

Abstract
Hydrogen sulphide (H2S), a chemical hazard in oil and gas production, has recently become a dreadful method of suicide, posing specific risks and challenges for the first responders. Currently, there is no proven effective treatment against H2S poisoning and its severe neurological, respiratory or cardiac after-effects. We have recently described that H2S is present in various compartments, or pools, in the body during sulphide exposure, which have different levels of toxicity. The general goals of our study were to (1) determine the concentrations and kinetics of the various pools of hydrogen sulphide in the blood, i.e., gaseous (CgH2S) versus total sulphide, i.e., reacting with monobromobimane (CMBBH2S), during and following H2S exposure in a small and large mammal and (2) establish the interaction between the pools of H2S and a methemoglobin (MetHb) solution or a high dose of hydroxocobalamin (HyCo). We found that CgH2S during and following H2S infusion was similar in sedated sheep and rats at any given rate of infusion/kg and provoked symptoms, i.e., hyperpnea and apnea, at the same CgH2S. After H2S administration was stopped, CgH2S disappeared within 1 min. CMBBH2S also dropped to 2–3μM, but remained above baseline levels for at least 30 min. Infusion of a MetHb solution during H2S infusion produced an immediate reduction in the free/soluble pool of H2S only, whereas CMBBH2S increased by severalfold. HyCo (70 mg/kg) also decreased the concentrations of free/soluble H2S to almost zero; CgH2S returned to pre-HyCo levels within a maximum of 20 min, if H2S infusion is maintained. These results are discussed in the context of a relevant scenario, wherein antidotes can only be administered after H2S exposure.
Hydrogen sulphide (H2S) intoxication is one of the most common causes of death following gas exposure in the workplace (Beauchamp et al., 1984; Fuller and Suruda, 2000; Guidotti, 1996; Reiffenstein et al., 1992). Although these intoxications remain rare, H2S is extremely toxic at relatively low concentrations (Fuller and Suruda, 2000) leading to a fatal apnea and cardiac arrest within a few minutes at only 0.1% (Beauchamp et al., 1984; Guidotti, 1996; Reiffenstein et al., 1992), corresponding to concentrations in the blood of only 5–8 μM of gaseous sulphide (Klingerman et al., 2013). H2S remains one of the primary chemical hazards in oil and gas production, well drilling and gas refining industries (Arnold et al., 1985; Costigan, 2003; EPA, 2003). H2S has also been more recently used as a method of suicide first in Japan and now in the United States, by mixing in a closed environment—typically in a car—a source of sulfur and various types of acidic solutions readily available in most discount stores. This method of suicide has increased over the past few years, creating new risks not only for those intent on harming themselves, but also for first responders and neighbors (Reedy et al., 2011; Truscott, 2008).
The toxicity of H2S/HS− is to be accounted for by its various effects on cellular function, the most important being its impediment of ATP formation via the inhibition of cytochrome c oxidase (Cooper and Brown, 2008; Dorman et al., 2002). Neurons, unable to oxidize H2S (Lagoutte et al., 2010), are particularly vulnerable (Almeida and Guidotti, 1999; Almeida et al., 2008; Greer et al., 1995; Tvedt et al., 1991).
Various agents have been proposed to treat H2S poisoning, some with the theoretical purpose of “oxidizing” H2S (e.g., hyperoxia) (Bitterman et al., 1986; Haouzi et al., 2011a; Smilkstein et al., 1985; Smith et al., 1976), trapping it with metallo-compounds (e.g., ferric iron produced by nitrite-induced methemoglobinemia or cobalt in vitamin B12) (Haouzi et al., 2011b; Smith, 1969; Smith et al., 1976; Truong et al., 2007; Van de Louw and Haouzi, 2012b) or removing it from cysteine residues using reducing agents (Warenycia et al., 1990). Finally, sodium bicarbonate and glucose have been shown to exert protective effects, based on an interesting series of observations (Almeida and Guidotti, 1999; Guidotti, 2010). There is still no consensus on the strategy to be used. More importantly, as described in the following paragraph, H2S is present in various forms in the blood and tissues, i.e., diffusible versus combined, which have very different toxic effects. Little is known about the interactions of these antidotes with the various pools of H2S responsible for sulphide toxicity. Perhaps more importantly, the efficacy of any antidotes used against H2S poisoning is eventually dictated by the fate of these various pools of H2S in the blood and tissues “after” H2S exposure, i.e., when patients are withdrawn from the source of intoxication and taken care of by paramedics or later on in the emergency room.
We have previously used a simple model to describe the various compartments of H2S in the blood of the rat during exogenous H2S administration (Klingerman et al., 2013). Briefly, in the rat, H2S infusion at a rate 2 μmol/min produces no visible effects on blood pressure or breathing, whereas at 10 μmol/min a terminal apnea occurs in minutes. Within this range, exogenous H2S is present in the blood in two different forms, displaying an exponential relationship with the dose administered (Klingerman et al., 2013). These two distinct pools of sulphide can be described as (1) a pool of “free” or diffusible H2S that includes (a) H2S in gaseous form (CgH2S), whose partial pressure in the arterial blood is in equilibrium with the alveolar gas and with all tissues (Carroll and Mather, 1989), and (b) the sulfhydryl anion, HS− (Almgren et al., 1976; Millero, 1986), and (2) a pool of combined H2S primarily composed of sulphide fixed on the ferrous iron of hemoglobin (Hb) and proteins in the plasma (Haggard, 1921; Smith, 1967; Smith and Gosselin, 1966; Wintner et al., 2010). The pool of gaseous H2S in the arterial blood can be estimated based on H2S alveolar partial pressure (Klingerman et al., 2013) or via direct measurement by amperometry (Wintner et al., 2010), whereas the total amount of H2S in the blood could be approached by a more global method such as the derivation of monobromobimane (MBB) (Wintner et al., 2010). H2S, being extremely soluble, in its gaseous form (Barrett et al., 1988; Carroll and Mather, 1989; De Bruyn et al., 1995; Douabul and Riley, 1979) diffuses with virtually no delay between the blood and the tissues and within the cells. HS− and the pool of combined sulphide are present at much higher concentrations than the gaseous form in the blood. Within the cells, H2S/HS− combines with various metallo-proteins (e.g., the ferrous iron of Hb) or proteins (e.g., cysteine residues) (Mustafa et al., 2009; Warenycia et al., 1990). The majority of H2S oxidation takes place in the mitochondria (Bouillaud and Blachier, 2011; Lagoutte et al., 2010), where H2S also expresses its main toxicity by blocking the cytochrome c activity and thus ATP formation. The rate of elimination of H2S in vivo is not established as far as these various pools of H2S are concerned. Yet this question is of major importance as it represents a prerequisite for offering a strategy of treatment for H2S poisoning. This question also resonates with the increasing number of studies, which have been using IV or IP injections of H2S in various animal models to reduce the inflammatory cascade and the oxidative stress produced by a hypoxic, ischemic or infectious insult (see Olson, 2011b, Szabo, 2007, and Whiteman and Winyard, 2011 for review and discussion).
The present study intends to address the following questions: (1) what is the rate of disappearance of the various forms (free, combined) of sulphides, after exogenous H2S administration in the rat, in keeping with the level of H2S infused and is there evidence for persistence of H2S (and under which form) after the cessation of exposure/injection? (2) To which extent can the data obtained in the rat on the various pools of H2S and their kinetics be transposed to a larger mammal more closely resembling the size of a human being? (3) How two potential H2S antidotes, i.e., a methemoglobin (MetHb) solution (600–1200 mg/kg) or a high dose (70 mg/kg) of hydroxocobalamin (HyCo), affect the pools of H2S, and could these interactions be reconciled with their use during and, more importantly, “following” H2S exposure?
Our main objective was to establish a rational frame of reference, which can be used to understand the short- and long-term effect, or the lack thereof, of an acute exposure to H2S and to define if a window of opportunity exists during which MetHb and/or HyCo could be effective as H2S antidotes in keeping with sulphide kinetics.
MATERIALS AND METHODS
1. Rat Experiments
Materials and Methods
The experimental procedures were performed on adult, male, Sprague Dawley rats (616 ± 0.14 g, 45 ± 2 weeks old). All procedures received prior approval from the Pennsylvania State University College of Medicine Institutional Care and Use Committee. Anesthesia was induced with 3.5% isoflurane in O2 followed by an intra-peritoneal injection of 1.2 g/kg urethane as previously described (Haouzi and Van de Louw, 2013). Rats were placed on a heating pad and body temperature (BT) was monitored with a rectal temperature probe (Thermalert TH-5, Physitemp, NJ) and kept between 36–38°C. The rats were tracheostomized and a catheter (14 g, 2.25 mm OD) was placed in the trachea. Catheters (PE-50 tubing) were introduced into the left external jugular vein and the right femoral artery. The jugular vein was used for NaHS infusion. At the end of the experiment, rats were euthanized with CO2 followed by aortic dissection.
Measurements
The tracheal catheter was connected to a Hans Rudolph low dead space two-way valve as previously described (Klingerman et al., 2013). The inspiratory port of the valve was connected to a pneumotachograph (1100 Series, Hans Rudolph, KS) to measure inspiratory flow. The expiratory valve was connected to two 5 ml “mixing chambers” placed in series. The outlet of the second chamber was connected to a filter containing activated charcoal. Mixed expired CO2 and H2S fractions were measured continuously from the second mixing chamber using CO2 infrared (Vacumed model 17630, Ventura, CA; 0–10%) and H2S (Interscan RM series, Simi Valley, CA; 0.001–1 ppm) analyzers. The gas analyzers were calibrated immediately before use with different gas mixtures containing a known concentration of CO2 in air or H2S in N2.
