

Abstract
Aims
About one-third of American adults and 20% of teenagers are obese. Obesity and its associated metabolic disturbances including hyperlipidaemia are risk factors for cardiovascular diseases including stroke. They can worsen neurological outcome after stroke. We determined whether obesity and hyperlipidaemia could induce cerebral vascular remodelling via matrix metalloproteases (MMP) and whether this remodelling affected neurological outcome after brain ischaemia.
Methods and results
Six-week-old male CD1, C57BL/6J, and MMP-9−/− mice were fed regular diet (RD) or high-fat diet (HFD) for 10 weeks. They were subjected to vascular casting or a 90 min middle cerebral arterial occlusion (MCAO). Mice on HFD were heavier and had higher blood glucose and lipid levels than those on RD. HFD-fed CD1 and C57BL/6J mice had an increased cerebral vascular tortuosity index and decreased inner diameters of the middle cerebral arterial root. HFD increased microvessel density in CD1 mouse cerebral cortex. After MCAO, CD1 and C57BL/6J mice on HFD had a bigger infarct volume, more severe brain oedema and blood–brain barrier damage, higher haemorrhagic transformation rate, greater haemorrhagic volume, and worse neurological function. HFD increased MMP-9 activity in the ischaemic and non-ischaemic brain tissues. Although HFD increased the body weights, blood glucose, and lipid levels in the MMP-9−/− mice on a C57BL/6J genetic background, the HFD-induced cerebral vascular remodelling and worsening of neurological outcome did not occur in these mice.
Conclusion
HFD induces cerebral vascular remodelling and worsens neurological outcome after transient focal brain ischaemia. MMP-9 activation plays a critical role in these HFD effects.
Keywords: Cerebral vascular remodelling, High fat diet, Ischemic stroke, Matrix metalloprotease, Mouse
1. Introduction
Stroke is a leading cause of death and neurological disability. Despite intensive research efforts, limited progress has been made in the development of pharmacological interventions to reduce ischaemic brain injury. Contributing factors for this situation include the frequent preclinical testing of prospective drugs in animals without risk factors for stroke and the fact that patients suffering from stroke frequently have co-morbidities and risk factors for vascular diseases.1
Obesity and its associated metabolic disturbances including hyperlipidaemia have been identified as risk factors for cardiovascular diseases, diabetes, and many other diseases.2 Obesity prevalence has increased over the years. About one-third of American adults and 20% of teenagers now are obese.3,4 A high-fat diet (HFD) has been considered a significant contributing factor for the pandemic of obesity in the USA5 and increased prevalence of obesity and hyperlipidaemia in young adults may be a major cause of the recent increase in stroke in this age group.6,7 Obesity and hyperlipidaemia also increase the severity of ischaemic brain injury.8,9 However, very little is known about the mechanism for this increase.
Cerebral vascular remodelling induced by various pathological stimuli may contribute to the neurological outcome after brain ischaemia. Diabetes can induce cerebral vascular remodelling.10 Metformin, a hypoglycaemic agent, and minocycline can attenuate cerebral vascular remodelling and the worsened neurological function outcome after transient focal brain ischaemia in diabetic rats. These two drugs also attenuate the increase in activity of matrix metalloprotease-9 (MMP-9) and matrix metalloprotease-2 (MMP-2) in the brains of the diabetic rats.11 HFD feeding increases cerebral blood vessel thickness, reduces cerebral vessel compliance, and increases brain infarct volumes in rats as well as increases blood–brain barrier (BBB) permeability and brain oedema after transient focal brain ischaemia in mice.12,13 However, the mechanisms for the cerebral vascular remodelling in the animals fed an HFD, and whether the remodelling contributes to the worsened neurological outcome after brain ischaemia, have not been investigated.
MMP-2 and MMP-9 are collagenases and gelatinases that exist in the blood vessels and are proposed to be involved in vascular remodelling.14 These MMPs have been implicated in haemorrhagic transformation (HT) after brain ischaemia.15–17 Their expression can be induced in microglia and astrocytes in ischaemic brain tissues.18 Increased MMP-9 activity is considered an important factor for HT after thrombolytic therapy. In fact, increased MMP-9 may be a biomarker for HT in patients after thrombolytic therapy.15 Hyperlipidaemia over-activates MMP-9 in the ischaemic brain tissues.13 Based on these observations, we hypothesize that a chronic HFD induces cerebral vascular remodelling and worsens neurological outcome after brain ischaemia via increases in MMP-9 and MMP-2 activity. To address this hypothesis, we subjected 6-week-old mice to an HFD for 10 weeks to simulate early onset of obesity in teenagers. Remodelling in the cerebral macrovessels and microvessels and the ischaemic tolerance of these mice were determined. The role of MMP-9 and MMP-2 in the vascular remodelling and ischaemic tolerance was assessed by measuring their activities in the brain tissues and using gene knockout mice.
2. Methods
2.1. Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Virginia (Charlottesville, VA, USA). All surgical and experimental procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications number 23–80) revised in 2011. All mice were housed in the vivarium under controlled conditions (23 ± 2°C; 12 h light/dark cycle) with free access to food and water. Male CD1 mice and C57BL/6J mice were purchased from Charles River (Wilmington, MA, USA). A pair of MMP-9 knockout (MMP-9−/−) mice (stock number 007084) from the Jackson laboratory (Bar Harbor, ME, USA) were bred in our vivarium to produce mice used in the study. These mice have a C57BL/6J gene background.
2.2. Feeding
Six-week-old male mice were allowed free access to either HFD (45% of calories supplied by fat; D12451, Research Diets Inc., New Brunswick, NJ, USA) or regular diet (RD; 4.5% of calories supplied by fat; Harland Laboratories, Dublin, VA, USA) for 10 weeks before they were used in experiments. Animals were weighed once a week. Non-invasive blood pressure measurement at the end of 10 weeks of feeding was performed using a CODA monitor (Kent Scientific Corporation, Torrington, CT, USA) as we have described before.19
2.3. Cerebral vasculature casting
Sixteen-week-old male CD1, C57BL/6J, or MMP-9−/− mice were deeply anaesthetized with 5% isoflurane and rapidly perfused via the aorta as described before12 with 37°C warm normal saline followed by 3 mL of 20% freshly made vascular casting resin (PU4ii, VasQtech, Zurich, Switzerland) mixed with blue dye. Brains were removed and fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) at 4°C overnight. Stereomicroscopic photos were taken from the top and from the bottom of the whole brain to determine the interior width of the middle cerebral artery (MCA) root and the collaterals between the MCA and the anterior cerebral artery (ACA) or between the MCA and the posterior cerebral artery (PCA). The vascular tortuosity index was assessed in the medium sized branches of the MCA. This index was calculated as the real vessel length divided by the straight-line distance between the two vessel ends. At least six determinations, each on a different vessel, were performed to calculate the average tortuosity index for each mouse. The left and right MCA root interior diameters were measured and averaged. These determinations were performed by a person who was blinded to the group assignment of the animals.
