

Abstract
The airway epithelium provides a barrier that separates inhaled air and its various particulates from the underlying tissues. It provides key physiological functions in both sensing the environment and initiating appropriate innate immune defenses to protect the lung. Protease-activated receptor-2 (PAR2) is expressed both apically and basolaterally throughout the airway epithelium. One consequence of basolateral PAR2 activation is the rapid, Ca2+-dependent ion flux that favors secretion in the normally absorptive airway epithelium. However, roles for apically expressed PAR2 activation have not been demonstrated, in part due to the lack of specific, high-potency PAR2 ligands. In the present study, we used the newly developed PAR2 ligand 2at-LIGRLO(PEG3-Pam)-NH2 in combination with well-differentiated, primary cultured airway epithelial cells from wild-type and PAR2−/− mice to examine the physiological role of PAR2 in the conducting airway after apical activation. Using digital imaging microscopy of intracellular Ca2+ concentration changes, we verified ligand potency on PAR2 in primary cultured airway cells. Examination of airway epithelial tissue in an Ussing chamber showed that apical activation of PAR2 by 2at-LIGRLO(PEG3-Pam)-NH2 resulted in a transient decrease in transepithelial resistance that was due to increased apical ion efflux. We determined pharmacologically that this increase in ion conductance was through Ca2+-activated Cl− and large-conductance K+ channels that were blocked with a Ca2+-activated Cl− channel inhibitor and clotrimazole, respectively. Stimulation of Cl− efflux via PAR2 activation at the airway epithelial surface can increase airway surface liquid that would aid in clearing the airway of noxious inhaled agents.
Keywords: allergic asthma, asthmagen, ion conductance, lipidated agonist, protease-activated receptor-2
the airway epithelium is the first site of cellular contact for inhaled allergens and other insults. It provides a barrier to protect underlying submucosa from inhalation threats and orchestrates critical aspects of airway innate immunity, such as inflammation, airway remodeling, and mucociliary clearance (16, 34). A subset of inhaled allergens (e.g., house dust mites, cockroaches, and various fungi) has been associated with the onset and pathogenesis of allergic asthma (36, 40). Allergic asthma is a chronic disease associated with airway remodeling that includes inflammation, secretory alterations, and airway hyperreactivity culminating in airway obstruction (20). Although allergens associated with asthma (i.e., asthmagens) are quite diverse, many of them contain proteases that can interact with the airway epithelium through a variety of mechanisms that include the activation of protease-activated receptor-2 (PAR2) (22).
PAR2 is a G protein-coupled receptor (GPCR) belonging to a family of GPCRs composed of four members (2). Unlike typical GPCRs, PAR ligand activation sequences are contained within the extracellular portion of the GPCR. Proteolytic cleavage of the NH2-terminus reveals the tethered ligand [SLIGKV for humans and SLIGRL for rodents (30)], which binds an activation pocket, leading to cellular signaling cascades. Exogenous asthmagen-associated proteases that have been demonstrated to activate PAR2 include those from the house dust mite, cockroaches, and fungi (22, 23, 32). Individuals with asthma are often sensitized to these protease-containing allergens; thus, understanding roles for PAR2 in normal and pathological airway physiology have been an intense research focus (2, 17, 21, 33, 41).
The full impact of PAR2 in allergic asthma is quite complex. PAR2 activation has been linked to long-term, detrimental airway inflammation and hypersecretion as well as beneficial bronchodilation (7, 8, 10, 12, 19, 38, 42). It has been suggested that these diametrically opposed outcomes may be separable by the activation of separate signaling pathways downstream of PAR2 (43). In studies examining more rapid responses in the airway epithelium, PAR2 has been linked to changes in airway epithelial ion conductance (5, 9, 25, 31, 35, 37). Interestingly, studies that have uncovered roles for PAR2 in ion conductance changes across the airway epithelium have focused on the basolateral activation of PAR2 (1, 24, 25, 35). This has occurred despite immunocytochemical evidence for apical expression of PAR2 in mouse and human tissues (6, 8, 25), where the receptor would most likely interact with inhaled proteases in healthy individuals. Further complicating the role of PAR2 in airway epithelial ion movement is the finding that, in some mouse strains, PAR2 ligands (i.e., SLIGRL-NH2) have been shown to activate ion conductance changes that appear independent of PAR2 (1).
In the present study, we evaluated the potency and specificity of a recently developed, lipidated PAR2 agonist, 2at-LIGRLO(PEG3-Pam)-NH2 (14), for use in well-differentiated, primary cultured airway epithelial cells from wild-type and PAR2−/− mice. We first examined PAR2 signaling with digital imaging microscopy of intracellular Ca2+ concentration ([Ca2+]i) changes. We next examined apical activation of PAR2 in Ussing chamber experiments designed to evaluate PAR2 effects on transepithelial resistance (TER) and alterations in ion conductance. We demonstrated that novel apical stimulation of PAR2 in the primary airway epithelium leads to the activation of Ca2+-regulated ion channels consistent with a subsequent apical efflux of Cl−. The availability of potent and specific PAR2 agonists suggests that apical PAR2 activation can contribute to mucociliary clearance and thus aid in the rapid clearing of asthmagens from healthy airways.
MATERIALS AND METHODS
Materials.
Cellgro DMEM:F-12 was from Mediatech (Manassas, VA). Lechner and LaVeck basal media, HBSS, glutamax, penicillin, and streptomycin were from InVitrogen (Carlsbad, CA). Fibronectin, collagen type I, and Nu-Serum were from Becton-Dickinson (Franklin Lakes, NJ). FBS and clotrimazole were from Sigma-Aldrich (St. Louis, MO). CaCCinh-A01 was from Tocris Bioscience (Bristol, UK). Semipermeable filters were Corning Costar 6.5-mm Transwell filters with a 0.4-μm Pore Polyester Membrane Insert (Lowell, MA). All other chemicals were from Sigma-Aldrich or Fisher Scientific (Pittsburgh, PA).
Tissue culture methods.
Animal protocols were approved by the Institutional Animal Care and Use Committee of The University of Arizona. Primary mouse tracheal epithelial (MTE) cells were cultured as previously described (39). C57Bl/6 wild-type and PAR2−/− mice on a C57Bl/6 background were used for these cultures. Isolated primary MTE cell cultures for digital imaging microscopy were seeded onto glass coverslips coated with rat tail collagen (11), cultured at 37°C with 5% CO2, and fed every other day in full culture medium. Although these cells establish cell junctions, they do not become well differentiated. MTE cells used in Ussing chamber experiments were seeded onto 6.5-mm semipermeable filters coated with a collagen-fibronectin-BSA matrix and cultured at 37°C with 5% CO2. MTE cells were grown in full culture medium to allow for confluent monolayers. After cell monolayers reached a TER of >500 Ω·cm2 (∼5 days), apical media were removed to establish an air interface, and media were changed to a 1:1 mixture of DMEM and Ham's F-12, 1% penicillin-streptomycin, and 2% Nu-Serum. These cells become a mixed population of well-differentiated cells with clearly established cilia.
