

Abstract
The epithelial Na+ channel (ENaC) plays a key role in the regulation of blood pressure and airway surface liquid volume. ERp29 is a 29-kDa thioredoxin-homologous endoplasmic reticulum (ER) protein that has only a single cysteine instead of the usual thioredoxin CXXC motif. Our group previously demonstrated that ERp29 promotes biogenesis of the cystic fibrosis transmembrane conductance regulator (CFTR). On the basis of similarities of CFTR and ENaC trafficking, we hypothesized that ERp29 would also regulate ENaC biogenesis and functional expression. In epithelial cells, overexpression of wild-type (wt) ERp29 increased ENaC functional expression [amiloride-sensitive short-circuit current (Isc)] in Ussing chamber experiments, as well as the abundance of the cleaved form of γ-ENaC in whole cell lysates. In contrast, siRNA-mediated depletion of ERp29 or overexpression of a mutant ERp29 lacking its single cysteine (C157S ERp29) decreased ENaC functional expression. Cells in which wt ERp29 was overexpressed had a smaller fractional increase in amiloride-sensitive Isc when trypsin was applied to the apical surface to activate uncleaved ENaC, while cells in which C157S ERp29 was overexpressed or ERp29 was depleted had a significantly greater fractional increase in amiloride-sensitive Isc in response to trypsin. Interestingly, these observations were not associated with altered expression of β-ENaC at the apical surface. Instead, ERp29 appeared to promote the interaction of β-ENaC with the Sec24D cargo recognition component of the coat complex II ER exit machinery. Together, these data support the hypothesis that ERp29 directs ENaC toward the Golgi, where it undergoes cleavage during its biogenesis and trafficking to the apical membrane.
Keywords: chaperone, biogenesis, trafficking, endoplasmic reticulum, epithelium
the epithelial sodium channel (ENaC) is found in the apical membrane of epithelia in a wide variety of tissues, including the airway, where it regulates airway surface liquid volume, and the distal nephron, where it regulates Na+ balance, blood volume, and blood pressure. ENaC gain-of-function mutations are associated with hypertension (11, 12, 29), while loss-of-function mutations cause hypotension (18). In the airway, functional overexpression of ENaC causes decreased mucociliary clearance and increased morbidity and mortality in mouse models (21). This may mimic the cystic fibrosis (CF) airway, which is characterized by relative hyperfunction of ENaC and decreased mucociliary clearance.
ENaC is a likely heterotrimer (19, 32) composed of three similar subunits, α, β, and γ (21). Each subunit maintains its respective NH2 and COOH termini in the cytoplasm, with large extracellular loops (6, 7). ENaC open probability (Po) is determined in large part by proteolysis of the extracellular loops of its α- and γ-subunits. Furin, a member of the pro-protein convertase family, resides in the trans-Golgi and can cleave ENaC's α-subunit at two sites and γ-subunit at one site in the luminal/extracellular loops during biogenesis (16); a second cleavage in γ-ENaC is required for ENaC Po to approach unity (1). ENaC can also bypass this furin proteolysis (and Golgi N-glycan modification) and reach the apical membrane in an uncleaved, low-Po form that has immature (endoglycosidase H-sensitive) N-glycans (17). These uncleaved, “near-silent” channels at the cell surface may serve as a “reserve” channel pool that can be proteolytically activated after delivery to the cell surface by endogenous cell surface proteases, such as channel-activating proteases (37, 38), or exogenous proteases, such as trypsin (3, 38). The mechanism(s) determining whether ENaC undergoes or bypasses furin cleavage during biogenesis is not known.
ERp29 is a 29-kDa endoplasmic reticulum (ER) luminal molecular chaperone that is expressed ubiquitously in mammalian tissues and is abundant in epithelia (31). ERp29 is homologous to the thioredoxins and the protein disulfide isomerases (PDIs). Interestingly, it lacks the characteristic thioredoxin CXXC motif but, instead, contains a single cysteine residue at position 157. Our group recently demonstrated that ERp29 has increased expression in epithelial cells treated with 4-phenylbutyrate, the prototype corrector of ΔF508 CFTR trafficking, and promotes wild-type (wt) and ΔF508 CFTR trafficking to the plasma membrane (35). ERp29 was thus the first ER luminal chaperone demonstrated to promote CFTR biogenesis. Because of similarities between CFTR and ENaC biogenesis and trafficking, we hypothesize that ERp29 would also regulate ENaC.1 Our data are consistent with a key role for ERp29 in directing ENaC's itinerary during biogenesis. In particular, our data suggest that ERp29 is a key determinant in regulating ENaC Po by directing whether ENaC undergoes cleavage en route to the apical epithelial surface.
MATERIALS AND METHODS
Cell culture.
Madin-Darby canine kidney (MDCK) type I cells that stably expressed COOH-terminal epitope-tagged murine ENaC subunits [HA-tagged α-, V5-tagged β-, and Myc-tagged γ-ENaC (MDCK αβγ-ENaC); a gift of Dr. T. Kleyman] were maintained in 50:50 Dulbecco's modified Eagle's medium (GIBCO)-Ham's F-12 (Cellgro) supplemented with 10% fetal bovine serum (Gemini), streptomycin (100 μg/ml; Invitrogen, Carlsbad, CA), and penicillin (100 U/ml; Invitrogen). The cells were maintained under antibiotic-selective pressure using hygromycin (Roche), blasticidin (Invitrogen), and G418 (Cellgro).
We generated additional stable cell lines with doxycycline (Dox)-inducible expression of wt or mutant (C157S) ERp29 using the pcDNA4-based Invitrogen T-REx system as we have done in previous work (8); the C157S mutation was introduced into ERp29 by the QuickChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). These cell lines were maintained under additional selective pressure with puromycin (Sigma) and Zeocin (Invitrogen).
Cells were grown on polarized monolayers on Snapwell inserts (Costar, Corning Life Sciences) for ion transport assays or on Transwell inserts for biochemical assays. Once they had achieved ≥400 Ω·cm2 resistance, the cells were treated with dexamethasone (1 μg/ml; Sigma-Aldrich) for 48 h prior to experimentation. Unless otherwise indicated, cell lines with inducible expression of wt or mutant ERp29 were treated with Dox (Sigma-Aldrich) for the final 8 h of the dexamethasone treatment.
Immortalized CF bronchial epithelial (CFBE41o−) parental cells, previously obtained from Dr. J. P. Clancy (then affiliated with the University of Alabama-Birmingham), were cultured as previously described by our group (25).
Transient overexpression of ERp29.