An external source of air was introduced into the first mixing chamber and gas flow (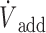 ) was continuously measured using a pneumotachograph (1100 Series). This approach has been described in detail elsewhere (Klingerman et al., 2013).
) was continuously measured using a pneumotachograph (1100 Series). This approach has been described in detail elsewhere (Klingerman et al., 2013).
Inspiratory flow, 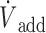 , arterial blood pressure (ABP), CO2 and H2S fractions, along with rectal temperature signals, were digitized at 200 Hz using an analog to digital data acquisition system (Powerlab 16/35, AD Instruments, Colorado Springs, CO). Data were displayed online and stored for later analysis. Breathing frequency (f) and tidal volume (VT) were determined using peak detection and integration of the inspiratory flow signal, respectively, and minute ventilation (
, arterial blood pressure (ABP), CO2 and H2S fractions, along with rectal temperature signals, were digitized at 200 Hz using an analog to digital data acquisition system (Powerlab 16/35, AD Instruments, Colorado Springs, CO). Data were displayed online and stored for later analysis. Breathing frequency (f) and tidal volume (VT) were determined using peak detection and integration of the inspiratory flow signal, respectively, and minute ventilation (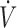 I) was computed as f × VT. The arterial line was connected to a pressure transducer (TA-100, CWE Inc., PA) to monitor ABP and was used for blood sampling.
I) was computed as f × VT. The arterial line was connected to a pressure transducer (TA-100, CWE Inc., PA) to monitor ABP and was used for blood sampling.
2. Sheep Experiments
Materials and Methods
Spontaneously breathing, sedated, adult Dorset sheep (Ovis aries, ewes, 1–2 years old) weighing 54 ± 13 kg were used in these experiments. The animals were pre-medicated with ketamine (20 mg/kg) IM and then anesthesia was induced with an IV loading dose of sodium thiopental (Pentothal 10 mg/kg). Anesthesia was subsequently maintained by an IV solution containing urethane (80 mg/kg) and alphachloralose (15 mg/kg), as previously described (Haouzi et al., 2011a). The infusion of urethane-chloralose was repeated every 2 h throughout the experiment.
The animals were tracheostomized and an inflatable-cuff tracheal cannula (Shilley No. 7) was inserted through the tracheostomy. Systemic ABP and heart rate were monitored by placing a catheter in one of the carotid arteries, which was also used for blood samples. Venous catheters (Plastimed 3F) were placed in one jugular and one femoral vein for injection of anesthetic agent or antidotes.
Measurements
Respiratory flow and the pulmonary gas exchange rate were recorded with a Hans-Rudolph pneumotachograph (Hans-Rudolph, Kansas City, MO) connected to the tracheostomy tube and to a differential pressure transducer. The expiratory valve was connected to a mixing chamber where the mixed expired O2 and CO2 fractions were measured continuously using O2 (Oxystar-100, CWE) and CO2 (model 17630, VacuMed, Ventura, CA) analyzers. H2S expired fractions were also measured continuously from the same mixing chamber (Interscan RM series, Simi Valley, CA; 1–200 ppm). The gas analyzers were calibrated immediately before use with different gas mixtures containing a known concentration of CO2 and O2 in air and H2S in N2. ABP and respiratory flow signals were digitized at 200 Hz (Power Lab system). The respiratory flow signal was integrated for breath-by-breath calculation of tidal volume and minute ventilation, oxygen consumption and CO2 production. At the end of the experiment, animals were euthanized by lethal injection of barbiturate (Pentothal 200 mg/kg) in the jugular catheter.
3. Determination of Dissolved Concentrations of H2S in the Arterial Blood
In the rat, expired H2S was determined as previously described (Klingerman et al., 2013). Briefly, the fraction of H2S was continuously measured from the second mixing chamber (FchH2S). Then, the mixed, expired fraction of H2S (FEH2S) was computed as FchH2S × (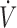 I +
I + 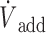 /
/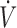 I). The partial pressure of expired H2S (PEH2S) was then calculated as FEH2S × (PB-PH2O), allowing the determination of the alveolar partial pressure of H2S (PAH2S) as PAH2S = PEH2S ×
I). The partial pressure of expired H2S (PEH2S) was then calculated as FEH2S × (PB-PH2O), allowing the determination of the alveolar partial pressure of H2S (PAH2S) as PAH2S = PEH2S × 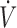 E/
E/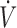 A, whereas
A, whereas 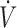 E/
E/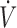 A was determined from the PECO2/PACO2 ratio.
A was determined from the PECO2/PACO2 ratio. 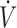 E was considered equal to
E was considered equal to 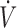 I, whereas PACO2 was estimated from arterial PCO2 (PaCO2) sampled at various times during the experiment, whereas PECO2 was continuously measured from the second mixing chamber. PaH2S was assimilated to PAH2S and the concentration of gaseous H2S in the blood was calculated as CgH2S = 0.00012 PaH2S, with 0.00012 being the coefficient of solubility of H2S (0.09 mole/L/760 mm Hg−1 at 37°C) (see Klingerman et al., 2013 for details and references). Following the assumption that H2S is under the form of H2S gas and HS− at a ratio of 30 and 70%, respectively, at a pH of 7 (Almgren et al., 1976.; Millero, 1986), the total concentration of dissolved H2S (CdH2S) was computed as 3 × CgH2S.
I, whereas PACO2 was estimated from arterial PCO2 (PaCO2) sampled at various times during the experiment, whereas PECO2 was continuously measured from the second mixing chamber. PaH2S was assimilated to PAH2S and the concentration of gaseous H2S in the blood was calculated as CgH2S = 0.00012 PaH2S, with 0.00012 being the coefficient of solubility of H2S (0.09 mole/L/760 mm Hg−1 at 37°C) (see Klingerman et al., 2013 for details and references). Following the assumption that H2S is under the form of H2S gas and HS− at a ratio of 30 and 70%, respectively, at a pH of 7 (Almgren et al., 1976.; Millero, 1986), the total concentration of dissolved H2S (CdH2S) was computed as 3 × CgH2S.
In the sheep, the same measurements were performed, but as no additional flow was used, FAH2S was simply determined as PAH2S = PEH2S × (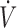 E/
E/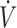 A).
A).
4. Measurement of H2S in the Blood Using Monobromobimane
Monobromobimane (MBB), 1,2-ethanedithiol, 4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES), solid phase extraction (SFE) columns (silica, 1 g/6 ml), and silica gel (200–400 mesh, 60 Å) were purchased from Sigma-Aldrich (St Louis, MO). Sodium sulphide was obtained from Alfa Aesar (Ward Hill, MA). Ethyl acetate and hydrochloric acid (0.1 N) were purchased from Fisher Scientific (Pittsburg, PA).
An extensive description of the preparation of the internal standard and various validations of this method has been presented in details elsewhere (Klingerman et al., 2013), following the method used by Wintner et al. (Wintner et al., 2010). Briefly, venous blood (200 μl) was added to a solution of MBB (20mM in 200 μl of acetonitrile) and 200 μl HEPES (50mM, pH 8.0) in a sealed vial. Samples were analyzed using a HPLC system (Shimadzu) with a Phenomenex (Torrance, CA) C-18 Bondclone (4.6 × 300 mm, 10 μm) column. Under these chromatographic conditions, sulphide-bimane eluted at 19.4 min. The levels of sulphide-bimane in rat blood were determined based on standard curves constructed for these analyses.
As a control, 200 μl of a MetHb solution (600 mg/ml saline) was reacted with MBB as described for the blood samples. HPLC analysis of this sample showed no interference with the detection of the sulphide-bimane. The components of H2S measured by these methods includes both the dissolved form of H2S and the part combined to the red cell components of the blood and plasma proteins (Klingerman et al., 2013; Olson, 2011a; Wintner et al., 2010).
5. Methemoglobin Concentration in the Blood
Arterial gas partial pressure in O2 and CO2 and MetHb concentrations were determined using a GEM Premier 4000 gas analyzer (Instrumentation Laboratory, Bedford, MA). The accuracy of the measurement of MetHb was previously validated using a spectrophotometric method in duplicate (Rahman et al., 2009). This technique was also used to determine the concentration of MetHb in the Hb solution, which was found to be about 98% (Van de Louw and Haouzi, 2012).
6. Protocol
Rat Experiments
NaHS infusion and recovery (12 rats): First, NaHS (sodium hydrosulphide hydrate, Sigma Aldrich, St Louis, MO) was dissolved in sterile saline at a concentration of 0.8 mg/ml, without exposure to air, assuming an equimolar conversion of NaHS into H2S/HS- (see last paragraph of method section for further details) after taking into account 25% hydration of NaHS. H2S was infused into the jugular vein using an infusion pump (Chemyx Inc., Model Fusion 100, Stafford, TX) at a rate ranging from 2.8 to 10.2 μmol/min corresponding to concentrations that ranged from levels below the threshold for ventilatory stimulation to levels producing apnea. These doses were chosen based on our previously published data (Klingerman et al., 2013). For the rates above 8 μmol/min, which produce an apnea after 2–3 min, all animals were ventilated and the circuit was modified to be able to collect the expiratory gases. CMBBH2S was measured before H2S infusion, at the end of infusion, then at 1, 3, 5, 10, 15 and 30 min following the cessation of exposure.
