

Abstract
Background
A comprehensive characterization of the effects of cigarette smoke on systemic soluble immune/inflammatory markers may provide insight into the mechanisms through which smoking causes disease.
Methods
Levels of 78 inflammation, immune, and metabolic markers were measured using multiplex immune assays in 1819 Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO) participants aged 55 to 74 years from three existing nested case-control studies. These data were made representative of the entire PLCO screening arm through reweighting with weights estimated in logistic regression models. We assessed associations between smoking status, cigarettes smoked per day, and time since quitting with dichotomized marker levels using adjusted weighted logistic regression models.
Results
Current smoking was associated with 10 inflammation markers after correcting for multiple testing, encompassing several components of the immune/inflammation response. Levels of seven of these markers (interleukin [IL]-15, IL-1RA, IL-1β, IL-16, stem cell factor, soluble interleukin 6 receptor, and soluble vascular endothelial growth factor receptor 3) were lower among current smokers (n = 414) when compared with never smokers (n = 548), with odds ratios (ORs) ranging from 0.44 to 0.27, while levels of CC motif ligand (CCL)/thymus and activation regulated chemokine (CCL17/TARC) (OR = 4.08, 95% confidence interval [CI] = 2.01 to 8.25), CCL11/EOTAXIN (OR = 2.57, 95% CI = 1.45 to 4.55), and C-reactive protein (CRP) (OR = 2.54, 95% CI = 1.29 to 4.98) were elevated. These markers were not associated with cigarettes per day among current smokers, but there were trends in IL-15, IL-1RA, IL-1β, CCL17/TARC, CCL11/EOTAXIN, and CRP levels across categories of years since quitting smoking.
Conclusions
Smoking is associated with a broad range of alterations in systemic immune and inflammation marker levels among older, long-term smokers. Smoking cessation may result in marker levels reverting back to those of never smokers over time.
Tobacco is a major cause of morbidity and mortality worldwide, causing one in five deaths overall and an estimated 160900 cancer deaths annually in the United States (1,2). Smoking causes cancer at 18 different tumor sites, and also causes a range of other chronic diseases, including coronary heart disease, stroke, and chronic obstructive pulmonary disease (3,4).
Tobacco contains numerous carcinogens, including polycyclic aromatic hydrocarbons, nitrosamines, aromatic amines, and N-nitrosodimethylamine (5). In addition, smoking-induced inflammation and immune modulation are emerging as potentially important mechanisms in the development of cancer and other systemic chronic diseases. Cigarette smoking leads to numerous pulmonary and systemic immunological changes. In the lung, smoking increases the number of macrophages, neutrophils, eosinophils, and mast cells, decreases the number of airway dendritic cells, and alters macrophage and neutrophil function (6,7). Systemically, smoking leads to elevated white blood cell counts, particularly neutrophils (8). Additionally, nicotine has been shown to be an immune suppressant (9). However, a broad evaluation of the spectrum of perturbations in host immune and inflammatory response caused by cigarette smoking is currently lacking. Most prior studies have focused on a select number of markers, such C-reactive protein (CRP), interleukin (IL)-6, and fibrinogen (10–15).
A broad characterization of the effects of cigarette smoke on systemic immune/inflammatory markers, proteins released by cells participating in the immune/inflammation process, may provide insight into the mechanisms through which tobacco smoking causes disease. Here, using data from 1819 individuals who participated in the population-based Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial, we investigated the association of cigarette smoking status, intensity and duration of smoking, and time since quitting with variation in systemic levels of 78 markers of immunity and inflammation.
Methods
Study Population
The PLCO Cancer Screening Trial recruited approximately 155000 55- to 74-year-old men and women from the general population from 1992 to 2001 (16). In addition to demographic, behavioral, and dietary information, blood samples were obtained at baseline and five subsequent annual visits from participants in the screening arm. Cancer diagnoses were ascertained through annual questionnaires and confirmed by medical chart abstraction and death certificate review (16,17). PLCO was approved by the Institutional Review Boards at each participating center, and all participants gave informed consent.
We combined data from three nested case-control studies (ie, studies of lung cancer [526 case and 592 matched control], ovarian cancer [150 case and 149 matched control] and non-Hodgkin’s lymphoma [NHL] [301 case and 301 matched control]) that were previously conducted in the screening arm (18–20). Details on the exclusion criteria, matching factors and inflammation markers measured in these studies are presented in Supplementary Table 1 (available online). The combined dataset was limited to non-Hispanic whites (n = 152 excluded). Six individuals were subjects in two of the case-control studies; only data from the first of the two studies were included in this data set. Individuals with a personal history of cancer prior to randomization (n = 31) and with incomplete smoking data (n = 11) were also excluded, for a total of 1819 individuals.
