimulated with the same pathogens. IL-16 from THP-1s was 18-fold higher than the media control after incubation with MRSA (p=0.0001) and 20-fold after infection with S. aureus 502A. Less IL-16 was measured in response to infection with Kp or PAK (p<0.05 compared to MRSA) (Figure 1b).
Figure 1. S. aureus induces IL-16 production in vivo and in vitro.
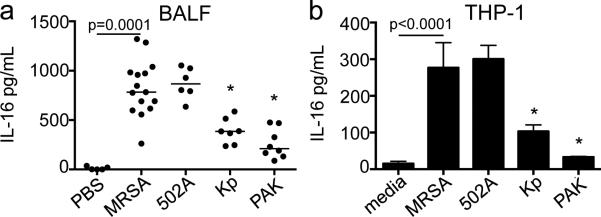
(a) C57BL/6J WT mice inoculated with 107 cfu of methicillin resistant S. aureus USA300 (MRSA), S. aureus 502A, K. pneumoniae ST 258 (Kp) or P. aeruginosa strain (PAK) or PBS. Bronchoalveolar lavage fluid(BALF) was obtained 24 hours after infection and quantitated by ELISA. Each dot represents an individual mouse; lines represent median values; data was compiled from 2 independent experiments. *p<0.05 as compared to MRSA, Student's t test, post hoc Dunnett's test. Lines represent median values. (b) THP-1s were stimulated with MRSA, S. aureus 502A, Kp or PAK (MOI 100) *p<0.05 compared to media, Student's t-test, post-hoc Dunnett's test. Graph shown is representative of 2 independent experiments.
IL-16 secretion is SpA dependent in specific cell types
The major S. aureus surface component SpA activates TNFR1 signaling10, a receptor expected to activate IL-16 processing. The ability of the WT MRSA, a spa null mutant, and purified SpA to stimulate IL-16 in several cell types was compared (Figure 2). Induction of IL-16 by the spa null mutant was significantly decreased as compared to MRSA in human bronchial epithelial cells (16HBEs) (p=0.028) (Figure 2a), human umbilical vein endothelial cells (HUVECs) (p=0.002) (Figure 2b) and T cells (Jurkats) (p=0.06) (Figure 2c); WT MRSA and the spa null mutant stimulated similar amounts of IL-16 in monocytes (THP-1s) (Figure 2d). SpA alone induced IL-16 release from 16HBEs, HUVECs, Jurkats and THP-1s (all p<0.05 as compared to media alone). Of note, LPS and TNF failed to stimulate IL-16 secretion in any of the cell types tested, with the exception of TNF stimulated Jurkats (p<0.05 as compared to media).
Figure 2. IL-16 secretion is SpA specific in multiple cell types.

(a) 16HBEs, (b) HUVECs, (c) Jurkats and (d) THP-1s were infected with wt MRSA or spa mutant (MOI 100, 4h for 16HBEs, 2h for HUVECs, Jurkats and THP-1s). SpA, LPS, and TNF were added at a concentration of 0.5 μM, 10 μg/mL, and 100 μg/mL respectively. IL-16 in culture supernatants was quantified by ELISA (*p<0.05 compared to media, Student's t-test, post-hoc Dunnett's test). Graphs represent data from at least 2 independent experiments.
TNFR1 participates in IL-16 secretion
TNFR1 ligation activates caspase-3 signaling23, important in the processing of pro-IL-16 to its active form22 and is stimulated by the IgG binding domain of SpA10. To establish the importance of TNFR1 mediated signaling in the induction of IL-16 production, we compared generation of IL-16 in WT and Tnfr1−/− mice (Figure 3). There was significantly less IL-16 in the BALF of infected Tnfr1−/− mice compared to WT controls (p<0.05) (Figure 3a). Protein content in the BALF was slightly lower in the KO mice (Figure 3b) with similar levels of KC (Figure 3c). Histopathology of Tnfr1−/− lungs infected with MRSA suggested less destruction of the normal architecture than WT mouse lung (Figure 3d), consistent with previous reports10. Numbers of bacteria recovered in BALF were similar (Figure 3e) and recruited innate immune cells into BALF and lung were not significantly different in the WT and Tnfr1−/− mice (Figure 3f-h). Participation of TNFR1 in IL-16 processing is evident, but incomplete attenuation of IL-16 production in Tnfr1−/− mice suggests that other pathways are involved.
Figure 3. TNFR1 is involved in IL-16 secretion.