Methemoglobin infusion (10 rats): A MetHb solution of 600 mg of human-derived Hb (Sigma Aldrich) in sterile saline (200 mg/ml) was infused slowly over about 1 min (30–90 s) into the femoral artery. It was determined in pilot experiments that the inhibitory effect of MetHb infusion on H2S-induced breathing simulation was similar whether it was administered into the femoral vein or artery. For this experiment, we chose to infuse into the artery to prevent a direct mixture with NaHS in the venous system. This dose was calculated based on an average endogenous Hb concentration (130 g/l) and blood volume (80 ml/kg) to reach a level of MetHb around 8%. This had been previously confirmed by pilot experiments in two rats, which were not included in the present data analysis. This protocol was performed in 10 rats. In three of these rats, CgH2S, CdH2S, and CMBBH2S were also measured 5, 10, and 15 min following the cessation of NaHS infusion.
Effect of withdrawal and infusion of whole blood versus methemoglobin solution on H2S concentration (3 rats): Because infusion of several milliliters of MetHb over 30–90 s alters the volemia and therefore could affect the rate of interaction between H2S and Hb, we sought to delineate the effects of changing blood volume (or Hb) per se. CgH2S was determined in four additional rats during (1) withdrawing and then injecting a 2 ml volume of arterial blood and (2) the injection of 2 ml of MetHb solution (150 mg/ml), corresponding to the baseline Hb concentration. We withdrew and then injected arterial blood twice before MetHb infusion and then two more times after.
Sheep Experiments
NaHS infusion and recovery (5 sheep): To limit the number of animals used, H2S was infused at five different levels in a ramp-like manner, following the same approach used previously in the rat (Klingerman et al., 2013). H2S was prepared a few minutes before the infusion and kept in airtight syringes. The solution was infused intravenously into the femoral vein using an infusion pump (Fusion 100; Chemyx Inc, Stafford, TX). The flow rate was increased from 135 to 665 μmol/min every 5 min by steps of 135 μmol/min, until an apnea occurred. This protocol was chosen based on our previous data and a series of pilot experiments, to cover a range of H2S exposure from no obvious symptoms to apnea, similar to the current experiments in the rat. H2S administration was stopped as soon as breathing ceased, and then the animals were mechanically ventilated (see the Results and Discussion sections for further details). The expiratory circuit allowed us to measure FEH2S continuously during and following exposure, whether during spontaneous or mechanical ventilation. During mechanical ventilation, the time constant of the changes in FEH2S was longer than during spontaneous breathing by 20 s, which was taken into account for the determination of FAH2S. Blood was sampled at the third minute into each infusion rate and then at 5, 10, and 15 min into recovery.
Methemoglobin and hydroxocobalamin infusion in the sheep: All of the sheep recovered from the apneic period, and minute ventilation was back to baseline within 10–15 min. Then a new infusion of a sub-lethal level of H2S (270–405 μmol/min) was initiated into the femoral vein for 3 min. Next, a solution of HyCo (Cyanokit 70 mg/kg) was infused slowly over about 2–3 min in a jugular vein and the level of FAH2S was monitored up to 25 min while H2S infusion was maintained.
Fifty to sixty minutes after the end of H2S infusion, when the ventilatory and circulatory parameters were back to baseline (including expired H2S, see the Result section), a second infusion of a sub-lethal level of H2S at the same level was resumed and MetHb solution was injected. Similar to the rat, the dose of MetHb was computed based on an average endogenous Hb concentration (130 g/l) and blood volume (80 ml/kg) to reach a level of MetHb around 8%. Then H2S was determined again following at the end of H2S infusion at 5, 10 and 15 min.
Injections were performed in the same animal, to minimize the number of sheep studied, assuming that all H2S in free forms had disappeared from the blood before a new trial (see the Discussion section for more details).
7. Data Analysis
All data are expressed as mean ± SD. The relationship between alveolar H2S (CgH2S) and total H2S in the blood (CMBBH2S) was established in keeping with the dose infused and the level of minute ventilation. Regression analyses using a linear, exponential, or power function were used to describe the relationship between CH2S and the rate of H2S infusion in both species and the best coefficients of regression are reported.
The concentration of H2S at which breathing was increased by 30% was chosen as the threshold for carotid body (CB) stimulation (H2S-induced respiratory symptoms). CgH2S as well as CMBBH2S at the different rates of infusion were compared using one-way analysis of variance (ANOVA); p < 0.05 was considered as significant. Kinetics of recovery of CgH2S and CMBBH2S were described as follows: the time constant of the response was established as the time required for CgH2S to drop by 64% after subtracting the time constant of the analyzer and the circuit. CgH2S and CMBBH2S during recovery were compared using Friedman Repeated Measures Analysis of Variance, followed by a post-hoc, pairwise, multiple comparison procedure (Bonferroni).
A paired t-test was used to compare CgH2S, CdH2S, and CMBBH2S before MetHb infusion and then 3 min after MetHb administration during NaHS infusion. A paired t-test was also used to compare the change in MetHb levels before and 3 min after MetHb infusion. The effects of hydroxocobalamin before, 3 min and 20 min after the end of injection were compared using a one-way ANOVA with repeated measurements. The effects of blood pressure on the H2S concentrations were compared by Friedman Repeated Measures Analysis of Variance, followed by a post-hoc, pairwise, multiple comparison procedure (Bonferroni). p < 0.05 was considered as significant.
8. Symbols and Conventions Used to Describe the Various Pools of Sulphide in the Blood and in Solution
H2S can refer to various and very different pools of sulphide: (1) its gaseous form, (2) H2S in solution (dissolved H2S), which at pH 7 comprises 70–80% HS− and 20–30% gaseous H2S, and (3) H2S present in combined forms (metallo-proteins or on cysteine residues) in a biological milieu, including the blood. For consistency and clarity, the symbol CgH2S will be used to refer to the concentration of the gaseous form of H2S in solution and CdH2S will refer to the concentration of dissolved H2S, comprising H2S in gaseous form and HS−. NaHS, when in a saline solution, dissociates into gaseous H2S and HS−, so that an infusion of NaHS will contain an equimolar concentration of H2S/HS− (with 70% of HS− and 30% gaseous H2S, the latter evaporating rapidly if syringes are not sealed or contain air). No combined forms of H2S are present in saline. Infusion of sulphide will be expressed as infusion of NaHS (and not H2S/HS−) in keeping with the way our solutions of sulphide were prepared. Total concentration of sulphide in the blood, determined by the MBB method, will be expressed as CMBBH2S corresponding to any sulphide reacting with MBB, comprising the soluble and combined forms of H2S.
RESULTS
NaHS Infusion and Recovery in the Rat
NaHS was infused at only one rate in any given rat (n = 12). The rate of infusion was randomly selected between 2.8 and 10.2 μmol/min and applied for 5 min (Fig. 1). As developed in the Materials and Methods section (Protocol), the lowest levels were below the threshold of breathing stimulation (no clinical signs), whereas the highest rate produced an apnea and a drop in blood pressure. Below 6 μmol/min, CgH2S remained stable during infusion suggesting that the rate of disappearance (oxidation) was similar to the rate of infusion; at higher levels, no steady state could be attained and CgH2S continued to rise until the cessation of infusion (Fig. 1). CgH2S was found to range between 0.4μM and 15.2μM in keeping with the level of H2S infused. These values corresponded to levels of total dissolved H2S concentrations (CdH2S) ranging from 1.2 to 50μM, while CMBBH2S ranged from 4.9 to 87.1μM. For clarity, concentrations are also displayed as a function of three levels of infusion in Figure 2, high (H: 8.9 ± 1.2 μmol/min), moderate (M: 7.9 ± 2 μmol/min), and low (L: 5.1 ± 2.4 μmol/min), whereas individual data are shown in Figure 3. The relationship between H2S concentrations and the rate of H2S infusion was very similar to the relationship established in another group of rats previously studied during a ramp-like increase in H2S infusion rate (Klingerman et al., 2013).
FIG. 1.

Example of recordings obtained in two rats showing the effect of intravenous H2S infusion for 5 min on FAH2S (proportional to CgH2S) at a dose producing no effect on breathing (4 μmol/min, upper panel) and at a level producing an apnea (8 μmol/min, lower panel). In the latter, the rat was mechanically ventilated. FAH2S, which was determined based on minute ventilation, FEH2S, and PaCO2 (see the Materials and Methods section for determination) increased as a function of the levels of H2S infused. Note the lack of a cardiovascular effect at the lowest infusion rate whereas at the highest level, FAH2S continues to rise until blood pressure starts to drop. Interruptions in the BP trace correspond to periods of blood sampling.
FIG. 2.

Concentrations of gaseous H2S in the rat (CgH2S, left panel) and total H2S (CMBBH2S, right panel) at various infusion rates. For clarity, concentrations were displayed as a function of three levels of infusion including high (H: 8.9 ± 1.2 μmol/min), moderate (M: 7.9 ± 2 μmol/min), and low (L: 5.1 ± 2.4 μmol/min). The relationships between CH2S and H2S rate of infusion (shown in dotted lines with the best exponential fits) are very similar to the relationship we previously established in another group of rats during a ramp-like increase in H2S infusion rate. CMBBH2S was determined by HPLC/fluorescence analysis of MBB-S-MBB formed from incubation of MBB reagent with the blood of the rat. CgH2S represents a small proportion of total CH2S, whereas both concentrations increase sharply as the rate increases. Increasing the rate of infusion in the low range by only 2 μmol/min (20–30%) results in an increase in the gaseous form of H2S and CMBBH2S by more than 2–3 times.