Laboratory Analysis
Serum specimens collected either at baseline (lung and NHL studies) or at a follow-up visit (ovary study) (processed at 1200xg for 15 minutes, frozen within two hours of collection, stored at -700C) were used to measure circulating levels of 86 markers (77 in the lung study, 60 in the ovary study, and 83 in the NHL study) (Supplementary Table 2, available online). These markers were selected based on methodologic work that evaluated the performance and reproducibility of multiplexed assays for measurement of inflammation markers in serum (21). Markers were measured using Luminex bead-based assays (EMD Millipore, Inc., Billerica, MA). Concentrations were calculated using either a four- or five-parameter standard curve. Serum samples were assayed in duplicate, and averaged to calculate concentrations. Blinded duplicates in the lung and NHL studies and duplicate measurements on study subjects in the ovary study were used to evaluate assay reproducibility through coefficients-of-variation (CVs) and intraclass correlation coefficients (ICCs) calculated on log-transformed values of the markers. ICCs were greater than 0.8 in 91% of these markers in the lung and NHL studies (18,19) and in 78% of these markers in the ovarian cancer study (20). Eight markers with greater than 90% of value below the lowest limit of detection (LLOD) were excluded from all analyses, resulting in 78 evaluable markers.
Statistical Analysis
The lung cancer, NHL, and ovarian cancer case-control studies had several different design features, including varying inclusion and matching criteria (Supplementary Table 1, available online). Therefore, the three studies as a whole were nonrepresentative of the PLCO. To combine data from the case-control studies, we developed sets of propensity-score adjusted sampling weights to ensure that our analysis accounted for the particular inclusion/exclusion criteria and sampling plan for each study (22,23) (Supplementary Table 1, available online). The sampling weights allowed us to include all participants with marker data (including cancer cases), and made our analysis as representative as possible of the non-Hispanic white PLCO screening arm (see the Supplementary Methods, available online for details). Cancer cases were included in this analysis, as they were cancer-free at the time of blood draw, and represented a small fraction of the data after weighting (2.8%). Sampling weights were derived from logistic regression models for the probability that an eligible screening arm participant would be selected into any given case-control study. Separate logistic regression models were conducted based on case-control status, study, and gender. Each logistic regression model had covariates of age, smoking history (ie, smoking status, years since quit, and pack-years) and vital status on December 31, 2009. Study-specific weights were then combined for each of the five combinations of case-control studies with a common subset of panels (all three studies, lung and NHL, lung and ovary, NHL and ovary, and lung alone). Simulations suggest that analyses using both weighting methods and additional regression adjustment for matching factors provide a good way to adjust for nonrepresentative sampling in nested case-control studies (24).
Information on smoking status, cigarettes smoked per day and number of years smoked among ever smokers, years since quitting among former smokers, and other covariates was collected at baseline for all participants. A number of markers had a sizeable fraction of values below the LLOD, which precluded analysis of these markers as continuous outcomes. Therefore, separately by study, inflammation marker levels were dichotomized as above or below the median value, or as detectable and undetectable if greater than 50% of the values were below the LLOD. Models categorizing marker levels into tertiles or quartiles produced similar results.
First, we assessed the association between current vs never smoking status and each of the 78 markers in weighted logistic regression models using standard survey regression analysis software (23). We applied a 5% false discovery rate (FDR) criterion, retaining only those markers that remained statistically significant for subsequent analyses. For the remaining analyses, a P value lower than .05 was considered to be statistically significant. These analyses included assessing associations between markers levels and: 1) former vs never and former vs current smoking status, 2) cigarettes smoked per day (1–10, 11–20, 21–30, and 31+ cigarettes per day), smoking duration (<40, 40–44, 45–49, and 50+ years) and pack-years smoked (<42, 42–52.24, 52.25–76.49, and 76.5+) among current smokers, and 3) time since quitting (current smoker, ≤5, 5.1–10, 10.1–20, 20+ years) among former smokers. All models were adjusted for age, sex, history of chronic bronchitis or emphysema, history of coronary heart disease or heart attack, body mass index, original study (ie, lung, ovary, or NHL), and year of serum collection. Spearman correlations were used to estimate unweighted correlations across markers.
All statistical tests were two-sided, and analyses were carried out in SAS 9.2 (Cary, NC).
Results
The current study included 548 never (weighted N = 27219), 857 former (weighted N = 25381) and 414 current smokers (weighted N = 5664). Compared with the entire eligible PLCO screening arm, the participants included in the present study were older and more likely to be current smokers (Table 1). Among current smokers, participants with marker measurements smoked more cigarettes per day with a longer duration, and among former smokers had fewer years since quitting than those in the full cohort. However, when weights were applied, the characteristics of the weighted population became very similar to the eligible PLCO screening arm population, showing that these weights help account for the nonrepresentative sampling (Table 1).
Table 1.