Age and sex matched cohorts of C57BL/6J WT and Tnfr1−/− mice were inoculated with 107 cfu of MRSA or PBS for 24 hours. BALF was quantitated for (a) IL-16 by ELISA, (b) total protein (c) and KC by ELISA. (d) H&E sections of fixed whole lungs. Magnification 100x. (e) Bacterial cfus were counted in BALF. (f) Neutrophils (PMNs) (Ly6G+/MHCII− ) and (g) Macrophages (CD11c+/MHCIIlow-mid) in the BALF, as well as CD4+ cells in lung homogenate (h) were enumerated by flow cytometry. Each dot represents an individual mouse and is compiled from 3 independent experiments. Lines represent median values.
Multiple receptors participate in MRSA induced IL-16 secretion
The direct association of SpA and IL-16 shown in stromal and T-cells, indicates that additional SpA receptors may be involved in the generation of IL-16. Accordingly, we tested the participation of EGF receptor (EGFR), a receptor for the IgG binding domain of SpA and directly activated in S. aureus infection25. In the presence of blocking antibody to EGFR, MRSA induction of IL-16 secretion in THP-1s was significantly diminished (p=0.023) as compared to an relevant IgG control (Figure 4a). Activation of FasL (CD95), another member of the TNFR superfamily23, also leads to pro-caspase-8 cleavage26, which in turn activates caspase-3 from its proform, by Ca2+ influx and calpain activation27. Pre-treatment of THP-1s with anti-FasL antibody prior to infection with MRSA also diminished the release of IL-16 (p=0.0007) (Figure 4b). As Ca2+ fluxes are generated by TLR2 signaling28, induced by S. aureus29, we monitored IL-16 production by THP-1s pre-treated with anti-TLR2 and found no diminution of IL-16 levels (Figure 4c). In addition, incubation of THP-1s with the TLR2 agonist Pam3Cys-Ser-Lys4 did not induce IL-16 production, indicating that TLR2 by itself is not responsible for IL-16 production. Thus, multiple TNFR effectors, as well as EGFR, participate in IL-16 secretion.
Figure 4. Multiple receptors are involved in MRSA induced IL-16 secretion in THP-1s.
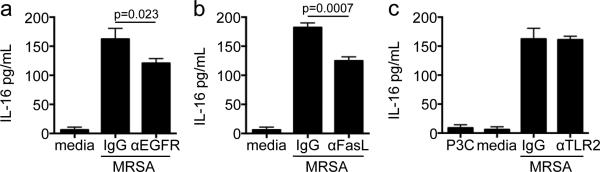
THP-1s were pretreated with neutralizing antibody or IgG isotype 1 hour prior to infection with MRSA at a MOI of 50. Neutralizing antibodies were (a) anti-EGF receptor (EGFR) (2 μg/mL), (b) anti-FasL (5 μg/mL) and (c) anti-TLR2 (2 μg/mL). Pam3Cys-Ser-Lys4 (P3C) (15μg/mL) was used as a control. p values as compared to isotype controls, Student's t-test. The above graphs represent data from at least 2 independent experiments.
Calcium, calpains and caspases are involved in IL-16 processing
Our results suggest that multiple ligands that activate caspase-8 and caspase-3 could participate in the induction of pro-IL-16 processing22. Given the participation of the Ca2+-dependent calpains in caspase activation, we examined the requirement of Ca2+ fluxes in the induction of IL-16. MRSA induced Ca2+ fluxes in Flo-4/AM loaded THP-1s within 10 min of exposure to MRSA with attenuation by pretreatment with BAPTA (Figure 5a and Supplementary Figure S1a-b). THP-1s in media alone showed no baseline Ca2+ fluxes and gave significant fluorescence when treated with thapsigargin (Figure 5b and Supplementary Figure S1c). IL-16 secretion was stimulated by ionomycin alone and significantly decreased in the presence of EDTA (p<0.05 compared to media) (Figure 5c). Pretreatment of THP-1s with calpeptin, a calpain inhibitor, resulted in diminished secretion of IL-16 (p<0.05 compared to DMSO) (Figure 5d), indicating that IL-16 processing in THP-1s is calpain, as well as calcium dependent. Similar differences in Jurkats with calcium and calpain inhibitors were not seen (data not shown). Caspase-3 involvement was demonstrated when THP-1s preloaded with Caspase-3/7 Green Detection Reagent were infected with MRSA, showing an increase in fluorescence over baseline (Figure 5e). We then observed decreased secretion of IL-16 when THP-1s were pretreated with pancaspase inhibitor Z-VAD-FMK (p<0.05 compared to DMSO with MRSA) or a caspase-3 inhibitor (p<0.05 compared to DMSO with MRSA) but not a caspase-1 inhibitor (Figure 5f). Similarly, IL-16 release was reduced in Jurkats pretreated with caspase-3 inhibitor (p<0.05 compared to DMSO with MRSA) (Figure 5g). Staurosporine control, an in vitro inducer of caspase activity30, showed induction of IL-16 secretion in both THP-1s and Jurkats.