FIG. 3.

(A) Relationship between CgH2S and CMBBH2S during H2S infusion obtained in all rats. (B) CMBBH2S (mean ± SD) before the cessation of H2S infusion and at 1, 3, 5, 10, 15 and 30 min into recovery. Note that within 1 min, CMBBH2S dropped to a level of H2S that averaged 3.1μM and continued to drop very slowly to reach 1.1μM at 30 min, still two to three times the baseline values. (C) CMBBH2S (mean ± SD) before the cessation of H2S infusion as a function of the dose infused including high (H: 8.9 ± 1.2 μmol/min, black bars), moderate (M: 7.9 ± 2 μmol/min, hatched bars), and low (L: 5.1 ± 2.4 μmol/min, gray bars) then 5, 10, and 15 min into recovery. Note that there was a difference between residual levels of CMBBH2S as a function of the dose infused. (D) Mean ± SD value of CgH2S before the cessation of H2S infusion and at 3, 5, 10, and 15 min into recovery (white bars). The expected values computed based on the total time constant of the response is shown in gray bars. Note that after the cessation of H2S exposure, CgH2S values were indistinguishable from those due to the expected time constant of our recording setting.
As illustrated in Figure 3A, CMBBH2S (‘total’ H2S) was significantly correlated with CgH2S. The level of CdH2S and CMBBH2S increased as a function of the rate of infusion but with an amount of non-soluble H2S (difference between CdH2S and CMBBH2S), which reached more than 50μM (about twice the total level of dissolved H2S present) at the highest concentrations versus 5μM (about six times the level of dissolved H2S present) at the lowest levels.
As soon as H2S infusion was stopped, expired/alveolar H2S partial pressure dropped with kinetics (Fig. 3B) indistinguishable from the time response of our analyzers and circuit (10–12 s at a flow of 500–600 ml/min). During the entire recovery period, CgH2S was undetectable.
CMBBH2S determined at 1 min dropped to a level ranging from 2 to 3μM (Fig. 3C), regardless of the level of H2S during sulphide infusion. For the highest level of H2S, sulphide concentrations dropped by more than 80μM within 1 min, whereas the 50μM of dissolved H2S (15μM of gaseous form) disappeared almost immediately (Fig. 3D). Following this abrupt drop, there was still a residual level of H2S above baseline measurable by the MBB, which was found to be 3–4 times the baseline level (0.88 ± 0.47μM). CMBBH2S subsided with a very slow kinetics in the 12 animals by about 1μM/15 min, in major contrast with the very fast off kinetics during the initial first minute of recovery. Only in the animals, which received the lowest level of H2S infusion, did CMBBH2S return to baseline at 30 min (Fig. 3B).
Effect of Methemoglobin Infusion on the Concentration of Gaseous and Total H2S
In a separate group of 10 rats, NaHS was infused at a rate ranging from 4.19 to 6.10 μmol/min. This caused a rise in CgH2S to 0.34 ± 0.10 or a CdH2S of 1.02 ± 0.30μM (p < 0.001). CMBBH2S increased to 7.87 ± 1.34μM (p < 0.01).
As illustrated in Figure 4, MetHb infusion decreased CgH2S by more than half, reaching 0.14 ± 0.06 (CdH2S = 0.42 ± 0.18μM) (p < 0.01). In major contrast, CMBBH2S rose by more than seven times from 7.87 ± 1.34μM to 51.78 ± 16.54μM (p < 0.01; Figs. 1–3). The kinetics of reduction of CgH2S in response to MetHb injection was very rapid with an exponential-like pattern. After correcting for the time constant of the circuit and analyzer, the response time constant following the onset of MetHb infusion was found to average 40 ± 21 s. Because the infusion of 600 mg of MetHb lasted about 1 min, less than half of MetHb was infused when CdH2S had already decreased by more than half. After a 600 mg MetHb infusion, the level of MetHb in the arterial blood increased to 8.02 ± 1.44% (p < 0.001) with a Hb concentration of 13.1 ± 1.5 g/100 ml.
FIG. 4.

(A) Recording from a rat receiving a continuous infusion of NaHS at a rate of 7.61 μmol/min. From top to bottom: respiratory flow ( ), alveolar fraction of H2S (FAH2S), minute ventilation (
), alveolar fraction of H2S (FAH2S), minute ventilation (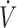 I), and additional air flow (
I), and additional air flow (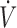 add) are shown. The dashed, vertical lines indicate the start and end of MetHb infusion. FAH2S (see the Methods section for determination) decreased dramatically during MetHb infusion and continued to decrease beyond the period of infusion. Note that small peaks in FAH2S are in each case preceded by an augmented breath (
add) are shown. The dashed, vertical lines indicate the start and end of MetHb infusion. FAH2S (see the Methods section for determination) decreased dramatically during MetHb infusion and continued to decrease beyond the period of infusion. Note that small peaks in FAH2S are in each case preceded by an augmented breath (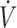 I). The HPLC/fluorescence analysis of the MBB-derived internal standard and MBB-S-MBB formed from incubation of MBB reagent with the blood of the rat is depicted at the top of the trace, prior to MetHb infusion and after MetHb infusion. Note the increase in MBB-S-MBB following MetHb infusion. (B) Concentrations of dissolved H2S (CdH2S, left panel) and total H2S (CMBBH2S, right panel), before and after MetHb infusion. The concentration of the diffusible fraction of H2S (CdH2S) decreased significantly following MetHb infusion, whereas the non-diffusible ‘sink’ of H2S (CMBBH2S) increased following MetHb. In one rat (solid circles; not included in the statistical analysis), NaHS was infused at 15.22 μmol/min before and after MetHb. The effect of MetHb on CdH2S and CMBBH2S was even more striking in this animal. (C) CMBBH2S (mean ± SD) before the cessation of H2S infusion (SS) and during recovery from H2S in control conditions (black bars) and following infusion of MetHb administered during sulphide infusion (open bars). Note that CMBBH2S remains significantly higher in the tests following MetHb infusion given during recovery after H2S than during control recovery. **Significantly different from before MetHb at p < 0.01.
I). The HPLC/fluorescence analysis of the MBB-derived internal standard and MBB-S-MBB formed from incubation of MBB reagent with the blood of the rat is depicted at the top of the trace, prior to MetHb infusion and after MetHb infusion. Note the increase in MBB-S-MBB following MetHb infusion. (B) Concentrations of dissolved H2S (CdH2S, left panel) and total H2S (CMBBH2S, right panel), before and after MetHb infusion. The concentration of the diffusible fraction of H2S (CdH2S) decreased significantly following MetHb infusion, whereas the non-diffusible ‘sink’ of H2S (CMBBH2S) increased following MetHb. In one rat (solid circles; not included in the statistical analysis), NaHS was infused at 15.22 μmol/min before and after MetHb. The effect of MetHb on CdH2S and CMBBH2S was even more striking in this animal. (C) CMBBH2S (mean ± SD) before the cessation of H2S infusion (SS) and during recovery from H2S in control conditions (black bars) and following infusion of MetHb administered during sulphide infusion (open bars). Note that CMBBH2S remains significantly higher in the tests following MetHb infusion given during recovery after H2S than during control recovery. **Significantly different from before MetHb at p < 0.01.
Three rats that received MetHb infusion during NaHS infusion (4.19–6.10 μmol/min) were studied following the cessation of NaHS infusion. CMBBH2S rose from 8.5 ± 3.61μM (NaHS alone) to 47.8 ± 20.7μM during MetHb and NaHS infusion. CMBBH2S remained elevated at 5 min (10.2 ± 7.2μM), 10 min (4.3 ± 1.8μM), and 15 min (3.4 ± 1μM). CMBBH2S at 10 min was even higher than during control NaHS infusion prior to MetHb infusion.
Blood Infusion and Withdrawal Versus Methemoglobin Infusion
In a separate group of four rats, we assessed whether withdrawing and infusing whole blood, and therefore changing volemia and cardiac output, would affect CgH2S in keeping with a ‘volume effect’ of the infusion of a MetHb solution. As illustrated in Figure 5, removing 2 ml of blood during a 7.6 μmol/min H2S infusion increased CgH2S from 0.43 ± 0.16μM to 0.64 ± 0.25μM (or CdH2S from 1.29 ± 0.48 to 1.92 ± 0.75μM) without reaching significance (p = 0.07), whereas infusing the same blood volume rapidly decreased CgH2S from 1.29 ± 0.51μM to 0.43 ± 0.17μM (p < 0.05). These changes in Cg/dH2S were therefore related to the change in volemia or cardiac output caused by the infusion or the withdrawal of blood, the latter likely offering less Hb to combine with H2S per unit of time. They were of short duration as illustrated in Figure 5. However, infusing a similar volume of MetHb in saline (300 mg, 150 mg/ml) caused a response that was different both quantitatively and qualitatively. The reduction in CgH2S was significantly more pronounced following MetHb infusion (CgH2S decreased from 0.84 ± 0.24μM to 0.28 ± 0.08μM). In addition, the most remarkable effect of the presence of MetHb was that further withdrawal or infusion of blood had only trivial effects on CgH2S (0.34 ± 0.14 and 0.28 ± 0.09μM respectively, not significantly different from each other [p = 0.88]). In some cases, these changes were undetectable.
