

Abstract
The development of nephrotoxicity limits the maximum achievable dosage and treatment intervals for cisplatin chemotherapy. Therefore, identifying mechanisms that regulate this toxicity could offer novel methods to optimize cisplatin delivery. MicroRNAs are capable of regulating many different genes, and can influence diverse cellular processes, including cell death and apoptosis. We previously observed miR-155 to be highly increased following ischemic or toxic injury to the kidneys and, therefore, sought to determine whether mice deficient in miR-155 would respond differently to kidney injury. We treated C57BL/6 and miR-155−/− mice with 20 mg/kg of cisplatin and found a significantly higher level of kidney injury in the miR-155−/− mice. Genome-wide expression profiling and bioinformatic analysis indicated the activation of a number of canonical signaling pathways relating to apoptosis and oxidative stress over the course of the injury, and identified potential upstream regulators of these effects. One predicted upstream regulator was c-Fos, which has two confirmed miR-155 binding sites in its 3′ UTR and, therefore, can be directly regulated by miR-155. We established that the miR-155−/− mice had significantly higher levels of c-Fos mRNA and protein than the C57BL/6 mice at 72 h after cisplatin exposure. These data indicate a role for miR-155 in the cisplatin response and suggest that targeting of c-Fos could be investigated to reduce cisplatin-induced nephrotoxicity.
Keywords: Kidney toxicity, cisplatin, miRNAs, microRNA-155
Cisplatin is a highly effective chemotherapeutic agent that mediates its anticancer activity by inducing cell cycle arrest and apoptosis. The anticancer activity of cisplatin is generally proportional to the dosage, but the development of nephrotoxicity, which also occurs in a dose-dependent manner, limits the achievable dosage and treatment intervals (Pabla and Dong, 2008; Wang and Lippard, 2005). The nephrotoxic injury induced by cisplatin is multifactorial, involving cell cycle arrest, cell death pathways, apoptotic signaling, the production of reactive oxygen species leading to activation of the oxidative stress response, and the effects of surrounding inflammatory cells. A number of targets relating to each of these factors have been investigated for their potential to protect the kidney from damage during cisplatin treatment (Pabla and Dong, 2008). Previous studies investigating the molecular mechanisms of cisplatin-induced cell death have identified microRNAs (miRNAs) that are involved in the regulation of apoptotic processes in cancer cells (Drayton, 2012). Therefore, we were interested to determine the role of miRNAs in the response to cisplatin-induced injury in the kidney.
We have previously shown miR-155 to be highly upregulated following bilateral renal ischemia reperfusion injury (IRI) or the administration of gentamicin at concentrations toxic to rat kidneys (Saikumar et al., 2012). Increased expression of miR-155 has also been found in solid tumors of diverse origin such as breast, lung, colon, and pancreas (Gironella et al., 2007; Volinia et al., 2006). In the kidneys, miR-155 expression has been observed in relation to renal malignancies, immunological events, as well as in response to injury. Normal and cancerous regions of tissues isolated from renal cell carcinoma patients could be distinguished by expression of miR-155, and some association with pathological stage was observed (Juan et al., 2010; Li et al., 2012). High levels of miR-155 expression in the kidneys have also been associated with IgA nephropathy, and renal allografts undergoing acute rejection (Anglicheau et al., 2009; Wang et al., 2011). More recently, it has been shown that miR-155 is upregulated in the kidneys of patients with glomerulonephritis and that miR-155−/− mice have a reduced onset of glomerulonephritis characterized by an attenuated Th17 response (Krebs et al., 2013).
Given that miR-155 has been implicated in the regulation of a number of pathways related to the induction of apoptosis and the control of inflammation, both of which are important for the progression of cisplatin-induced kidney toxicity, we investigated whether mice deficient in the expression of miR-155 would have differences in the development of kidney toxicity following the administration of cisplatin. We found an increased severity of kidney injury in the miR-155−/− mice in response to cisplatin, with higher levels of apoptosis observed, and gene expression analysis indicated that the miR-155 target, c-Fos, may be dysregulated in these mice. The research presented here demonstrates a role for miR-155 in the regulation of the injury response in kidneys exposed to cisplatin, and may indicate novel targets in cisplatin-induced nephrotoxicity.
MATERIALS AND METHODS
Mice
Prof. Howard Weiner (Brigham and Women's Hospital) kindly provided breeding pairs of miR-155−/− mice (B6.Cg-Mir155tm1.1Rsky/J [Thai et al., 2007] originally purchased from The Jackson Laboratory). Age and weight matched C57BL/6 (wild type) mice were used as controls. All mice were maintained under standard housing conditions in accordance with NIH guidelines for the care and use of laboratory animals. All animal experiments were performed with the approval of the Institutional Animal Care and Use Committees (IACUC) of Harvard Medical School.
Kidney injury and collection of tissues
One group of male mice was injected with a single 20 mg/kg dose of cisplatin as described previously, with saline-injected mice used as controls (Hoffmann et al., 2012). Another group of male mice were subjected to bilateral renal IRI surgery for 30 min as described previously (Ajay et al., 2012). Animals that received sham surgery underwent anesthesia and laparotomy only and were sacrificed after 24 h to be used as controls. The unilateral ureteral obstruction (UUO) studies were conducted in female mice. These mice were anesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg of body weight), the left kidney was exposed via a flank incision, and a 3.0 silk suture used to tie off the ureter at the lower pole. The contralateral kidneys were used as controls.