[Ca2+]i measurements.
Cell monolayers were washed with modified HBSS (1.3 mM CaCl2, 5.0 mM KCl, 0.3 mM KH2PO4, 0.5 mM MgCl2, 0.4 mM MgSO4, 137.9 mM NaCl, 0.3 mM Na2PO4, and 1% glucose additionally buffered with 25 mM HEPES, pH 7.4) and loaded for 45–60 min with 5 μM fura 2-AM in HBSS. Cells were removed from fura 2-AM loading solution and placed back in matching HBSS for at least 20 min before Ca2+ imaging. Fura-2 fluorescence was observed on an Olympus IX70 microscope with a ×40 oil-immersion objective after alternating excitation between 340 and 380 nm by a 75-W Xenon lamp linked to a Delta Ram V illuminator (PTI, London, ON, Canada) and a gel optic line. Images of emitted fluorescence above 505 nm were recorded by an ICCD camera (PTI) or an EVO-512 camera (Roper Scientific, Tucson, AZ) and simultaneously displayed on a color monitor. The imaging system was under software control (ImageMaster, PTI) and collected a ratio approximately every 0.6 s. [Ca2+]i was calculated by ratiometric analysis of fura-2 fluorescence using equations originally published in Ref. 18. A typical field of view contained 30–40 cells at a resting [Ca2+]i estimated to be ∼75 nM. A change in [Ca2+]i was considered positive if the cell increased its [Ca2+]i twofold or more.
Ussing chamber experiments.
MTE cells on 6.5-mm Transwell filters were mounted in an EasyMount Ussing chamber system (Physiologic Instruments, San Diego, CA) and bathed on both sides with Krebs-Ringer buffer containing (in mM) 115 NaCl, 25 NaHCO3, 0.4 KH2PO4, 2.4 K2HPO4, 1.2 CaCl2, and 1.2 MgCl2 with 10 glucose. Bath solutions were continuously circulated with a gas lift by bubbling with 95% air and 5% CO2 at 37°C (pH 7.4). MTE cell monolayers were either current clamped and observed for changes in TER or voltage clamped and monitored for changes in short circuit current (Isc) and TER with a multichannel voltage/current-clamp VCC MC8 (Physiologic Instruments). For current-clamp experiments, MTE monolayers were clamped to 0 μA, and a 5-μA pulse of 200-ms duration was imposed every 10 s to monitor TER. Changes in Isc from those experiments were calculated using Ohm's law. Changes in TER (ΔTER) were calculated from the difference between the TER measurement at baseline and the peak measurement change after the addition of agonist. In voltage-clamp experiments, MTE cell monolayers were clamped to 0 mV, and a 5-mV pulse of 200-ms duration was imposed every 10 s. Changes in Isc (ΔIsc) were calculated from the difference between the initial Isc measurement at baseline and the peak measurement change after the addition of agonist. Data were analyzed using Acquire and Analyze software (version 2.3, Physiologic Instruments).
Statistics.
An unpaired Student's t-test was used for comparisons between two data sets. One-way ANOVA with Tukey's multiple-comparison test was used for data sets with more than two comparisons. P values of <0.05 were used to establish significant differences between data sets. Values are expressed as means ± SE unless otherwise noted.
RESULTS
High-potency lipidated ligand specifically activates PAR2 in primary cultured airway cells.
We initially tested the specificity and concentration-response relationship of the PAR2 synthetic tethered ligand 2at-LIGRLO(PEG3-Pam)-NH2 (14) in the primary MTE cell model using digital imaging microscopy. In each experiment, MTE cells from either wild type or PAR2−/− mice were exposed to varying concentrations of 2at-LIGRLO(PEG3-Pam)-NH2, and [Ca2+]i was monitored over time. In a typical experiment with wild-type MTE cells, 2at-LIGRLO(PEG3-Pam)-NH2 elicited a rise in [Ca2+]i with a population average peak of ∼200 nM that recovered to baseline within 90 s (Fig. 1, A and D). MTE cells from PAR2−/− mice, however, showed no measureable change in [Ca2+]i after the addition of 2at-LIGRLO(PEG3-Pam)-NH2 (Fig. 1B). To confirm that the loss of [Ca2+]i signaling was due to lack of PAR2 in MTE cells, 5 μM ATP was applied 3 min after the addition of 2at-LIGRLO(PEG3-Pam)-NH2. The ATP response was similar in both wild-type and PAR2−/− MTE cells (Fig. 1, A and B, respectively). A concentration-response relationship was established for the percentage of cells that responded with a change in [Ca2+]i to 2at-LIGRLO(PEG3-Pam)-NH2 in wild-type MTE cells (EC50: 835 nM, 95% confidence interval: 261 nM–2.68 μM; Fig. 1C).
Fig. 1.

2at-LIGRLO(PEG3-Pam)-NH2 activates protease-activated receptor-2 (PAR2) in mouse tracheal epithelial (MTE) cells. Average intracellular Ca2+ concentration ([Ca2+]i) was plotted over time. A: wild-type MTE cells increased [Ca2+]i after 2at-LIGRLO(PEG3-Pam)-NH2 (1 μM) and responded to ATP (5 μM) with a second rise in [Ca2+]i. B: 2at-LIGRLO(PEG3-Pam)-NH2 (1 μM) evoked no change in [Ca2+]i in PAR2−/− MTE cells; however, their response to ATP (5 μM) was similar to wild-type cells. C: concentration-response curve for 2at-LIGRLO(PEG3-Pam)-NH2 in wild-type MTE cells [EC50: 835 nM, 95% confidence interval (CI): 261 nM–2.68 μM]. D: color panels of [Ca2+]i from wild-type MTE cells after 2at-LIGRLO(PEG3-Pam)-NH2. White lines depict cell borders. The time after application is noted. A color bar indicating approximate [Ca2+]i is given in the bottom left of D. Traces in A and B represent average [Ca2+]i of >35 cells in a single experiment and are means ± SE; n ≥ 3 for each experiment protocol.
Apically applied lipidated PAR2 agonist alters TER and Isc in polarized primary airway epithelial cells.