Participant characteristic in subjects with measured inflammation marker data, after weights were applied, and in eligible participants in the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial screening arm
| Characteristic | No. (%) | Weighted, No. (%)* | PLCO screening arm, No. (%) |
|---|---|---|---|
| Total | 1819 | 58264 | 58264 |
| Sex | |||
| Female | 814 (44.8) | 28331 (48.6) | 28331 (48.6) |
| Male | 1005 (55.3) | 29933 (51.4) | 29933 (51.4) |
| Age group, y | |||
| ≤59 | 399 (21.9) | 18950 (32.5) | 20056 (34.4) |
| 60–64 | 533 (29.3) | 19911 (34.2) | 17954 (30.8) |
| 65–69 | 537 (29.5) | 11250 (19.3) | 12877 (22.1) |
| ≥70 | 350 (19.2) | 8153 (14.0) | 7377 (12.7) |
| BMI category, kg/m2 | |||
| <25 | 639 (35.1) | 18009 (30.9) | 18914 (32.4) |
| 25–30 | 792 (43.5) | 26939 (46.2) | 24753 (42.5) |
| ≥30 | 367 (20.2) | 12473 (21.4) | 14015 (24.1) |
| Missing | 21 (1.2) | 843 (1.4) | 582 (1.0) |
| Smoking status | |||
| Never | 548 (30.1) | 27219 (46.7) | 27389 (47.0) |
| Former | 857 (47.1) | 25381 (43.6) | 25032 (43.0) |
| Current | 414 (22.8) | 5664 (9.7) | 5843 (10.0) |
| Cigarettes per day in current smokers | |||
| 1–10 | 49 (11.8) | 836 (14.8) | 1007 (17.2) |
| 11–20 | 165 (39.9) | 1875 (33.1) | 2411 (41.3) |
| 21–30 | 109 (26.3) | 1760 (31.1) | 1452 (24.9) |
| 31–40 | 67 (16.2) | 495 (8.7) | 721 (12.3) |
| 41–60 | 22 (5.3) | 674 (11.9) | 221 (3.8) |
| 61+ | 2 (0.5) | 25 (0.4) | 31 (0.53) |
| Smoking duration in current smokers, y | |||
| <40 | 67 (16.2) | 1453 (25.6) | 1788 (30.6) |
| 40–44 | 126 (30.4) | 2461 (43.4) | 1903 (32.6) |
| 45–49 | 124 (30.0) | 1124 (19.8) | 1271 (21.8) |
| 50+ | 97 (23.4) | 627 (11.1) | 881 (15.1) |
| Years since quit in former smokers, y | |||
| <10 | 284 (33.1) | 4718 (18.6) | 5417 (21.6) |
| 10–19.9 | 233 (27.2) | 6603 (26.0) | 6480 (25.9) |
| 20–29.9 | 178 (20.8) | 7243 (28.5) | 6453 (25.8) |
| 30–39.9 | 127 (14.8) | 5193 (20.5) | 5223 (20.9) |
| 40+ | 35 (4.1) | 1625 (6.4) | 1459 (5.8) |
| Original case-control study | |||
| Lung cancer study | 998 (54.9) | 24410 (41.9) | --- |
| NHL study | 572 (31.5) | 24410 (41.9) | --- |
| Ovarian cancer study | 249 (13.7) | 9444 (16.2) | --- |
* Weighted estimates were calculated using the combined lung, non-Hodgkin’s lymphoma, and ovary weight. BMI = body mass index; NHL = non-Hodgkin’s lymphoma.
Compared with never smokers, nine of 78 markers were statistically significantly different among current smokers after a 5% FDR criterion was applied to the P values (Table 2; 19 markers had a P value less than .05; see Supplementary Table 3, available online, for all markers). In addition, we considered CRP to be statistically significant, as it had a small P value (P = .007), and there is strong evidence for its association with cigarette smoking from prior studies. Study-specific odds ratios (ORs) for these markers were similar to those from our pooled weighted analysis (Supplementary Table 4, available online). These 10 markers included proteins involved in chemotaxis of T-cells ([C-C motif] ligand CCL 17/thymus and activation regulated chemokine [CCL17/TARC]), recruitment of eosinophils and allergic inflammation ([CCL11]/EOTAXIN), chemotaxis and T-cell activation (interleukin [IL]-16), increased inflammation (IL-1β and CRP), anti-inflammatory response (IL-1Ra), T-cell and natural killer–cell regulation (IL-15), regulators of cell growth and differentiation (soluble interleukin 6 receptor [sIL-6R]), hematopoiesis (stem cell factor [SCF]), and angiogenesis (soluble vascular endothelial growth factor receptor 3 [sVEGFR3]). For a majority of these markers, all of which were either cytokines or soluble receptors (IL-15, IL-1Ra, IL-1β, IL-16, SCF, sIL-6R and sVEGFR3), levels were lower among current smokers when compared with never smokers, with odds ratios ranging from 0.44 to 0.27. In contrast, current smokers had elevated levels of the chemokines CCL17/TARC (OR = 4.08, 95% CI = 2.01 to 8.25) and CCL11/EOTAXIN (OR = 2.57, 95% CI = 1.45 to 4.55), and the acute phase protein CRP (OR = 2.54, 95% CI = 1.29 to 4.98). These 10 markers were not highly correlated, with the exception of IL-1Ra and IL-15 (r = 0.54, P < .0001), IL-15 and IL-1B (r = 0.61, P < .0001), and IL-1Ra and IL-1B (r = 0.94, P < .0001) (Supplementary Table 5, available online).
Table 2.