2.4. Microvessel density and MCA wall thickness determination
Sixteen-week-old male CD1 mice under deep isoflurane anaesthesia were perfused with normal saline followed by ice-cold 4% paraformaldehyde in PBS. Brains were harvested and immersed in 4% paraformaldehyde at 4°C overnight. A brain block containing the MCA root was dissected out, dehydrated, and paraffin embedded. Cross-sections of 5 μm thickness were obtained to assess the wall thickness of the MCA root. At least six random determinations, each at different locations of the vessel in the section, were performed to calculate the average thickness of the MCA for each mouse. Also, 5 μm-thick coronal sections at Bregma −1.18 mm from the left and right hemispheres were obtained to assess the microvessel density. The sections were incubated with a rabbit polyclonal anti-mouse von Willebrand factor (vWF) antibody (1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) followed by an anti-rabbit IgG secondary antibody conjugated with NL-637 (1:200 dilution; R&D Systems, Minneapolis, MN, USA). Microscopic photos of the cerebral cortex were taken with 1/12 s exposure time and ×100 magnification. The mean grey values in five randomly selected 250 μm2 areas in each animal were determined with Image J (National Institutes of Health, Bethesda, MD, USA). The mean value of the five determinations was calculated as the microvessel density index for each animal. These determinations were performed by a researcher blinded to the group assignment of the mice.
2.5. Transient middle cerebral arterial occlusion and neurological outcome assessment
Middle cerebral arterial occlusion (MCAO) in the mice was achieved by an intraluminal filament technique as we described previously.20 Briefly, mice were anaesthetized with 1.4–1.6% isoflurane carried by pure oxygen. A small mid-line neck incision was made. The right common carotid artery and the external carotid artery were dissected. The external carotid artery was cut open gently and a suture (#1622 for CD1 mice, #1419 for C57BL/6J and MMP9-/- mice; Beijing Sunbio Biotechnology Co. Ltd, Beijing, China) was advanced through the cut into the internal carotid artery until a slight resistance was felt to achieve MCAO. The incision was infiltrated with 0.2% ropivacaine and the animal was allowed to awaken. Ninety minutes after the onset of MCAO, the animal was re-anaesthetized briefly and the suture was retracted into the external carotid artery. During the surgery to create MCAO, the rectal temperatures of mice were maintained strictly at 37 ± 0.2°C with a warming blanket. Their heart rates and pulse oximeter oxygen saturation (SpO2) were monitored continuously and non-invasively using a MouseOX Murine Plus Oximeter System (Starr Life Sciences Corporation, Oakmont, PA, USA).
Motor coordination was evaluated just before and 3 days after the MCAO for CD1 and C57BL/6J mice and 1 day after MCAO for MMP-9−/− mice as we described before.20 Mice were placed on a rotarod with the speed accelerated from 4 to 40 rpm within 5 min. The latency and speed were recorded when a tested mouse fell off the rod. The speed-latency index, which is latency (s) × speed (rpm), was calculated. The ratio of this index obtained after MCAO and before MCAO was calculated to reflect the change in coordinate function of each mouse.
After determination of the coordinate function, mice were anaesthetized deeply with 5% isoflurane and intracardially perfused with normal saline. Brains were cut into 1-mm thick coronal slices. Slices were inspected for gross HT in the brain parenchyma and then stained with 1% 2, 3, 5-triphenyl-tet razolium chloride solution to evaluate the oedema index and infarct volume. The infarct area and the areas of the left and right cerebral hemispheres in each brain slice were quantified using Image J (National Institutes of Health). Oedema index = right hemisphere volume/left hemisphere volume. Corrected brain infarct volume in percentage = [left hemisphere volume − (right hemisphere volume − right infarction volume)] × 100 /left hemisphere volume.
Haemorrhagic volume in the brain was determined in the same way as for measuring infarct volume. Brains were harvested at 24 h after the MCAO and cut into 1 mm coronal slices. The slices were photographed with a standard bar in the view. Haemorrhagic volume in each slice was determined after the haemorrhagic area in the right hemisphere was delineated by using a Wacom Bamboo drawing pad. The sum of haemorrhagic volumes in the right hemisphere slices was calculated. The percentage value of this sum of volumes relative to the left hemisphere volume was then determined for each mouse.
2.6. Blood glucose, lipid profile, and haemoglobin A1c measurements
The random and 6 h fasting blood glucose levels of 16-week-old mice that had been on RD or HFD for 10 weeks were measured with a glucose strip method using blood from the tails. Blood collected from CD1 and C57Bl/6J mice just before sacrifice at 72 h after the MCAO and from MMP-9−/− mice before sacrifice for cerebral vasculature casting were tested for the concentrations of blood lipids and haemoglobin A1c (HbA1c) by the central laboratory of the University of Virginia Hospital. Plasma insulin levels were determined by enzyme-linked immunosorbent assay (mouse insulin kit; Shibayagi, Gunma, Japan).
2.7. IgG extravasation evaluation
Sixteen-week-old C57BL/6J mice or MMP-9−/− mice fed with RD or HFD for 10 weeks were subjected to sham surgery or a 90 min right MCAO. Sham surgery was performed by making the neck incision and dissecting the common carotid artery but without the MCAO. Mice under deep anaesthesia at 24 h after brain ischaemia or sham surgery were perfused with normal saline followed by ice-cold 4% paraformaldehyde. Tissue preparation was the same as for microvessel density evaluation. Five-micrometre thick coronal sections at Bregma −0.98 were obtained and incubated with anti-mouse IgG antibody conjugated with horseradish peroxidase (1:1000 dilution; Santa Cruz, Biotechonology). The sections were incubated with diaminobenzidine and mounted. Pictures were taken in the right frontal cerebral cortex area 1 (Fr1) and the right striatum. Quantification of IgG extravasation was done as we have described before.21 Briefly, three independent microscopic fields were acquired randomly in the right Fr1 (an ischaemic penumbral region) and the right striatum (an ischaemic core)22–24 of each section. Three sections per mouse were imaged. Positively stained areas were quantified by the number of pixels per image with intensity above a predetermined threshold level as determined using Image J 1.47n software and expressed as the percentage of the positively stained area in the total area of interest in the microscopic field. All quantitative analyses were performed in a blinded fashion.
2.8. MMP activity assessment
Sixteen-week-old CD1 mice fed RD or HFD for 10 weeks were subjected to sham surgery or the 90 min right MCAO. Animals were perfused with ice-cold normal saline at 6 h after the onset of ischaemia and brains were harvested. The right hemisphere Fr1, separated from the rest of cerebral cortex, and the striatum were stored at −80°C until use. Total, cytoplasmic and nuclear protein were extracted from these brain tissues according to Abcam's nuclear fractionation protocol except that dithiothreitol was omitted from buffer A in the cytoplasmic protein extraction to preserve MMP activity. Sixty micrograms of cytoplasmic protein were used in the MMP zymographical assay. Total cellular and nuclear fractions were adjusted to 2 μg protein/μL, denatured by boiling in loading buffer and saved at −20°C for western blotting.
Gelatin zymography for measuring MMP-9 and MMP-2 activity was performed as described before.25 Sixty micrograms of protein in 30 μL were mixed with 5 × sample buffer. Forty-five-micrograms of protein per lane were loaded onto 10% polyacrylamide gels containing 0.1% gelatin and subjected to electrophoresis. Gelatinolytic activity was revealed after incubating the gels with re-naturing buffer (2.5% Triton x-100), developing buffer (0.5% Coomassie blue G-250 in 30% methanol and 10% acetic acid), then de-staining solution. MMP activity was visualized as clear bands in the gel at appropriate molecular weights.
2.9. Western blotting
The samples for western blotting of MMP-9 were prepared as described in Section 2.8 (MMP activity assessment). For western blotting of endothelial nitric oxide synthase (eNOS) and phospho-eNOS, the right Fr1 and striatum were harvested 24 h after the MCAO and homogenized in Pierce RIPA buffer (#89901; Thermo Scientific, Worcester, MA, USA) containing a protease inhibitor cocktail (#P2714; Sigma, St Louis, MO, USA) and a phosphatase inhibitor cocktail (#04906845001; Roche Applied Science, Indianapolis, IN, USA). Homogenates were centrifuged at 13 000 rpm at 4°C for 20 min and the supernatants were collected for western blotting.