FIG. 5.

Effects of an acute change in volemia on CgH2S. (A) Recording during withdrawal (W) or infusion (I) of 2 ml of blood before and after infusing 2 ml of MetHb solution. Respiratory flow (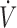 ), alveolar fraction of H2S (FAH2S), and additional air flow (
), alveolar fraction of H2S (FAH2S), and additional air flow (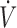 add) signals are shown. (B) Effect of blood versus MetHb infusion on mean CgH2S, th esymbol B related the basleine values before withdrawal (W) or infusion (I) of blood. (C) Change in CgH2S after withdrawal or infusion of blood or MetHb compared with values obtained before each treatment. Withdrawing and infusing blood produced a small rise and decrease in CdH2S, respectively. Infusing MetHb, instead of blood, produced a dramatic decrease in CgH2S (p < 0.01) that persisted, and in most cases prevented, any other effects of volemia on CgH2S. *Significantly different at p < 0.05, **Significantly different at p < 0.01.
add) signals are shown. (B) Effect of blood versus MetHb infusion on mean CgH2S, th esymbol B related the basleine values before withdrawal (W) or infusion (I) of blood. (C) Change in CgH2S after withdrawal or infusion of blood or MetHb compared with values obtained before each treatment. Withdrawing and infusing blood produced a small rise and decrease in CdH2S, respectively. Infusing MetHb, instead of blood, produced a dramatic decrease in CgH2S (p < 0.01) that persisted, and in most cases prevented, any other effects of volemia on CgH2S. *Significantly different at p < 0.05, **Significantly different at p < 0.01.
CgH2S and CMBBH2S During and Following NaHS Infusion in the Sheep
Figure 6 is an illustration of the change in the alveolar fraction of H2S (gaseous) at various rates of NaHS infusion in one animal, along with the averaged response (Fig. 7A). H2S was undetectable in its soluble form in baseline conditions whereas baseline CMBBH2S was found to average 0.61 ± 0.28μM. The relationship between CgH2S, CMBBH2S, and the rates of infusion is displayed in Figure 7C along with the levels of H2S at which breathing was stimulated and an apnea occurred: breathing was stimulated at an infusion rate of 270 μmol/min, leading to values of CgH2S, CdH2S, and CMBBH2S that averaged 0.07 ± 0.03, 0.21 ± 0.90, and 9.59 ± 3.53μM, respectively. A terminal apnea occurred at CgH2S, CdH2S, and CMBBH2S concentrations, which averaged 4.93 ± 2.78, 14.79 ± 8.34, and 106.69 ± 22.99μM, respectively. The rates of infusion leading to hyperventilation or to apnea were very close, resulting in a very steep dose-toxic effect relationship as soon as symptoms occurred (Fig. 7C). The relationship between CgH2S and CMBBH2S revealed that the ability of the blood to combine with H2S was much larger in the rat at any given level of CgH2S or infusion rate. Another way to express this notion is illustrated in Figure 8; the concentration of sulphide as a function of the rate of infusion per body weight was higher in the combined form but not in the soluble form in the sheep when compared with the rat.
FIG. 6.

Example of the effects of infusing NaHS on breathing in an adult sheep at incremental levels. Note that in this example, breathing started to rise between an infusion rate of 270 and 405 μmol/min whereas an apnea occurred at 675 μmol/min.
FIG. 7.

(A) Concentration (mean ± SD) of the gaseous form of H2S in the arterial blood (CgH2S; open bars) and of H2S measured in the blood using MBB (CMBBH2S; black bars). Increasing the rate of NaHS infusion caused an increase in CgH2S and CMBBH2S. The majority of H2S found in the blood was not in the dissolved form. CgH2S increased from zero to a value of 5.6μM when a terminal apnea occurred, corresponding to CMBBH2S that averaged 106.8μM, up to 20 times the gaseous form. As shown in (B), the H2S concentration increased exponentially with the rate of infusion and the ratio between CgH2S (open circles) and CMBBH2S (black circles) decreased as H2S concentrations increased. (C) CMBBH2S in baseline conditions and following H2S infusion at 5, 10, and 15 min into recovery; note that CMBBH2S dropped dramatically after H2S infusion but remained higher than baseline for at least 15 min.
FIG. 8.

Relationship between partial pressure of H2S and the various pools of H2S in the arterial blood during H2S exposure in the sheep and the rat. These graphs have been created based on the exponential regressions obtained in the current and previous studies on the rat and the present results for the sheep. Note that the ability of the blood to transport H2S is much higher in the sheep than in the rat. In the left-hand panel, the values of CgH2S, CdH2S, and CMBBH2S for the sheep and the rat are presented at levels of infusion wherein hyperpnea (Hyp) and apnea occurred. Note that the major part of H2S in the blood is not in gaseous form and that the ratio between CgH2S and CMBBH2S decreases as the level of H2S increases.
At the cessation of NaHS infusion, CgH2S dropped with virtually no delay, the kinetics of this response was indiscernible from the off-transient of the analyzer, like in the rat. CMBBH2S also dropped very rapidly reaching a residual level of about 2μM at 5, 10, and 15 min but remained significantly higher than baseline values (Fig. 7C).
Effects of Hydroxocobalamin and Methemoglobin in the Sheep
HyCo (70 mg/kg in 200 ml) was injected over a 5 min period in each sheep whereas NaHS infusion (270–405 μmol/min) was maintained for at least 15 additional min. As illustrated in Figure 9A in one animal and in Figure 9C for the average response, HyCo infusion decreased CgH2S from an average of 1.01 ± 0.4μM to 0.07 ± 0.09μM within a few minutes (p < 0.01). In contrast, CMBBH2S did not change significantly (37.2 ± 13.1 vs. 26.1 ± 14.9μM). The kinetics of the reduction of CgH2S in response to HyCo injection was very rapid (Fig. 9A). CgH2S remained low for 10–12 min during the infusion, and then increased returning to pre-Hyco levels within 20 min.
FIG. 9.

(A) Recording obtained in one sheep receiving a continuous infusion of NaHS at a rate of 405 μmol/min. From top to bottom: systemic blood pressure and mixed expired fraction of H2S are shown. The dashed, vertical lines indicate the start and end of HyCo solution injection. The gaseous form of H2S in the blood decreased dramatically during HyCo infusion reaching almost zero despite the persistence of sulphide infusion. (B) Recording obtained in one sheep receiving a continuous infusion of NaHS at a rate of 405 μmol/min, same traces as (A). The two dashed, vertical lines indicate the start and end of MetHb infusion. FAH2S decreased dramatically during MetHb infusion and continued to decrease beyond the period of MetHb infusion. Note the increase in ABP that was systematically observed in each test following MetHb injection. (C) Mean ± SD relationship between the concentrations of gaseous H2S (CgH2S) and total CMBBH2S, before (b) and after (a) MetHb infusion (black symbols) and HyCo infusion (open symbols). CgH2S significantly decreased following MetHb infusion just like in the rat with a fourfold increase in CMBBH2S associated with an increase of MetHb to 6.34 ± 0.73%. The decrease in CgH2S was even more significant following HyCo infusion with a small decrease in CMBBH2S, suggesting a different mode of action of these two antidotes on the pools of combined H2S. (D) CMBBH2S (mean ± SD) before H2S infusion (baseline) and during recovery from H2S following infusion of MetHb administered during sulphide infusion. Note that CMBBH2S is 15–20 times higher in the tests following MetHb infusion given during recovery from H2S infusion than during control recovery as shown in Figure 7.
Infusion of MetHb lasted about 2–3 min. Like in the rat, CgH2S decreased by half from 1.61 ± 0.34 to 0.83 ± 0.44μM (p < 0.01), whereas CMBBH2S rose by more than threefold, from 54.9 ± 11.4 to 163 ± 28.1μM (p < 0.01) as illustrated in Figures 9B and 9C. The level of MetHb in the arterial blood increased to 6.34 ± 0.73% (p < 0.001). In four of the five sheep, CMBBH2S was measured following the cessation of NaHS infusion (after having received a MetHb infusion) at 5, 10, and 15 min. As shown in Figure 9D, CMBBH2S remained much higher than during control recovery, suggesting that H2S combined to ferric iron remains present in the blood following the cessation of sulphide exposure.
DISCUSSION
We found that, in the rat, infusion of NaHS at a constant rate ranging from 4 to 9 μmol/min produced an increase in the concentration of dissolved and combined H2S, similar to those we previously obtained during a ramp-like increase in H2S in the same species (Klingerman et al., 2013). At the lowest rate of infusion, CgH2S and CdH2S represent a small portion of the total pool of H2S present in the blood as measured by the MBB method (Fig. 8). This proportion increased as the rate of infusion and CgH2S concentrations increased. At the cessation of sulphide exposure, dissolved H2S dropped with virtually no delay to undetectable levels, whatever the rate of infusion of H2S, whereas CMBBH2S dropped to levels ranging from 2 to 3μM in less than 1 min, but remained higher than baseline for at least 30 min. Very similar results were obtained in the sheep. MetHb infusion increased dramatically the ability of the blood to combine with free H2S, reducing, in turn, CgH2S. Finally, HyCo decreased the concentration of the gaseous form of H2S to almost zero and thus displaced H2S combined with the Hb, but for a limited period of time if infusion is maintained. These results and their implications are discussed in the following paragraphs.