Statistically significant associations between current smoking status and circulating inflammation markers
| Marker | Never | Former | Current | P* |
|---|---|---|---|---|
| (n = 548; weighted N = 27219) | (n = 857; weighted N = 25381) | (n = 414; weighted N = 5664) | ||
| OR (95% CI) | OR (95% CI) | |||
| Acute phase protein | ||||
| CRP | 1.0 | 1.29 (0.72 to 2.31) | 2.54 1.29 to 4.98 | .007 |
| Chemokines | ||||
| CCL17/TARC | 1.0 | 1.14 (0.72 to 1.79) | 4.08 (2.01 to 8.25) | <.001 |
| CCL11/EOTAXIN | 1.0 | 1.34 (0.90 to 2.01) | 2.57 (1.45 to 4.55) | .001 |
| Cytokines | ||||
| IL-15† | 1.0 | 0.78 (0.47 to 1.28) | 0.27 (0.13 to 0.59) | <.001 |
| IL-1RA† | 1.0 | 0.57 (0.36 to 0.89) | 0.29 (0.14 to 0.61) | .001 |
| IL-1Β | 1.0 | 0.76 (0.49 to 1.18) | 0.37 (0.19 to 0.74) | .005 |
| IL-16 | 1.0 | 0.53 (0.34 to 0.83) | 0.31 (0.17 to 0.55) | <.001 |
| SCF | 1.0 | 0.62 (0.38 to 0.99) | 0.37 (0.21 to 0.66) | <.001 |
| Soluble receptors | ||||
| sIL-6R | 1.0 | 0.53 (0.36 to 0.79) | 0.43 (0.24 to 0.77) | .005 |
| sVEGFR3 | 1.0 | 0.74 (0.50 to 1.10) | 0.44 (0.25 to 0.76) | .003 |
* Estimated with weighted logistic regression. All models adjusted for age, sex, history of chronic bronchitis or emphysema, history of coronary heart disease or heart attack, body mass index, case-control study of origin, and year of serum collection. P value comparing odds of above median marker level in current vs never smokers was estimated with a two-sided Wald Test. CCL = C-C motif ligand; TARC = thymus and activation regulated chemokine; CI = confidence interval; CRP = C-reactive protein; IL = interleukin; OR = odds ratio; SCF = stem cell factor; sIL-6R = soluble interleukin 6 receptor; sVEGFR3 = soluble vascular endothelial growth factor receptor 3.
† Categorized as detectable/undetectable.
In sensitivity analyses 1) restricting the analysis to controls only (Supplementary Table 6, available online) and 2) restricting the analysis to subjects from the lung and NHL studies only (where the collection of smoking information and serums was contemporaneous, data not shown), similar associations with current smoking were observed. Additionally, when we excluded markers with ICCs under 0.80, the same markers were identified as meeting the FDR criterion for statistical significance, with the exception of IL-1Ra, which was excluded because of an ICC under 0.8 in the ovary study alone (data not shown).
Notably, smoking intensity (Table 3) and pack-years smoked (data not shown) were not associated with marker levels among current smokers. Additionally, smoking duration was associated with increased levels of SCF (50+ vs <40 years: OR = 6.61, 95% CI = 1.00 to 4.39, P trend = .03) and sIL-6R (OR = 8.76, 95% CI = 1.30 to 58.9, P trend = .01) (Table 4), despite having lower levels than never smokers.
Table 3.
Associations between smoking intensity and markers levels among current smokers
| Markers | Cigarettes per day | P trend* | |||
|---|---|---|---|---|---|
| 1–10 | 11–20 | 21–30 | 31 | ||
| (n = 49; weighted N = 836) | (n = 165; weighted N = 1875) | (n = 109; weighted N = 1760) | (n = 91; weighted N = 894) | ||
| OR (95% CI) | OR (95% CI) | OR (95% CI) | |||
| Acute phase protein | |||||
| CRP | 1.0 | 0.91 (0.27 to 3.13) | 1.00 (0.28 to 3.58) | 2.76 (0.69 to 11.0) | .16 |
| Chemokines | |||||
| CCL17/TARC | 1.0 | 1.29 (0.35 to 4.72) | 0.58 (0.14 to 2.51) | 2.24 (0.52 to 9.64) | .57 |
| CCL11/ EOTAXIN | 1.0 | 1.27 (0.35 to 4.65) | 2.40 (0.57 to 10.1) | 1.69 (0.31 to 9.17) | .38 |
| Cytokines | |||||
| IL-15† | 1.0 | 0.20 (0.04 to 1.02) | 0.08 (0.01 to 0.91) | 1.33 (0.29 to 6.08) | .33 |
| IL-1RA† | 1.0 | 0.22 (0.05 to 1.06) | 0.10 (0.02 to 0.59) | 0.72 (0.17 to 3.12) | .82 |
| IL-1Β | 1.0 | 0.24 (0.06 to 0.99) | 0.11 (0.02 to 0.61) | 0.76 (0.19 to 3.06) | .87 |
| IL-16 | 1.0 | 1.97 (0.55 to 7.11) | 1.18 (0.33 to 4.27) | 1.25 (0.31 to 5.12) | .84 |
| SCF | 1.0 | 1.97 (0.59 to 6.60) | 1.49 (0.47 to 4.77) | 0.95 (0.28 to 3.21) | .57 |
| Soluble receptors | |||||
| sIL-6R | 1.0 | 0.19 (0.05 to 0.67) | 0.33 (0.09 to 1.19) | 1.17 (0.31 to 4.41) | .36 |
| sVEGFR3 | 1.0 | 0.99 (0.27 to 3.55) | 0.74 (0.17 to 3.14) | 2.25 (0.47 to 10.9) | .34 |
* Estimated with weighted logistic regression. All models adjusted for age, sex, history of chronic bronchitis or emphysema, history of coronary heart disease or heart attack, body mass index, case-control study of origin, and year of serum collection. P trend calculated by including categories of cigarettes per day in the model as a continuous variable with a two-sided Wald Test. CCL = C-C motif ligand; TARC = thymus and activation regulated chemokine; CI = confidence interval; CRP = C-reactive protein; IL = interleukin; OR = odds ratio; SCF = stem cell factor; sIL-6R = soluble interleukin 6 receptor; sVEGFR3 = soluble vascular endothelial growth factor receptor 3.