At the time of sacrifice, blood was transferred into heparinized tubes and centrifuged at 10000 × g for 10 min at 4°C to separate the plasma, which was collected and stored at −80°C for further analysis. The kidneys collected for histology were cut along the frontal plane and fixed in 10% neutral buffered formalin overnight before being transferred to 70% ethanol. After being processed and embedded in paraffin, 6 μm sections were prepared. The remaining kidney tissue was snap frozen in liquid nitrogen for RNA and protein analysis.
Clinical chemistry and histology scoring
Serum creatinine (SCr) and blood urea nitrogen (BUN) were measured as described previously (Hoffmann et al., 2012; Krishnamoorthy et al., 2011). An independent pathologist evaluated H&E stained sections under a light microscope (only the time point for each group of samples was disclosed). A kidney injury score grading scale from 0 to 5 was used to assess the severity of the injury: 0 = no lesions; 1 = minimal injury characterized by occurrence of necrosis and debris; 2 = mild injury with single cell necrosis, pyknotic cells, and apoptosis; 3 = moderate injury characterized by tubular distension, vacuolation, and some cellular debris; 4 = severe injury with occasional hyaline casts observed, patchy epithelial necrosis, and attenuated tubular epithelial lining; and 5 = very severe injury characterized by extensive tubular epithelial necrosis in all segments, loss of epithelial layer from many tubules with widespread intraluminal cellular debris, and frequent hyaline casts particularly prominent in the medullary region. The injury was evenly distributed throughout the kidneys, and the percentage of tubules exhibiting damage was incorporated into the scoring with higher scores correlating with larger areas of affected tissue. Representative images demonstrating the histological changes observed for each acute tubular injury score level are shown in Supplementary figure 1.
RNA isolation and qRT-PCR
RNA was isolated from kidney tissue using Trizol Reagent (Life Technologies), with the quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) carried out as described previously (Krishnamoorthy et al., 2011). The miRNA was analyzed using a TaqMan MicroRNA Reverse Transcription Kit and TaqMan Universal Master Mix II with TaqMan MicroRNA Assays for mmu-miR-155-5p and U87 (Life Technologies).
Microarray and data analysis
RNA was extracted from kidney tissues using miRNeasy Mini Kits (Qiagen). Microarray analysis was conducted using 500 ng of total RNA and Mouse GE 4×44K v2 Microarrays (Agilent). Sample labeling, microarray hybridization, and washing were done following the One-Color Microarray-Based Gene Expression Analysis Protocol (Agilent, v6.5). The microarrays were scanned with the Agilent DNA Microarray Scanner (5 μm resolution) and the resulting images analyzed by Agilent Feature Extraction Software (v10.5). Array data were normalized using 75th percentile normalization (TargetValue = 1000) within USFDA's ArrayTrack (Fang et al., 2009; Tong et al., 2003) and differentially expressed genes were identified using limma (Smyth, 2004, 2005). Genes that were differentially expressed (p < 0.0001) were used for further analysis with Ingenuity Pathway Analysis (IPA). Heatmaps were generated using the MultiExperiment Viewer software. MicroRNA-155 targets were identified using the microRNA.org database (Betel et al., 2008, 2010). The complete dataset is available in the GEO database (GSE57292).
Analysis of apoptosis
TUNEL staining was carried out using 6 μm Formalin-Fixed, Paraffin-Embedded (FFPE) sections and an In Situ Cell Death Detection Kit with Fluorescein detection (Roche). Ten fields of view were randomly captured for each sample without knowledge of the sample's group and TUNEL positive nuclei were determined using ImageJ.
Protein and western blotting
Protein was extracted from tissue homogenates using RIPA buffer, denatured with protein loading dye, and resolved on a 4–12% Bis-Tris gel (Life Technologies). The proteins were transferred to nitrocellulose membrane and probed for c-Fos (Santa Cruz), and β-actin (Cell Signaling Technology), and imaged using a ChemiDoc (BioRad). The images were collected and analyzed using ImageLab software (BioRad).
Statistical analyses
Results are expressed as mean ± SEM. Statistical analyses were performed using t-tests with a statistical significance of p < 0.05 (GraphPad Prism software).
RESULTS
MicroRNA-155 Deficient Mice Demonstrate a Higher Susceptibility to Cisplatin Nephrotoxicity
Given our previous identification of miR-155 as a highly upregulated miRNA in response to ischemic and toxic insult to the kidney, we sought to determine the role of miR-155 in cisplatin-induced kidney injury. We found that miR-155−/− (knockout) mice treated with a single dose of 20 mg/kg cisplatin displayed a considerably higher level of kidney injury than C57BL/6 (wild type) controls. At 72 h, the level of SCr was 6.7-fold higher in the miR-155−/− mice as compared with C57BL/6 mice (p = 0.048; Fig. 1A), whereas BUN was 1.4-fold higher in the knockout at 72 h (p = 0.036; Fig. 1B). The expression of Kim-1 mRNA in the kidneys of miR-155−/− mice was 2.7-fold higher than C57BL/6 mice at 72 h (p < 0.001; Fig. 1C). A histological examination of H&E stained kidneys demonstrated a severe progressive kidney injury that was characterized by widespread necrosis and tubular distension (Figs. 1D and E). Both the wild-type and knockout mice showed similar histological features at 24 h, with minimal frequency of single cell necrosis and occasional karyopyknosis. However, from 48 h onward, the miR-155−/− mice had a significantly higher degree of injury as demonstrated by diffuse degenerative epithelial changes including low or absent epithelial lining with prominent epithelial necrosis in all segments and widespread intraluminal cellular debris. In contrast, these changes were rarely observed in the C57BL/6 mice and they displayed a milder injury, with only focal tubular distension and loss of epithelial lining, in addition to less evident necrosis, epithelial vacuolation, and only occasional collections of intraluminal cellular debris (p < 0.05 from 48 h onward; Figs. 1D and E). These findings demonstrated that the miR-155−/− mice developed a higher level of kidney injury following the administration of cisplatin.