Approximately 30 μg of total cellular protein per lane were loaded onto 10% polyacrylamide gels. After electrophoresis, proteins were transferred onto a polyvinylidene difluoride membrane and then incubated overnight at 4°C with primary antibodies. The primary antibodies were rabbit polyclonal anti-MMP-9 antibody (1:500 dilution; Aviva Systems Biology, San Diego, CA, USA), rabbit polyclonal anti-eNOS antibody (1:500 dilution; Cell Signaling Technology, Danvers, MA, USA), rabbit polyclonal anti-phospho-eNOS (ser1177) antibody (1:500 dilution; Cell Signaling Technology), and rabbit polyclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:5000 dilution; Sigma-Aldrich, St. Louis, MO). Blots were washed with PBS-Tween, and incubated with appropriate secondary antibodies (1:5000; Santa Cruz Biotechnology, Inc.). Protein bands were revealed using the enhanced chemiluminescence method and analysed with a gel imaging analysis system (G-Box, Syngene, Frederic, MD, USA).
2.10. Statistical analysis
Parametrical data are presented as means ± SD. Statistical analysis of these results was carried out by t-test or two-way analysis of variance followed by the Tukey test as appropriate. The HT frequencies were analysed by the Fisher exact method. A P ≤ 0.05 was accepted as significant. All statistical analyses were performed with SigmaStat (SYSTAT Software Inc., Point Richmond, CA, USA).
3. Results
3.1. HFD-induced obesity and hyperlipidaemia
Although the starting body weights of the two groups of CD1 wild-type male mice were similar, the mice fed HFD were heavier, and had higher levels of random and fasting blood glucose and blood HbA1c than did those fed RD. The HFD also increased blood cholesterol, triglycerides, and low-density lipoprotein (LDL). Although high-density lipoprotein (HDL) also was increased by feeding the HFD, the ratio of LDL/HDL and the ratio of cholesterol/HDL were higher than those in mice fed the RD (Table 1). These results suggest that HFD feeding for 10 weeks induces obesity, hyperglycaemia, and hyperlipidaemia in the CD1 mice. The homeostatic model assessment (HOMA) of the insulin resistance index was increased from 0.591 ± 0.249 (n = 8) in mice fed with RD to 0.949 ± 0.377 (n = 8) in mice fed HFD (P = 0.042), suggesting that HFD feeding induces insulin resistance. The mean arterial blood pressures were 82 ± 9 mmHg (n = 7) or 87 ± 11 mmHg (n = 6), respectively, for the mice fed RD or HFD for 10 weeks (P = 0.413), suggesting that HFD feeding for 10 weeks does not change significantly the blood pressure.
Table 1.
Mouse body weight, blood glucose, and lipid profiles
| CD1 mice |
C57BL/6J |
MMP-9−/− mice |
||||
|---|---|---|---|---|---|---|
| Regular diet | High-fat diet | Regular diet | High-fat diet | Regular diet | High-fat diet | |
| Weight (g, 6-week old) | 25.8 ± 1.4 | 26.2 ± 1.2 | 19.3 ± 1.1 | 19.4 ± 1.1 | 19.7 ± 1.7 | 20.2 ± 2.3 |
| Weight (g, 16-week old) | 39.8 ± 2.8 | 50.4 ± 5.5* | 26.7 ± 2.0 | 35.2 ± 3.8* | 24.7 ± 2.3 | 40.0 ± 5.5* |
| Random BG (mg/dL) | 161 ± 49 | 229 ± 57* | 150 ± 21 | 203 ± 58* | ||
| Fasting BG (mg/dL) | 98 ± 12 | 124 ± 22* | 57.4 ± 8.4 | 75.2 ± 9.0* | 132 ± 17 | 169 ± 22* |
| HbA1c (%) | 3.1 ± 0.8 | 4.1 ± 1.0* | 3.3 ± 0.5 | 3.9 ± 0.5* | 4.0 ± 0.2 | 4.3 ± 0.2* |
| Cholesterol (mg/dL) | 149 ± 26 | 258 ± 63* | 151 ± 12.3 | 195 ± 14.4* | 99 ± 9 | 159 ± 40* |
| Triglycerides (mg/dL) | 66.8 ± 30.1 | 99.9 ± 29.8* | 52.7 ± 5.1 | 72.1 ± 5.9* | 62.6 ± 17.7 | 90 ± 12.4* |
| HDL (mg/dL) | 77.3 ± 14.6 | 105.0 ± 18.3* | 90.7 ± 11.3 | 119.4 ± 8.5* | 62.0 ± 4.2 | 76.3 ± 15.8* |
| LDL (mg/dL) | 61.0 ± 15.2 | 136.4 ± 48.0* | 35.9 ± 8.7 | 53.0 ± 10.9* | 26.9 ± 7.5 | 67.7 ± 30.8* |
| LDL/HDL | 0.82 ± 0.20 | 1.31 ± 0.40* | 0.40 ± 0.11 | 0.45 ± 0.09 | 0.44 ± 0.14 | 0.88 ± 0.28* |
| Cholesterol/HDL | 1.97 ± 0.18 | 2.47 ± 0.45* | 1.67 ± 0.10 | 1.64 ± 0.04 | 1.60 ± 0.14 | 2.08 ± 0.26* |
Initial body weights just before they were placed on different diets are listed in the first line of the table. The rest of data are from 16-week-old mice after they had been on different diets for 10 weeks. Mice were fasted 6 h before blood was harvested for measuring fasting blood glucose (BG). Results are means ± SD (n = 4–19).
LDL, low-density lipoprotein; HDL, high-density lipoprotein.
*P < 0.05 compared with corresponding regular diet-fed mice.
3.2. HFD-induced cerebral vascular remodelling
HFD feeding increased the tortuosity index of the MCA medium size branches but did not affect the number of collaterals between MCA and ACA or between MCA and PCA in the CD1 mice (Figure 1). HFD feeding also decreased the inner width of the MCA root (Figure 1) and increased the wall thickness of the MCA root (Figure 2). Mice fed HFD had an increased microvessel density as determined by the expression of vWF (Figure 2), an endothelial cell marker. These results indicate that HFD feeding causes remodelling of both cerebral macrovessels and microvessels.
Figure 1.

MMP-9 knockout abolishes HFD-induced remodelling in cerebral macrovessels. Cerebral vascular casting was produced by infusing PU4ii with blue dye. (A) Representative images of the dorsal surface of the cerebrum. (B) Tortuosity index of the MCA medium size branches. (C) Number of collaterals between MCA and ACA or between MCA and PCA. (D) Representative images of brain area at the MCA root. An arrow in the first panel indicates the MCA. (E) Quantification of the inner width of the MCA root. Results in B, C, and E are means ± SD (n = 7–9). *P < 0.05 compared with the corresponding results from mice fed regular diet.
Figure 2.

HFD induces remodelling in cerebral macrovessels and microvessels. Cerebral vessels in brain sections of CD1 and MMP-9−/− mice were revealed after immunostaining for vWF. (A and B) Representative images of brain area at the MCA root of CD1 mice. (C and D) Representative images of CD1 mouse cerebral cortex. Inserts in these two panels are ×200 magnifications. (E) Wall thickness of the MCA root of CD1 mice. (F) Microvessel density in the CD1 mouse cerebral cortex. (G and H) Representative images of brain area at the MCA root of MMP-9−/− mice. (I and J) Representative images of MMP-9−/− mouse cerebral cortex. (K) Wall thickness of the MCA root of MMP-9−/− mice. (L) Microvessel density in the MMP-9−/− mouse cerebral cortex. Scale bar in each panel represents 200 µm. Results in E, F, K, and L are means ± SD (n = 4–7). *P < 0.05 compared with the corresponding results of mice fed regular diet.