In healthy individuals, asthmagen exposure in the airway lumen first contacts the apical membrane of the airway epithelium. Thus, we evaluated physiological responses of well-differentiated primary MTE cells after apical exposure of PAR2-specific agonists in Ussing chamber experiments. To better mimic endogenous airway epithelial physiology, MTE cell cultures were first current clamped and measured for TER changes after exposure to 10 μM 2at-LIGRLO(PEG3-Pam)-NH2; Isc was subsequently calculated. In primary MTE cell cultures from wild-type mice, a transient decrease in TER was observed (141.2 ± 42.15 Ω·cm2, n = 6; Fig. 2, A and B). However, in MTE cell cultures from PAR−/− mice, changes in TER in response to 2at-LIGRLO(PEG3-Pam)-NH2 were negligible (13.5 ± 12.84 Ω·cm2, n = 4; Fig. 2, A and B). The observed loss in TER after the addition of 2at-LIGRLO(PEG3-Pam)-NH2 could not be explained by a substantial breakdown in the epithelial barrier as fluorescently labeled permeability markers as small as 600 Da did not significantly penetrate the epithelial barrier of wild-type cells (data not shown). The loss in TER, however, was concurrent with a transient increase in calculated Isc in wild-type epithelia (6.8 ± 1.5 μA/cm2, n = 6; Fig. 2C and D). In contrast, the calculated change in Isc in response to 2at-LIGRLO(PEG3-Pam)-NH2 from PAR2−/− epithelia was negligible (1.0 ± 0.18 μA/cm2, n = 4; Fig. 2, C and D).
Fig. 2.

2at-LIGRLO(PEG3-Pam)-NH2 activation of PAR2 alters transepithelial resistance (TER) and short circuit current (Isc) in MTE cells. Ussing chamber experiments were conducted under current-clamp conditions and apical application of 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM). A: representative TER traces in wild-type MTE cells and PAR2−/− MTE cells. B: quantified peak changes in TER in wild-type MTE cells (−141.2 ± 42.15 Ω·cm2, n = 6) compared with PAR2−/− MTE cells (13.5 ± 12.84 Ω·cm2, n = 4). C: representative traces of Isc in wild-type MTE cells and PAR2−/− cells. D: quantification of peak changes in Isc from wild-type MTE cells (6.8 ± 1.5 μA/cm2, n = 6) and PAR2−/− MTE cells (1.0 ± 0.18 μA/cm2, n = 4). *Significant difference (P < 0.05 by Student's t-test).
Since our initial experiments demonstrated apical PAR2-specific activation, we next compared the sensitivity of wild-type MTE cell cultures to an activating peptide [SLIGRL-NH2 (3)], a more potent nonlipidated peptidomimetic PAR2 agonist [2at-LIGRL-NH2 (15)], and the most potent, lipidated PAR2 agonist [2at-LIGRLO(PEG3-Pam)-NH2] in Ussing chamber experiments. Under voltage-clamp conditions, 10 μM apical addition of SLIGRL-NH2 resulted in a minimal increase in Isc and a correspondingly minimal decrease in TER (ΔIsc: 2.91 ± 0.285 μA/cm2 and ΔTER: −25.27 ± 2.85 Ω·cm2, n = 3; Fig. 3, A–C). A slightly larger but not significantly different response from SLIGRL-NH2 was observed after the apical addition of 2at-LIGRL-NH2 (ΔIsc: 5.27 ± 0.394 μA/cm2 and ΔTER: −42.00 ± 14.0 Ω·cm2, n = 3; Fig. 3, A–C). However, the lipidated PAR2 ligand 2at-LIGRLO(PEG3-Pam)-NH2 evoked a change in Isc and TER (ΔIsc: 17.09 ± 1.88 μA/cm2 and ΔTER: −270.3 ± 51.1 Ω·cm2, n = 6; Fig. 3, A–C) that was significantly different from SLIGRL-NH2 and 2at-LIGRL-NH2 changes. In contrast to the results observed after apical addition of PAR2 agonists, basolateral application of SLIGRL-NH2, 2at-LIGRL-NH2, or 2at-LIGRLO(PEG3-Pam)-NH2 to wild-type MTE cells all yielded full PAR2 activation [SLIGRL-NH2: ΔIsc 25.05 ± 2.31 μA/cm2 and ΔTER −428.3 ± 54.7 Ω·cm2, n = 3; 2at-LIGRL-NH2: ΔIsc 26.37 ± 2.86 μA/cm2 and ΔTER −517.5 ± 19.6 Ω·cm2, n = 3; and 2at-LIGRLO(PEG3-Pam)-NH2: ΔIsc 17.27 ± 0.43 μA/cm2 and ΔTER −326.4 ± 35 Ω·cm2, n = 3; Fig. 4, A–C].
Fig. 3.