FIG. 1.
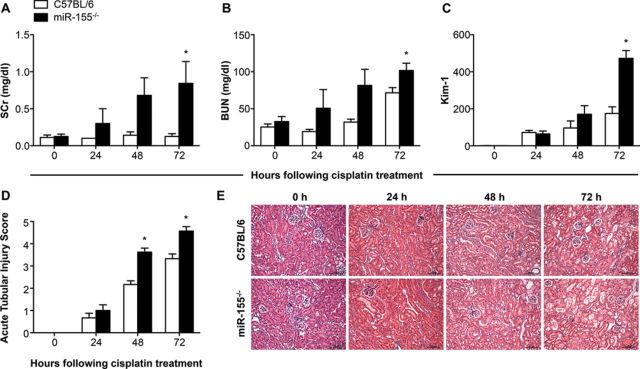
Analysis of kidney injury in miR-155−/− mice treated with cisplatin. The levels of serum creatinine (SCr; A) and blood urea nitrogen (BUN; B) were measured at 0, 24, 48, and 72 h following the administration of cisplatin. Kim-1 mRNA from kidney lysates was assessed by qRT-PCR, normalized to Gapdh, and is shown as fold change relative to the 0 h C57BL/6 group (C). An independent pathologist scored the histological injury in a blinded manner (D), and representative images from each group at each time point are shown (E; scale bar = 100 μm). Data are represented as mean ± SEM and *p < 0.05 in comparison to the C57BL/6 group at the same time point (n = 4–8 mice/group).
The Responses to Ischemic and Fibrotic Kidney Injuries Are Not Affected by miR-155 Expression
To determine whether the increased severity of kidney injury was specific to cisplatin-induced kidney toxicity, we also investigated the response of miR-155−/− mice to bilateral renal IRI and kidney fibrosis induced by UUO. The mice subjected to bilateral renal IRI demonstrated a peak of SCr and BUN at 24 and 48 h, and significantly increased expression of Kim-1 mRNA in the kidneys at 24 h when compared with mice that had been subjected to sham surgery (Supplementary figs. 2A–C). However, comparison of wild-type and knockout mice at each time point did not show any differences in the level of injury. Similarly, mice subjected to UUO surgery exhibited high levels of Kim-1 expression in kidneys at 7 and 14 days post-surgery (Supplementary fig. 2E), in addition to sustained increases in the expression of fibrosis-associated genes such as α-smooth muscle actin, collagen 1A1, and fibronectin (Supplementary figs. 2F–H), but no differences were observed in either the injury response or the level of fibrosis when the knockout and the wild type were directly compared. This would suggest that miR-155 may be regulating a protein or pathway that is central to the development of cisplatin-induced nephrotoxicity, whereas any proteins or pathways that may be targeted by miR-155 in the IRI or UUO models are not essential for the development of ischemic or fibrotic kidney injury.
We hypothesized that miR-155 expression would be increased over the course of the cisplatin injury, but not in mice subjected to IRI or UUO where the C57BL/6 and miR-155−/− mice showed the same response to injury. To establish whether miR-155 expression differed between each of these models, and therefore might explain why the increased injury was only observed in the miR-155−/− mice treated with cisplatin, expression of miR-155 in the kidneys of C57BL/6 mice was measured using Taqman miRNA assays and normalized to U87. The expression of miR-155 did not change over the course of the cisplatin injury or bilateral renal IRI in C57BL/6 mice (Supplementary figs. 3A and B), but at 7 and 14 days following UUO surgery, miR-155 was 1.8- and 3.5-fold higher, respectively (p < 0.01; Supplementary fig. 3C). These results indicate that mice and rats have different miRNA expression in response to ischemic and toxic injuries to the kidneys, as we have shown here that mice subjected to bilateral renal IRI or the administration of cisplatin showed no change in miR-155 expression, whereas we had previously seen that miR-155 expression was increased in rats exposed to similar injuries (Saikumar et al., 2012).
Gene Expression Profiling Indicates Higher Apoptotic and Oxidative Stress Signaling in miR-155−/− Mice Following Cisplatin Treatment
To investigate the molecular mechanisms responsible for increased cisplatin-induced toxicity in the miR-155−/− mice, we performed genome-wide expression profiling on kidneys isolated from wild-type and knockout mice at 0, 24, 48, and 72 h. We began by analyzing the gene expression changes that occurred over the course of the injury using limma (Smyth, 2004, 2005) with a significance threshold of p < 0.0001. Of the 2243 genes that were observed to be differentially expressed during at least one time point in the C57BL/6 mice, 1178 were upregulated and 1065 were downregulated (Fig. 2A). In contrast, we observed 3266 genes to have a significant change in expression in the miR-155−/− mice, with 1791 genes upregulated and 1475 genes downregulated (Fig. 2A). We used the calculated fold changes and p values for these genes to conduct an analysis using IPA software. There were a number of pathways related to apoptotic signaling and oxidative stress that were commonly upregulated across the time course of injury in both the knockout and the wild-type mice. Of the top five pathways identified, the NRF2-Mediated Oxidative Stress Response and p53 Signaling were more strongly associated with the 24 h time point for both the knockout and the wild-type mice, whereas Apoptosis Signaling, Production of Nitric Oxide and Reactive Oxygen Species by Macrophages, and Acute Phase Response Signaling showed a stronger association at 48 and 72 h (Fig. 2B). In comparing the two strains of mice, we found that a higher ratio of the genes comprising each canonical pathway were found in the dataset of differentially expressed genes from the miR-155−/− mice, especially at later time points (Fig. 2C).