3.3. HFD worsened neuropathological and functional outcome and increased MMP-9 activity in brain tissues
To determine the effects of HFD on brain ischaemic tolerance, we subjected the CD1 mice to a 90 min right MCAO. There was no significant difference in heart rates, SpO2, and body temperature at the onset of brain ischaemia between the CD1 mice fed RD and those fed HFD (Figure 3). HFD feeding increased the brain oedema index, infarct volumes, HT frequency, and haemorrhagic volume, and worsened the neurological functions as assessed by the performance on a rotarod (Figures 4 and 5). These results suggest that HFD worsens neurological outcome after transient focal brain ischaemia.
Figure 3.

Physiological data. Body weights were determined before mice were subjected to a 90 min right MCAO. The heart rates, SpO2 and rectal temperatures were readings at 5 min after the onset of the MCAO. Results are means ± SD (n = 7–14). *P < 0.05 compared with the corresponding results of the mice fed regular diet.
Figure 4.
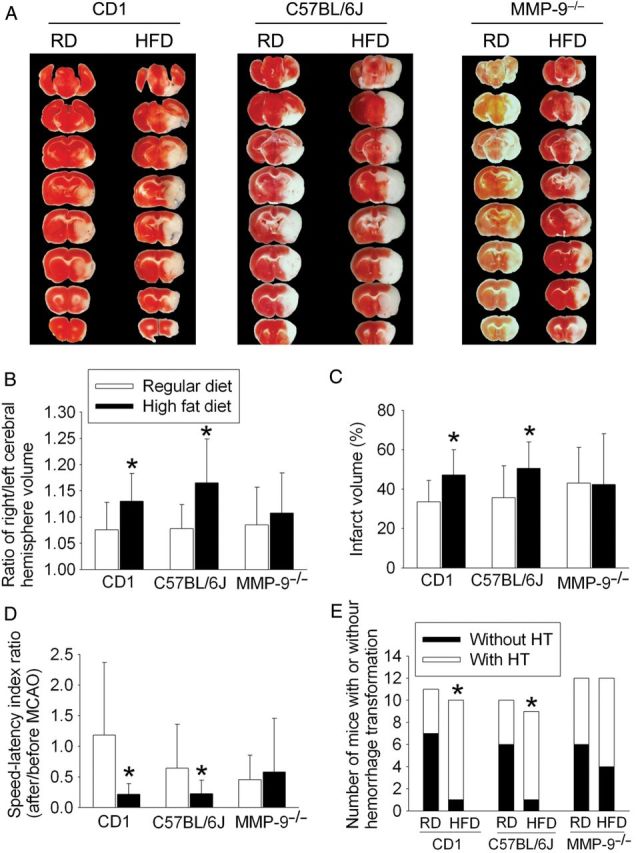
MMP-9 knockout abolishes HFD-induced worsening of neurological outcome after transient focal brain ischaemia. Mice were subjected to a 90 min right MCAO. (A) Representative brain slices stained with 2,3,5-triphenyl-tetrazolium chloride. (B–E) Quantitative results. Results are means ± SD (n = 9–12). *P < 0.05 compared with the corresponding results from the mice fed regular diet.
Figure 5.
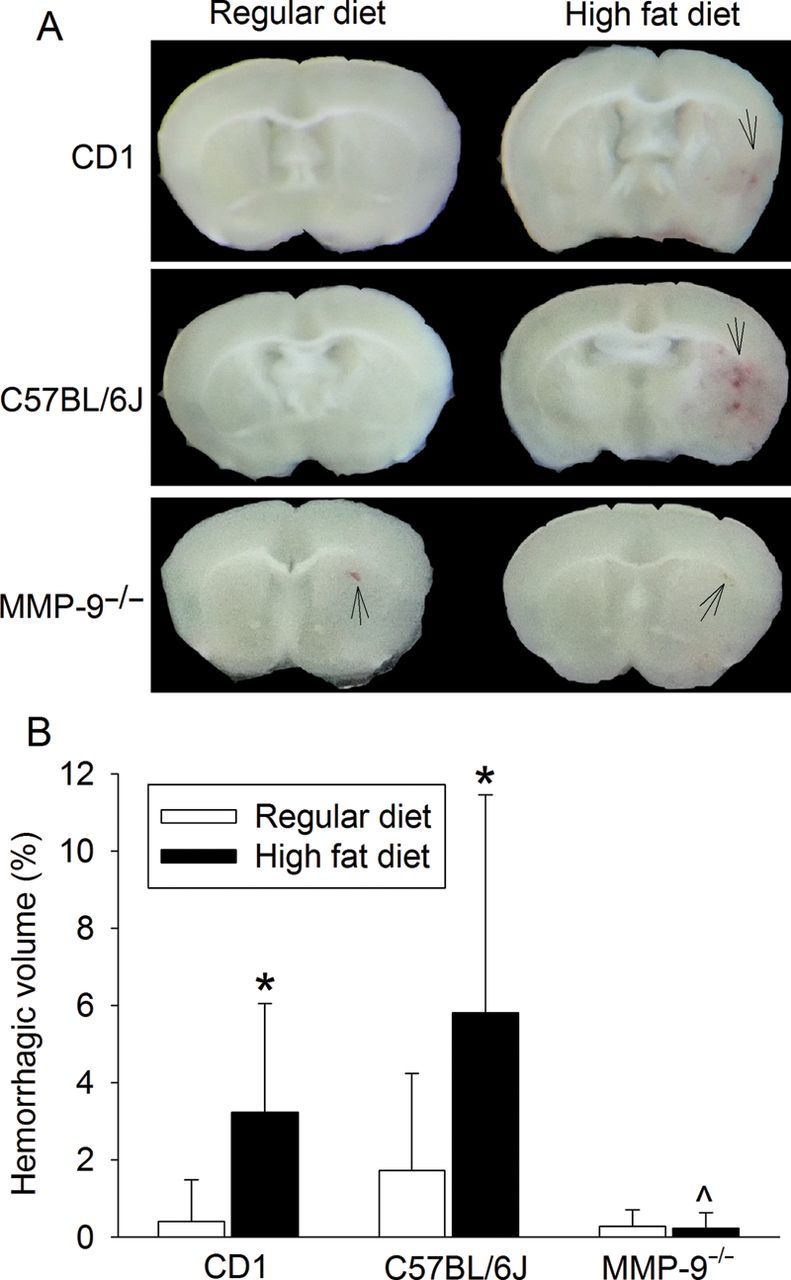
MMP-9 knockout abolishes HFD-induced increase of haemorrhagic volumes in the brain. Mice were subjected to a 90 min right MCAO and haemorrhagic volumes were evaluated at 24 h after the MCAO. (A) Representative images of fresh brain slices. Arrow indicates haemorrhage. (B) Quantitative results. Results are means ± SD (n = 8–14). *P < 0.05 compared with the corresponding results of mice fed regular diet. ^P < 0.05 compared with the corresponding results of C57BL/6J mice fed HFD.
The MMP-9 activity was increased in the ischaemic brain tissues. This increase was enhanced by HFD feeding in the ischaemic cerebral cortex. HFD also increased MMP-9 activity in the non-ischaemic brain tissues. Although MMP-2 activity was increased in the ischaemic core tissues (striatum), MMP-2 activity in the ischaemic and non-ischaemic brain tissues was not changed by HFD (Figure 6). These results suggest that MMP-9 may be more important than MMP-2 in mediating the HFD-induced cerebral vascular remodelling and worsening of neurological outcome after brain ischaemia.