† Categorized as detectable/undetectable.
Table 4.
Associations between smoking duration and markers levels among current smokers
| Markers | Smoking duration, y | P trend* | |||
|---|---|---|---|---|---|
| ≤40 | 40–44 | 45–49 | 50 | ||
| (n = 67; weighted N = 1453) | n = 126; weighted N = 2461) | (n = 124; weighted N = 1124) | (n = 97; weighted N = 627) | ||
| OR (95% CI) | OR (95% CI) | OR (95% CI) | |||
| Acute phase Protein | |||||
| CRP | 1.0 | 0.43 (0.14 to 1.36) | 0.28 (0.08 to 1.00) | 0.42 (0.07 to 2.67) | .15 |
| Chemokines | |||||
| CCL17/TARC | 1.0 | 3.22 (0.73 to 13.2) | 3.10 (0.73 to 13.2) | 5.01 (0.93 to 26.9) | .09 |
| CCL11/ EOTAXIN | 1.0 | 1.05 (0.29 to 3.86) | 0.74 (0.16 to 3.35) | 2.56 (0.27 to 24.1) | .69 |
| Cytokines | |||||
| IL-15† | 1.0 | 1.12 (0.14 to 8.92) | 1.55 (0.15 to 16.1) | 3.22 (0.14 to 75.4) | .61 |
| IL-1RA† | 1.0 | 0.64 (0.10 to 4.20) | 1.31 (0.18 to 9.54) | 0.87 (0.05 to 14.2) | 1.00 |
| IL-1Β | 1.0 | 0.51 (0.09 to 2.89) | 0.94 (0.16 to 5.67) | 1.37 (0.11 to 16.6) | .94 |
| IL-16 | 1.0 | 0.53 (0.14 to 1.96) | 0.36 (0.08 to 1.71) | 0.56 (0.08 to 4.01) | .37 |
| SCF | 1.0 | 1.88 (0.67 to 5.29) | 3.46 (0.86 to 14.0) | 6.61 (1.00 to 43.9) | .03 |
| Soluble receptors | |||||
| sIL-6R | 1.0 | 4.64 (1.22 to 17.6) | 5.20 (0.98 to 27.7) | 8.76 (1.30 to 58.9) | .01 |
| sVEGFR3 | 1.0 | 2.82 (0.91 to 8.74) | 3.48 (0.86 to 14.0) | 1.61 (0.21 to 12.5) | .39 |
* Estimated with weighted logistic regression. All models adjusted for age, sex, history of chronic bronchitis or emphysema, history of coronary heart disease or heart attack, body mass index, case-control study of origin, year of serum collection. P trend calculated by including categories of years smoked in the model as a continuous variable with a two-sided Wald Test. CCL = C-C motif ligand; TARC = thymus and activation regulated chemokine; CI = confidence interval; CRP = C-reactive protein; IL = interleukin; OR = odds ratio; SCF = stem cell factor; sIL-6R = soluble interleukin 6 receptor; sVEGFR3 = soluble vascular endothelial growth factor receptor 3.
† Categorized as detectable/undetectable.
With the exception of sIL-6R, levels of each of the remaining nine markers differed between former and current smokers (Supplementary Table 7, available online). As former smokers represent a heterogeneous group with a varying number of years since smoking cessation, we further investigated the association between time since quitting and marker levels. Among former smokers, a trend was observed between years since quitting (0 years [ie, current smokers], ≤5, 5.1–10, 10.1–20, 20+ years, P trend < .05) and marker levels for CCL17/TARC, CCL11/EOTAXIN, IL-15, IL-1β, IL-1Ra, and CRP (Figure 1A). No statistically significant trend was observed between years since quitting and sVEGFR3, IL-16, sIL-6R, and SCF (Figure 1B), and three of these markers (ie, IL-16, SCF, and sIL-6R) were statistically significantly lower among former smokers compared with never smokers.
Figure 1.