A pcDNA4 plasmid encoding wt ERp29 was previously described (35). C157S ERp29 was generated using a PCR-based mutagenesis technique as we have done previously (27, 39), and its sequence was confirmed by automated DNA sequence analysis performed at sequencing facilities of the Children's Hospital of Philadelphia. Transient transfections of αβγ-ENaC-expressing MDCK cells were performed with the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. For transfections of cells on tissue culture plates, plasmid (4 μg) was used; for transfections of cells on Snapwell or Transwell inserts, plasmid (0.1 μg/cm2) plus carrier DNA (0.5 μg/cm2) was used. As cells transiently overexpressing ERp29 (wt or C157S) behaved similarly to the stably transfected cells with inducible overexpression of ERp29, these approaches were used interchangeably, and data were combined for analysis.
Depletion of ERp29 by siRNA.
ERp29 expression was depleted using an ERp29 small interfering RNA (siRNA; Dharmacon/Thermo Scientific, Rockford, IL) as we previously described (35). Briefly, 60 pmol of ERp29 or control siRNA were delivered to MDCK or CFBE41o− cells by transfection with Lipofectamine 2000 according to the manufacturer's protocol. Cells were assayed 48 h after transfection, as described below.
Antibodies.
Rabbit anti-ERp29 and mouse anti-ERp29 (used at 1:2,500–1:5,000 dilution) were purchased from Abcam (Cambridge, MA); rabbit anti-γ-ENaC (used at 1:2,500–1:5,000 dilution) from Abcam (Cambridge, MA) or Thermo Fisher Scientific; rabbit anti-HA epitope (to detect α-ENaC, used at 1:2,500 dilution) from Pharmingen (San Diego, CA) or Roche (Indianapolis, IN); mouse anti-V5 epitope (to detect β-ENaC, used at 1:2,500–1:5,000 dilution) from Invitrogen; mouse anti-Sec24D (used at 1:1,000 dilution) from AbNova (Taiwan); mouse monoclonal anti-GAPDH (used at 1:10,000 dilution) from Calbiochem; and horseradish peroxidase (HRP)-conjugated secondary antibodies [goat anti-rabbit-HRP (1:2,000–1:5,000 dilution) and goat anti-mouse-HRP (1:10,000 dilution)] from Millipore.
Immunoblotting.
Whole cell lysates were prepared by incubating cells on ice for 30 min in RIPA buffer (150 mM NaCl, 50 mM Tris·HCl, pH 8, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing a 1:1,000 dilution of protease inhibitor mixture (Sigma-Aldrich). The lysates were collected, passaged through a 21-gauge needle, and cleared by centrifugation (14,000 g for 15 min at 4°C). Protein content in the lysate supernatants was determined using DC protein assay reagents (Bio-Rad) and bovine serum albumin (BSA) as a standard. Samples were denatured using 6× Laemmli sample buffer (125 mM Tris, pH 6.8, 4% SDS, 10% glycerol, 0.006% bromophenol blue, 1.8% 2-mercaptoethanol; final concentration 1–2×), and equal amounts of protein (25 μg) were resolved using SDS-PAGE and transferred to nitrocellulose using semidry techniques (Bio-Rad). Nonspecific protein binding was diminished by incubating the membrane in 5% BSA or 5% nonfat milk in Tris-buffered saline (10 mM Tris·HCl, pH 8, 150 mM NaCl) with 0.1% Tween 20. Primary antibodies and HRP-conjugated secondary antibodies were applied in Tris-buffered saline with 0.1% Tween 20 with 5% nonfat milk or 3% BSA. Immunoreactivity was detected by chemiluminescence (SuperSignal, Thermo Fisher, Waltham, MA) and fluorography. Densitometry was performed using the AlphaImager 2200 system (AlphaInnotech, Santa Clara, CA) (8, 35) or ImageJ 1.48 software (downloaded from http://rsbweb.nih.gov/ij/).
Coimmunoprecipitation.
For coimmunoprecipitation experiments, cells were lysed under nondenaturing conditions in RIPA buffer without SDS, and protein content was determined as described above. Protein A-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) that had been preincubated with primary antibody for 1 h were incubated overnight with cell lysate proteins (500 μg of total protein) at 4°C. Precipitated proteins were released by heating the samples for 3.5 min at 90°C in 2× Laemmli sample buffer, resolved by SDS-PAGE, and revealed by immunoblotting.
Surface biotinylation.
MDCK αβγ-ENaC cells were transiently transfected with ERp29 and grown on Transwell inserts until transepithelial resistance reached ≥500 Ω·cm2. The cells were placed on ice for 30 min and washed with PBS containing Ca2+ and Mg2+, and their apical surface was exposed to 1 mg/ml sulfo-NHS-SS-biotin (Thermo Fisher Scientific) in the biotinylation buffer (10 mM H3BO4, 137 mM NaCl, 1 mM CaCl2, pH 8.0) twice for 25 min on ice. The biotinylation reaction was terminated by washing the cells with a quenching buffer (192 mM glycine, 25 mM Tris·HCl, pH 8.3) followed by 20 min of incubation with quenching buffer. Biotinylated proteins were precipitated using NeutrAvidin beads (Invitrogen), resolved by SDS-PAGE, and revealed by immunoblotting. We routinely assessed that GAPDH was present in whole cell lysates, but not in the NeutrAvidin-precipitated proteins, as a control for cellular integrity and lack of labeling of intracellular proteins in these experiments.
Transepithelial ion transport measurements in Ussing chambers.
MDCK αβγ-ENaC or CFBE41o− cells were grown as polarized epithelial monolayers on Snapwell inserts as described above. When transepithelial resistance was ≥300 Ω·cm2 as assessed by an epithelial voltohmmeter (World Precision Instruments, Sarasota, FL), cells were transfected with control or ERp29-directed siRNA or with wt ERp29 pcDNA4 or C157S ERp29 pcDNA4 or mock-transfected. After 48 h and when transepithelial resistance was ≥400 Ω·cm2, cells were mounted in a vertical Ussing chamber (Physiologic Instruments, San Diego, CA) and underwent continuous voltage clamping for determination of short-circuit current (Isc). The bath solutions (mM: 115 NaCl, 25 NaHCO3, 2.4 KH2PO4, 1.24 K2HPO4, 1.2 MgCl2, 1.2 CaCl2, 10 glucose, pH 7.4, at 37°C) were symmetrical. Isc was analyzed using Acquire & Analyze data acquisition software (Physiologic Instruments). Resistance was monitored and calculated by Ohm's law using 2-mV bidirectional pulses every 90 s. Apical application of 10 μM amiloride was used to define ENaC-mediated currents. Trypsin (10 μg/ml final concentration; Sigma-Aldrich) was added to the apical bath as indicated. Because of greater day-to-day variability in baseline Isc in the siRNA transfection experiments, data were normalized by the average amiloride-sensitive Isc at baseline in control cells for a given day's experiment prior to analysis. In experiments using MDCK αβγ-ENaC cells with inducible expression of wt or C157S ERp29, cells were treated with Dox (as described above) and studied in Ussing chambers after transepithelial resistance reached ≥400 Ω·cm2.