Figure 6.
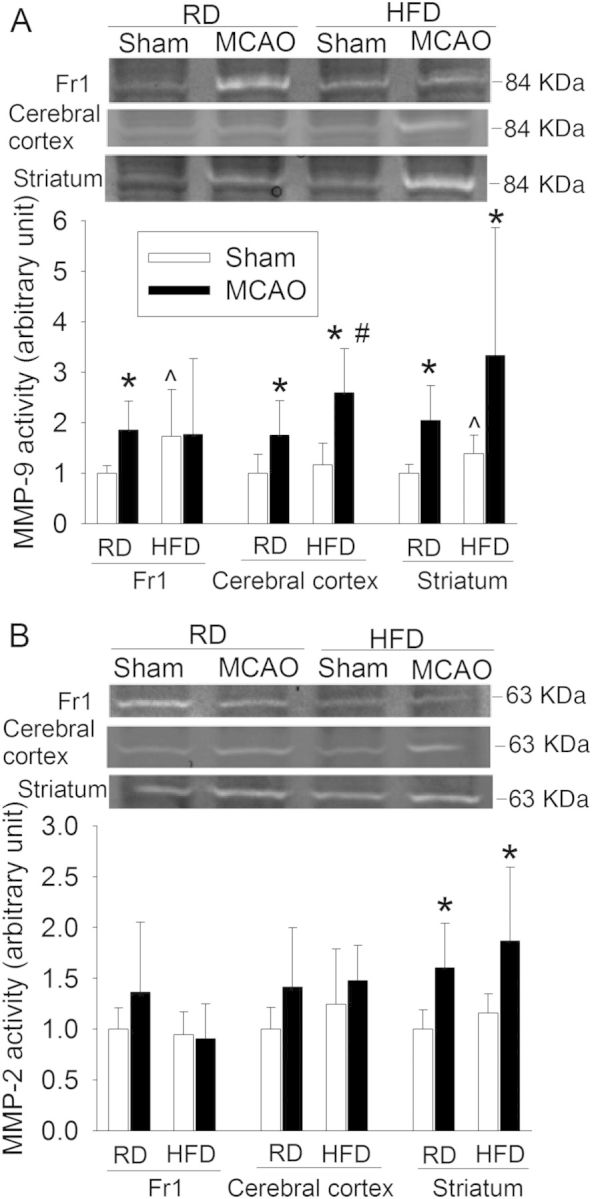
HFD increases MMP-9 activity in ischaemic and non-ischaemic brain tissues. (A) MMP-9 activity. (B) MMP-2 activity. Representative zymographical images are presented in the top panel. A graphic presentation of MMP-9 and MMP-2 activity quantified by integrating the volume of autoradiograms is shown in the bottom panel. Values in graphs are the means ± SD (n = 6–11). *P < 0.05 compared with corresponding sham-operated mice. ^P < 0.05 compared with corresponding mice that were fed regular diet and but were not subjected to the MCAO. #P < 0.05 compared with corresponding mice that were fed regular diet and subjected to the MCAO.
The MMP-9 protein expression in the ischaemic striatum was significantly decreased in both RD- and HFD-fed mice. MMP-9 expression in the cerebral cortex was not changed by brain ischaemia or HFD (Figure 7).
Figure 7.
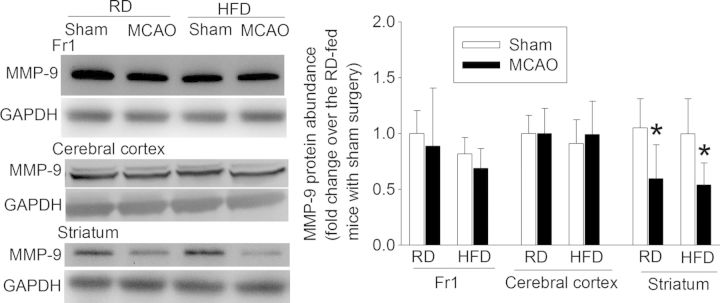
Effects of brain ischaemia and HFD feeding on MMP-9 protein expression. Representative western blotting images are presented in the left panel and the graphic presentation of protein expression is shown in the right panel. Values in graphs are the means ± SD (n = 7–9). *P < 0.05 compared with the corresponding sham-operated mice.
3.4. MMP-9 knockout attenuated HFD-induced vascular remodelling and worsening of neurological outcome
Since our results implicated a role of MMP-9 in the HFD-induced cerebral vascular remodelling, we used MMP-9−/− mice to further determine this role. Since these mice have a C57BL/6J mouse gene background, we first confirmed that HFD-induced cerebral vascular remodelling in the C57BL/6J mice as indicated by increased tortuosity index and smaller inner width of the MCA root in the mice fed HFD than those in the mice fed RD (Figure 1). C57BL/6J mice on the HFD also were heavier and had a higher level of blood HbA1c, cholesterol, triglycerides, LDL (Table 1 and Figure 3), and HOMA insulin resistance index (0.402 ± 0.1 of the mice fed RD vs. 0.716 ± 0.198 of the mice fed the HFD, n = 7, P = 0.003) than those on RD. HFD-fed mice also had poorer neurological outcome after brain ischaemia than the mice on RD (Figures 4 and 5). HFD feeding also increased damage of the BBB in the Fr1 as assessed by IgG extravasation (Figure 8). These results suggest that pathological changes similar to those in the CD1 mice were induced by HFD in the C57BL/6J mice.
Figure 8.
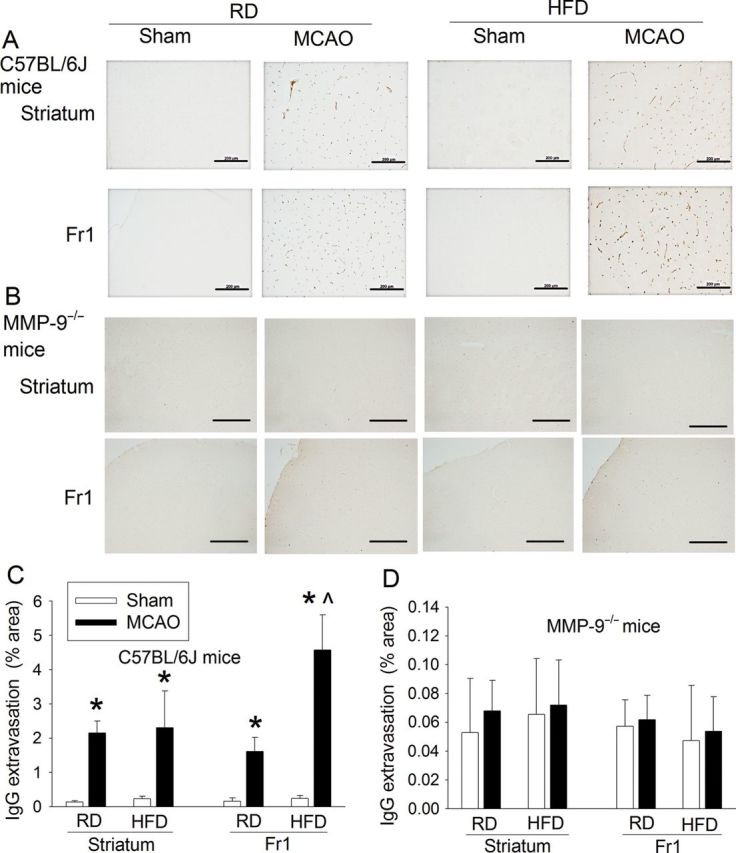
HFD increases IgG extravasation in the ischaemic brain tissues. C57BL/6J and MMP-9−/− mice were subjected to a 90 min right MCAO or sham surgery. Their brains were harvested at 24 h after the MCAO or surgery. Coronal brain sections at Bregma −0.98 were immunostained for IgG (in brown). (A and B) Microscopic photos in the right striatum and frontal cortex area 1 (Fr1). Scale bar in each panel represents 200 µm. (C and D) quantification of IgG extravasation. Results are means ± SD (n = 3–5). *P < 0.05 compared with the corresponding sham-operated mice.