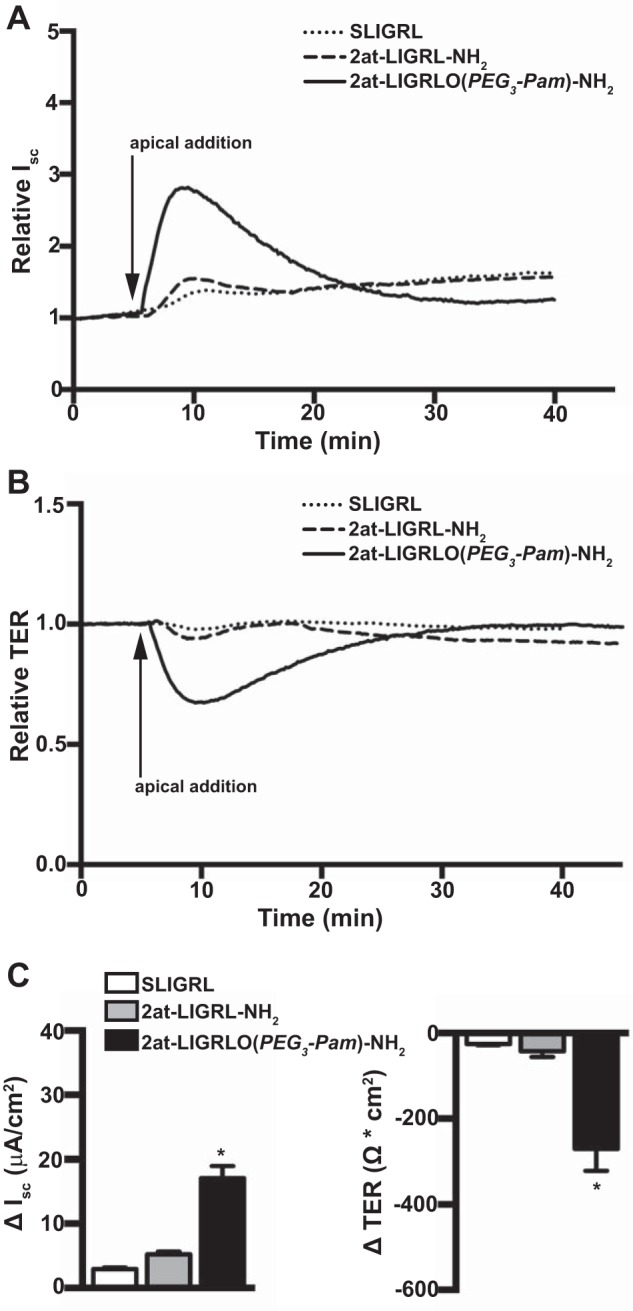
2at-LIGRLO(PEG3-Pam)-NH2 activates apical PAR2 in MTE cells. All traces are from Ussing chamber experiments conducted under voltage-clamp conditions. A: representative relative Isc traces of apical addition of 10 μM PAR2 agonists demonstrating that only 2at-LIGRLO(PEG3-Pam)-NH2 was sufficiently potent to evoke a robust PAR2 response. B: representative relative TER traces of apical addition of PAR2 agonists demonstrating that only 2at-LIGRLO(PEG3-Pam)-NH2 was sufficiently potent to evoke a robust PAR2 response. C: quantification of apical addition of PAR2 agonists showing a minimal response from SLIGRL-NH2 (ΔIsc: 2.91 ± 0.285 μA/cm2 and ΔTER: −25.27 ± 2.85 Ω·cm2, n = 3) and a slightly higher but still minimal response from 2at-LIGRL-NH2 (ΔIsc: 5.27 ± 0.394 μA/cm2 and ΔTER: −42.00 ± 14.0 Ω·cm2, n = 3) in contrast to 2at-LIGRLO(PEG3-Pam)-NH2, which showed a robust response (ΔIsc: 17.09 ± 1.88 μA/cm2 and ΔTER −270.3 ± 51.1 Ω·cm2, n = 6). *Significant difference (P < 0.05 by one-way ANOVA).
Fig. 4.
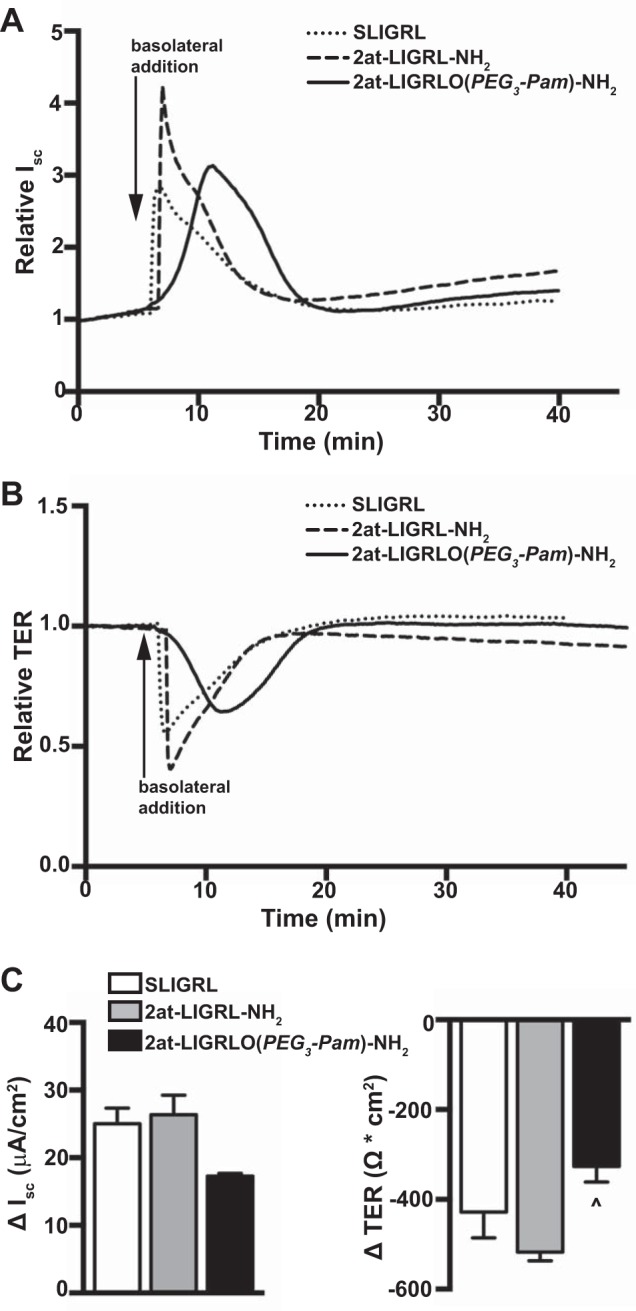
Basolateral activation of PAR2 in MTE cells is similar among PAR2 agonists. Ussing chamber experiments were conducted under voltage clamp. A: representative relative Isc traces showing that PAR2 agonists (10 μM) all evoke a similar and robust increase in Isc when applied basolaterally. B: representative relative traces of TER after basolateral addition of PAR2 agonists showing a similar transient reduction in TER. C: quantification of responses to PAR2 agonists after basolateral application was similar [SLIGRL-NH2: ΔIsc 25.05 ± 2.31 μA/cm2 and ΔTER −428.3 ± 54.7 Ω·cm2, n = 3; 2at-LIGRL-NH2: ΔIsc 26.37 ± 2.86 μA/cm2 and ΔTER −517.5 ± 19.6 Ω·cm2, n = 3; and 2at-LIGRLO(PEG3-Pam)-NH2: ΔIsc 17.27 ± 0.43 μA/cm2 and ΔTER −326.4 ± 35 Ω·cm2, n = 3]. ^Significant difference from 2at-LIGRL-NH2 (P < 0.05 by one-way ANOVA).
Apical activation of PAR2 alters primary airway epithelial cell ion conductance via Ca2+-regulated ion channels.