FIG. 2.
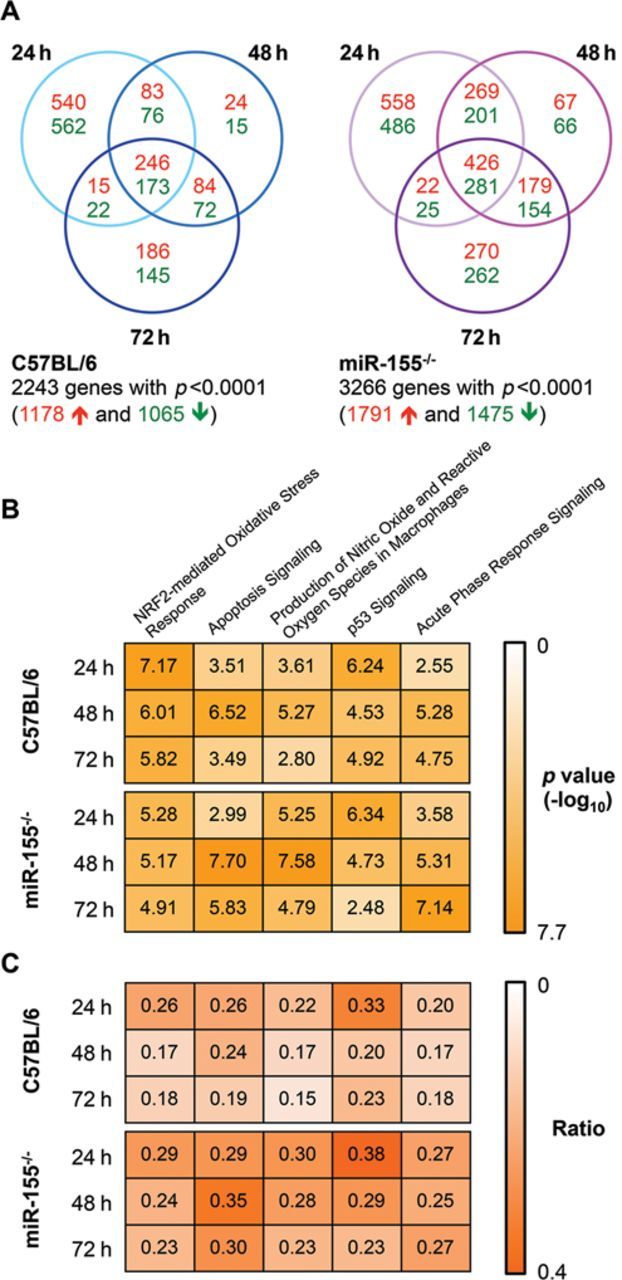
Microarray analysis of cisplatin treated mice identified the most significantly associated canonical pathways. Genes with differential expression following the administration of cisplatin were determined using limma with a significance threshold of p < 0.0001. The number of genes significantly upregulated (red text) or downregulated (green text) at each time point is shown for both C57BL/6 and miR-155−/− mice (A). Ingenuity Pathway Analysis was used to determine canonical signaling pathways associated with these genes. The associated p values for the top five pathways are shown (B; the values are shown as –log10(p value); the maximum value of 7.70 corresponds to the lowest p value of 2 × 10−08). The ratio for each of these pathways is shown (C) and represents the proportion of genes in that pathway that are significantly differentially expressed in each data set.
When we visualized apoptosis occurring in the kidneys using TUNEL staining, we found a 1.8-fold increase in apoptotic nuclei in the knockout mice at 72 h with over 100 TUNEL positive nuclei observed for each field of view in the knockout (p = 0.033; Figs. 3A and B). In contrast, generally fewer than 65 TUNEL positive nuclei were observed per field of view for the C57BL/6 mice (Figs. 3A and B). Investigation of a few selected inflammatory and injury response cytokines associated with the identified canonical pathways demonstrated 2.1- and 3.7-fold higher levels of TNFα and IL-1β in the miR-155−/− mice at 72 h (p = 0.027 and 0.023; Figs. 3C and D). IL-6 expression was noticeably increased over the course of the injury in both wild-type and knockout mice, and was 8.0-fold higher in the miR-155−/− mice at 72 h when compared with the wild-type mice (p = 0.059; Fig. 3E). This indicates a higher level of kidney damage in the miR-155−/− mice following cisplatin administration, demonstrated by increased levels of injury-associated cytokines and apoptotic cells in the knockout mice.
FIG. 3.
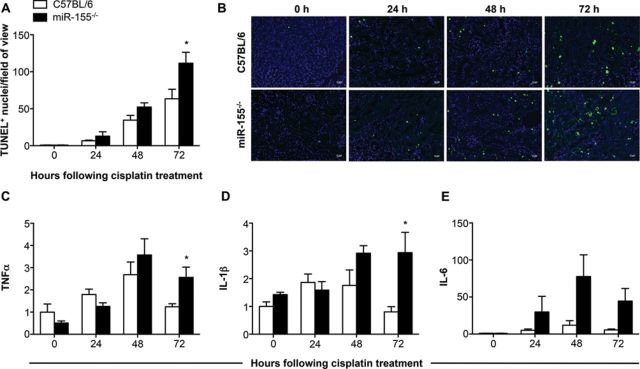
Measurement of apoptosis and inflammatory cytokines in the kidneys of mice following cisplatin injury. The number of TUNEL positive nuclei per field of view was quantified (A), with representative images of each group at each time point shown (B; scale bar = 20 μm). The transcript levels of TNF-α, IL-1β, and IL-6 were measured, normalized to Gapdh, and are shown as fold change relative to the 0 h C57BL/6 group (C, D, and E, respectively). Data are represented as mean ± SEM and *p < 0.05 in comparison with the C57BL/6 group at the same time point (n = 4–8 mice/group).