Similar to the CD1 and C57BL/6J wild-type mice, HFD feeding increased the body weights as well as blood glucose, HbA1c, and lipid levels in the MMP-9−/− mice (Table 1). However, the tortuosity index of the MCA branches, inner width at the MCA root, wall thickness of MCA root, microvessel density in the cerebral cortex, and neurological outcome, including BBB damage after transient focal brain ischaemia were not affected by HFD feeding in the MMP-9−/− mice (Figures 1, 2, 4, and 8). Also, the haemorrhagic volumes in the MMP-9−/− mice fed HFD after MCAO were smaller than those in the C57BL/6J mice fed HFD (Figure 5). These results suggest that MMP-9 plays a critical role in the HFD-induced cerebral vascular remodelling and worsening of brain damage after focal brain ischaemia–reperfusion. Interestingly, phospho-eNOS was increased in the ischaemic brain tissues in the RD-fed C57BL/6J mice. Activation/phosphorylation of eNOS is a protective mechanism.26 The increase of phospho-eNOS in the ischaemic brain tissues was inhibited by HFD feeding. A similar pattern of changes occurred in the MMP-9−/− mice, suggesting that loss of MMP-9 has minimal impact on this protective mechanism. The expression of eNOS was not significantly affected by HFD feeding in either C57BL/6J or MMP-9−/− mice (Figure 9).
Figure 9.

HFD inhibits phosphorylation of eNOS in the ischaemic brain tissues. C57BL/6J and MMP-9−/− mice were subjected to a 90 min right MCAO or sham surgery. Their brains were harvested at 24 h after the MCAO or sham surgery. (A and B) phospho-eNOS in the frontal cortex area 1 (Fr1) and striatum of the C57BL/6J and MMP-9−/− mice. (C and D) eNOS in the Fr1 and striatum of the C57BL/6J and MMP-9−/− mice. Results are means ± SD (n = 4–6). *P < 0.05 compared with the corresponding sham-operated mice.
4. Discussion
Vascular complications of obesity, including stroke, are increasing as obesity has become pandemic, with onset in younger patients. An HFD is considered to be a major contributor to the obesity.5 To closely simulate early onset obesity and hyperlipidaemia in humans, we fed outbred CD1 mice an HFD starting when the mice were 6 weeks old. Ten weeks of HFD-induced obesity, hyperglycaemia, hyperlipidaemia, and insulin resistance in the mice. These features capture very well the presentations of obesity in humans.
There is a lack of knowledge about cerebral vascular remodelling in patients with obesity and hyperlipidaemia, especially in young adult patients with these pathological conditions. Feeding 3-week-old rats an HFD for 10 weeks has been shown to increase the wall thickness of the MCA and to decrease the compliance of this vessel.12 Our study extends these findings by showing that HFD increased the tortuosity of major MCA branches, decreased the inner width of the MCA root, and increased the microvessel density in the cerebral cortex of the CD1 mice, indicating that remodelling in both cerebral macrovessels and microvessels is induced by an HFD. Hypertension also can induce vascular remodelling and brain pathology.27 However, consumption of an HFD for 10 weeks did not increase significantly the blood pressure in the CD-1 mice, suggesting that the HFD may not induce cerebral vascular remodelling through hypertension.
Mechanisms for HFD-induced cerebral vascular remodelling have not been studied; however, previous studies have shown that there is significant cerebral remodelling in diabetic rats;10,11 and that diabetic rats have increased MMP-9 activity in the cerebral macrovessels. MMP-2 activity is not changed in the cerebral vessels of diabetic rats. Chronic use of metformin, a hypoglycaemic drug, and minocycline, an anti-inflammatory agent that also can inhibit MMP, reduces the cerebral vascular remodelling and also attenuates MMP-9 activity in the cerebral vessels of the diabetic rats.11 These results suggest a significant association between cerebral vascular remodelling and MMP-9 activity under diabetic conditions. MMP-9 and MMP-2 are the major gelatinases.14 They can degrade and stimulate matrix deposition, leading to restructuring of blood vessels and the surrounding matrix.14
A major finding of our study is that MMP-9 plays a critical role in the HFD-induced cerebral vascular remodelling. Our results showed that the HFD-induced obesity, hyperglycaemia, and hyperlipidaemia in MMP-9−/− mice, as it did in CD1 mice. However, the HFD did not induce cerebral vascular remodelling in the MMP-9−/− mice as it did in the CD1 mice. HFD also induced cerebral vascular remodelling in the wild-type C57BL/6J mice that share genetic background with the MMP-9−/− mice. These results argue strongly that HFD-induced cerebral vascular remodelling requires MMP-9. Consistent with this finding, our study also showed that HFD increased MMP-9 activity but did not affect MMP-2 activity in the brain tissues.
Our results suggest that the HFD-induced increase in MMP-9 activity and the subsequent cerebral vascular remodelling contribute to the worsened neurological outcome after brain ischaemia. Hyperlipidaemia has been shown to increase ischaemic brain injury,9,12,28 although controversial findings on the effects of hyperlipidaemia on brain ischaemic tolerance have been reported.29,30 Direct evidence for a contribution of diabetes- or HFD-induced cerebral vascular remodelling to neurological outcome after brain ischaemia has not been reported in humans or animals. Our results showed that the HFD worsened the neurological outcome after transient focal brain ischaemia in both CD1 and C57BL/6J mice but did not change the neurological outcome in the MMP-9−/− mice. Since HFD also did not induce cerebral vascular remodelling in the MMP-9−/− mice, the results would suggest that the HFD-induced cerebral vascular remodelling contributes to the worse neurological outcome after brain ischaemia. However, since MMP-9 knockout can reduce ischaemic brain injury in mice,31,32 it is possible that the lack of worsening of neurological outcome in the MMP-9−/− mice fed HFD is due to MMP-9 knockout alone and is not the result of the lack of cerebral vascular remodelling in these mice. Nevertheless, our study showed that the neuropathological and functional outcomes, including brain infarct volumes, oedema index, IgG extravasation, coordination, and HT frequency, were similar between the C57BL/6J mice and MMP-9−/− mice fed the RD. This finding argues for the involvement of HFD-induced cerebral vascular remodelling in the worsened neurological outcome after brain ischaemia for the wild-type mice fed the HFD.