Because PAR2 activation in airway epithelia leads to an increase in [Ca2+]i, we examined the potential contribution of the rise in [Ca2+]i to observed PAR2 changes in ion conductance. MTE cell cultures from wild-type mice were voltage clamped and measured for changes in Isc and TER after the apical addition of 2at-LIGRLO(PEG3-Pam)-NH2 with or without a 30-min preincubation with the Ca2+ chelator BAPTA-AM (Fig. 5, A–C). Under normal conditions, apical addition of 2at-LIGRLO(PEG3-Pam)-NH2 resulted in ΔIsc of 17.09 ± 1.88 μA/cm2 and ΔTER of −270.3 ± 51.1 Ω·cm2 (n = 6). However, after intracellular Ca2+ chelation with BAPTA, apical application of 2at-LIGRLO(PEG3-Pam)-NH2 resulted in an attenuated ΔIsc of 2.54 ± 0.346 μA/cm2 and ΔTER of −84.07 ± 3.44 Ω·cm2 (n = 3).
Fig. 5.
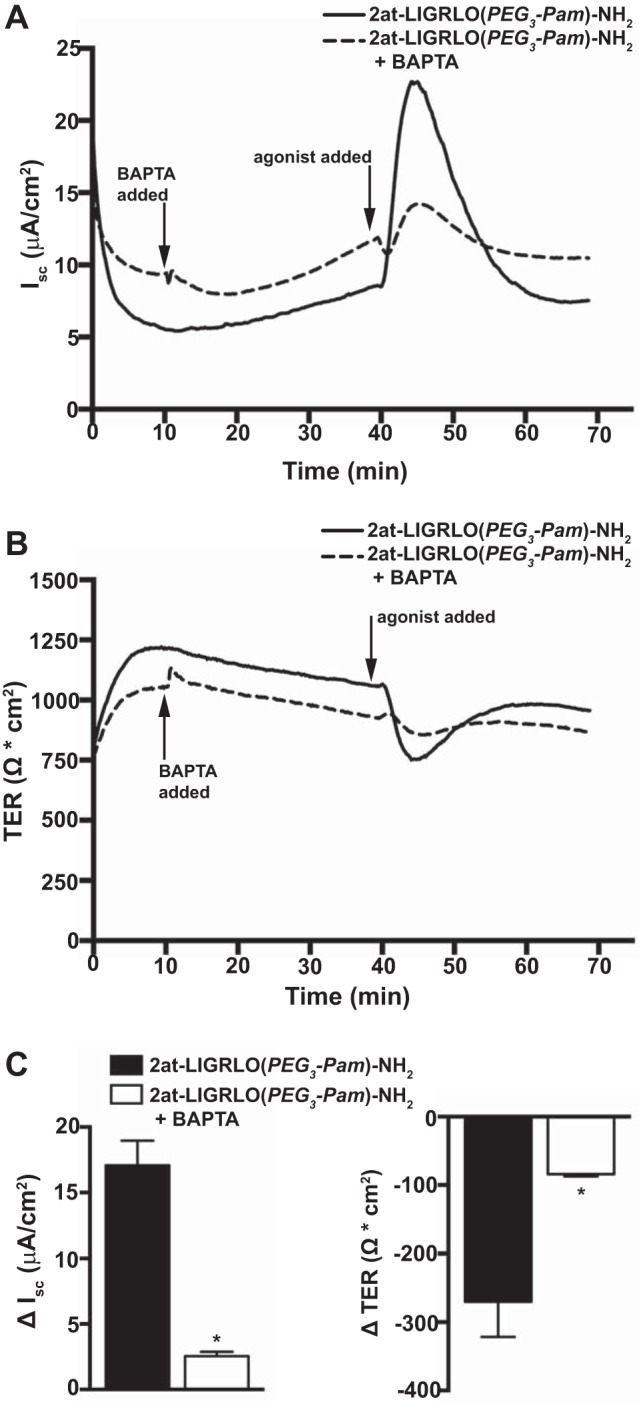
PAR2 alterations in airway epithelial ion conductance are largely mediated by [Ca2+]i increases. Ussing chamber experiments were conducted under voltage-clamp conditions. A: representative Isc traces showing that apical addition of 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM) alone evoked a robust PAR2 response that was attenuated with BAPTA (50 μM) preincubation. B: MTE cells pretreated with BAPTA showed a diminished loss of TER after the apical application of 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM) compared with cells treated with 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM) alone. C: quantification of Isc and TER demonstrating a significant reduction in the PAR2 response to 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM) in MTE cells pretreated with BAPTA (50 μM) (BAPTA treatment: ΔIsc 2.54 ± 0.346 μA/cm2 and ΔTER −84.07 ± 3.44 Ω·cm2, n = 3; BAPTA free: ΔIsc 17.09 ± 1.88 μA/cm2 and ΔTER −270.3 ± 51.1 Ω·cm2, n = 6). *Significant difference (P < 0.05 by unpaired Student's t-test).
To elucidate the mechanism(s) by which a PAR2-evoked rise in [Ca2+]i increased airway epithelial ion conductance, we examined the role of Ca2+-regulated ion channels in the apical membrane of primary MTE cells. Primary MTE cell cultures were voltage clamped and pretreated apically with 10 μM Ca2+-activated Cl− channel (CaCC) inhibitor for 3 min followed by the apical addition of 10 μM 2at-LIGRLO(PEG3-Pam)-NH2 (Fig. 6, A and B). CaCC inhibitor significantly reduced 2at-LIGRLO(PEG3-Pam)-NH2-evoked changes in Isc and TER (ΔIsc: 3.76 ± 0.677 μA/cm2 and ΔTER: −32.25 ± 2.09 Ω·cm2, n = 4; Fig. 6G).
Fig. 6.

PAR2 activates Ca2+-regulated ion channels in MTE cells. Ussing chamber experiments were conducted under voltage clamp-conditions. A and B: representative traces of Isc (A) and TER (B) showing that the apical addition of 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM) alone evoked a robust PAR2 response that was largely attenuated by Ca2+-activated Cl− channel inhibitor (CaCC-inh; 10 μM). C and D: representative traces of Isc (C) and TER (D) showing that clotrimazole (30 μM) reduced the apical 2at-LIGRLO(PEG3-Pam)-NH2-evoked PAR2 response compared with agonist alone. E and F: representative traces of Isc (E) and TER (F) showing that combined apical treatment of CaCC-inh (10 μM), clotrimazole (30 μM), and 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM) completely diminished the PAR2 activation seen with 2at-LIGRLO(PEG3-Pam)-NH2 (10 μM) alone. G: quantification of changes in Isc (left) and TER (right) showing that compared with 2at-LIGRLO(PEG3-Pam)-NH2 alone (ΔIsc: 17.09 ± 1.88 μA/cm2 and ΔTER: −270.3 ± 51.1 Ω·cm2, n = 6), inhibition with CaCC-inh (ΔIsc: 3.76 ± 0.677 μA/cm2 and ΔTER: −32.25 ± 2.09 Ω·cm2, n = 4) or clotrimzole (ΔIsc: 3.21 ± 0.73 μA/cm2 and ΔTER: −69.63 ± 8.149 Ω·cm2, n = 3) significantly reduced the PAR2 response. The two inhibitors combined completely abolished the apical PAR2 response to 2at-LIGRLO(PEG3-Pam)-NH2 (ΔIsc: 0.017 ± 0.017 μA/cm2 and ΔTER: −0.533 ± 0.393 Ω·cm2, n = 3). *Significant difference (P < 0.05 by one-way ANOVA).