Pools of H2S and Their Kinetics in the Rat and Sheep During and Following H2S Exposure
Based on the methodology used to measure H2S, one can distinguish, for the sake of convenience, different pools of H2S in the blood. First, there is a gaseous pool which diffusion and rate of exchange can be understood just like for any other gas; however, due to the extremely high solubility of H2S, compared with O2 or even CO2, very small partial pressure can maintain a fair amount of H2S allowing diffusion to persist. By comparison, a partial pressure of about 0.01 Torr is required to maintain a concentration of about 0.01μM of dissolved O2, whereas a partial pressure of only 0.0001 Torr can achieve the same effects for H2S. We found that in both rats and sheep, these concentrations represent a small portion of the total H2S present in the blood (Fig. 8). The second component is represented by the anion HS−, as for every molecule of H2S in gaseous form, 3–4 molecules of HS− are present at physiological pH (Almgren et al., 1976; Millero, 1986), increasing the capacity of the plasma to transport H2S by 3–4 times at any given H2S partial pressure. These two components represent the soluble/dissolved pool of sulphide. The second pool is to be accounted for by the discrepancy between the concentration of the pool of exchangeable sulphide—measured based on expired gas or by amperometry (Wintner et al., 2010)—and the total amount of H2S in the blood measured by the MBB technique (Klingerman et al., 2013; Wintner et al., 2010). This difference is likely due to the presence of a pool of H2S combined with Hb and plasma proteins. Increasing the level of MetHb in the blood dramatically increased CMBBH2S.
The concentrations of gaseous H2S required to stimulate the chemoreceptors, or stop breathing in the sheep, were not very different from the level required to produce symptoms in the rat (Klingerman et al., 2013) (Fig. 8). Indeed, despite a weight of about 100 times larger than the rat, any given rate of infusion/kg led to similar levels of CgH2S that were previously found in the rat when hyperventilation or apnea was produced (Klingerman et al., 2013), i.e., less than 1 μM for breathing stimulation and around 5–8μM for apnea. The only difference found in the sheep was a higher concentration of total H2S for any given level of gaseous H2S as compared with levels in the rat (about 2×). It is quite intriguing that the same rate of H2S infused per kilogram produced the same effects and led to similar concentrations of gaseous H2S in both species, whereas we would have anticipated that a much lower rate of H2S would produce greater effects in larger mammals. This suggests that the rate of disappearance of the dissolved form of H2S, per gram of tissue, is very similar in these two species. As CdH2S remains stable throughout a period of several minutes of infusion at a rate of 8–12 μmol/kg/min, both in the rat and in the sheep, the rate of elimination/oxidation of H2S must be at least 8–12 μmol/kg/min. This level of oxidation is perfectly compatible with the disappearance of H2S during recovery, wherein for a concentration of 80μM of CMBBH2S, 6μM of CgH2S vanishes within a minute. Indeed, it did not take more than 1 min for gaseous H2S present at these lethal concentrations to disappear. Incidentally, the rate of elimination of H2S by the lungs represents only a small proportion of the rate of H2S infused in both species (Klingerman et al., 2013). CMBBH2S values obtained in humans during low levels of sulphide infusion (Toombs et al., 2010) fit with the effects observed in our sheep and suggest that large mammals including humans may possess a higher ability to combine H2S than the rat.
The most intriguing observation is certainly the baseline, as well as the persistence of a very small pool of H2S in the blood with a very slow time constant. The levels of CMBBH2S, both in the baseline condition and during recovery, were indistinguishable between the sheep and the rat. H2S fixed on the Hb or proteins certainly accounts for this small H2S pool that is not rapidly exchangeable. The much greater level of CMBBH2S after MetHb infusion supports this contention. This also suggests that H2S inside the cells may also remain combined with other metallo-compounds (Haouzi and Klingerman, 2013), the main “targets” of sulphide toxicity, beyond the period of exposure. Similarly, H2S combined with cysteine residues, if part of this pool in the intracellular compartment, would be likely to persist as well (Mustafa et al., 2009; Warenycia et al., 1990).
Methemoglobin Solution and Hydroxocobalamin as Antidotes During H2S Poisoning
As presented in the Introduction section, various agents have been proposed to treat H2S poisoning: hyperoxia (Bitterman et al., 1986; Haouzi et al., 2011a; Smilkstein et al., 1985, 1976), metallo-compounds (ferric iron using nitrite-induced methemoglobinemia or cobalt in vitamin B12) (Haouzi et al., 2011b; Smith, 1969; Smith et al., 1976; Truong et al., 2007; Van de Louw and Haouzi, 2012), reducing agents (Warenycia et al., 1990), sodium bicarbonate, or high level of glucose (Almeida and Guidotti, 1999; Guidotti, 2010).
Sodium nitrite-induced methemoglobinemia has long been proposed as a treatment of H2S poisoning in humans (Hall and Rumack, 1997; Smith, 1981; Smith and Gosselin, 1979); animal studies have demonstrated a protective effect against H2S-induced lethality in rodents when injected “prior” to the toxic exposure (Smith and Gosselin, 1979; Smith et al., 1976). The oxidation of the ferrous (Fe2+) to ferric iron (Fe3+) present in Hb, produced by sodium nitrite, allows the combination of H2S into a non-soluble, i.e., non-diffusible, form (Smith and Gosselin, 1966) and seems also to be able to catalyze the oxidation of H2S (Beck et al., 1981).
Twenty milligram per kilogram of sodium nitrite (Kohn et al., 2002) has been previously used intravenously, resulting in the production of 10–25% MetHb 20 min after injection (Kohn et al., 2002). We have recently shown that much lower levels of nitrite (5 mg/kg) and MetHb (3%) were sufficient to prevent the ventilatory stimulation produced by an IV bolus of H2S (Van de Louw and Haouzi, 2012). Twenty minutes following the injection of low levels of sodium nitrite, we found that the response to a bolus injection of a 2.4mM (0.5 ml) solution of H2S was virtually abolished (Van de Louw and Haouzi, 2012). It has been proposed that 2–4 moles of sulphide can be inactivated in vivo for each available ferric heme group in MetHb (Smith, 1967). Kohn and colleagues have reported that 30 min were needed to maximize MetHb concentrations in the blood after IV infusion of 20 mg/kg of nitrite to rats (Kohn et al., 2002). Besides its delay of action, the major limitations of using nitrite-induced methemoglobinemia to counteract H2S intoxication are (1) the deleterious effects of nitrite-induced vasodilation (Hall et al., 1989), (2) the reduction in the amount of functional Hb and thus a reduction in O2 transport, and (3) the negative interaction between nitrite and O2 in the production of MetHb (Beck et al., 1981). These limitations could be overcome by the use of a solution of free MetHb. The lack of reductase in a free Hb solution, normally present in red cells, allows the spontaneous oxidation of ferrous iron into stable MetHb. We took advantage of this usually undesirable natural oxidation to produce a solution of crystalline Hb containing 98% of MetHb. Rapid infusion of such a solution was able to increase the MetHb level to 8% and to dramatically reduce gaseous H2S concentrations in the blood via a rise in the CMBBH2S sink by more than seven times during sub-lethal infusion of H2S. A similar effect could be produced using a much lower level of MetHb as suggested by the kinetics of the response, wherein CgH2S dropped whereas only a small portion of total MetHb had been infused. These effects, which remained present following H2S exposure although MetHb was injected during H2S infusion, were similar in the rat and in the sheep.
There was a large discrepancy between the decrease in the CgH2S, which dropped by half (and by less than 1μM), and the rise in CMBBH2S, which increased by more than 40μM. This suggests that not only a very large portion of the soluble form of H2S (CgH2S) was combined with MetHb, but also H2S that should have diffused into the tissues and been oxidized was trapped in the process. There are clinical reports supporting the use of nitrite-induced methemoglobinemia after H2S intoxication (Hall and Rumack, 1997). For obvious reasons, these observations remain anecdotal, as control case studies in humans cannot be performed.
The combination of H2S and its “transporter,” ferric iron molecules, was almost immediate. Changing the cardiac output by withdrawing and infusing 2 ml of blood affected the concentration of dissolved H2S with an inverse relationship. This suggests that the level of Hb available to interact with H2S per unit of time does influence the ratio between combined and dissolved H2S. The rate of complexation of H2S in the blood is in part controlled by the rate at which Hb circulates. This phenomenon is therefore magnified dramatically by the presence of MetHb that abolished the volemia-induced CgH2S changes. In other words, the infusion of MetHb not only had a persistent effect on CgH2S, but the presence of MetHb made the ability of the blood to combine with H2S almost independent of volemia/cardiac output.