Expression and electrophysiological analyses of ENaC in Xenopus oocytes.
The use of adult female Xenopus laevis as a source for oocytes was approved by the Institutional Animal Care and Use Committee of Children's Hospital of Philadelphia. Murine αβγ-ENaC and human ERp29 were expressed in Xenopus oocytes as previously described (20, 33, 34). Briefly, ERp29 and α-, β-, and γ-ENaC cRNAs were prepared with a cRNA synthesis kit (mMESSAGE mMACHINE, Ambion, Austin, TX) according to the manufacturer's protocol. cRNA concentrations were determined spectroscopically. Oocytes obtained from adult female X. laevis [NASCO (Fort Atkinson, WI) or Xenopus Express (Plant City, FL)] were enzymatically defolliculated and maintained at 18°C in modified Barth's saline [mM: 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.3 Ca(NO3)2, 0.41 CaCl2, 0.82 MgSO4, 15 HEPES, pH 7.6] supplemented with 10 μg/ml sodium penicillin, 10 μg/ml streptomycin sulfate, and 100 μg/ml gentamicin sulfate. A Nanoject II microinjector (Drummond Scientific, Broomall, PA) was used to inject each batch of oocytes (50 nl/oocyte) obtained from an individual frog with murine α-, β-, and γ-ENaC (0.33 ng/subunit) or ERp29 (10 ng) cRNAs alone or both αβγ-ENaC and ERp29 cRNAs dissolved in RNase-free water.
Whole cell current was measured in Xenopus oocytes 24–48 h after injection with a two-electrode voltage-clamp method (20, 33, 34). Oocytes were placed in a 1-ml chamber containing modified ND96 (mM: 96 NaCl, 1 KCl, 0.2 CaCl2, 5.8 MgCl2, 10 HEPES, pH 7.4) and impaled with 0.5- to 5-MΩ resistance micropipettes filled with 3 M KCl. The whole cell currents were measured by voltage clamping the oocytes in 20-mV steps between −140 and +60 mV adjusted for baseline transmembrane potential (transmembrane potential of the oocyte under nonclamped conditions). Whole cell currents were digitized at 200 Hz during the voltage steps, recorded directly onto a hard disk, and analyzed with pCLAMP 8 software (Axon Instruments, Foster City, CA). The difference in whole cell currents measured in the absence and presence of 10 μM amiloride was used to define the amiloride-sensitive Na+ current that was carried by ENaC. Whole cell currents were recorded at −100 mV for comparisons. All measurements were performed at room temperature.
Statistical analysis.
Statistical significance was determined by a two-tailed Student's t-test or a one-way ANOVA as appropriate. Statistical analysis was performed using SigmaStat software (version 2.03, Aspire, Ashburn, VA). P ≤ 0.05 was considered significant.
RESULTS
ERp29 regulates whole cell ENaC expression in MDCK epithelial cells.
We developed an epithelial model for mechanistic studies of ERp29's regulation of ENaC by selecting MDCK cells that stably express epitope-tagged ENaC subunits (COOH-terminal HA-tagged α-, V5-tagged β-, and Myc-tagged γ-subunits) and that also had Dox-inducible expression of wt ERp29. Previous studies suggest that these COOH-terminal epitope tags on the ENaC subunits do not interfere with ENaC trafficking or function (13, 36) but do increase sensitivity of many biochemical assays. We also hypothesized that ERp29's single cysteine residue (C157) would be critical for ERp29 function; therefore, we also selected an αβγ-ENaC-expressing MDCK cell line with Dox-inducible expression of an ERp29 where this residue was mutated to serine (C157S ERp29).
As shown in Fig. 1, wt and C157S ERp29-expressing cells had greater ERp29 expression in response to Dox. At 50 ng/ml Dox, wt ERp29 expression increased ∼6-fold (Fig. 1A, representative of n = 5, P = 0.02), while C157S ERp29 expression increased ∼6.6 fold (Fig. 1A, representative of n = 5, P = 0.02) compared with controls. In parallel, ∼3.3- and ∼2.8-fold increases in expression of wt and C157S ERp29 were obtained when αβγ-ENaC-expressing MDCK cells were transiently transfected with plasmids encoding wt and C157S ERp29, respectively, compared with cells transfected with control plasmid [Fig. 1B, representative of n = 5 (P = 0.04) and n = 5 (P = 0.03), respectively].
Fig. 1.

Influence of 29-kDa endoplasmic reticulum (ER) protein (ERp29) overexpression on whole cell epithelial Na+ channel (ENaC) expression in Madin-Darby canine kidney (MDCK) epithelial cells. A: MDCK cells stably expressing murine αβγ-ENaC and with doxycycline (Dox)-inducible expression of wt ERp29 or C157S ERp29 were treated for 8 h with Dox (50 ng/ml). B: MDCK cells stably expressing murine αβγ-ENaC were transiently transfected with control, wt ERp29, or C157S ERp29 plasmid. Whole cell lysates were prepared, and ERp29, HA-tagged α-ENaC, V5-tagged β-ENaC, γ-ENaC, and GAPDH (A) and ERp29 and GAPDH (B) expression was determined by immunoblotting. Immunoblots (IB) are representative of >3 independent experiments; those in A are juxtapositions taken from noncontiguous lanes of the same experimental gels. *P < 0.05 versus control.
Overexpression of wt ERp29 increased β-ENaC whole cell expression (∼2.5 fold, n = 8, P = 0.01), as well as whole cell expression of the lower-molecular-weight, presumably cleaved γ-ENaC (∼1.6-fold, n = 6, P = 0.002). Expression of the higher-molecular-weight, presumably uncleaved γ- and α-ENaC was not changed with wt ERp29 overexpression (Fig. 1A). In contrast, expression of C157S ERp29 caused ∼40% decreased expression of lower-molecular-weight, presumably cleaved γ-ENaC (n = 5, P = 0.008), without significantly altering expression of α-, β-, and presumably uncleaved γ-ENaC (Fig. 1A). Similar results were obtained when αβγ-ENaC-expressing MDCK cells were transiently transfected with wt or C157S ERp29. As in our previous work using αβγ-ENaC-expressing MDCK cells as a model system, we did not routinely detect the ∼65-kDa, presumably cleaved form of α-ENaC in immunoblots of whole cell lysates (8, 9).
Treatment of αβγ-ENaC-expressing MDCK cells with ERp29-directed siRNA caused an ∼60% diminution of ERp29 expression compared with the cells transfected with a nontargeting control siRNA (n = 4, P = 0.003; Fig. 2). This depletion of ERp29 expression was associated with an ∼60% decrease in β-ENaC expression (n = 4, P = 0.02), while expression of α-ENaC, full-length γ-ENaC, and cleaved γ-ENaC was not significantly changed (Fig. 2).