Previous studies have shown that MMP-9 activity in ischaemic brain tissues is increased in the early phase after transient focal brain ischaemia. MMP-9 knockout mice have smaller infarct volumes and attenuated damage to BBB integrity. These findings establish the damaging role of MMP-9 in ischaemic brain injury.31,32 Our study showed that the C57BL/6J and MMP-9−/− mice fed the RD had similar magnitudes of brain infarct volume, oedema index, IgG extravasation, diminished co-ordination, and HT frequency after brain ischaemia. These results do not suggest that MMP-9 plays a major role in the ischaemia-induced brain cell death and BBB damage in the RD-fed mice. The reasons for the discrepant findings between our studies and the previous studies are not known. Similar brain ischaemia models created with a suture technique were used in our study and the previous studies.31,32 However, our MMP-9−/− mice have a C57BL/6J gene background and the MMP-9−/− mice used in the previous studies had a CD1 gene background.31,32 Also, we used 16-week-old mice and the age of the mice used in the previous studies was not specified in the reports.31,32
HT is a complication of ischaemic stroke. MMPs, especially MMP-9, have been implicated in the development of HT.15–17 Our results showed that the HT frequency in the C57BL/6J and MMP-9−/− mice fed RD was similar. A similar result was reported in a recent study, although MMP-9−/− mice had smaller haemorrhagic volumes in the cerebral cortex than the wild-type mice after a suture-induced MCAO. The haemorrhagic volumes in the basal ganglia was similar between the MMP-9−/− and wild-type mice.33 On the other hand, increased haemorrhage and brain oedema in the MMP-9−/− mice have been reported in a collagenase-induced intracerebral haemorrhage model.34 Nevertheless, our results showed that HFD feeding activates MMP-9, consistent with a previous study.13 Haemorrhagic volumes after MCAO were increased in the C57BL/6J mice fed HFD but not in the MMP-9−/− mice fed HFD when compared with the corresponding RD-fed mice. The haemorrhagic volumes in the HFD-fed MMP-9−/− mice were smaller than those in the HFD-fed C57BL/6J mice. These results suggest that MMP-9 plays a critical role in haemorrhage after brain ischaemia in the mice with hyperlipidaemia.
Our study suggests that the HFD-induced increase in MMP-9 activity in the ischaemic and non-ischaemic brain tissues is not due to the increased MMP-9 protein expression. HFD did not increase MMP-9 protein in the brain tissues. Instead, MMP-9 protein expression was decreased in the ischaemic striatum of HFD- and RD-fed mice. This decrease may be due to the cell death in the ischaemic core tissues. These results underscore the importance of measuring the activity of MMPs in the ischaemic brain tissues. Various factors, such as tissue plasminogen activators, can activate MMPs.35–37 Future studies are needed to determine how an HFD can activate MMP-9 in the brain.
We focused on determining the role of MMP-9 but not MMP-2 in the HFD-induced cerebral remodelling and worsening of ischaemic brain injury because HFD increased MMP-9 activity in the ischaemic and non-ischaemic brain tissues. Although MMP-2 activity in the ischaemic striatum was higher than that in the non-ischaemic striatum, HFD did not affect the MMP-2 activity in the ischaemic and non-ischaemic brain tissues. These results do not suggest a role for MMP-2 in the HFD effects. In support of this suggestion, MMP-9 knockout abolished the HFD-induced cerebral vascular remodelling and worsening of ischaemic brain injury.
Activation of eNOS has been shown to be protective against ischaemic brain injury.26 Hyperlipidaemia can attenuate the activation of eNOS.38,39 Consistent with these previous findings, our study showed that phospho-eNOS was increased in the ischaemic brain tissues of C57BL/6J mice and that this increase was inhibited in the mice fed with HFD. A similar pattern of changes in the phospho-eNOS level occurred in the MMP-9−/− mice, suggesting that eNOS activation may not be affected significantly in these knockout mice. Therefore, the ineffectiveness of HFD on worsening ischaemic brain injury in the MMP-9−/− mice may not be due to altered eNOS activation in these mice.
Our study has limitations. MMP-9 is expressed not only in the blood vessels but also in brain cells, such as neurons.40,41 We used brain tissues for measuring MMP-9 activity and expression, so the results reflect MMP-9 activity and expression in all cells in the tissues. Thus, we do not know from our study the cell origin of the MMP-9 that is important for the HFD-induced cerebral vascular remodelling and worsening of ischaemic injury after brain ischaemia. MMP-9 secreted from one type of cell may move around and should work on extracellular matrix around it. Thus, MMP-9 from all types of cells may contribute to the cerebral vascular remodelling (macrovessels and microvessels) and the BBB damage in the ischaemic brain tissues. Also, we did not specifically use any drugs to maximally dilate all blood vessels before vascular casting. It is possible that different wall thicknesses and diameters of MCA root between the RD- and HFD-fed mice are due to different vascular tones of mice fed these two diets. However, the mice were anaesthetized with 5% isoflurane, a very powerful vascular dilator,42 before the vascular casting; and the blood pressure of RD- and HFD-fed awake mice was similar. Finally, the degree of increase in some lipids, such as LDL, in the MMP-9−/− mice may be smaller than that in the CD-1 mice after HFD feeding. This smaller increase may contribute to the ineffectiveness of the HFD on cerebral vascular remodelling and ischaemic brain injury in MMP-9−/− mice. However, the degree of increase of those lipids in the HFD-fed C57BL/6J was even smaller than those in the MMP-9−/− mice and HFD still induced cerebral vascular remodelling and more severe ischaemic brain injury in C57BL/6J mice.
In summary, we have shown that HFD feeding of 6-week-old mice for 10 weeks increased the body weights, blood glucose, and lipid levels in the CD1, C57BL/6J, and MMP-9−/− mice. The HFD feeding also induced cerebral vascular remodelling and worsening of neurological outcome after brain ischaemia in the CD1 and C57BL/6J mice but not in the MMP-9−/− mice. These results suggest that MMP-9 plays a critical role in the HFD-induced cerebral vascular remodelling and decrease of brain ischaemic tolerance.
Authors’ contributions
J.D.: research design, performance of experiments, data analysis, and writing of the methods and materials section; C.F.: performance of experiments and data analysis; J.Z.: performance of experiments and data analysis; L.X.: research design; Z.Z.: project concept inception, research design, data analysis, and manuscript writing.
Conflict of interest: none declared.
Funding
This study was supported by grants (R01 GM065211 and R01 GM098308 to Z.Z.) from the National Institutes of Health, Bethesda, MD, by a grant from the International Anesthesia Research Society (2007 Frontiers in Anesthesia Research Award to Z.Z.), Cleveland, OH, by a Grant-in-Aid from the American Heart Association Mid-Atlantic Affiliate (10GRNT3900019 to Z.Z.), Baltimore, MD, by a grant from Voices Against Brain Tumor (Investigator-initiated Project to Z.Z.), New York, NY, the Robert M. Epstein Professorship endowment (to Z.Z.), University of Virginia, Charlottesville, VA, by a grant from the Minister of Education (IRT1053, Program for Changjiang Scholars and Innovative Research Team in University to L.X.), Beijing, China, and a grant from the Nature Science Foundation of China (81300989 to J.D.), Beijing, China.