Large doses of HyCo have also been shown to reduce the hepatotoxicity of H2S in a rat model, prevent H2S-induced mortality in mice (Mihajlovic, 1999; Truong et al., 2007), and magnify the ability of the blood or tissue of rats exposed to HyCo to oxidize hydrogen sulphide (Van de Louw and Haouzi, 2012). In addition, vitamin B12 is currently used as an antidote for cyanide intoxication at a dose 106 times the normal daily intake (Fortin et al., 2006; Hall et al., 2009; Shepherd and Velez, 2008). At this dose (5 g in adult), concentrations of vitamin B12 in the blood increase from a few pM to several mM range in humans (Forsyth et al., 1993) and more than 500μM in the rat. In contrast to MetHb, which remains in the blood, vitamin B12 at this concentration diffuses into the cells and into the mitochondria via non-transcobalamin mechanisms (Gimpert et al., 1975; Hall et al., 1984), where cobalt could complex H2S and catalyze its oxidation. We found that vitamin B12 injection during H2S infusion in the sheep was able to drop the level of dissolved H2S with a much larger magnitude than MetHb, whereas total H2S was much higher than expected from the reduction in soluble H2S. As CMBBH2S follows the drop in CgH2S during recovery, CMBBH2S seems to constitute, except for few residual μM, a pool exchangeable almost immediately with the diffusible part. This would suggest that H2S combined with HyCo is accessible to the MBB measurement and may be able to displace H2S fixed on Hb.
Of note is that, to reduce the number of sheep used in the study, the infusion of MetHb was performed in animals that had previously received HyCo. The effects of MetHb were tested when the effects of Hyco had already vanished at least on sulphide concentrations; still, results were identical to the rat.
These two antidotes had a very rapid effect on H2S concentrations, occurring within tens of seconds, but seem to act against H2S poisoning very differently on the various pools of H2S and thus may have a very different impact on what could have happened whenever injected “after” exposure. Indeed, the putative efficacy of an antidote to be used when a patient is withdrawn from the source of H2S intoxication relies on two main prerequisites: (1) H2S must still be present in the body in a form that contributes, per se, to the persistence of toxic effects following H2S exposure, and (2) the recovery kinetics of the different forms of H2S in the blood and tissues must be compatible with a timeline allowing rescuers to administer an antidote. In other words, if all gaseous H2S is already gone, the very reason for using an antidote becomes obsolete. Although gaseous H2S is probably not present for more than few minutes, at best, after the cessation of exposure, there is still no clear answer about how long non-soluble forms of H2S, such as with Cu, Zn, Fe, or Co containing metallo-proteins or sulfane sulphur, could persist. This makes the use of MetHb, which remains in the intravascular compartment and which acts by increasing the H2S sink in the blood, an approach that might be very limited at a time when all dissolved H2S has already been oxidized or eliminated. A similar reasoning can be applied for vitamin B12. The only difference with MetHb is that vitamin B12 can diffuse into the cells and into the mitochondria via non-transcobalamin mechanisms (Gimpert et al., 1975; Hall et al., 1984). Whether cobalt compounds can interact with the form of H2S combined with metallo-proteins and displace some of the H2S present on cytochrome c while no free H2S is present in the blood and in the tissues remains an outstanding question.
Finally, it is possible to imagine clinical scenarios wherein the fast kinetics of gaseous sulphide recovery could be slowed, during a very severe reduction in cardiac output, wherein the rate of pulmonary exchange is decreased. A patient is found in cardiac arrest or in extremely severe shock, could present such a phenomenon. Nevertheless, we found that the fast disappearance of the soluble form of H2S is still present in the most severe forms of intoxication, wherein blood pressure was reduced. The extreme diffusibility of H2S in its gaseous form will also allow the H2S concentration to drop in the alveolar gas at a very fast rate, even during an apnea, as long as some cardiac output/pulmonary blood flow is maintained whether H2S is inhaled instead of infused. For all practical purposes, one could speculate that any improvement of hemodynamics should by itself improve the ability of H2S to be exchanged and oxidized in the tissues, a strategy that may prove to be even more effective than using any trapping molecules. Of note is that the blood and the plasma are able to continue to oxidize H2S at a very fast rate (Klingerman et al., 2013; Olson, 2011a; Wintner et al., 2010).
In conclusion, H2S metabolism during H2S administration is very similar in a large and a small mammal. H2S presence in its free, soluble form cannot be sustained in a durable manner in the blood and tissues beyond a minute in both species even at levels producing a terminal apnea. Whereas MetHb and HyCo solutions are very potent antidotes against H2S, the efficacy of administering metallo-compounds following H2S exposure can be challenged on the basis of their interaction with free H2S and kinetics described in both animal models.
Acknowledgments
The authors would to thank Neil Trushin from the Department of Pharmacology at Penn State University College of Medicine for his technical assistance in H2S determination with HPLC.
FUNDING
CounterACT Program, National Institutes of Health Office of the Director (NIH OD); National Institute of Neurological Disorders and Stroke (NINDS) [1R21NS080788-02].
REFERENCES
- Almeida A. F., Guidotti T. L. Differential sensitivity of lung and brain to sulphide exposure: A peripheral mechanism for apnea. Toxicol. Sci. 1999;50:287–293. doi: 10.1093/toxsci/50.2.287. [DOI] [PubMed] [Google Scholar]
- Almeida A. F., Nation P. N., Guidotti T. L. Mechanism and treatment of sulphide-induced coma: A rat model. Int. J. Toxicol. 2008;27:287–293. doi: 10.1080/10915810802210166. [DOI] [PubMed] [Google Scholar]
- Almgren T., Dyrssen D., Elgquist B., Johannsson O. Dissociation of hydrogen sulphide in seawater and comparison of pH scales. Marine Chem. 1976;4:289–297. [Google Scholar]
- Arnold I. M., Dufresne R. M., Alleyne B. C., Stuart P. J. Health implication of occupational exposures to hydrogen sulphide. J. Occup. Med. 1985;27:373–376. doi: 10.1097/00043764-198505000-00018. [DOI] [PubMed] [Google Scholar]
- Barrett T. J., Anderson G. M., Lugowski J. T. The solubility of hydrogen sulphide in 0–5 m NaCl solutions at 25–95 C and one atmosphere. Geochim. Cosmochim. Acta. 1988;52:807–811. [Google Scholar]
- Beauchamp R. O., Jr, Bus J. S., Popp J. A., Boreiko C. J., Andjelkovich D. A. A critical review of the literature on hydrogen sulphide toxicity. Crit. Rev. Toxicol. 1984;13:25–97. doi: 10.3109/10408448409029321. [DOI] [PubMed] [Google Scholar]
- Beck J. F., Bradbury C. M., Connors A. J., Donini J. C. Nitrite as antidote for acute hydrogen sulphide intoxication? Am. Ind. Hyg. Assoc. J. 1981;42:805–809. doi: 10.1080/15298668191420738. [DOI] [PubMed] [Google Scholar]
- Bitterman N., Talmi Y., Lerman A., Melamed Y., Taitelman U. The effect of hyperbaric oxygen on acute experimental sulphide poisoning in the rat. Toxicol. Appl. Pharmacol. 1986;84:325–328. doi: 10.1016/0041-008x(86)90140-7. [DOI] [PubMed] [Google Scholar]
- Bouillaud F., Blachier F. Mitochondria and sulphide: A very old story of poisoning, feeding, and signaling? Antioxid. Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- Carroll J. J., Mather A. E. The solubility of hydrogen sulphide in water from 0 to 90°C and pressures to 1 MPa. Geochim. Cosmochim. Acta. 1989;53:1163–1170. [Google Scholar]
- Cooper C. E., Brown G. C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulphide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- Costigan M. G. Hydrogen sulphide: UK occupational exposure limits. Occup. Environ. Med. 2003;60:308–312. doi: 10.1136/oem.60.4.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruyn W. J., Swartz E., Hu J. H., Shorter J. A., Davidovits P., Worsnop D. R., Zahniser S., Kolb C. E. Henry's law solubilities and Setchenow coefficients for biogenic reduced sulfur species obtained from gas-liquid uptake measurements. J. Geophys. Res. 1995;100:7245–7251. [Google Scholar]
- Dorman D. C., Dautrebande L., Moulin F. J., McManus B. E., Mahle K. C., James R. A., Struve M. F. Cytochrome oxidase inhibition induced by acute hydrogen sulphide inhalation: Correlation with tissue sulphide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol. Sci. 2002;65:18–25. doi: 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- Douabul A. A., Riley J. P. The solubility of gases in distilled water and seawater—V. Hydrogen sulphide. Deep Sea Res. A. 1979;26:259–268. [Google Scholar]
- EPA. Toxicological Review of Hydrogen Sulphide (CAC No 7783-06-04) Washington, DC: United States Environmental Protection Agency; 2003. [Google Scholar]