Figure 5. MRSA induction of IL-16 release is calcium and calpain dependent in THP-1s.

Calcium fluxes were measured by fluorescence activation on an inverted microscope with a GFP mercury laser. THP-1s loaded with AM/Flo-4 (1mM) and PowerLoad concentrate (1mM) 2 hours prior to stimulation with (a) MRSA (MOI 100) with and without BAPTA (6μM), or (b) media alone followed by thapsigargin (1μM) (T) as a positive control. Representative cells were chosen and fluorescence plotted over time. (c) IL-16 levels in supernatants of THP-1s pretreated 1 hour prior to infection with EGTA (0.5 or 1mmol) with MRSA (MOI 50) was measured 2 hours after infection. (d) IL-16 levels in supernatants of THP-1s pretreated 1 hour prior to infection with calpeptin (CPEP) (20 or 200 μM) with MRSA (MOI 50) was measured 2 hours after infection. Ionomycin (iono) in DMSO was 5 mM. (e) THP-1s were preloaded with Caspase-3/7 Green Detection agent (5μM) 30 minutes prior to infection and then infected with MRSA (MOI 100). Fluorescence was then plotted over time. (f) IL-16 levels in supernatants of THP-1s were pretreated 1 hour prior to infection with respective inhibitor with MRSA (MOI 50) for 2 hours; caspase 1 inhibitor (C1 inh) 50 μM, caspase-3 inhibitor (C3 inh) 50 μM, and pan-caspase inhibitor (ZVAD) 100 μM. Staurosporine in DMSO was 1μM. (g) IL-16 levels in supernatants of Jurkats were pretreated 1 hour prior to infection with respective inhibitor with MRSA (MOI 50) for 2 hours; caspase 1 inhibitor (C1 inh) 50 μM and caspase-3 inhibitor (C3 inh) 50 μM. Staurosporine in DMSO was 1μM. *p<0.05 value as compared to MRSA infected samples, Student's t-test, post-hoc Dunnett's test for multiple comparisons. The above graphs represent data from at least 2 independent experiments.
T cells are a major source of IL-16
Our in vitro data suggest that multiple cells types can produce IL-16. To determine the major source of IL-16 in the setting of S. aureus pneumonia, immunohistochemistry was performed on lung sections of WT mice infected with MRSA. IL-16 staining was observed in discrete cells (black arrows), and not a generalized response of the respiratory epithelium in MRSA infection (Figure 6a). Since CD4 itself is a receptor for IL-16, we predicted that CD4+ cells would participate in an autocrine loop and were a likely source of the cytokine in the lung. Cd4−/− and WT mice were infected with MRSA, and at 24 hours post inoculation, IL-16 in the BALF was nearly half in the airway of the Cd4−/− mouse compared to WT counterparts (p=0.0043) (Figure 6b). The apparent correlation between the abundance of CD4+ cells and IL-16 production was further confirmed in MRSA infected mice with increased numbers of CD4+ cells. We noted that mice treated with clodronate, depleted of alveolar macrophages had a significant increase in the numbers of pulmonary CD4+ cells (Figure 6c), but not other T-cell populations12. These mice had a corresponding increase in IL-16 in the setting of MRSA pneumonia (Figure 6d), suggesting CD4+ cells are stimulated by, as well as generate IL-16 in an autocrine manner.
Figure 6. IL-16 is produced by CD4+ cells.

(a) Immunohistochemistry by standard methods with anti-IL-16 antibody on fixed whole lungs of C57BL/6J WT given PBS or infected with 107 MRSA for 24 hours. Magnification 100x. Black arrows point to areas of IL-16 staining (brown). (b) Age and sex-matched cohorts of C57BL/6J WT or Cd4−/− mice were inoculated intranasally with 107 MRSA and IL-16 was quantified in BALF collected by ELISA. Clodronate (Clod) or PBS liposome treated mice were given 107 MRSA for 24 hours. BALF stained for (c) CD4+ populations were analyzed by flow cytometry and (d) IL-16 was measured by ELISA. Each dot represents an individual mouse and data is combined from 2 separate experiments. Lines represent median values.