References
- 1.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatnagar D, Soran H, Durrington PN. Hypercholesterolaemia and its management. BMJ. 2008;337:a993. doi: 10.1136/bmj.a993. [DOI] [PubMed] [Google Scholar]
- 3.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 4.Ogden CL, Carroll MD, Curtin LR, Lamb MM, Flegal KM. Prevalence of high body mass index in US children and adolescents, 2007–2008. JAMA. 2010;303:242–249. doi: 10.1001/jama.2009.2012. [DOI] [PubMed] [Google Scholar]
- 5.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.von Sarnowski B, Putaala J, Grittner U, Gaertner B, Schminke U, Curtze S, Huber R, Tanislav C, Lichy C, Demarin V, Basic-Kes V, Ringelstein EB, Neumann-Haefelin T, Enzinger C, Fazekas F, Rothwell PM, Dichgans M, Jungehulsing GJ, Heuschmann PU, Kaps M, Norrving B, Rolfs A, Kessler C, Tatlisumak T. Lifestyle risk factors for ischemic stroke and transient ischemic attack in young adults in the Stroke in Young Fabry Patients Study. Stroke. 2013;44:119–125. doi: 10.1161/STROKEAHA.112.665190. [DOI] [PubMed] [Google Scholar]
- 7.George MG, Tong X, Kuklina EV, Labarthe DR. Trends in stroke hospitalizations and associated risk factors among children and young adults, 1995–2008. Ann Neurol. 2011;70:713–721. doi: 10.1002/ana.22539. [DOI] [PubMed] [Google Scholar]
- 8.Yatsuya H, Folsom AR, Yamagishi K, North KE, Brancati FL, Stevens J. Race- and sex-specific associations of obesity measures with ischemic stroke incidence in the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2010;41:417–425. doi: 10.1161/STROKEAHA.109.566299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langdon KD, Clarke J, Corbett D. Long-term exposure to high fat diet is bad for your brain: exacerbation of focal ischemic brain injury. Neurosci. 2011;182:82–87. doi: 10.1016/j.neuroscience.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Prakash R, Kelly-Cobbs AI, Ogbi S, Kozak A, El-Remessy AB, Schreihofer DA, Fagan SC, Ergul A. Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes. 2010;59:228–235. doi: 10.2337/db09-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elgebaly MM, Prakash R, Li W, Ogbi S, Johnson MH, Mezzetti EM, Fagan SC, Ergul A. Vascular protection in diabetic stroke: role of matrix metalloprotease-dependent vascular remodeling. J Cereb Blood Flow Metab. 2010;30:1928–1938. doi: 10.1038/jcbfm.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deutsch C, Portik-Dobos V, Smith AD, Ergul A, Dorrance AM. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvasc Res. 2009;78:100–106. doi: 10.1016/j.mvr.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ElAli A, Doeppner TR, Zechariah A, Hermann DM. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke. 2011;42:3238–3244. doi: 10.1161/STROKEAHA.111.615559. [DOI] [PubMed] [Google Scholar]
- 14.Olszynski K, Zimowska M. Structure and function of matrix metalloproteinases. Postepy Biochem. 2009;55:76–84. [PubMed] [Google Scholar]
- 15.Montaner J, Molina CA, Monasterio J, Abilleira S, Arenillas JF, Ribo M, Quintana M, Alvarez-Sabin J. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- 16.Montaner J, Alvarez-Sabin J, Molina CA, Angles A, Abilleira S, Arenillas J, Monasterio J. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 2001;32:2762–2767. doi: 10.1161/hs1201.99512. [DOI] [PubMed] [Google Scholar]
- 17.Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–836. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- 18.del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, Koziol JA. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007;38:646–651. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- 19.Deng J, Li J, Li L, Feng C, Xiong L, Zuo Z. Glutamate transporter type 3 knockout leads to decreased heart rate possibly via parasympathetic mechanism. Transgenic Res. 2013;22:757–766. doi: 10.1007/s11248-012-9680-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Zuo Z. Glutamate transporter type 3 knockout reduces brain tolerance to focal brain ischemia in mice. J Cereb Blood Flow Metab. 2011;31:1283–1292. doi: 10.1038/jcbfm.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Jiang W, Zuo Z. Pyrrolidine dithiocarbamate attenuates surgery-induced neuroinflammation and cognitive dysfunction possibly via inhibition of nuclear factor kappaB. Neurosci. 2014;261:1–10. doi: 10.1016/j.neuroscience.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of p38 mitogen-activated protein kinase. Mol Pharmacol. 2004;65:1172–1180. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Sheng W, Feng C, Zuo Z. Pyrrolidine dithiocarbamate attenuates brain Abeta increase and improves long-term neurological outcome in rats after transient focal brain ischemia. Neurobiol Dis. 2012;45:564–572. doi: 10.1016/j.nbd.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Yin J, Li L, Deng J, Feng C, Zuo Z. Isoflurane postconditioning reduces ischemia-induced nuclear factor-kappaB activation and interleukin 1beta production to provide neuroprotection in rats and mice. Neurobiol Dis. 2013;54:216–224. doi: 10.1016/j.nbd.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 26.Li Q, Atochin D, Kashiwagi S, Earle J, Wang A, Mandeville E, Hayakawa K, d'Uscio LV, Lo EH, Katusic Z, Sessa W, Huang PL. Deficient eNOS phosphorylation is a mechanism for diabetic vascular dysfunction contributing to increased stroke size. Stroke. 2013;44:3183–3188. doi: 10.1161/STROKEAHA.113.002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carnevale D, Mascio G, D'Andrea I, Fardella V, Bell RD, Branchi I, Pallante F, Zlokovic B, Yan SS, Lembo G. Hypertension induces brain beta-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension. 2012;60:188–197. doi: 10.1161/HYPERTENSIONAHA.112.195511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim E, Tolhurst AT, Qin LY, Chen XY, Febbraio M, Cho S. CD36/fatty acid translocase, an inflammatory mediator, is involved in hyperlipidemia-induced exacerbation in ischemic brain injury. J Neurosci. 2008;28:4661–4670. doi: 10.1523/JNEUROSCI.0982-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He K, Merchant A, Rimm EB, Rosner BA, Stampfer MJ, Willett WC, Ascherio A. Dietary fat intake and risk of stroke in male US healthcare professionals: 14 year prospective cohort study. BMJ. 2003;327:777–782. doi: 10.1136/bmj.327.7418.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arvanitidis AP, Corbett D, Colbourne F. A high fat diet does not exacerbate CA1 injury and cognitive deficits following global ischemia in rats. Brain Res. 2009;1252:192–200. doi: 10.1016/j.brainres.2008.11.058. [DOI] [PubMed] [Google Scholar]
- 31.Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- 32.Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suofu Y, Clark JF, Broderick JP, Kurosawa Y, Wagner KR, Lu A. Matrix metalloproteinase-2 or -9 deletions protect against hemorrhagic transformation during early stage of cerebral ischemia and reperfusion. Neurosci. 2013;212:180–189. doi: 10.1016/j.neuroscience.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Lee SR, Arai K, Tsuji K, Rebeck GW, Lo EH. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 36.Tsuji K, Aoki T, Tejima E, Arai K, Lee SR, Atochin DN, Huang PL, Wang X, Montaner J, Lo EH. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36:1954–1959. doi: 10.1161/01.STR.0000177517.01203.eb. [DOI] [PubMed] [Google Scholar]
- 37.Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamashiro K, Milsom AB, Duchene J, Panayiotou C, Urabe T, Hattori N, Ahluwalia A. Alterations in nitric oxide and endothelin-1 bioactivity underlie cerebrovascular dysfunction in ApoE-deficient mice. J Cereb Blood Flow Metab. 2010;30:1494–1503. doi: 10.1038/jcbfm.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lubrano V, Vassalle C, Blandizzi C, Del Tacca M, Palombo C, L'Abbate A, Baldi S, Natali A. The effect of lipoproteins on endothelial nitric oxide synthase is modulated by lipoperoxides. Eur J Clin Invest. 2003;33:117–125. doi: 10.1046/j.1365-2362.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- 40.Szklarczyk A, Lapinska J, Rylski M, McKay RD, Kaczmarek L. Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J Neurosci. 2002;22:920–930. doi: 10.1523/JNEUROSCI.22-03-00920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- 42.Iida H, Ohata H, Iida M, Watanabe Y, Dohi S. Isoflurane and sevoflurane induce vasodilation of cerebral vessels via ATP-sensitive K+ channel activation. Anesthesiology. 1998;89:954–960. doi: 10.1097/00000542-199810000-00020. [DOI] [PubMed] [Google Scholar]