Fig. 2.
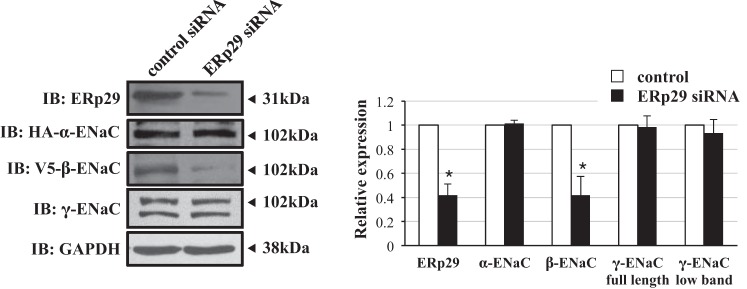
Influence of ERp29 depletion on whole cell ENaC expression in MDCK epithelial cells. MDCK αβγ-ENaC-expressing cells were transfected with nontargeting or ERp29-directed siRNA. Whole cell lysates were prepared, and ERp29, HA-taggged α-ENaC, V5-tagged β-ENaC, γ-ENaC, and GAPDH expression was determined by immunoblotting. Representative immunoblots (of ≥3 independent experiments) are taken from noncontiguous lanes of the same experimental gels. *P < 0.05 versus control.
Together, these data suggest that ERp29 can modulate the whole cell expression of the ENaC subunits and that C157 may be a critical determinant of this regulation.
ERp29 regulates ENaC functional expression in Xenopus oocytes, MDCK cells, and CFBE41o− cells.
In Xenopus oocytes injected with cRNAs encoding αβγ-ENaC, coinjection of ERp29 cRNA increased ENaC functional expression (whole oocyte current at −100-mV holding potential that was inhibited by 10 μM amiloride in 2-electrode voltage-clamp experiments) approximately twofold compared with oocytes injected with αβγ-ENaC alone (Fig. 3; P = 0.046).
Fig. 3.
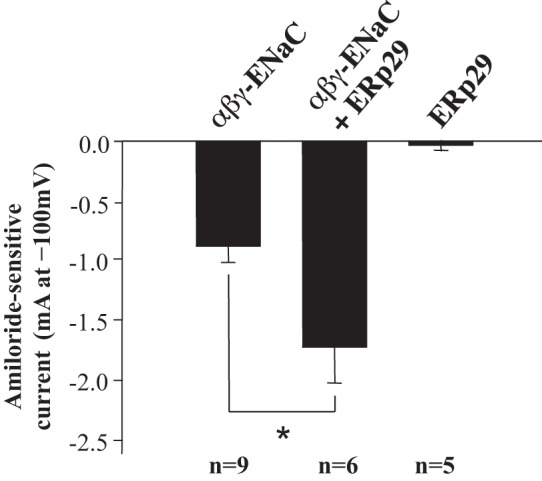
ERp29 promotes ENaC functional expression in Xenopus oocytes. Xenopus oocytes were injected with cRNA encoding murine αβγ-ENaC (0.33 ng/subunit) or 10 ng of ERp29 cRNA or coinjected with αβγ-ENaC and ERp29 cRNAs. Whole oocyte current at −100-mV holding potential that was sensitive to inhibition by 10 μM amiloride was determined by 2-electrode voltage clamp 28–48 h after injection. Values are means ± SE. *P < 0.05 (by t-test).
In MDCK αβγ-ENaC cells with inducible expression of wt ERp29 or in MDCK αβγ-ENaC cells transiently transfected by wt ERp29, overexpression of wt ERp29 was associated with an ∼1.7-fold increase in amiloride-sensitive Isc at baseline (corresponding to the already cleaved and active ENaC) in Ussing chambers (Fig. 4A, baseline; n = 9, P = 0.003). In contrast, overexpression of ERp29 C157S (Fig. 4B, baseline; n = 9, P = 0.039) or siRNA-mediated depletion of ERp29 (Fig. 4C, baseline; n = 9, P = 0.001) caused significant decreases in ENaC functional expression under baseline conditions.
Fig. 4.

ERp29 regulates ENaC functional expression and the fraction of cleaved vs. uncleaved ENaC at the apical surface in MDCK and CF bronchial epithelial (CFBE41o−) cells. A–D: MDCK αβγ-ENaC wt ERp29 cells treated with 50 ng of Dox (to induce ERp29 expression) and MDCK αβγ-ENaC cells transiently transfected with control or wt ERp29 expression plasmids (A), MDCK αβγ-ENaC C157S ERp29 cells treated with 50 ng of Dox (to induce ERp29 C157S expression) and MDCK αβγ-ENaC cells transiently transfected with control or C157S ERp29 expression plasmids (B), and MDCK αβγ-ENaC cells (C) and CFBE41o− parental cells (D) transfected with nontargeting or ERp29 siRNA were grown as monolayers on permeable supports and mounted in Ussing chambers. Short-circuit current (Isc) was determined at baseline, after application of trypsin (10 mg/ml) to the apical surface for 5 min to activate the uncleaved, lower-open probability (Po) ENaC, and after application of amiloride. In A and B, data are presented as baseline amiloride-sensitive Isc, change in Isc following trypsin treatment, and total amiloride-sensitive Isc after trypsin treatment (total). In C and D, data are expressed relative to baseline control Isc to account for day-to-day variability in these reference currents. Black bars: control (A–D); gray bars: overexpressed wt ERp29 (A), overexpressed CS ERp29 (B), and ERp29 (C and D) siRNA. Values are means ± SE. *P < 0.05 (by t-test).
To ensure that overexpression of multiple proteins (α-, β-, and γ-ENaC and ERp29) in the MDCK model system did not confound these data, we performed similar experiments using CFBE41o− cells, which were recently demonstrated by our group to endogenously express functional ENaC (25). Notably, siRNA-mediated depletion of ERp29 in CFBE41o− cells was also associated with a decrease in amiloride-sensitive (ENaC-mediated) Isc (Fig. 4D, baseline; n = 7, P = 0.001).
Together, these data are consistent with modulation of ENaC functional expression by ERp29. We performed additional experiments to test whether these alterations in ENaC functional expression were due to changes in ENaC's Po or in the quantity of ENaC at the apical surface in these epithelial models.
ERp29 regulates the cleavage state of ENaC at the apical surface.