Neutralization of IL-16 improves clearance of MRSA and pathology
The exuberant host response to infection is a hallmark of S. aureus pneumonia and we postulated that neutralization of IL-16 would dampen its harmful consequences. By directly affecting CD4+ cells, we hypothesized that we might limit the excessive proinflammatory response, without compromising the recruitment of phagocytes essential to clear infection. As mice lacking IL-16 production are not available, we pretreated mice with anti-IL-16 before giving our standard intranasal inoculum of MRSA, documenting the expected reduction in the amount of IL-16 in the airway as compared to IgG control (p=0.04) (Figure 7a). A major effect on MRSA clearance was observed with a 6-fold reduction in cfus recovered from the airways (p=0.0042) (Figure 7b) and better preservation of the normal lung architecture, than in the IgG-treated control (Figure 7c). Decreased amounts of KC (p=0.016) were seen in the neutralization group (Figure 7d), but the cellular populations recruited to the BALF and lung were unaffected (Supplemental Figure S2a-h). TNF, IL 6 and protein levels were similar (Figure 7e-g). Thus, IL-16 contributes to lung pathology in MRSA pneumonia.
Figure 7. Neutralization of IL-16 improves clearance of MRSA.

Anti-IL-16 antibody or IgG isotype control was injected i.p. (100 μg) 2 hours prior to infection with 107 cfu MRSA. Effect of IL-16 neutralization was documented by quantifying (a) IL-16 by ELISA and (b) bacterial clearance was measured by cfus in the BALF after 18 hours. (c) H&E sections of fixed whole lungs. Magnification 100x, inset box 400x. Cytokines (d) KC, (e) IL 6 and (f) TNF, as well as (g) protein were measured in the BALF. p values are as shown, as compared to IgG isotype, by Student's t-test. Each dot represents an individual mouse and data is combined from 2 separate experiments. Lines represent median values.
DISCUSSION
IL-16 has been described in a number of inflammatory and oncologic conditions since its description by Cruikshank in 198231. However, despite its importance in the development of TH1-mediated inflammation32, relatively little is known about its role in the pathogenesis of bacterial infection, including S. aureus pneumonia33. The biology of murine and human IL-16 is highly conserved34, thus the analysis of IL-16 in murine models of infection is likely applicable in human tissues as well. IL-16, as a ligand for CD4+ cells, functions in T cell activation as well as proliferation35 and has biologic activity in both its pro-form and mature processed form36. We demonstrate that MRSA induces IL-16 acutely in the lung, where it participates in the exuberant and potentially damaging inflammatory response to infection.
Multiple cell types contribute to the proinflammatory signaling produced in response to S. aureus with the production of NF-κB dependent cytokines and chemokines37. Epithelial, endothelial and immune cells can all produce IL-16 as part of the immediate response to MRSA. However, in contrast to the NF-κB dependent cytokines, IL-16 production appears to be specifically linked to the TNF cascade and dependent upon both TNF and specific bacterial ligands. LPS and TNF alone failed to stimulate IL-16 production, whereas MRSA and SpA alone were potent stimuli in most of the cell types examined. The central role of the TNF cascade in the induction of IL-16 processing, as demonstrated by diminished IL-16 production in infected mice lacking TNFR1, suggests an explanation for the enhanced ability of MRSA to activate this cytokine. In addition to stimulating TNF indirectly through production of PAMPs and pattern recognition receptors on immune cells, SpA directly ligates both TNFR1 and EGFR on multiple cell types to initiate signaling10,25 through redundant signaling pathways. IL-16 secretion into the airway of infected mice lacking TNFR1 was not fully attenuated and signaling by multiple receptors, including EGFR and FasL, were found to initiate the processing of IL-16 to its active form.
A second stimulus, the generation of a Ca2+ flux sufficient to activate calpain activity was also required for IL-16 production. We demonstrated this response in THP-1s, representative of alveolar macrophages, the predominant immune cell to initially encounter respiratory pathogens. We were unable to show this calcium dependence in Jurkats, as Ca2+ fluxes are readily activated in T cells38, which also possess the CD4 receptor for IL-16. In vivo, these CD4+ cells appear to be the most responsive to the multiple stimuli apparently required to initiate IL-16 processing. In vitro, we and others18 have demonstrated that multiple cell types can produce IL-16. However, the requirement for TNF signaling or ligation of a TNFR family receptor plus the need for calpain activity likely limits the in vivo production of the cytokine to specific cell types. This requirement for multiple stimuli may also help to explain why LPS alone, or the Gram negative airway pathogens tested were much less capable in stimulating IL-16 production.