Cisplatin Injury Induces Higher Expression of the miR-155 Target c-Fos in the Knockout Mice
To identify genes that may be responsible for the increased severity of kidney injury in the miR-155−/− mice, we identified transcription factors that were predicted to be upstream regulators of the observed gene expression (using IPA). Two transcription factors central to the canonical pathway predictions, p53 and Nrf2, were both identified as potential upstream regulators in addition to c-Fos, Hnf4a, and Trim24. Using the microRNA.org resource, we identified miR-155 binding sites within the 3′ UTR for four of the top five predicted upstream regulators (Fig. 4). The mirSVR score uses miRanda sequence binding predictions calibrated to correlate with target gene downregulation and can be interpreted as an empirical probability of target inhibition (Betel et al., 2010). This database also calculates a PhastCons score for each predicted binding site that corresponds to the phylogenetic conservation of the binding site and may imply a conservation of the miRNA-mRNA interaction (Betel et al., 2008). The authors of this resource recommend a maximum of −0.1 for the mirSVR score to increase the likelihood of identifying a binding pair with a meaningful interaction, and a cut-off of 0.57 to select for target site conservation among mammals (Betel et al., 2008, 2010). The single site predicted for Hnf4a does not meet the recommended criteria for the scoring of the binding site, or mammalian conservation (Fig. 4C), whereas Trim24 has a single predicted binding site that narrowly meets the recommended mirSVR and PhastCons score limits (Fig. 4D). The 3′ UTR of Nrf2 has a predicted miR-155 binding site that has a PhastCons score that would not indicate sequence conservation, but this site has a very high mirSVR score suggesting a high probability of functional binding (Fig. 4A). Of the three miR-155 binding sites predicted to exist in the c-Fos 3′ UTR, the two binding sites closest to the 3′ end both have high mirSVR and PhastCons scores, indicating mammalian conservation and a high probability of functional interactions (Fig. 4B). Of these transcription factors, c-Fos has previously been shown to be regulated by miR-155, with luciferase reporter assays confirming a functional interaction for both the human and mouse c-Fos 3′ UTR (Dunand-Sauthier et al., 2011).
FIG. 4.

miR-155 binding sites in the 3′ UTR of upstream regulators. The 3′ UTR of upstream regulators were assessed to determine if there were potential binding sites for miR-155 using the microRNA.org resource. Each binding site is predicted using the miRanda algorithm and a mirSVR score is assigned to represent the likelihood of target downregulation occurring, and the PhastCons score determines the degree of phylogenetic conservation of the interaction. Potential binding sites were identified for Nrf2 (A), c-Fos (B), Hnf4a (C), and Trim24 (D). For each gene, the position of the binding site in the 3′ UTR and the predicted binding interaction with miR-155 is shown (the | symbol represents Watson-Crick base pairing, and the : symbol represents G-U wobble base pairing).
Results from the microarray experiment indicated the expression of both Nrf2 and c-Fos to be increased over the course of cisplatin injury in the C57BL/6 and miR-155−/− mice, whereas the expression of Trim24 was unchanged in response to cisplatin. The expression of Hnf4a was decreased (data not shown). The expression changes of Nrf2 and c-Fos were confirmed by qRT-PCR. No significant difference in Nrf2 expression was observed between the knockout and the wild type at any of the time points (Fig. 5A), but the expression of c-Fos was 2.9-fold higher in the miR-155−/− mice when compared with the C57BL/6 mice at 72 h (p = 0.019; Fig. 5B). Western blot analysis of the 72 h samples demonstrated a 5.6-fold higher expression of a 66 kDa protein recognized by an anti-c-Fos antibody in the knockout mice, indicating that the protein levels of c-Fos were also affected by the absence of miR-155 regulation (p = 0.042; Figs. 5C and D). To gain a better understanding of the gene expression changes that might have occurred in response to increased expression of c-Fos, we used the list of c-Fos regulated genes identified with IPA to create a subset of the microarray data. We retained all genes that showed at least a twofold change in either direction during at least one time point, separated the list based on whether the gene was upregulated or downregulated during the injury in the C57BL/6 mice, and performed hierarchal clustering using MultiExperiment Viewer software (Figs. 5E and F). Overall, the c-Fos regulated genes showed similar expression patterns, but there were a number of gene clusters showing a heightened expression in the miR-155−/− mice, including genes such as Socs3, Mapk6, Tgf-β1, and Mmp10 that had markedly higher expression in the knockout at 72 h (Fig. 5F). Additionally, c-Fos binding partners such as Mafk and c-Jun were also noticeably upregulated in the miR-155−/− mice at 72 h (Fig. 5F). These results demonstrate the increased expression of c-Fos in the knockout mice, which are unable to mediate regulation of gene expression via miR-155, and suggest that unregulated production of c-Fos in response to cisplatin may increase the level of apoptosis in the kidneys.
FIG. 5.