ENaC's Po is regulated in large part by cleavage of the extracellular loops of its α- and γ-subunits; uncleaved channels are near-silent and have a Po approximating 0, while fully cleaved channels have a Po approximating 1 (1, 3–5). Others have demonstrated that both cleaved and uncleaved channels are present at the surface of epithelial cells (17) and that the uncleaved channels can be acutely activated by treatment of the apical surface with exogenous trypsin. We thus used this technique to test whether ERp29 modulates the fraction of uncleaved, low-Po vs. cleaved, higher-Po ENaC present at the surface as the mechanism by which it influences ENaC functional expression by exposing the apical surface of these cells to trypsin in Ussing chambers, as we and others have previously described (5, 8, 9, 25). Briefly, after a baseline Isc was determined, trypsin was added to the apical surface to cleave and fully activate all apical ENaC; then cells were treated with amiloride to determine amiloride-sensitive Isc (schematic in Fig. 4). In this paradigm, the amiloride-sensitive Isc at baseline represents an apical surface channel that is already cleaved, while the trypsin-stimulated change in amiloride-sensitive Isc represents a channel at the apical surface that is uncleaved and near-silent at the beginning of the experiment and is acutely activated by trypsin cleavage. The total amiloride-sensitive Isc is then the sum of the baseline and trypsin-stimulated Isc and is reflective of the total amount of ENaC at the apical surface (Fig. 4, total).
When wt ERp29 was overexpressed in αβγ-ENaC-expressing MDCK cells, trypsin caused a higher baseline ENaC-mediated Isc (see above) and a smaller increase in amiloride-sensitive Isc than in controls (Fig. 4A; n = 9, P = 0.025). These data suggest that there were more cleaved, higher-Po channels and fewer near-silent, low-Po channels at the cell surface when ERp29 was overexpressed. Interestingly, the total amiloride-sensitive Isc was similar for control and ERp29-overexpressing cells (Fig. 4A, total), suggesting that the number of fully activated ENaCs at the apical surface after trypsin was similar. If it is assumed that all apical surface ENaC is fully activated by trypsin in this protocol, these data suggest that ERp29 overexpression does not alter ENaC surface expression.
In contrast, expression of C157S ERp29 (Fig. 4B, trypsin; n = 9, P = 0.009) and siRNA-mediated depletion of ERp29 (Fig. 4C, trypsin; n = 9, P = 0.033) resulted in lower baseline ENaC-mediated Isc (see above) and an increase in the trypsin-stimulated amiloride-sensitive Isc, suggesting that there were fewer cleaved, higher-Po channels and more near-silent, uncleaved channels at the apical surface under these conditions. Furthermore, neither of these perturbations altered the total amiloride-sensitive Isc (Fig. 4, B and C, total), which is again consistent with the concept that ERp29 does not regulate ENaC apical surface expression.
We performed similar experiments in CFBE41o− cells to test the effect of ERp29 depletion on the cleavage state of endogenously expressed ENaC. As shown in Fig. 4D, in CFBE41o− cells, ERp29 depletion resulted in a decrease in endogenous ENaC-mediated current at baseline (see above) and a significant increase in the trypsin-stimulated amiloride-sensitive Isc (n = 7, P = 0.026). These data again suggest that ERp29 promotes the cleavage/higher-Po state of ENaC without significantly influencing the total amount of ENaC at the apical surface. We were not able to effectively overexpress ERp29 or C157S ERp29 in the CFBE41o− cells, perhaps because the whole cell expression of endogenous ERp29 is significantly greater in CFBE41o− than MDCK cells (Fig. 5; n = 3, P = 0.033). This is consistent with work of others demonstrating that ERp29 is especially abundant in lung and brain (31).
Fig. 5.
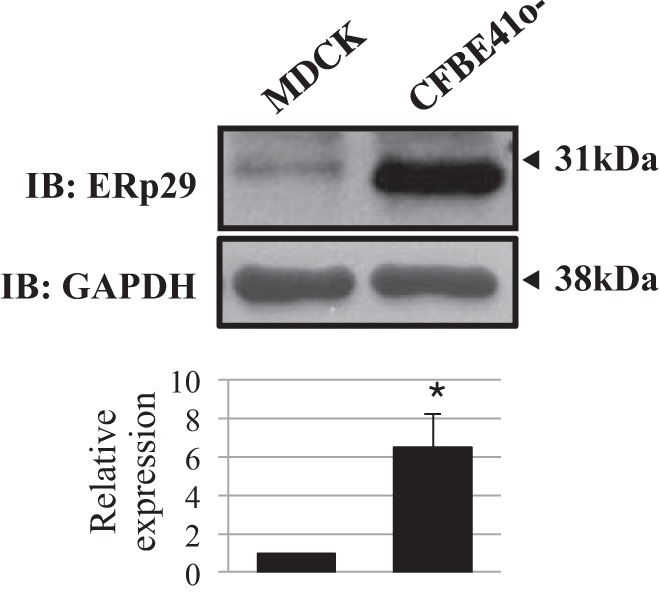
Differential expression of ERp29 in MDCK and CFBE41o− cells. MDCK cells stably expressing murine αβγ-ENaC and CFBE41o− cells were grown as polarized monolayers on permeable supports. Whole cell lysates were prepared, and ERp29 and GAPDH expression was determined by immunoblotting. Data are representative of 3 independent experiments. *P < 0.05.
Taken together, these data support the hypothesis that ERp29 regulates ENaC functional expression in epithelial cells by regulating the cleavage of ENaC and, therefore, its Po. Importantly, these data also support the hypothesis that ERp29's single cysteine residue C157 is important in ERp29 function and that our data in MDCK cells are likely not confounded by artifacts of overexpression.
ERp29 does not modulate ENaC surface expression.
We performed surface biotinylation experiments to directly confirm that ERp29 does not modulate the amount of ENaC at the apical surface (Fig. 6). We focused on the β-ENaC subunit, as this subunit is not subject to proteolytic cleavage and, therefore, its surface expression reflects that of both uncleaved and cleaved αβγ-ENaC heterotrimers. β-ENaC's V5 epitope was also the most robust for detection in this assay. As predicted by the data of Fig. 4, β-ENaC expression at the apical surface was similar when wt or C157S ERp29 was overexpressed compared with control conditions (Fig. 6; n = 5 independent experiments, P = not significant). Furthermore, and consistent with our group's published data (35) and the data of others (14), ERp29 was also present at the MDCK cell surface, while the lack of detection of biotinylated GAPDH suggests that intracellular proteins were not nonspecifically labeled in these experiments. These data thus suggest that ERp29 does not significantly influence ENaC abundance in apical membrane cells and further support the hypothesis that ERp29 primarily alters ENaC functional expression by altering ENaC cleavage and Po.
Fig. 6.