- Forsyth J. C., Mueller P. D., Becker C. E., Osterloh J., Benowitz N. L., Rumack B. H., Hall A. H. Hydroxocobalamin as a cyanide antidote: Safety, efficacy and pharmacokinetics in heavily smoking normal volunteers. J. Toxicol. Clin. Toxicol. 1993;31:277–294. doi: 10.3109/15563659309000395. [DOI] [PubMed] [Google Scholar]
- Fortin J. L., Giocanti J. P., Ruttimann M., Kowalski J. J. Prehospital administration of hydroxocobalamin for smoke inhalation-associated cyanide poisoning: 8 years of experience in the Paris Fire Brigade. Clin. Toxicol. 2006;44(Suppl. 1):37–44. doi: 10.1080/15563650600811870. [DOI] [PubMed] [Google Scholar]
- Fuller D. C., Suruda A. J. Occupationally related hydrogen sulphide deaths in the United States from 1984 to 1994. J. Occup. Environ. Med. 2000;42:939–942. doi: 10.1097/00043764-200009000-00019. [DOI] [PubMed] [Google Scholar]
- Gimpert E., Jakob M., Hitzig W. H. Vitamin B12 transport in blood. I. Congenital deficiency of transcobalamin II. Blood. 1975;45:71–82. [PubMed] [Google Scholar]
- Greer J. J., Reiffenstein R. J., Almeida A. F., Carter J. E. Sulphide-induced perturbations of the neuronal mechanisms controlling breathing in rats. J. Appl. Physiol. 1995;78:433–440. doi: 10.1152/jappl.1995.78.2.433. [DOI] [PubMed] [Google Scholar]
- Guidotti T. L. Hydrogen sulphide. Occup. Med. (Lond.) 1996;46:367–371. doi: 10.1093/occmed/46.5.367. [DOI] [PubMed] [Google Scholar]
- Guidotti T. L. Hydrogen sulphide: Advances in understanding human toxicity. Int. J. Toxicol. 2010;29:569–581. doi: 10.1177/1091581810384882. [DOI] [PubMed] [Google Scholar]
- Haggard H. W. The fate of sulphides in the blood. J. Biol. Chem. 1921;49:519–529. [Google Scholar]
- Hall A. H., Kulig K. W., Rumack B. H. Suspected cyanide poisoning in smoke inhalation: Complications of sodium nitrite therapy. J. Toxicol. Clin. Exp. 1989;9:3–9. [PubMed] [Google Scholar]
- Hall A. H., Rumack B. H. Hydrogen sulphide poisoning: An antidotal role for sodium nitrite? Vet. Hum. Toxicol. 1997;39:152–154. [PubMed] [Google Scholar]
- Hall A. H., Saiers J., Baud F. Which cyanide antidote? Crit. Rev. Toxicol. 2009;39:541–552. doi: 10.1080/10408440802304944. [DOI] [PubMed] [Google Scholar]
- Hall C. A., Begley J. A., Green-Colligan P. D. The availability of therapeutic hydroxocobalamin to cells. Blood. 1984;63:335–341. [PubMed] [Google Scholar]
- Haouzi P., Bell H., Philmon M. Hydrogen sulphide oxidation and the arterial chemoreflex: Effect of methemoglobin. Respir. Physiol. Neurobiol. 2011a;177:273–283. doi: 10.1016/j.resp.2011.04.025. [DOI] [PubMed] [Google Scholar]
- Haouzi P., Bell H., Van de Louw A. Hypoxia-induced arterial chemoreceptor stimulation and hydrogen sulphide: Too much or too little? Respir. Physiol. Neurobiol. 2011b;179:97–102. doi: 10.1016/j.resp.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Haouzi P., Klingerman C. M. Fate of intracellular H2S/HS(-) and metallo-proteins. Respir. Physiol. Neurobiol. 2013;188:229–230. doi: 10.1016/j.resp.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P., Van de Louw A. Uncoupling mitochondrial activity maintains body [Formula: see text] during hemorrhage-induced O2 deficit in the anesthetized rat. Respir. Physiol. Neurobiol. 2013;186:87–94. doi: 10.1016/j.resp.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Klingerman C. M., Trushin N., Prokopczyk B., Haouzi P. H2s concentrations in the arterial blood during H2s administration in relation to its toxicity and effects on breathing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013;305:630–638. doi: 10.1152/ajpregu.00218.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn M. C., Melnick R. L., Ye F., Portier C. J. Pharmacokinetics of sodium nitrite-induced methemoglobinemia in the rat. Drug Metab. Dispos. 2002;30:676–683. doi: 10.1124/dmd.30.6.676. [DOI] [PubMed] [Google Scholar]
- Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., Bouillaud F. Oxidation of hydrogen sulphide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Mihajlovic A. Antidotal mechanisms for hydrogen sulphide toxicity. Toronto. 1999:69. [Google Scholar]
- Millero F. J. The thermodynamics and kinetics of hydrogen sulphide system in natural waters. Marine Chem. 1986;18:121–147. [Google Scholar]
- Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., Snyder S. H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson K. R. A practical look at the chemistry and biology of hydrogen sulphide. Antioxid. Redox Signal. 2011a;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson K. R. The therapeutic potential of hydrogen sulphide: Separating hype from hope. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011b;301:R297–R312. doi: 10.1152/ajpregu.00045.2011. [DOI] [PubMed] [Google Scholar]
- Rahman M. M., Kim S. J., Kim G. B., Hong C. U., Lee Y. U., Kim S. Z., Kim J. S., Kang H. S. Nitrite-induced methemoglobinaemia affects blood ionized and total magnesium level by hydrolysis of plasma adenosine triphosphate in rat. Basic Clin. Pharmacol. Toxicol. 2009;105:294–300. doi: 10.1111/j.1742-7843.2009.00450.x. [DOI] [PubMed] [Google Scholar]
- Reedy S. J., Schwartz M. D., Morgan B. W. Suicide fads: Frequency and characteristics of hydrogen sulphide suicides in the United States. West. J. Emerg. Med. 2011;12:300–304. [PMC free article] [PubMed] [Google Scholar]
- Reiffenstein R. J., Hulbert W. C., Roth S. H. Toxicology of hydrogen sulphide. Annu. Rev. Pharmacol. Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- Shepherd G., Velez L. I. Role of hydroxocobalamin in acute cyanide poisoning. Ann. Pharmacother. 2008;42:661–669. doi: 10.1345/aph.1K559. [DOI] [PubMed] [Google Scholar]
- Smilkstein M. J., Bronstein A. C., Pickett H. M., Rumack B. H. Hyperbaric oxygen therapy for severe hydrogen sulphide poisoning. J. Emerg. Med. 1985;3:27–30. doi: 10.1016/0736-4679(85)90216-1. [DOI] [PubMed] [Google Scholar]
- Smith R. P. The oxygen and sulphide binding characteristics of hemoglobins generated from methemoglobin by two erythrocytic systems. Mol. Pharmacol. 1967;3:378–385. [PubMed] [Google Scholar]
- Smith R. P. Cobalt salts: Effects in cyanide and sulphide poisoning and on methemoglobinemia. Toxicol. Appl. Pharmacol. 1969;15:505–516. doi: 10.1016/0041-008x(69)90052-0. [DOI] [PubMed] [Google Scholar]
- Smith R. P. Nitrite treatment for hydrogen sulphide poisoning. Ann. Intern. Med. 1981;95:782. doi: 10.7326/0003-4819-95-6-782_1. [DOI] [PubMed] [Google Scholar]
- Smith R. P., Gosselin R. E. On the mechanism of sulphide inactivation by methemoglobin. Toxicol. Appl. Pharmacol. 1966;8:159–172. doi: 10.1016/0041-008x(66)90112-8. [DOI] [PubMed] [Google Scholar]
- Smith R. P., Gosselin R. E. Hydrogen sulphide poisoning. J. Occup. Med. 1979;21:93–97. doi: 10.1097/00043764-197902000-00008. [DOI] [PubMed] [Google Scholar]
- Smith R. P., Kruszyna R., Kruszyna H. Management of acute sulphide poisoning. Effects of oxygen, thiosulfate, and nitrite. Arch. Environ. Health. 1976;31:166–169. doi: 10.1080/00039896.1976.10667212. [DOI] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nature reviews. Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Toombs C. F., Insko M. A., Wintner E. A., Deckwerth T. L., Usansky H., Jamil K., Goldstein B., Cooreman M., Szabo C. Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br. J. Clin. Pharmacol. 2010;69:626–636. doi: 10.1111/j.1365-2125.2010.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong D. H., Mihajlovic A., Gunness P., Hindmarsh W., O'Brien P. J. Prevention of hydrogen sulphide (H2S)-induced mouse lethality and cytotoxicity by hydroxocobalamin (vitamin B(12a)) Toxicology. 2007;242:16–22. doi: 10.1016/j.tox.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Truscott A. Suicide fad threatens neighbours, rescuers. CMAJ. 2008;179:312–313. doi: 10.1503/cmaj.080878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tvedt B., Skyberg K., Aaserud O., Hobbesland A., Mathiesen T. Brain damage caused by hydrogen sulphide: A follow-up study of six patients. Am. J. Ind. Med. 1991;20:91–101. doi: 10.1002/ajim.4700200109. [DOI] [PubMed] [Google Scholar]
- Van de Louw A., Haouzi P. Ferric Iron and Cobalt (III) Compounds to Safely Decrease Hydrogen Sulphide in the Body? Antioxid Redox Signal. 2012b;19:510–516. doi: 10.1089/ars.2012.4513. [DOI] [PubMed] [Google Scholar]
- Warenycia M. W., Goodwin L. R., Francom D. M., Dieken F. P., Kombian S. B., Reiffenstein R. J. Dithiothreitol liberates non-acid labile sulphide from brain tissue of H2S-poisoned animals. Arch. Toxicol. 1990;64:650–655. doi: 10.1007/BF01974693. [DOI] [PubMed] [Google Scholar]
- Whiteman M., Winyard P. G. Hydrogen sulphide and inflammation: The good, the bad, the ugly and the promising. Expert Rev. Clin. Pharmacol. 2011;4:13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- Wintner E. A., Deckwerth T. L., Langston W., Bengtsson A., Leviten D., Hill P., Insko M. A., Dumpit R., VandenEkart E., Toombs C. F., Szabo C. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br. J. Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]