We next investigated the potential involvement of large-conductance Ca2+-activated K+ channels in PAR2-dependent alterations in ion conductance. Voltage-clamped MTE cell cultures were pretreated for 3 min with 30 μM clotrimazole followed by the apical addition of 10 μM 2at-LIGRLO(PEG3-Pam)-NH2 (Fig. 6, C and D). Pretreatment with clotrimazole significantly reduced 2at-LIGRLO(PEG3-Pam)-NH2-evoked changes in Isc and TER (ΔIsc: 3.21 ± 0.73 μA/cm2 and ΔTER: −69.63 ± 8.149 Ω·cm2, n = 3; Fig. 6G).
To evaluate if the two channel types above accounted for the observed PAR2-dependent transcellular ion conductances, primary MTE cell cultures were voltage clamped and pretreated apically with a combination of 10 μM CaCC inhibitor and 30 μM clotrimazole for 3 min followed by the apical addition of 10 μM 2at-LIGRLO(PEG3-Pam)-NH2 (Fig. 6, E and F). In these experiments, the response to 2at-LIGRLO(PEG3-Pam)-NH2 was almost completely abolished (ΔIsc: 0.017 ± 0.017 μA/cm2 and ΔTER: −0.533 ± 0.393 Ω·cm2, n = 3; Fig. 6G).
DISCUSSION
In the present study, we show, for the first time in primary airway epithelial cells, the robust apical activation of PAR2 leading to a transient decrease in TER accompanied by an increase in Ca2+-regulated Cl− and K+ conductances. These changes could only be observed with the recently developed, high-potency, lipidated PAR2 ligand 2at-LIGRLO(PEG3-Pam)-NH2 (14). Apically positioned PAR2 in the conducting airway epithelium is ideally located to detect endogenous proteases that can accompany allergen inhalation. We postulated that the transient changes in ion conductance could provide a temporary increase in airway surface liquid. This would be complemented by Ca2+-induced increases in ciliary beat (13). Thus, initial activation of PAR2 in the lumenal airway could aid innate immune function via rapid, local upregulation of mucociliary clearance.
PAR2 is expressed throughout the airway epithelium. Its activation after asthmagen exposure has been associated with both protective affects via bronchorelaxation (7, 8, 10, 19) and detrimental effects (e.g., cytokine release and subsequent chronic inflammation) in animal models of asthma (12, 38, 42). It has come to light in recent work that this dichotomy in PAR2 activation effects may be due to dual signaling pathways downstream of PAR2, namely, Gq-dependent Ca2+ signaling and G protein-independent β-arrestin signaling (29). Similar signaling has been reported for another GPCR targeted in asthma treatment, β-adrenergic receptor (βAR) signaling (43). Because of the diametrically opposed responses, continued development of novel, potent, efficacious, and (potentially) biased signaling drugs that affect PAR2 or βAR are highly desirable. Recent progress in improving PAR2 ligand potency and efficacy has occurred through the lipidation of the known PAR2 agonists (4, 14). Such lipidated compounds, which include 2at-LIGRLO(PEG3-Pam)-NH2 used in this study, have orders of magnitude increased potency with improved selectivity for PAR2 over traditionally used peptides (e.g., SLIGRL-NH2) or peptidomimetics (e.g., 2at-LIGRL-NH2 or 2-furoyl-LIGRLO-NH2).
In the present study, we characterized the use of lipidated PAR2 ligands in the activation of primary cultured MTE cells. Interestingly, apical application of the lipidated PAR2 agonist to cultured MTE cells initiated a Ca2+ flux with a markedly higher EC50 (835 nM) and wider 95% confidence interval (261 nM–2.68 μM) than those observed in an immortalized human bronchial airway cell line (16HBE14o- cells; EC50: 3.95 nM, 95% confidence interval: 3.43–4.55 nM). The need for higher concentrations of lipidated PAR2 ligands in primary cultured cells was not unexpected as it is not uncommon to observe differences in immortalized and primary cultured cells. However, this change in activity emphasizes the need for potent and efficacious ligands to evaluate roles for apically expressed PAR2 in the airway. As a measure of specificity, PAR2−/− cells did not respond to the lipidated ligand with a transient change in [Ca2+]i or ion flux.
Roles for PAR2 in airway ion conductance in the mouse or human airway epithelium have traditionally been evaluated using basolateral application, in part due to the lack of response to activating peptides and/or peptidomimetics when applied apically (9, 25, 28, 31, 35, 37, 45). Our results examining basolaterally activated PAR2 were similar to previous findings in that all of the PAR2 ligands tested [SLIGRL-NH2, 2at-LIGRL-NH2, and 2at-LIGRLO(PEG3-Pam)-NH2 at 10 μM] evoked rapid, transient changes in TER and Isc. It was somewhat surprising to us that a more robust response with the lipidated compound compared with traditional activating peptide and/or peptidomimetic was not observed after basolateral activation. We also noted a slightly different shape in the representative traces from those experiments with 2at-LIGRLO(PEG3-Pam)-NH2, which required more time to develop a full response in Ussing chamber experiments. It is likely these changes represent the lipid sequence interacting with the tissue culture filter and/or collagen during application and thus hindering presentation of the agonist to the receptor on the basolateral membrane.
Proteases in asthmagens have been shown to alter the barrier function of healthy and asthmatic airways (22, 27, 44). Furthermore, remodeled airway epithelium in asthmatics includes a reduction in barrier function (20). Thus, basolateral activation of PAR2 could be relevant after extended asthmagen exposure or in diseased or compromised airways. However, basolateral activation of PAR2 by inhaled asthmagens would not necessarily be expected in a healthy individual. A further caveat of interpreting basolateral PAR2 activation is the demonstration in model epithelial cells of distinct pools of PAR2 that display differential signaling when activated at apical or basolateral membranes (26).