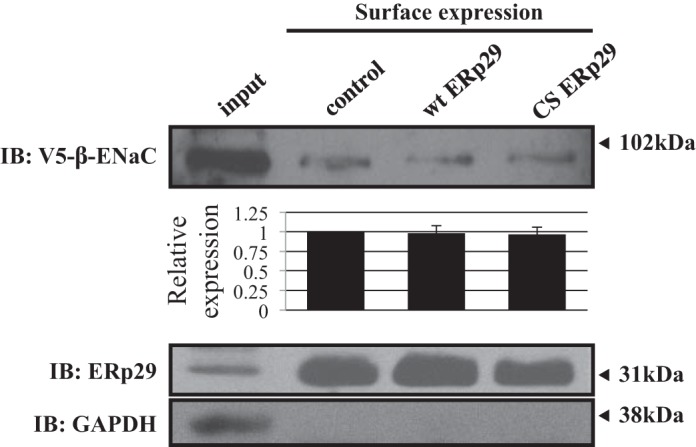
ERp29 does not affect ENaC expression at the apical surface. MDCK αβγ-ENaC cells transiently transfected with control, wt ERp29, or ERp29 C157S (CS) plasmids underwent biotinylation of surface proteins. Whole cell lysates were prepared, and biotinylated proteins were precipitated with NeutrAvidin beads. ERp29, V5-tagged β-ENaC, and GAPDH in the captured, biotinylated proteins were revealed by immunoblotting (representative of 4 independent experiments). GAPDH was present in whole cell lysates, but not in the captured, biotinylated proteins, suggesting that only surface proteins were labeled.
ERp29 interacts with ENaC and regulates ENaC interaction with Sec24D.
The interaction of β-ENaC and ERp29 was tested. Figure 7 demonstrates recovery of β-ENaC after immunoprecipitation of ERp29. β-ENaC recovery increased significantly when wt ERp29 was overexpressed (n = 3, P = 0.032) and trended toward an increase when C157S ERp29 was overexpressed (n = 3, P = 0.086), suggesting that ERp29 interacts with ENaC and that C157 is not a determinant of this interaction. Notably, this is consistent with the crystal structure of ERp29, where C157 is found outside the client binding site (2).
Fig. 7.
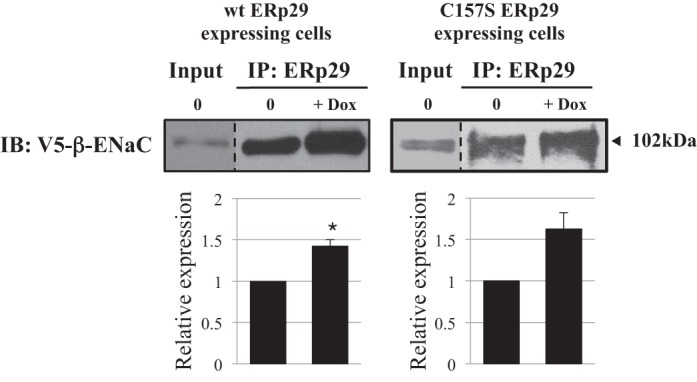
β-ENaC interacts with ERp29. MDCK αβγ-ENaC wt or C157S ERp29 cells were treated with Dox for 8 h. Whole cell lysates were prepared under nondenaturing conditions. ERp29 and its interacting proteins were precipitated using anti-ERp29, and V5-tagged β-ENaC associated with ERp29 was revealed by immunoblotting. Input lane represents 10% of the total protein subject to immunoprecipitation (IP). Data are representative of 3 independent experiments. *P = 0.032.
Previous data suggest that coat complex II (COP II) vesicles may transport ENaC from the ER to the Golgi. α-ENaC has an ER exit signal in its COOH terminus that is consistent with a COP II interaction motif. We recently demonstrated that ENaC interacts with the Sec24D cargo recognition component of COP II and that this interaction, as well as ENaC biogenesis, is promoted by the cytoplasmic chaperone heat shock protein 70 (Hsp70) (8). Using a coimmunoprecipitation approach (Fig. 8), we confirmed our previous finding that β-ENaC interacts with Sec24D (8) and found that the interaction of β-ENaC and Sec24D was enhanced by overexpression of wt ERp29 (n = 4, P = 0.013). In contrast, overexpression of C157S ERp29 inhibited the association of β-ENaC with Sec24D, even though β-ENaC still interacts with C157S ERp29 (Fig. 7; n = 3, P = 0.017). These changes in β-ENaC-Sec24D association were not due to a change in the whole cell expression of Sec24D (Fig. 8). Finally, these data are consistent with ERp29 promoting the association of ENaC with COP II and, therefore, likely its movement from ER to Golgi via COP II.
Fig. 8.
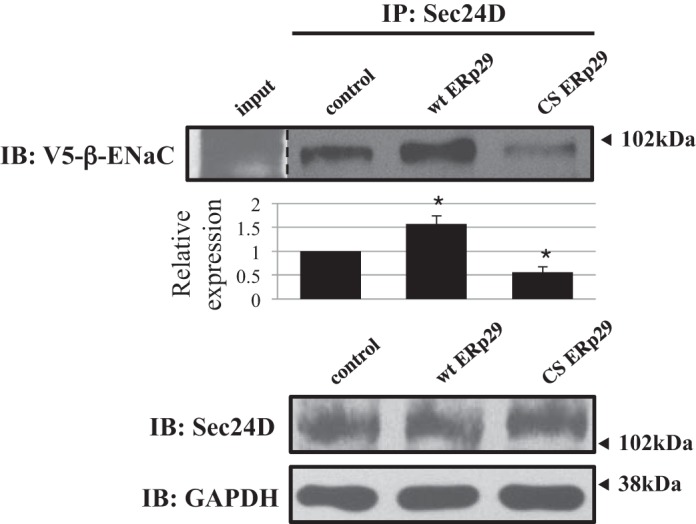
ERp29 modulates ENaC interaction with Sec24D. MDCK αβγ-ENaC cells were transiently transfected with control, wt ERp29, or ERp29 C157S (CS) plasmids. Sec24D and its interacting proteins were precipitated using anti-Sec24D, and V5-tagged β-ENaC associated with Sec24D was revealed by immunoblotting. Immunoblots of Sec24D and GAPDH in whole cell lysate are also presented. Data are representative of 3 independent experiments. *P < 0.05.
DISCUSSION
ENaC plays a critical role in the regulation of blood volume and blood pressure, as well as airway liquid surface volume. Its regulation is therefore directly relevant to hypertension and CF. On the basis of findings that the prototype ΔF508 CFTR corrector 4-phenylbutyrate (24) increased ERp29 expression (35) and promoted ENaC activity in epithelial cells (22), we interrogated whether ERp29 would also regulate ENaC.
Our data not only suggest that ERp29 modulates ENaC expression (Figs. 1 and 2) but also that it does, indeed, interact with ENaC in epithelial cells (Fig. 7) and promotes ENaC functional expression in Xenopus oocytes (Fig. 3) and in MDCK and CFBE41o− epithelial cells (Fig. 4). In considering the mechanism(s) underlying these effects, we interrogated two factors that determine ENaC activity/functional expression: “Po,” the channel's open probability, and “N,” the total number of channels present at the cell surface. In the case of ENaC, Po is, in large part, determined by cleavage of the α- and γ-subunits in their luminal/extracellular loops; uncleaved channels have a Po that approximates 0, while fully cleaved channels have a Po approximating 1 (Fig. 9). Interestingly, our Ussing chamber and surface biotinylation data suggest that ERp29 does not modulate N (Figs. 4 and 5) but, rather, alters the fraction of ENaC at the apical surface in its cleaved/higher-Po form (Fig. 4).