Once T cells are recruited, IL-16 CD4 interactions initiate an autocrine loop to further induce T cell production of more IL-16. The ability of S. aureus to directly initiate a T cell cytokine response may contribute to virulence. The improved MRSA clearance in mice with decreased IL-16 suggests that T cell recruitment and activation may contribute to lung damage in this setting. In our murine model system, we were unable to detect differences in numbers of the major immune cells recruited in mice treated with anti-IL-16 or the isotype control. Thus, the effects of the anti-IL-16 may be in blocking further T-cell activity and their contribution to cytokine production.
In several other murine models of bacterial pneumonia, the ability to clear infection and preserve lung architecture correlated with decreased cytokine expression and not with the absolute numbers of inflammatory cells recruited into the lung39,40. There are multiple potential sources of neutrophil and macrophage chemokines37,41. Limiting the participation of IL-16 and its T cell targets did not limit phagocytic influx, but resulted in decreased production of KC. Of note, KC is a major neutrophil chemokine that has been linked to lung injury in other models of sepsis 42,43 consistent with the hypothesis that cytokines themselves, may be directly associated with lung damage.
In the absence of an Il16−/− mouse it is difficult to fully evaluate the role of this cytokine in the acute response to MRSA pneumonia. Our data indicate that the capability of MRSA, as opposed to commensal or opportunistic Gram negative airway pathogens, to activate IL-16 and initiate this amplification of CD4+ T cell participation further contributes to its virulence in the setting of pneumonia.
METHODS
Cell culture, bacterial strains, and purified proteins
Human airway epithelial cells (16HBEs, D. Gruenert, California Pacific Medical Center Research Institute, San Francisco, CA), human umbilical vein endothelial cells in primary culture (HUVECs, B. Hopkins, Columbia College of Physicians and Surgeons, New York, NY), THP-1 human monocytic cells (THP-1s, ATCC, Manassas, VA), and Jurkat human T-cells (Jurkats, E6-1, ATCC, Manassas, VA) were grown at 37°C with 5% CO2. THP-1s were activated with 1 μM phorbol 12-myristate 13-acetate (PMA) and Jurkats were activated with 0.1 μM PMA + 0.1 μM ionomycin, both for 24 hours prior to infection. Methicillin resistant S. aureus USA300 (MRSA)44, isogenic spa mutant7, S. aureus 502A, K. pneumoniae ST 258 and P. aeruginosa PAK were grown in LB and resuspended in antibiotic free media (16HBEs – Bronchiolife, Lifeline Technologies, HUVECs – Vasculife, Lifeline Technologies or THP-1s/Jurkats – RPMI, Corning, with 10% heat inactivated fetal bovine serum, Benchmark) for infections in vitro. Purified SpA (Calbiochem) and TNF (Peprotech) were added at differential concentrations based on the assay. Pam3Cys-Ser-Lys4 (EMC Microcollections), thapsigargin (Invitrogen), ionomycin (Calbiochem) and staurosporine (Cell Signaling) were used as positive controls in respective assays.
Antibodies and Inhibitors
Antibodies used in vitro were anti-EGFR (1005; Santa Cruz), anti-FasL (NOK1; BD Biosciences), anti-TLR2 (H 175; Santa Cruz), anti-TNFR1 (H-271; Santa Cruz) and anti-IL-16 (ab9563; Abcam). Respective IgG isotype controls were used for comparison; rabbit (2027; Santa Cruz) or mouse (2025; Santa Cruz). Inhibitors used were pancaspase inhibitor ZVAD-FMK (Santa Cruz), caspase-3 inhibitor (Calbiochem), caspase 1 inhibitor (Calbiochem), calpeptin (Calbiochem), BAPTA (Calbiochem) and EDTA (Sigma).
Mouse studies
In vivo experiments were performed using age and sex-matched WT, Tnfr1−/−, and Cd4−/− C57BL/6J mice (Jackson Laboratories). Mice were anaesthetized with 100 mg/kg ketamine and 5 mg/kg xylazine, infected intranasally with MRSA, S. aureus 502A, K. pneumoniae ST 258 or P. aeruginosa PAK (107 cfu in 50 QL of PBS), and sacrificed 18-24 hours after infection. To deplete macrophages, 75 QL of clodronate liposomes or PBS liposome controls were inoculated intranasally 24 hours prior to infection with MRSA as previously described12. For the neutralization of IL-16, 100 μg of anti-IL-16 antibody (ab9563; Abcam) or IgG isotype (Jackson ImmunoResearch) was injected intraperitoneally 2 hours prior to infection as previously described45. Animal experiments were performed in accordance with the guidelines of the IACUC at Columbia University (protocol number AAAE5252 and AAAD0624).