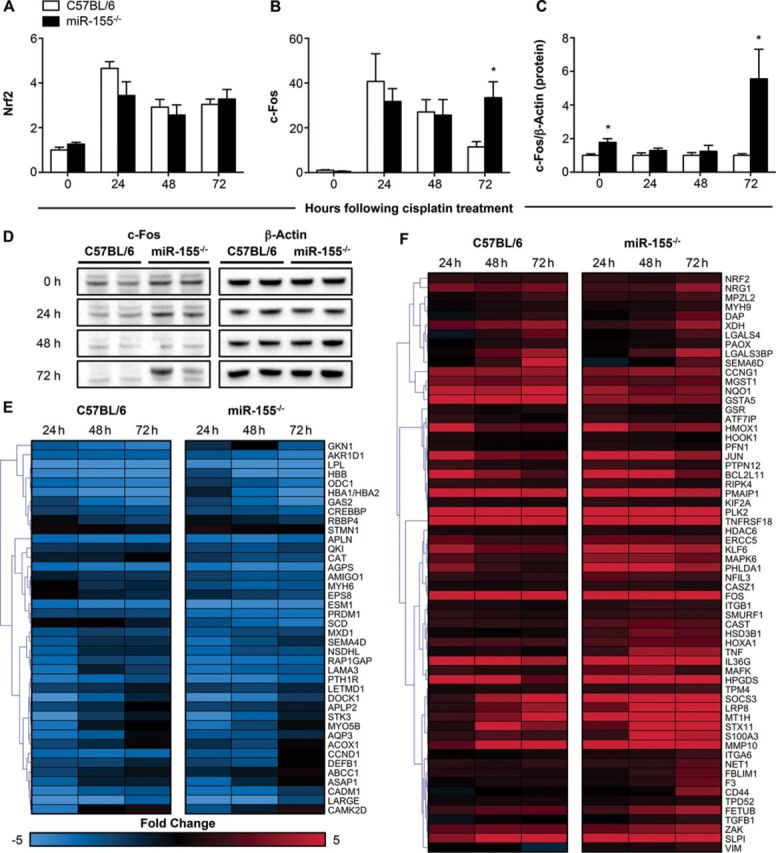
Expression of potential upstream regulators and c-Fos target genes. Upstream regulators identified by IPA included Nrf2 and c-Fos, which are predicted targets of miR-155. The gene expression levels of Nrf2 (A) and c-Fos (B) in the kidneys were measured by qRT-PCR, normalized to Gapdh, and are shown as fold change relative to the untreated group of C57BL/6 mice (n = 4–8 mice/group). The increased presence of c-Fos protein in kidney lysates was assessed by immunoblotting, normalized to β-actin, quantified, and expressed as fold change relative to the C57BL/6 mice at the same time point (C; n = 4 mice/group). Representative blots for c-Fos and β-actin are shown for two C57BL/6 and two miR-155−/− mice at each time point (D). The expression of c-Fos regulated genes was determined from the microarray, and both downregulated (E) and upregulated (F) c-Fos target genes are shown (expression represented as fold change in comparison to each 0 h group). Data are represented as mean ± SEM and *p < 0.05 in comparison with the C57BL/6 group at the same time point.
DISCUSSION
In this paper, we have shown that miR-155 knockout mice experience a greater degree of kidney injury following the administration of cisplatin. When we expanded our study to include diverse kidney injury models such as bilateral renal IRI and UUO, representing ischemic and fibrotic injuries respectively, we found no difference in the development of injury between the miR-155−/− and C57BL/6 mice, suggesting that miR-155 regulation is specifically important for cisplatin-induced damage in the kidneys. Genome-wide expression profiling of kidneys from the wild-type and knockout mice injected with cisplatin indicated significant upregulation of genes involved in apoptosis signaling, and measurement of apoptosis by TUNEL staining demonstrated increased numbers of apoptotic nuclei in the miR-155−/− mice. Among the top five transcription factors identified as potential upstream regulators we found potential miR-155 binding sites for four of the genes, but only observed differential expression for c-Fos. Our data indicate that c-Fos may be dysregulated in the miR-155−/− mice, and that the increased expression of c-Fos contributes to apoptosis. c-Fos functions as one part of the dimeric transcription factor, Activator Protein 1 (AP-1), in combination with Jun proteins such as c-Jun or JunB, or proteins of the ATF or Maf families (Eferl and Wagner, 2003).
AP-1 complexes have established roles in processes essential for cell survival, such as proliferation and differentiation, but can also act to induce apoptosis depending on the combination of proteins composing the AP-1 dimer, as well as the cell type and context (Fleischmann et al., 2003; Ishihara et al., 2011; Zhang et al., 2007). In particular, the combination of c-Fos with JunB or JunD is understood to inhibit anti-apoptotic proteins such as Bcl2 and Bcl-xL, resulting in increased apoptosis (Eferl and Wagner, 2003; Passegue et al., 2001). Analysis of the microarray data indicates that the expression of JunB and JunD is both higher in the miR-155−/− mice at 72 h, but to understand how c-Fos may be linked to the increased levels of apoptosis seen in the miR-155−/− mice, it will be necessary to establish the binding partner of c-Fos in the AP-1 complex in this model. The increased expression of c-Fos has previously been observed in human primary proximal tubule cells treated with cisplatin, and a functional AP-1 complex can be detected as soon as 2 h following treatment of the cells (Wainford et al., 2009). Additionally, the ectopic expression of c-Fos in serum-starved human colorectal cancer cell lines was sufficient to induce apoptosis, whereas in human hepatoma cells the induced expression of a dominant negative form of c-Fos protected the cells from apoptosis (Kalra and Kumar, 2004; Preston et al., 1996). These previous findings provide evidence for the increased expression of c-Fos resulting in the induction of apoptosis, and indicate mechanisms by which the higher levels of c-Fos we have observed in the miR-155−/− mice may lead to more apoptosis.