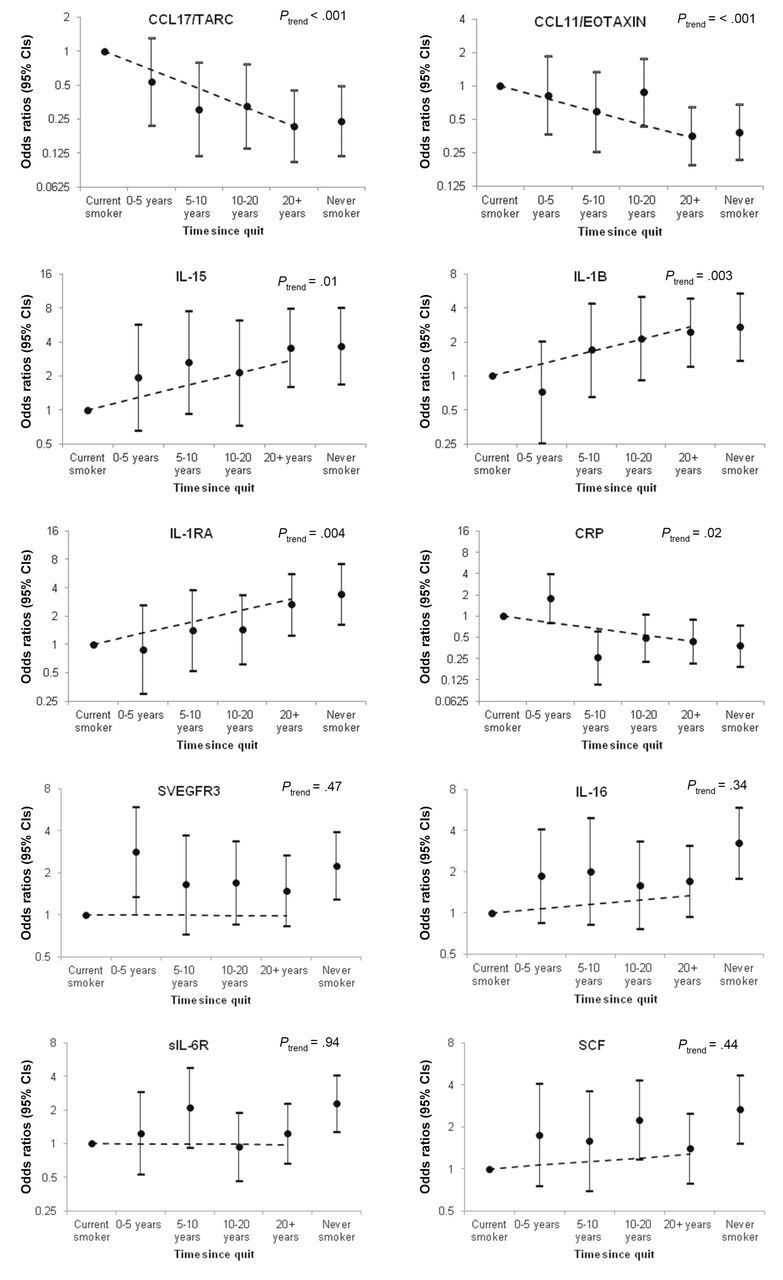
Association between years since smoking cessation and inflammation marker levels among former smokers. Marker levels were dichotomized at the median or at the lowest limit of detection. Points represent odds ratios, solid lines represent 95% confidence intervals, and dashed lines represent the trend across categories. A) includes markers with statistically significant trends across categories of time since quitting (CCL17/TARC, CCL11/EOTAXIN, IL-15, IL-1β, IL-1RA, and CRP). B) includes markers without statistically significant trends across categories of time since quitting sVEGFR3, IL-16, sIL-6R, and SCF). *P trend calculated by including categories of years since quitting in the model as a continuous variable with a two-sided Wald Test.
Discussion
In a broad investigation of the effects of cigarette smoking on systemic immune and inflammation marker levels in 55- to 74-year-old men and women, we found substantial differences in several immune markers between current and never smokers. These markers mediate various mechanisms of the immune/inflammation response, such as chemotaxis of T-cells, eosinophils, and other cells, inflammation, anti-inflammation processes, as well as markers involved in cell development/differentiation, cell growth and activation, angiogenesis, and hematopoiesis. These observations provide evidence that smoking may have a wide range of effects on systemic immunity and inflammation. Importantly, for many of these markers, levels in former smokers approached those of never smokers over time, suggesting that cessation may result in a reversal of smoking-associated alterations in immunity and inflammation.
Cigarette smoking is known to cause several pulmonary and systemic immune alterations pertaining to both the number of immune cells, such as increases in macrophages, neutrophils, eosinophils, and mast cells and functionality of various immune cells (8). For example, among smokers, polymorphonuclear neutrophils express depressed migration and chemotaxis, alveolar macrophages are less mature and are hyperdense with condensed cytoplasms and elevated expression of the monocyte marker, CD14, and, in dendritic cells, endocytic and phagocytic activities and the stimulation of T-cell responses are reduced (8). Consistent with observations of broad alterations in immunity, we found that current smoking was associated with differences in systemic levels of 10 immune/inflammation markers (CCL17/TARC, CCL11/EOTAXIN, IL-15, IL-1B, IL-1Ra, CRP, SVEGFR3, IL-16, sIL-6R, and SCF), which are produced by a range of cell types, including innate immune cells, adaptive immune cells, endothelial cells, epithelial cells, stromal cells, and fibroblasts and represent several components of the immune/inflammation response (25). CCL17/TARC, IL-1Ra, and CRP have each been previously associated with lung cancer (19); thus, in addition to being associated with cigarette smoking, some of these systemic alterations may additionally be relevant for disease risk.