Bronchoalveolar Lavage Fluid (BALF) Assays
BALF was obtained by instilling 1 mL aliquots of sterile PBS with calcium and magnesium into a cannulated trachea three times. Serial dilutions for bacterial enumeration were performed on the BALF prior to centrifuging. IL-16 mouse ELISA (R&D Systems), TNF mouse ELISA (eBioscience), KC mouse ELISA (R&D systems), IL 6 mouse ELISA (Biolegend), and Bradford Assay (Thermo Scientific) for protein content were performed on the BALF supernatant.
Analysis of immune cell populations
Analysis of cell populations in BALF or single cell suspension of lung homogenate was conducted as before46. Cells were labeled with a combination of phycoerythrin (PE)-labeled anti-NK 1.1 (PK136; eBioscience), fluorescein (FITC)-labeled anti-Ly6G (RB6-8C5; eBioscience), allophycocyanin (APC)-labeled anti-MHCII (M5/114.15.2; eBioscience), PerCP-Cy5.5-labelled anti-CD11c (N418; Biolegend), or phycoerythrin (PE)-labeled CD4 (RM4-5; Biolegend) alone. Neutrophils (PMNs) were defined as Ly6G+/MHCII− , Macrophages as CD11c+/MHCIIlow-mid and Dendritic cells (DCs) as CD11c+/MHCIIhigh. Fc block (anti-mouse CD16/32) was also added to each sample (93; eBioscience). Uniform dyed microspheres (Bangs Laboratories, Inc) were added to calculate the concentration of cellular components. Samples were run on FACS Calibur and analyzed on FlowJo (ver 10.0.6).
Histopathology and Immunohistochemistry
Whole mouse lung was fixed with 4% paraformaldehyde for 24 hours, 70% ethanol for 24 hours, and then prepared in paraffin blocks. H&E staining was performed on 5 mm sections for gross pathology. For immunohistochemistry (IHC), 5 mm sections on glass slides were rehydrated with xylene alternative (Safe Clear, Protocol) and ethanol. Antigens were presented using an acidic buffer (sodium citrate 10mM, 0.05% Tween 20, pH 6) at 60°C overnight. Sections were additionally permeabilized with Tween 20 0.5% for 10 minutes and quenched with 0.3% H2O2 for 10 minutes. Detection of IL-16 by IHC with peroxidase staining was performed using the Immunocruz ABC Staining kit (Santa Cruz). Controls were performed with secondary antibody only.
Calcium Imaging
THP-1s were loaded with AM/Flo-4 (Invitrogen) with PowerLoad concentrate (Invitrogen) prior to imaging and stimulation. Cells were imaged on an Olympus 1X81 inverted microscope with a GFP mercury laser. Images were analyzed and videos made using MetaMorph software (Ver.7.5.3.0, April, 2008).
Caspase-3/7 Detection Assay
THP-1s were preloaded with CellEvent Caspase-3/7 Green Detection Reagent (Invitrogen) 30 minutes prior to infection. Cells were stimulated and then read at absorption/emission maxima of 502/530 nm every 10 minutes for 2 hours on a Tecan Infinite 200 (V2.11, April, 2008).
Statistics
All statistical analysis was performed using GraphPad Prism Version 6.0c (March 21, 2013). Samples with normal distributions, were analyzed by Student's t test. Mouse samples were compared using the nonparametric Mann-Whitney test. For multiple comparisons, an ANOVA was then followed by a post hoc Dunnett's test to compare to the control group. Differences in groups were considered significant if p < 0.05.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by R01HL079395 to AP. Fluorescence microscopy was performed in the laboratory of Jahar Bhattacharya with the gracious assistance of Kristen Westphalen at the Columbia University College of Physicians and Surgeons, New York, New York, USA.