Whereas we have identified an association between miR-155 and c-Fos in our study, miRNAs are able to regulate multiple genes simultaneously, and it is therefore possible that other pro-apoptotic genes could also be inhibited by miR-155 during the cisplatin response. Apart from the regulation of c-Fos (Dunand-Sauthier et al., 2011), miR-155 has demonstrated functional relationships with a number of other apoptosis-related genes. In Capan2 cells, inhibition of miR-155 was shown to increase the expression of Tp53inp1, and exposure to gamma radiation led to higher levels of apoptosis in cells without miR-155 (Gironella et al., 2007). An inverse correlation between miR-155 and the tumor suppressor Foxo3a has been observed in breast cancer cell lines and tissues, and the inhibition of miR-155 led to increased apoptosis in cells treated with doxorubicin, paclitaxel, or VP16 (Kong et al., 2010). Higher levels of apoptosis in response to cisplatin have also been observed in A549 cells with inhibited expression of miR-155, and a relationship between miR-155 and the apoptosis activator Apaf-1 was suggested as the increased susceptibility to apoptosis was reduced when Apaf-1 was also inhibited (Zang et al., 2012). Other genes that have previously been associated with apoptosis and are confirmed targets of miR-155 include Bach1, Map3k7ip2, Pmaip1, Tspan14, and Lpin1 (Ceppi et al., 2009; Koch et al., 2012). These results in combination with our findings provide evidence for the ability of miR-155 to regulate the apoptotic response to DNA-damaging compounds such as cisplatin. Investigation of cisplatin-induced kidney toxicity in mice deficient for both c-Fos and miR-155 would indicate whether the regulation of c-Fos is the primary mediator of the effect we have observed here, or if additional apoptotic proteins are regulated in the kidneys following the administration of cisplatin.
The findings presented here demonstrate that the inhibition of miR-155 can affect the development of cisplatin-induced kidney toxicity, leading to increased apoptosis and cell death. We identified c-Fos as a potential target of miR-155 during the cisplatin response, and confirmed differential expression of c-Fos mRNA and protein in the miR-155−/− mice at 72 h. A greater understanding of the various molecular mechanisms responsible for cisplatin-induced kidney toxicity is essential in the development of targeted therapeutics to reduce the effect of nephrotoxicity in clinical practice without sacrificing anti-tumor efficacy.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institute of Environmental Health Sciences (Outstanding New Environmental Scientist Award [ES017543] to V.S.V.).
Supplementary Material
Disclaimer: The findings and conclusions presented in this article are those of the authors and do not necessarily reflect those of the Food and Drug Administration.
REFERENCES
- Ajay A. K., Saikumar J., Bijol V., Vaidya V. S. Heterozygosity for fibrinogen results in efficient resolution of kidney ischemia reperfusion injury. PLoS One. 2012;7:e45628. doi: 10.1371/journal.pone.0045628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglicheau D., Sharma V. K., Ding R., Hummel A., Snopkowski C., Dadhania D., Seshan S. V., Suthanthiran M. MicroRNA expression profiles predictive of human renal allograft status. Proc. Natl. Acad. Sci. U.S.A. 2009;106:5330–5335. doi: 10.1073/pnas.0813121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D., Koppal A., Agius P., Sander C., Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D., Wilson M., Gabow A., Marks D. S., Sander C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceppi M., Pereira P. M., Dunand-Sauthier I., Barras E., Reith W., Santos M. A., Pierre P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2735–2740. doi: 10.1073/pnas.0811073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayton R. M. The role of microRNA in the response to cisplatin treatment. Biochem. Soc. Trans. 2012;40:821–825. doi: 10.1042/BST20120055. [DOI] [PubMed] [Google Scholar]
- Dunand-Sauthier I., Santiago-Raber M. L., Capponi L., Vejnar C. E., Schaad O., Irla M., Seguin-Estevez Q., Descombes P., Zdobnov E. M., Acha-Orbea H., et al. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood. 2011;117:4490–4500. doi: 10.1182/blood-2010-09-308064. [DOI] [PubMed] [Google Scholar]
- Eferl R., Wagner E. F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Fang H., Harris S. C., Su Z., Chen M., Qian F., Shi L., Perkins R., Tong W. ArrayTrack: An FDA and public genomic tool. Methods Mol. Biol. 2009;563:379–398. doi: 10.1007/978-1-60761-175-2_20. [DOI] [PubMed] [Google Scholar]
- Fleischmann A., Jochum W., Eferl R., Witowsky J., Wagner E. F. Rhabdomyosarcoma development in mice lacking Trp53 and Fos: Tumor suppression by the Fos protooncogene. Cancer Cell. 2003;4:477–482. doi: 10.1016/s1535-6108(03)00280-0. [DOI] [PubMed] [Google Scholar]
- Gironella M., Seux M., Xie M. J., Cano C., Tomasini R., Gommeaux J., Garcia S., Nowak J., Yeung M. L., Jeang K. T., et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc. Natl. Acad. Sci. U.S.A. 2007;104:16170–16175. doi: 10.1073/pnas.0703942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann D., Bijol V., Krishnamoorthy A., Gonzalez V. R., Frendl G., Zhang Q., Goering P. L., Brown R. P., Waikar S. S., Vaidya V. S. Fibrinogen excretion in the urine and immunoreactivity in the kidney serves as a translational biomarker for acute kidney injury. Am. J. Pathol. 2012;181:818–828. doi: 10.1016/j.ajpath.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara Y., Ito F., Shimamoto N. Increased expression of c-Fos by extracellular signal-regulated kinase activation under sustained oxidative stress elicits BimEL upregulation and hepatocyte apoptosis. FEBS J. 2011;278:1873–1881. doi: 10.1111/j.1742-4658.2011.08105.x. [DOI] [PubMed] [Google Scholar]