Our results suggest that one effect of current smoking may be the suppression of systemic immune marker levels. Current smoking was associated with reduced systemic levels of seven immune/inflammation markers (IL-1β, IL-1Ra, sIL-6R, IL-15, IL16, SVEGFR3, and SCF) when compared with levels among never smokers. Of note, markers with lower systemic levels among current smokers included both proinflammatory (IL-1β) and anti-inflammatory cytokines (IL-1Ra). Our results are consistent with studies showing associations between cigarette smoking and increased levels of CRP in epidemiologic studies and decreased levels of IL-1β in vitro and in animal models (12,13,26,27). However, prior literature regarding the effect of cigarette smoking on local (ie, in the lung evaluated through bronchioloalveolar lavages) and systemic immune responses has been inconsistent, with reports of both increased as well as decreased levels of cytokines (8). Nevertheless, our results may reflect an immunosuppressive effect of cigarette smoking on a number of important cytokines in inflammation and immunity, consistent with an overall immune suppressive effect of nicotine (9).
We found no association between intensity of smoking (ie, cigarettes smoked per day) and alterations in systemic immune/inflammation marker levels among current smokers. Although surprising to us, these results suggest a low threshold (ie, one to 10 cigarettes per day) for the effect of smoking on circulating immune markers. Thus, even a low degree of exposure to cigarette smoke may alter systemic levels of immune/inflammation markers. This is consistent with a study showing a single cigarette decreased IL-1β production by greater than 90% in peripheral blood mononuclear cells (26). It is possible that a potential dose-response relationship with smoking intensity, particularly at low levels of exposure, was masked in our study, given that the lowest category of information on cigarettes smoked per day was one to 10 cigarettes, and detailed information on intermittent smoking was not available.
Additionally, associations with smoking duration were only observed for SCF and sIL-6R. However, we note that the range of smoking duration in our study was quite narrow (interquartile range = 38.6–46.1 years), given the older age of the PLCO population and the relatively uniform age at smoking initiation (median = 16.9 years, interquartile range = 14.5–19.0 years). Therefore, our results reflect years of chronic long-term exposure, and we cannot entirely exclude dose-response relationships of duration of smoking with systemic immune/inflammation marker levels.
Our results suggest substantial differences in circulating immune markers with smoking cessation, as most of the markers differed between current and former smokers. Among former smokers, levels of CRP, chemokines (CCL17/TARC, CCL11/EOTAXIN), and most cytokines (IL-15, IL-1β, IL-1RA) approached levels observed among never smokers with increasing time since smoking cessation. Therefore, active smoking may induce changes in these markers that gradually revert back to “normal” once exposure to cigarette smoking is removed. In contrast, levels of sVEGFR3, IL-16, SCF, and sIL-6R were not related to time since quit and were statistically significantly different among former smokers compared with never smokers. Alterations in these markers may occur shortly after smoking cessation, remaining at an intermediate level between never and current smokers for many years. CRP levels were substantially increased among the most recent quitters, before declining toward levels observed in never smokers among those with at least five years since cessation. This unique pattern may reflect an increased prevalence of cardiovascular disease and other inflammatory conditions among recent quitters. Additional longitudinal studies that assess changes over time are needed to further understand the trajectories of marker levels following smoking cessation.
The main strengths of our study include the comprehensive investigation of the effects of tobacco smoking on alterations in circulating immune and inflammation markers measured using a well-characterized technology and a novel two-stage design to reweight analyses to the population-based PLCO screening arm cohort.
We also note our study’s limitations. We could not measure all possible markers of systemic immunity and inflammation, nor could we measure alterations occurring locally in the lung or other organs. Importantly, our analyses were cross-sectional, thus we were unable to examine changes in marker levels prospectively with changes in smoking behaviors. Our measures of smoking exposure relied on self-report and did not include information on nicotine dependency or the recency of smoking. Importantly, our study was limited to older individuals with long-term histories of cigarette smoking; and, thus, may not be generalizable to younger populations and more recent initiators. Our results need replication in other cohorts and prospective settings. Finally, residual confounding is a possibility, given that we were only able to adjust for those factors and conditions where information was collected in PLCO.
In conclusion, our results show that current smokers have broadly different levels of systemic immune/inflammation marker levels, and that, while some of these marker levels were also different among former smokers, many seemed to approximate those of never smokers after years of cessation. Our results have important research implications. Systemic alterations in the levels and profile of soluble immune markers of inflammation may reflect the overall immune/inflammatory cancer-promoting microenvironment, and may inform possible etiologic mechanisms involved in smoking-induced chronic diseases. Future studies that incorporate both smoking behaviors and smoking-related immune and inflammatory markers can provide insight into the etiologic mechanisms of tobacco-related diseases and potentially aid in the identification of high-risk individuals for preventive interventions.
Funding
This work was supported by the Intramural Research Program of the National Cancer Institute.
The authors acknowledge Mr. Craig Williams and Mr. Michael Curry of Information Management Services, Inc., who were compensated for statistical programming.
The sponsor reviewed and approved final submission, but did not have a role in design and conduct of the study, the collection, management, analysis, or interpretation of the data, preparation of the manuscript, nor the decision to submit for publication.
The information in this article is not a formal dissemination of information by the FDA and does not represent agency position or policy. The contents are the responsibility of the authors alone.
References
- 1. Smoking-attributable mortality, years of potential life lost, and productivity losses--United States, 2000–2004. MMWR Morb Mortal Wkly Rep. 2008;57(45):1226–1228 [PubMed] [Google Scholar]
- 2. American Cancer Society. Smoking Cancer Mortality Table. September 18, 2009.