As mentioned above, there exists limited data using traditional activating peptides (e.g., SLIGRL-NH2) and peptidomimetics (e.g., 2at-LIGRL-NH2) applied to the apical airway epithelial membrane. Although Cl− current was observed after high concentrations (30 μM) of SLIGRL-NH2 to the mouse (BALB/c) trachea, these changes were attributed to nonspecific neurokinin-1 receptor activation (1). Using the potent and selective lipidated compound 2at-LIGRLO(PEG3-Pam)-NH2, we were able to demonstrate, for the first time, apical PAR2 activation in primary mouse tracheal epithelia that resulted in transient changes in TER and Isc. To rule out the possibility that the lipidated agonist disrupted the epithelium and gained access to the basolateral side for activation, we repeated our recordings with 10 μM concentrations of SLIGRL-NH2 and O(PEG3-Pam)-NH2. As with 10 μM SLIGRL-NH2 alone, no epithelial response was observed.
Apical activation of PAR2-dependent Ca2+-regulated Cl− and K+ currents that could drive secretion in our well-differentiated MTE cells is consistent with the basolateral activation of the whole mouse trachea previously reported (25). Basolateral activation of primary human cells from absorptive airway also displayed Ca2+-regulated Cl− and K+ currents that were supplemented with cAMP-dependent ion flow (9, 24, 25). These differences can be attributed to known differences in ion channel and transporter expression in the mouse and human airway (25). We conclude from these data that although PAR2 is differentially sensitive in the apical and basolateral absorptive airway epithelium, activation results in similar, transient Ca2+ dependent Cl− and K+ currents that temporarily provide a secretory phenotype (i.e., net Cl− efflux). It would be interesting to use the high-potency lipidated PAR2 agonist to determine if apical activation of secretory airway epithelial model cells [e.g., Calu-3 cells (31, 37, 45)] would result in similar signaling as we observed above or differential signaling, as observed in human colon epithelial models (26).
In summary, we have shown apical activation of PAR2 on primary airway epithelial cells using the highly potent and specific lipidated peptidomimetic 2at-LIGRLO(PEG3-Pam)-NH2. To our knowledge, this is the first time activation of PAR2 has been demonstrated at the apical surface of airway epithelial cells. Using highly potent and specific agonists to PAR2 we can isolate receptor-specific contributions to airway epithelial function not previously possible. A plausible physiological implication for PAR2 activation at the apical airway surface that results in Cl− efflux is an increase in airway surface liquid. Increases in airway surface liquid would serve to aid in flushing out noxious stimuli such as inhaled asthmagens in a healthy individual.
GRANTS
This work was primarily supported by National Institutes of Health (NIH) Grant R01-NS-073664 (multi-principal investigator; to S. Boitano, T. J. Price, and J. Vagner), NIH Grant R01-AI-083403 (to M. O. Daines), and NIH Training Grant ES-007091 (to C. L. Sherwood). This work was additionally supported in part by NIH Superfund Research Grant ES-04940 (to S. Boitano).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.L.S., M.O.D., T.J.P., J.V., and S.B. conception and design of research; C.L.S. and S.B. performed experiments; C.L.S. and S.B. analyzed data; C.L.S., T.J.P., J.V., and S.B. interpreted results of experiments; C.L.S. and S.B. prepared figures; C.L.S. and S.B. drafted manuscript; C.L.S., M.O.D., T.J.P., J.V., and S.B. edited and revised manuscript; C.L.S., M.O.D., T.J.P., J.V., and S.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Terry E. Machen for discussions on the development and execution of the experiments, Rhea Pereira and Dipti V. Tillu for help with animal care, and Daniel X. Sherwood for software that allowed for Ca2+ signaling evaluations.
Present address of T. J. Price: School of Behavioral and Brain Sciences, University of Texas at Dallas, 800 W. Campbell Road, Richardson, TX.
REFERENCES
- 1.Abey HT, Fairlie DP, Moffatt JD, Balzary RW, Cocks TM. Protease-activated receptor-2 peptides activate neurokinin-1 receptors in the mouse isolated trachea. J Pharmacol Exp Ther 317: 598–605, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, Hooper JD. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther 130: 248–282, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, Sundelin J, Scarborough RM. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem 271: 16466–16471, 1996 [DOI] [PubMed] [Google Scholar]
- 4.Boitano S, Hoffman J, Tillu DV, Asiedu MN, Zhang Z, Sherwood CL, Wang Y, Dong X, Price TJ, Vagner J. Development and evaluation of small peptidomimetic ligands to protease-activated receptor-2 (PAR2) through the use of lipid tethering. PLOS ONE 9: e99140, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho HJ, Choi JY, Yang YM, Hong JH, Kim CH, Gee HY, Lee HJ, Shin DM, Yoon JH. House dust mite extract activates apical Cl− channels through protease-activated receptor 2 in human airway epithelia. J Cell Biochem 109: 1254–1263, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Chow JM, Moffatt JD, Cocks TM. Effect of protease-activated receptor (PAR)-1, -2 and -4-activating peptides, thrombin and trypsin in rat isolated airways. Br J Pharmacol 131: 1584–1591, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cocks TM, Fong B, Chow JM, Anderson GP, Frauman AG, Goldie RG, Henry PJ, Carr MJ, Hamilton JR, Moffatt JD. A protective role for protease-activated receptors in the airways. Nature 398: 156–160, 1999 [DOI] [PubMed] [Google Scholar]
- 8.Cocks TM, Moffatt JD. Protease-activated receptor-2 (PAR2) in the airways. Pulm Pharmacol Ther 14: 183–191, 2001 [DOI] [PubMed] [Google Scholar]
- 9.Danahay H, Withey L, Poll CT, van de Graaf SF, Bridges RJ. Protease-activated receptor-2-mediated inhibition of ion transport in human bronchial epithelial cells. Am J Physiol Cell Physiol 280: C1455–C1464, 2001 [DOI] [PubMed] [Google Scholar]
- 10.De Campo BA, Henry PJ. Stimulation of protease-activated receptor-2 inhibits airway eosinophilia, hyperresponsiveness and bronchoconstriction in a murine model of allergic inflammation. Br J Pharmacol 144: 1100–1108, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dirksen ER, Felix JA, Sanderson MJ. Preparation of explant and organ cultures and single cells from airway epithelium. Methods Cell Biol 47: 65–74, 1995 [DOI] [PubMed] [Google Scholar]