- Juan D., Alexe G., Antes T., Liu H., Madabhushi A., Delisi C., Ganesan S., Bhanot G., Liou L. S. Identification of a microRNA panel for clear-cell kidney cancer. Urology. 2010;75:835–841. doi: 10.1016/j.urology.2009.10.033. [DOI] [PubMed] [Google Scholar]
- Kalra N., Kumar V. c-Fos is a mediator of the c-myc-induced apoptotic signaling in serum-deprived hepatoma cells via the p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 2004;279:25313–25319. doi: 10.1074/jbc.M400932200. [DOI] [PubMed] [Google Scholar]
- Koch M., Mollenkopf H. J., Klemm U., Meyer T. F. Induction of microRNA-155 is TLR- and type IV secretion system-dependent in macrophages and inhibits DNA-damage induced apoptosis. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E1153–E1162. doi: 10.1073/pnas.1116125109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W., He L., Coppola M., Guo J., Esposito N. N., Coppola D., Cheng J. Q. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J. Biol. Chem. 2010;285:17869–17879. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Krebs C. F., Kapffer S., Paust H. J., Schmidt T., Bennstein S. B., Peters A., Stege G., Brix S. R., Meyer-Schwesinger C., Muller R. U., et al. MicroRNA-155 drives TH17 immune response and tissue injury in experimental crescentic GN. J. Am. Soc. Nephrol. 2013;24:1955–1965. doi: 10.1681/ASN.2013020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy A., Ajay A. K., Hoffmann D., Kim T. M., Ramirez V., Campanholle G., Bobadilla N. A., Waikar S. S., Vaidya V. S. Fibrinogen beta-derived Bbeta(15–42) peptide protects against kidney ischemia/reperfusion injury. Blood. 2011;118:1934–1942. doi: 10.1182/blood-2011-02-338061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Chen T., Zhong Z., Wang Y., Li Y., Zhao X. microRNA-155 silencing inhibits proliferation and migration and induces apoptosis by upregulating BACH1 in renal cancer cells. Mol. Med. Rep. 2012;5:949–954. doi: 10.3892/mmr.2012.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabla N., Dong Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- Passegue E., Jochum W., Schorpp-Kistner M., Mohle-Steinlein U., Wagner E. F. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell. 2001;104:21–32. doi: 10.1016/s0092-8674(01)00188-x. [DOI] [PubMed] [Google Scholar]
- Preston G. A., Lyon T. T., Yin Y., Lang J. E., Solomon G., Annab L., Srinivasan D. G., Alcorta D. A., Barrett J. C. Induction of apoptosis by c-Fos protein. Mol. Cell. Biol. 1996;16:211–218. doi: 10.1128/mcb.16.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikumar J., Hoffmann D., Kim T. M., Gonzalez V. R., Zhang Q., Goering P. L., Brown R. P., Bijol V., Park P. J., Waikar S. S., et al. Expression, circulation, and excretion profile of microRNA-21, -155, and -18a following acute kidney injury. Toxicol. Sci. 2012;129:256–267. doi: 10.1093/toxsci/kfs210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Smyth G. K. Limma: Linear models for microarray data. In: Gentleman R., Carey V., Dudoit S., Irizarry R., Huber W., editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York, NY: Springer; 2005. pp. 397–420. [Google Scholar]
- Thai T. H., Calado D. P., Casola S., Ansel K. M., Xiao C., Xue Y., Murphy A., Frendewey D., Valenzuela D., Kutok J. L., et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- Tong W., Cao X., Harris S., Sun H., Fang H., Fuscoe J., Harris A., Hong H., Xie Q., Perkins R., et al. ArrayTrack–supporting toxicogenomic research at the U.S. Food and Drug Administration National Center for Toxicological Research. Environ. Health Perspect. 2003;111:1819–1826. doi: 10.1289/ehp.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S., Calin G. A., Liu C. G., Ambs S., Cimmino A., Petrocca F., Visone R., Iorio M., Roldo C., Ferracin M., et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainford R. D., Weaver R. J., Hawksworth G. M. The immediate early genes, c-fos, c-jun and AP-1, are early markers of platinum analogue toxicity in human proximal tubular cell primary cultures. Toxicol. In Vitro. 2009;23:780–788. doi: 10.1016/j.tiv.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Wang D., Lippard S. J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- Wang G., Kwan B. C., Lai F. M., Chow K. M., Li P. K., Szeto C. C. Elevated levels of miR-146a and miR-155 in kidney biopsy and urine from patients with IgA nephropathy. Dis. Markers. 2011;30:171–179. doi: 10.3233/DMA-2011-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang Y. S., Zhong Y. F., Fang Z., Li B., An J. MiR-155 inhibits the sensitivity of lung cancer cells to cisplatin via negative regulation of Apaf-1 expression. Cancer Gene Ther. 2012;19:773–778. doi: 10.1038/cgt.2012.60. [DOI] [PubMed] [Google Scholar]
- Zhang X., Zhang L., Yang H., Huang X., Otu H., Libermann T. A., DeWolf W. C., Khosravi-Far R., Olumi A. F. c-Fos as a proapoptotic agent in TRAIL-induced apoptosis in prostate cancer cells. Cancer Res. 2007;67:9425–9434. doi: 10.1158/0008-5472.CAN-07-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.