- 3. Centers for Disease Control and Prevention. Smoking & Tobacco Use: Health Effects of Cigarette Smoking. August 1, 2013. Atlanta, GA, Centers for Disease Control and Prevention [Google Scholar]
- 4. Secretan B, Straif K, Baan R, et al. A review of human carcinogens--Part E: tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 2009;10(11):1033–1034 [DOI] [PubMed] [Google Scholar]
- 5. U.S.Department of Health and Human Services. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease. A Report of the Surgeon General. 2010. Atlanta, GA. [PubMed]
- 6. Mehta H, Nazzal K, Sadikot RT. Cigarette smoking and innate immunity. Inflamm Res. 2008;57(11):497–503 [DOI] [PubMed] [Google Scholar]
- 7. Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2(5):372–327 [DOI] [PubMed] [Google Scholar]
- 8. Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34(3):J258–J265 [DOI] [PubMed] [Google Scholar]
- 9. Cui WY, Li MD. Nicotinic modulation of innate immune pathways via alpha7 nicotinic acetylcholine receptor. J Neuroimmune Pharmacol. 2010;5(4):479–488 [DOI] [PubMed] [Google Scholar]
- 10. Wannamethee SG, Lowe GD, Shaper AG, et al. Associations between cigarette smoking, pipe/cigar smoking, and smoking cessation, and haemostatic and inflammatory markers for cardiovascular disease. Eur Heart J. 2005;26(17):1765–1773 [DOI] [PubMed] [Google Scholar]
- 11. O’Connor MF, Irwin MR. Links between behavioral factors and inflammation. Clin Pharmacol Ther. 2010;87(4):479–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frost-Pineda K, Liang Q, Liu J, et al. Biomarkers of potential harm among adult smokers and nonsmokers in the total exposure study. Nicotine Tob Res. 2011;13(3):182–193 [DOI] [PubMed] [Google Scholar]
- 13. Liu J, Liang Q, Frost-Pineda K, et al. Relationship between biomarkers of cigarette smoke exposure and biomarkers of inflammation, oxidative stress, and platelet activation in adult cigarette smokers. Cancer Epidemiol Biomarkers Prev. 2011;20(8):1760–1769 [DOI] [PubMed] [Google Scholar]
- 14. Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation between markers of systemic vascular inflammation and smoking in women. Am J Cardiol. 2002;89(9):1117–1119 [DOI] [PubMed] [Google Scholar]
- 15. Helmersson J, Larsson A, Vessby B, Basu S. Active smoking and a history of smoking are associated with enhanced prostaglandin F(2alpha), interleukin-6 and F2-isoprostane formation in elderly men. Atherosclerosis. 2005;181(1):201–207 [DOI] [PubMed] [Google Scholar]
- 16. Prorok PC, Andriole GL, Bresalier RS, et al. Design of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials. 2000;21(6 Suppl):273S–309S [DOI] [PubMed] [Google Scholar]
- 17. Oken MM, Hocking WG, Kvale PA, et al. Screening by chest radiograph and lung cancer mortality: the Prostate, Lung, Colorectal, and Ovarian (PLCO) randomized trial. JAMA. 2011;306(17):1865–1873 [DOI] [PubMed] [Google Scholar]
- 18. Purdue MP, Hofmann JN, Kemp TJ, et al. A prospective study of 67 serum immune & inflammation markers and risk of non-Hodgkin lymphoma. Blood. 2013;122(6):951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shiels MS, Pfeiffer RM, Hildesheim A, et al. Circulating inflammation markers and prospecitve risk of lung cancer. J Natl Cancer Inst. 2013;105(24):1871–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trabert B, Pinto L, Hartge P, et al. Pre-diagnostic serum levels of inflammation markers and risk of ovarian cancer in the Prostate, Lung, Colorectal and Ovarian Cancer (PLCO) Screening Trial. Gynecol Oncol. 2014; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chaturvedi AK, Kemp TJ, Pfeiffer RM, et al. Evaluation of multiplexed cytokine and inflammation marker measurements: a methodologic study. Cancer Epidemiol Biomarkers Prev. 2011;20(9):1902–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Austin PC. An Introduction to Propensity Score Methods for Reducing the Effects of Confounding in Observational Studies. Multivariate Behav Res. 2011;46(3):399–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Korn EL, Graubard BI. Analysis of Health Surveys. 1 ed. New York, NY: John Wiley & Sons; 1999 [Google Scholar]
- 24. Stoer NC, Samuelsen SO. Inverse probability weighting in nested case-control studies with additional matching--a simulation study. Stat Med. 2013;32(30):5328–5339 [DOI] [PubMed] [Google Scholar]
- 25. Murphy K. Janeway’s Immunobiology. 8th ed. New York, NY: Garland Science; 2012 [Google Scholar]
- 26. Ouyang Y, Virasch N, Hao P, et al. Suppression of human IL-1beta, IL-2, IFN-gamma, and TNF-alpha production by cigarette smoke extracts. J Allergy Clin Immunol. 2000;106(2):280–287 [DOI] [PubMed] [Google Scholar]
- 27. Kim TH, Kim SJ, Lee SM. Stimulation of the alpha7 Nicotinic Acetylcholine Receptor Protects Against Sepsis by Inhibiting Toll-like Receptor via Phosphoinositide 3-Kinase Activation. J Infect Dis. 2014;209(10):1668–1677 [DOI] [PubMed] [Google Scholar]