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1.Klevens RM, et al. Invasive Methicillin-Resistant Staphylococcus aureus Infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Guidelines for the Management of Adults with Hospital-acquired, Ventilator-associated, and Healthcare associated Pneumonia. Am. J. Respir. Crit. Care Med. 2005;171:388–416. doi: 10.1164/rccm.200405-644ST. [DOI] [PubMed] [Google Scholar]
- 3.Morens DM, Taubenberger JK, Fauci AS. Predominant Role of Bacterial Pneumonia as a Cause of Death in Pandemic Influenza: Implications for Pandemic Influenza Preparedness. J. Infect. Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kollef MH, et al. Epidemiology and Outcomes of Health-care–Associated Pneumonia Results From a Large US Database of Culture-Positive Pneumonia. Chest. 2005;128:3854–3862. doi: 10.1378/chest.128.6.3854. [DOI] [PubMed] [Google Scholar]
- 5.Inoshima I, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011;17:1310–1314. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diep BA, et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. U. S. A. 2010;107:5587–5592. doi: 10.1073/pnas.0912403107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin FJ, Gomez MI, Wetzel DM, Memmi G, O'Seaghdha M, Soong G, Schindler C, Prince A. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Invest. 2009;119:1931–1939. doi: 10.1172/JCI35879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kebaier C, et al. Staphylococcus aureus α-Hemolysin Mediates Virulence in a Murine Model of Severe Pneumonia Through Activation of the NLRP3 Inflammasome. J. Infect. Dis. 2012;205:807–817. doi: 10.1093/infdis/jir846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomez MI, et al. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat. Med. 2004;10:842–848. doi: 10.1038/nm1079. [DOI] [PubMed] [Google Scholar]
- 10.Gomez MI. Staphylococcus aureus Protein A Activates TNFR1 Signaling through Conserved IgG Binding Domains. J. Biol. Chem. 2006;281:20190–20196. doi: 10.1074/jbc.M601956200. [DOI] [PubMed] [Google Scholar]
- 11.Ichikawa A, et al. CXCL10-CXCR3 enhances the development of neutrophil mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 2013;187:65–77. doi: 10.1164/rccm.201203-0508OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin FJ, Parker D, Harfenist BS, Soong G, Prince A. Participation of CD11c(+) leukocytes in methicillin resistant Staphylococcus aureus clearance from the lung. Infect. Immun. 2011;79:1898–1904. doi: 10.1128/IAI.01299-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLoughlin RM, et al. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc. Natl. Acad. Sci. U. S. A. 2006;103:10408–10413. doi: 10.1073/pnas.0508961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Köckritz-Blickwede von, M., et al. Immunological Mechanisms Underlying the Genetic Predisposition to Severe Staphylococcus aureus Infection in the Mouse Model. Am. J. Pathol. 2008;173:1657–1668. doi: 10.2353/ajpath.2008.080337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson GJ, et al. A Novel Core Genome-Encoded Superantigen Contributes to Lethality of Community-Associated MRSA Necrotizing Pneumonia. PLoS Pathog. 2011;7:e1002271. doi: 10.1371/journal.ppat.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang D, et al. Long-term exposure of chemokine CXCL10 causes bronchiolitis-like inflammation. Am. J. Respir. Cell Mol. Biol. 2012;46:592–598. doi: 10.1165/rcmb.2011-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamza T, Barnett JB, Li B. Interleukin 12 a Key Immunoregulatory Cytokine in Infection Applications. Int. J. Mol. Sci. 2010;11:789–806. doi: 10.3390/ijms11030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruikshank WW, Kornfeld H, Center DM. Interleukin-16. J. Leukoc. Biol. 2000;67:757–766. doi: 10.1002/jlb.67.6.757. [DOI] [PubMed] [Google Scholar]
- 19.Cruikshank W, Center DM. Modulation of lymphocyte migration by human lymphokines. II. Purification of a lymphotactic factor (LCF). J. Immunol. 1982;128:2569–2574. [PubMed] [Google Scholar]
- 20.Cruikshank W, Little F. lnterleukin-16: the ins and outs of regulating T-cell activation. Crit. Rev. Immunol. 2008;28:467–483. doi: 10.1615/critrevimmunol.v28.i6.10. [DOI] [PubMed] [Google Scholar]