Fig. 9.

A model of ERp29's role in ENaC biogenesis. wt ERp29 regulates ENaC biogenesis by promoting the interaction of ENaC with Sec24D/coat complex II (COP II) and delivery of ENaC to the Golgi, where it is processed; this diminishes the proportion of ENaC that bypasses Golgi processing. C157S ERp29 disfavors the interaction of ENaC with Sec24D/COP II and, in turn, its delivery to and processing in the Golgi. This increases the proportion of ENaC that bypasses Golgi processing. ENaC, epithelial Na+ channel; ER, endoplasmic reticulum; ERp29, 29-kDa ER protein; wt, wild-type, C157S, C157S ERp29.
ERp29 is homologous to the thioredoxins and the PDIs. However, the CXXC active site motif characteristic of the thioredoxins and PDIs is not present in ERp29 (23). Instead, ERp29 contains a single cysteine at position 157 (C125 in the mature protein) that, according to the crystal structure, is not within the client binding site (2). Because this cysteine is strictly conserved, and with no evidence that ERp29 has PDI-like activity (15), we also hypothesized that C157 plays a critical role in ERp29 function and demonstrated that overexpression of C157S ERp29 decreased ENaC functional expression (Fig. 4B) without decreasing its interaction with β-ENaC (Fig. 7). The effect of C157S ERp29 on ENaC functional expression was, in fact, similar to that of siRNA-mediated depletion of ERp29 expression, which further suggests that C157 is critical for ERp29 function. ERp29, while primarily thought of as an ER luminal chaperone, is now known to traverse the secretory pathway and have effects distal to the ER. ERp29 is secreted into thyrocyte growth medium (28), and we previously demonstrated that ERp29 is present at the cell surface and in the growth medium of IB3-1 CF bronchiolar epithelial cells (35), and in the present study we show that ERp29 is present at the cell surface of MDCK cells (Fig. 5). Furthermore, we and others have suggested that ERp29 may “escort” client proteins to and/or through post-ER compartments. Indeed, the role of ERp29 as an escort is supported by its regulation of connexin 43 hemichannel assembly. Connexin 43 monomers do not assemble into hexameric hemichannels until they reach the Golgi; correct localization of this assembly is regulated by ERp29 (10). Similarly, the Drosophila paralog of ERp29, windbeutel, escorts the glycosaminoglycan-modifying enzyme Pipe to the Golgi, where Pipe acts to direct embryonic ventralization (30).
A major gap in our understanding of ENaC biogenesis is the mechanism underlying why some channels undergo furin cleavage and N-glycan maturation in the Golgi while other bypass this Golgi processing and reach the apical surface in an uncleaved, low-Po form that has immature N-glycans (17). We recently demonstrated that ENaC interacts with the Sec24D cargo recognition component of COP II ER-to-Golgi transport machinery and that the cytosolic chaperone Hsp70 promotes ENaC biogenesis and functional expression and its interaction with Sec24D (8). We showed that Hsp70 promoted ENaC functional expression by increasing its surface expression (N), rather than by altering the fraction of ENaC at the surface that had undergone cleavage (8), suggesting that Hsp70 equally promoted delivery of both cleaved and uncleaved ENaC to the apical surface. This contrasts the effects we observe with ERp29, where N is not altered but the fraction of channel that is cleaved/active is increased (Figs. 4 and 5), suggesting that ERp29 may promote delivery of Golgi-processed ENaC while diminishing the proportion of ENaC that bypasses Golgi processing (Fig. 9).
Interestingly, wt ERp29 overexpression promoted the ENaC-Sec24D interaction, while C157S ERp29 overexpression diminished this interaction (Fig. 8). These data lead us to suggest that ERp29 interacts with ENaC to promote its association with Sec24D/COP II and delivery from the ER to the Golgi, positioning it for proteolysis by furin and N-glycan maturation prior to delivery to the apical membrane. These data also may suggest that ENaC, which bypasses Golgi processing (furin proteolysis and N-glycan maturation), exits the ER and is delivered to the plasma membrane via a Sec24D/COP II-independent pathway (Fig. 9). In this hypothetical mechanism, ERp29 thus serves as a molecular switch to determine ENaC's itinerary to the plasma membrane. As Hsp70 increases both cleaved and uncleaved ENaC at the plasma membrane, its action to facilitate ENaC biogenesis must occur prior to the influence of ERp29 on biogenesis.
In summary, our data are consistent with ERp29 increasing ENaC functional expression by promoting its ER exit and delivery to the Golgi through a Sec24D/COP II-mediated itinerary and, thus, facilitating the proteolytic cleavage of its extracellular loops during biogenesis. This effect is dependent on the presence of ERp29's single cysteine, C157. These data also suggest that uncleaved ENaC at the plasma membrane avoids Golgi processing by trafficking via a non-COP II-mediated ER exit pathway. Finally, these data are the first demonstration of an ER luminal chaperone that regulates ENaC.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants T32 DK-07748 (R. A. Chanoux), R01 DK-58046 (R. C. Rubenstein), and R01 DK-73185 (R. C. Rubenstein).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.G., Y.B., L.S., R.A.C., and R.C.R. are responsible for conception and design of the research; Y.G., Y.B., R.A.C., and R.C.R. performed the experiments; Y.G., Y.B., L.S., and R.C.R. analyzed the data; Y.G., Y.B., L.S., R.A.C., and R.C.R. interpreted the results of the experiments; Y.G., Y.B., and R.C.R. prepared the figures; Y.G., Y.B., and R.C.R. drafted the manuscript; Y.G., Y.B., L.S., R.A.C., and R.C.R. edited and revised the manuscript; Y.G., Y.B., L.S., R.A.C., and R.C.R. approved the final version of the manuscript.
Footnotes
This article is the topic of an Editorial Focus by My N. Helms and Phi Trac (13a).