- 12.Ebeling C, Forsythe P, Ng J, Gordon JR, Hollenberg M, Vliagoftis H. Proteinase-activated receptor 2 activation in the airways enhances antigen-mediated airway inflammation and airway hyperresponsiveness through different pathways. J Allergy Clin Immunol 115: 623–630, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Evans JH, Sanderson MJ. Intracellular calcium oscillations regulate ciliary beat frequency of airway epithelial cells. Cell Calcium 26: 103–110, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Flynn AN, Hoffman J, Tillu DV, Sherwood CL, Zhang Z, Patek R, Asiedu MN, Vagner J, Price TJ, Boitano S. Development of highly potent protease-activated receptor 2 agonists via synthetic lipid tethering. FASEB J 27: 1498–1510, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn AN, Tillu DV, Asiedu MN, Hoffman J, Vagner J, Price TJ, Boitano S. The protease-activated receptor-2-specific agonists 2-aminothiazol-4-yl-LIGRL-NH2 and 6-aminonicotinyl-LIGRL-NH2 stimulate multiple signaling pathways to induce physiological responses in vitro and in vivo. J Biol Chem 286: 19076–19088, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganesan S, Comstock AT, Sajjan US. Barrier function of airway tract epithelium. Tissue Barriers 1: e24997, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs)–focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal 11: 86, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450, 1985 [PubMed] [Google Scholar]
- 19.Henry PJ. The protease-activated receptor2 (PAR2)-prostaglandin E2-prostanoid EP receptor axis: a potential bronchoprotective unit in the respiratory tract? Eur J Pharmacol 533: 156–170, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Holgate ST. Pathogenesis of asthma. Clin Exp Allergy 38: 872–897, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP, Ramachandran R. Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease. Br J Pharmacol 171: 1180–1194, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacquet A. Interactions of airway epithelium with protease allergens in the allergic response. Clin Exp Allergy 41: 305–311, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Kauffman HF. Interaction of environmental allergens with airway epithelium as a key component of asthma. Curr Allergy Asthma Rep 3: 101–108, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Kunzelmann K, Schreiber R, Konig J, Mall M. Ion transport induced by proteinase-activated receptors (PAR2) in colon and airways. Cell Biochem Biophys 36: 209–214, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Kunzelmann K, Sun J, Markovich D, Konig J, Murle B, Mall M, Schreiber R. Control of ion transport in mammalian airways by protease activated receptors type 2 (PAR-2). FASEB J 19: 969–970, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Lau C, Lytle C, Straus DS, DeFea KA. Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am J Physiol Cell Physiol 300: C113–C123, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leino MS, Loxham M, Blume C, Swindle EJ, Jayasekera NP, Dennison PW, Shamji BW, Edwards MJ, Holgate ST, Howarth PH, Davies DE. Barrier disrupting effects of alternaria alternata extract on bronchial epithelium from asthmatic donors. PLOS ONE 8: e71278, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin KW, Park J, Crews AL, Li Y, Adler KB. Protease-activated receptor-2 (PAR-2) is a weak enhancer of mucin secretion by human bronchial epithelial cells in vitro. Int J Biochem Cell Biol 40: 1379–1388, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El-Mays T, Vliagoftis H, Hollenberg MD, Wilson EH, Walker JK, DeFea KA. β-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci USA 109: 16660–16665, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nystedt S, Emilsson K, Larsson AK, Strombeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem 232: 84–89, 1995 [DOI] [PubMed] [Google Scholar]
- 31.Palmer ML, Lee SY, Maniak PJ, Carlson D, Fahrenkrug SC, O'Grady SM. Protease-activated receptor regulation of Cl− secretion in Calu-3 cells requires prostaglandin release and CFTR activation. Am J Physiol Cell Physiol 290: C1189–C1198, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Park MK, Cho MK, Kang SA, Park HK, Kim DH, Yu HS. Acanthamoeba protease activity promotes allergic airway inflammation via protease-activated receptor 2. PLOS ONE 9: e92726, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peters T, Henry PJ. Protease-activated receptors and prostaglandins in inflammatory lung disease. Br J Pharmacol 158: 1017–1033, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev 242: 186–204, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Rievaj J, Davidson C, Nadeem A, Hollenberg M, Duszyk M, Vliagoftis H. Allergic sensitization enhances anion current responsiveness of murine trachea to PAR-2 activation. Pflügers Arch 463: 497–509, 2012 [DOI] [PubMed] [Google Scholar]
- 36.Salazar F, Ghaemmaghami AM. Allergen recognition by innate immune cells: critical role of dendritic and epithelial cells. Front Immunol 4: 356, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato S, Ito Y, Kondo M, Ohashi T, Ito S, Nakayama S, Shimokata K, Kume H. Ion transport regulated by protease-activated receptor 2 in human airway Calu-3 epithelia. Br J Pharmacol 146: 397–407, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidlin F, Amadesi S, Dabbagh K, Lewis DE, Knott P, Bunnett NW, Gater PR, Geppetti P, Bertrand C, Stevens ME. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J Immunol 169: 5315–5321, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Sherwood CL, Liguori AE, Olsen CE, Lantz RC, Burgess JL, Boitano S. Arsenic compromises conducting airway epithelial barrier properties in primary mouse and immortalized human cell cultures. PLOS ONE 8: e82970, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sokol K, Sur S, Ameredes BT. Inhaled environmental allergens and toxicants as determinants of the asthma phenotype. Adv Exp Med Biol 795: 43–73, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sokolova E, Reiser G. A novel therapeutic target in various lung diseases: airway proteases and protease-activated receptors. Pharmacol Ther 115: 70–83, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Takizawa T, Tamiya M, Hara T, Matsumoto J, Saito N, Kanke T, Kawagoe J, Hattori Y. Abrogation of bronchial eosinophilic inflammation and attenuated eotaxin content in protease-activated receptor 2-deficient mice. J Pharmacol Sci 98: 99–102, 2005 [DOI] [PubMed] [Google Scholar]
- 43.Walker JK, DeFea KA. Role for β-arrestin in mediating paradoxical betaAR and PAR signaling in asthma. Curr Opin Pharmacol 16: 142–147, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wan H, Winton HL, Soeller C, Stewart GA, Thompson PJ, Gruenert DC, Cannell MB, Garrod DR, Robinson C. Tight junction properties of the immortalized human bronchial epithelial cell lines Calu-3 and 16HBE14o-. Eur Respir J 15: 1058–1068, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Winter MC, Shasby SS, Ries DR, Shasby DM. PAR2 activation interrupts E-cadherin adhesion and compromises the airway epithelial barrier: protective effect of β-agonists. Am J Physiol Lung Cell Mol Physiol 291: L628–L635, 2006 [DOI] [PubMed] [Google Scholar]