- 21.Little FF, Lynch E, Fine G, Center DM, Cruikshank WW. Tumor Necrosis Factor-α–Induced Synthesis of Interleukin 16 in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2003;28:354–362. doi: 10.1165/rcmb.2002-0043OC. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, et al. Processing and activation of pro-interleukin-16 by caspase 3. J. Biol. Chem. 1998;273:1144–1149. doi: 10.1074/jbc.273.2.1144. [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 24.Wang P, et al. IL-16 Induces Intestinal Inflammation via PepT1 Upregulation in a Pufferfish Model: New Insights into the Molecular Mechanism of Inflammatory Bowel Disease. J. Immunol. 2013;191:1413–27. doi: 10.4049/jimmunol.1202598. [DOI] [PubMed] [Google Scholar]
- 25.Gomez MI, Seaghdha MO, Prince AS. Staphylococcus aureus protein A activates TACE through EGFR-dependent signaling. EMBO J. 2007;26:701–709. doi: 10.1038/sj.emboj.7601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2011;19:36–41. doi: 10.1038/cdd.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaisid T, Barnoy S, Kosower NS. Calpain activates caspase-8 in neuron-like differentiated PC12 cells via the amyloid-beta-peptide and CD95 pathways. Int. J. Biochem. Cell Biol. 2009;41:2450–2458. doi: 10.1016/j.biocel.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Chun J, Prince A. Activation of Ca2+-dependent signaling by TLR2. J. Immunol. 2006;177:1330–1337. doi: 10.4049/jimmunol.177.2.1330. [DOI] [PubMed] [Google Scholar]
- 29.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 30.Chae H. Molecular mechanism of staurosporine-induced apoptosis in osteoblasts. Pharmacol. Res. 2000;42:373–381. doi: 10.1006/phrs.2000.0700. [DOI] [PubMed] [Google Scholar]
- 31.Center DM, Cruikshank W. Modulation of lymphocyte migration by human lymphokines. I. Identification and characterization of chemoattractant activity for lymphocytes from mitogen stimulated mononuclear cells. J. Immunol. 1982;128:2563–2568. [PubMed] [Google Scholar]
- 32.Lynch EA, Heijens CAW, Horst NF, Center DM, Cruikshank WW. Cutting edge: IL-16/CD4 preferentially induces Th1 cell migration: requirement of CCR5. J. Immunol. 2003;171:4965–4968. doi: 10.4049/jimmunol.171.10.4965. [DOI] [PubMed] [Google Scholar]
- 33.Glass WG, Sarisky RT, Vecchio AMD. Not-So-Sweet Sixteen: The Role of IL 16 in Infectious and Immune-Mediated Inflammatory Diseases. J. Interferon Cytokine Res. 2006;26:511–520. doi: 10.1089/jir.2006.26.511. [DOI] [PubMed] [Google Scholar]
- 34.Keane J, et al. Conservation of structure and function between human and murine IL 16. J. Immunol. 1998;160:5945–5954. [PubMed] [Google Scholar]
- 35.Wilson KC, Center DM, Cruikshank WW. The effect of interleukin-16 and its precursor on T lymphocyte activation and growth. Growth Factors. 2004;22:97–104. doi: 10.1080/08977190410001704679. [DOI] [PubMed] [Google Scholar]
- 36.Ren F, et al. Pro-IL-16 regulation in activated murine CD4+ lymphocytes. J. Immunol. 2005;174:2738–2745. doi: 10.4049/jimmunol.174.5.2738. [DOI] [PubMed] [Google Scholar]
- 37.Parker D, Prince A. Immunopathogenesis of Staphylococcus aureus pulmonary infection. Semin. Immunopathol. 2012;34:281–297. doi: 10.1007/s00281-011-0291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen TS, Prince AS. Bacterial pathogens activate a common inflammatory pathway through IFNλ regulation of PDCD4. PLoS Pathog. 2013;9:e1003682. doi: 10.1371/journal.ppat.1003682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J. Clin. Invest. 2013;123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Archer NK, Harro JM, Shirtliff ME. Clearance of Staphylococcus aureus Nasal Carriage Is T Cell Dependent and Mediated through Interleukin-17A Expression and Neutrophil Influx. Infect. Immun. 2013;81:2070–2075. doi: 10.1128/IAI.00084-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahuja N, et al. Circulating IL-6 mediates lung injury via CXCL1 production after acute kidney injury in mice. Am. J. Physiol. Renal Physiol. 2012;303:F864–72. doi: 10.1152/ajprenal.00025.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neumann B, et al. Mechanisms of acute inflammatory lung injury induced by abdominal sepsis. Int. Immunol. 1999;11:217–227. doi: 10.1093/intimm/11.2.217. [DOI] [PubMed] [Google Scholar]
- 44.Diep BA, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 45.Meagher C, et al. Neutralization of interleukin-16 protects nonobese diabetic mice from autoimmune type 1 diabetes by a CCL4-dependent mechanism. Diabetes. 2010;59:2862–2871. doi: 10.2337/db09-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parker D, et al. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio. 2011;2:e00016–11. doi: 10.1128/mBio.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.