REFERENCES
- 1.Adebamiro A, Cheng Y, Johnson JP, Bridges RJ. Endogenous protease activation of ENaC: effect of serine protease inhibition on ENaC single channel properties. J Gen Physiol 126: 339–352, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barak NN, Neumann P, Sevvana M, Schutkowski M, Naumann K, Malesevic M, Reichardt H, Fischer G, Stubbs MT, Ferrari DM. Crystal structure and functional analysis of the protein disulfide isomerase-related protein ERp29. J Mol Biol 385: 1630–1642, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J Biol Chem 282: 6153–6160, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol 288: L813–L819, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Caldwell RA, Boucher RC, Stutts MJ. Serine protease activation of near-silent epithelial Na+ channels. Am J Physiol Cell Physiol 286: C190–C194, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Canessa CM, Merillat AM, Rossier BC. Membrane topology of the epithelial sodium channel in intact cells. Am J Physiol Cell Physiol 267: C1682–C1690, 1994 [DOI] [PubMed] [Google Scholar]
- 7.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994 [DOI] [PubMed] [Google Scholar]
- 8.Chanoux RA, Robay A, Shubin CB, Kebler C, Suaud L, Rubenstein RC. Hsp70 promotes epithelial sodium channel functional expression by increasing its association with coat complex II and its exit from endoplasmic reticulum. J Biol Chem 287: 19255–19265, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chanoux RA, Shubin CB, Robay A, Suaud L, Rubenstein RC. Hsc70 negatively regulates epithelial sodium channel trafficking at multiple sites in epithelial cells. Am J Physiol Cell Physiol 305: C776–C787, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das S, Smith TD, Sarma JD, Ritzenthaler JD, Maza J, Kaplan BE, Cunningham LA, Suaud L, Hubbard MJ, Rubenstein RC, Koval M. ERp29 restricts connexin43 oligomerization in the endoplasmic reticulum. Mol Biol Cell 20: 2593–2604, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel γ-subunit: genetic heterogeneity of Liddle syndrome. Nat Genet 11: 76–82, 1995 [DOI] [PubMed] [Google Scholar]
- 12.Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. A de novo missense mutation of the β-subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci USA 92: 11495–11499, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanwell D, Ishikawa T, Saleki R, Rotin D. Trafficking and cell surface stability of the epithelial Na+ channel expressed in epithelial Madin-Darby canine kidney cells. J Biol Chem 277: 9772–9779, 2002 [DOI] [PubMed] [Google Scholar]
- 13a.Helms MN, Trac P. ERp29, a chaperone protein ushering in new insights on ion transport regulation. Focus on “ERp29 regulates epithelial sodium channel functional expression by promoting channel cleavage.” Am J Physiol Cell Physiol July 9, 2014. 10.1152/ajpcell.00217.2014 [DOI] [PubMed] [Google Scholar]
- 14.Holbrook LM, Watkins NA, Simmonds AD, Jones CI, Ouwehand WH, Gibbins JM. Platelets release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br J Haematol 148: 627–637, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Hubbard MJ, Mangum JE, McHugh NJ. Purification and biochemical characterization of native ERp29 from rat liver. Biochem J 383: 589–597, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Hughey RP, Bruns JB, Kinlough CL, Kleyman TR. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem 279: 48491–48494, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B, Gatzy J, Burnier M, Horisberger JD, Beermann F, Boucher R, Rossier BC. A mouse model for the renal salt-wasting syndrome pseudohypoaldosteronism. Proc Natl Acad Sci USA 94: 11710–11715, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Jiang Q, Li J, Dubroff R, Ahn YJ, Foskett JK, Engelhardt J, Kleyman TR. Epithelial sodium channels regulate cystic fibrosis transmembrane conductance regulator chloride channels in Xenopus oocytes. J Biol Chem 275: 13266–13274, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10: 487–493, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Pruliere-Escabasse V, Planes C, Escudier E, Fanen P, Coste A, Clerici C. Modulation of epithelial sodium channel trafficking and function by sodium 4-phenylbutyrate in human nasal epithelial cells. J Biol Chem 282: 34048–34057, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Rainey-Barger EK, Mkrtchian S, Tsai B. Dimerization of ERp29, a PDI-like protein, is essential for its diverse functions. Mol Biol Cell 18: 1253–1260, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing ΔF508-CFTR. J Clin Invest 100: 2457–2465, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubenstein RC, Lockwood SR, Lide E, Bauer R, Suaud L, Grumbach Y. Regulation of endogenous ENaC functional expression by CFTR and ΔF508-CFTR in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 300: L88–L101, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samaha FF, Rubenstein RC, Yan W, Ramkumar M, Levy DI, Ahn YJ, Sheng S, Kleyman TR. Functional polymorphism in the carboxyl terminus of the α-subunit of the human epithelial sodium channel. J Biol Chem 279: 23900–23907, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Sargsyan E, Baryshev M, Szekely L, Sharipo A, Mkrtchian S. Identification of ERp29, an endoplasmic reticulum lumenal protein, as a new member of the thyroglobulin folding complex. J Biol Chem 277: 17009–17015, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J 15: 2381–2387, 1996 [PMC free article] [PubMed] [Google Scholar]
- 30.Sen J, Goltz JS, Konsolaki M, Schupbach T, Stein D. Windbeutel is required for function and correct subcellular localization of the Drosophila patterning protein Pipe. Development 127: 5541–5550, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Shnyder SD, Hubbard MJ. ERp29 is a ubiquitous resident of the endoplasmic reticulum with a distinct role in secretory protein production. J Histochem Cytochem 50: 557–566, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Stewart AP, Haerteis S, Diakov A, Korbmacher C, Edwardson JM. Atomic force microscopy reveals the architecture of the epithelial sodium channel (ENaC). J Biol Chem 286: 31944–31952, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suaud L, Carattino M, Kleyman TR, Rubenstein RC. Genistein improves regulatory interactions between G551D-cystic fibrosis transmembrane conductance regulator and the epithelial sodium channel in Xenopus oocytes. J Biol Chem 277: 50341–50347, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Suaud L, Li J, Jiang Q, Rubenstein RC, Kleyman TR. Genistein restores functional interactions between ΔF508-CFTR and ENaC in Xenopus oocytes. J Biol Chem 277: 8928–8933, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Suaud L, Miller K, Alvey L, Yan W, Robay A, Kebler C, Kreindler JL, Guttentag S, Hubbard MJ, Rubenstein RC. ERp29 regulates ΔF508 and wild-type cystic fibrosis transmembrane conductance regulator (CFTR) trafficking to the plasma membrane in cystic fibrosis (CF) and non-CF epithelial cells. J Biol Chem 286: 21239–21253, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valentijn JA, Fyfe GK, Canessa CM. Biosynthesis and processing of epithelial sodium channels in Xenopus oocytes. J Biol Chem 273: 30344–30351, 1998 [DOI] [PubMed] [Google Scholar]
- 37.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 389: 607–610, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J Gen Physiol 120: 191–201, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan W, Samaha FF, Ramkumar M, Kleyman TR, Rubenstein RC. Cystic fibrosis transmembrane conductance regulator differentially regulates human and mouse epithelial sodium channels in Xenopus oocytes. J Biol Chem 279: 23183–23192, 2004 [DOI] [PubMed] [Google Scholar]